
Introduction
Gastric cancer is the third leading cause of cancer death, and the fifth most common malignancy in the world [1]. The underlying pathogenesis of gastric cancer still remains unclear. It is important to develop an authentic animal model to imitate process of human gastric carcinogenesis. Genomic sequencing studies demonstrated that the protein-coding genes of both mice and humans shown high similarity [2]. Since mice disclosed the superiority on short inter-generation interval, high reproducibility as well as similar genetic background and formula experimental protocols, compared to other species, they have attracted considerable attentions as useful models to uncover the mechanisms of diseases and cancer, and to use for preventive and therapeutic interventions [3].
However, mice rarely develop gastric cancer spontaneously. To get a cancerous phenotype, researchers must feed them with helicobacter or chemical carcinogens. Along with the development of genomic manipulation, peoples could quickly create genetically engineered mice. The appearance of gene modified mouse models enabled us to explore the association of unique genotype with the specialized phenotypes. The creation of genetically engineered mice is mainly based on gene transfer technologies. In early stage, researchers studied the association of disease with mouse genetics by means of spontaneous or induced mutation models [4-6]. The technology of gene transfer is also called ‘‘reverse genetic’’ approach. In this approach, an identified gene which involved in human disease is selected as a target of gene manipulation in mice [3]. This approach is proper for uncovering the relevance of unique cell types or genetic pathways in pathogenesis of some diseases. The mouse with exogenous genome sequences is called transgenic mice, and the mouse with lost or altered endogenous gene is called knockout/knockin mouse. If the expression of targeted gene is controlled in specific time or organs/cells, we call it conditional transgenic or knockout/knockin mice. Generally, transgenic mouse was created to investigate the consequences of gene over-expression, and the knockout/knockin mouse was used to study the impacts of gene low-expression or mutation.
Construction of genetically engineered mice
Typically, there are two basic approaches to produce genetically engineered mice, one is carried out through microinjection of DNA into the pronucleus of zygotes, the other is based on manipulation at embryonic stem cells. Generation of traditional transgenic mice depends on pronuclear microinjection of DNA, while generation of traditional knockout mice depends on embryonic stem cells and homologous recombination techniques, which get a higher success compared to other strategies. However, the complete process requires more than a year to generate a genetically modified mouse. The technology of site-specific nucleases provided a new choice for rapid generation of transgenic models, including zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeat (CRISPR) systems. Compared to ZFNs and TALENs, CRISPR-mediated genome engineering is easy and efficient. Scientists could directly inject CRISPR systems into zygotes and haploid ES cells instead of ES cells to shorten the breeding time. In this paper, we introduce the generation of transgenic mice, ES cells targeted knockout mice and the latest CRISPR-Cas knockout mice briefly.
Transgenic mice are strains of mice, which express exogenous genes or DNA sequences. One of the most commonly used methods to create transgenic mice is DNA pronucleus microinjection, by which, exogenous DNA is injected into one of the fertilized pronucleus eggs. Exogenous DNA will be integrated into the genome of mouse fertilized eggs and pass on to offsprings stably. Fertilized eggs usually come from C57BL/6 mice or FVB mice. The former stain is the source of published mouse genome sequencing. The latter stain has the advantage of easy pronucleus injection due to its big and clear pronucleus. By different DNA sequences injection, researchers will get transgenic mice with expression of human genes, microRNAs, small interfering RNAs or BAC/YAC gene sequences.
Gene target mice are divided into complete knockout mice, conditional knockout mice and gene knockin mice. Complete knockout mice are mice with deletion of target gene in whole tissues and cells of body. These mice can be used to study the function of target gene in the physiology and pathology of the whole body. Since complete knockout mice disclosed higher embryonic lethal rate, the utility of complete knockout mice is limited. To avoid the problem of embryonic death, another kind of knockout mice, conditional or inducible knockout mice are developed. These mice are generated using the LoxP-Cre system. Two LoxP loci are inserted into one or several important exons of targeted gene to produce floxed mice. These mice express targeted gene normally when its genotype is wild type. After crossing floxed mice with Cre mice, the floxed exons will be deleted, and resulted in knockout of targeted gene. The exogenous Cre expression is depended on control of proper promoter in Cre mice. That is, different promoters drive Cre expressed in different tissues and cells. For example, Onoyama and coworkers generated mice with liver-specific null mutations of Fbxw7 by means of two different Cre-loxP systems (Mx1-Cre and Alb-Cre) [7]. Cre expression occurs in Alb-Cre mice embryos and maintains lifelong, while Mx1 gene promoter was activated only after injection of poly(I)-poly(C) into mice. Up-to-date, there are several kinds of Cre mice expressing CRE recombinase in stomach, including Atp4b-Cre (express CRE in parietal cells), Capn8-Cre (express CRE in glandular pit cells), Pgc-Cre (express CRE in chief cells), K19-Cre (express CRE in glandular isthmus cells), Villin-Cre (express CRE in glandular progenitor cells). The details of Cre mice and floxed mice can be viewed in MGI website (http://www.informatics.jax.org/). The following flowchart is an example of creation of Shh (Sonic Hedgehog) conditional knockout mice in gastric parietal cells (Figure 1). The method of genotyping detection is summarized in (Figure 2) [8]. Based on literature, the mouse models with conditionally targeted gene knocked out at stomach are listed in Table 1.
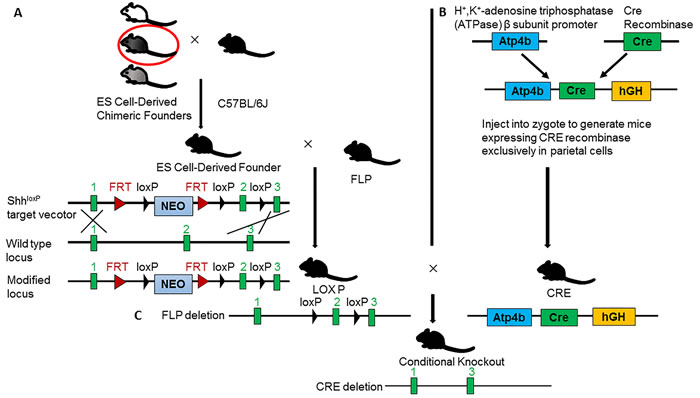
Figure 1: The flowchart of creating conditional knockout mice with specific ablation of Shh in gastric parietal cells. A. Crossing high degree chimeric founders with C57BL/ 6J mice. Targeted ES cells are transferred to the blastocoel cavities of 3.5 day blastocyst embryos, and then, the embryos are transferred to surrogate mothers to complete gestation. The offspring are chimeric mice with two sets of genome. The chimeric degree is judged by coat color. Since the ES cells are come from black mice, while blastocyst embryos are from white mice, so, the greater the share of black, the higher degree of chimeric. B. Deletion of selected gene Neo. The FLP can recognize FRT sequence and remove the fragment between two FRT. Crossing the ES cell-derived founder with FLP mice to obtain F1 heterozygous mice (Shhfl/+) without Neo gene. Further crossing F1 heterozygous mouse to generate F2 homozygous (Shhfl/fl) mice, which carry loxP-Shh-loxP in chromosomes. C. Creation of conditional knockout of Shh in gastric parietal cells. Since CRE recombinase is specifically expressed under the control of Atp4b promoter in gastric parietal cells, researchers should cross Shhfl/fl mice with Atp4b-Cre mice, and get the conditional knockout mice with exon2 deletion of Shh gene in gastric parietal cells (Shhfl/fl; Atp4b-Cre). In Shh conditional knockout mice, Shh is not expressed in gastric parietal cells, but normally expressed in other cells.
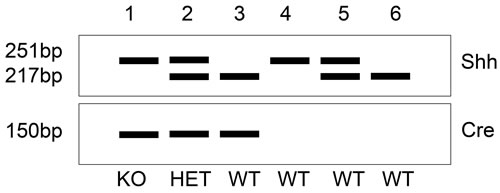
Figure 2: Schematic figure of genotyping for evaluating conditional knockout mice (Shhfl/fl; Atp4b-Cre). Evaluation of mouse genotyping is based on polymerase chain reaction (PCR). Primers are designed to modified Shh gene and Cre recombinase. The size of PCR product of floxed Shh is 251bp, while the wild type is 217bp. Heterozygous genotype show two products of both 251bp and 217bp. The size of Cre product is located at 150bp. The samples DNA from mouse 1, 2 and 3 are Cre positive. Mouse 1 represents homozygous knockout mouse (KO). Mouse 2 is heterozygous genotype (HET), and mouse 3, 4, 5 and 6 are wild type mice (WT).
CRISPR-Cas is a new technology that has emerged recently, and obtained widespread attention. In CRISPR-Cas system, a sequence-specific guide RNA is used to guide nuclease to create a nick on targeted location. A new sequence is inserted by homologous recombination. CRPSPR-Cas system consists of clustered regularly interspaced palindromic repeats (CRISPR) and the CRISPR-associated proteins (Cas), which have the structure of nucleic acid enzyme activity. The CRISPR-Cas was divided into several subtypes. Each CRISPR-Cas has its unique features in biochemistry and molecular genetics. CRISPR-Cas9 is widely used in generation of genetically engineered mouse [9].
Table 1: The targeted genes are conditionally knocked out in stomach as well as other organs on mouse models*
Model name |
Stomach |
Other organs |
References |
Foxa3-Cre |
Glandular stomach cells |
Liver, pancreas, intestine, ovary, testis, heart and adipose tissue |
|
Capn8-Cre |
Gastric pit cells |
Liver and skin |
[15] |
Pdx1-Cre |
Endocrine cells of gastric antrum (such as gastrin, somatostatin and serotonin) |
Endocrine cells of duodenum and pancreatic islet cells |
|
Villin-Cre |
Gastric progenitor cells |
Progenitor cells of intestine and kidney proximal tubules |
|
Edn2-iCre |
Stomach cells |
Cells of periovulatory ovary and intestine |
[20] |
Tff1-CreERT2 |
Epithelium of the glandular stomach |
ND |
[21] |
Atp4b-Cre |
Parietal cell |
N |
[22] |
Lgr5-CreERT2 |
Base cells of glands in gastric pylorus and corpus region |
Base cells of intestine and hair follicle |
[23] |
Tff2-CreERT2 |
Parietal cell, mucous neck cell and chief cells |
ND |
[24] |
H(+), K(+)-ATPase β-subunit promoter |
Parietal cell |
N |
[25] |
*N, No; ND, Not detectable
Pathogenic study of transgenic/knockout/knockin mice
INS-GAS mice
Gastrin is a crucial hormone produced by antrum G cells of gastric mucosa, and responsible for gastric acid secretion and oxyntic cells differentiation. Insulin-gastrin (INS-GAS, FVB/N background) transgenic mice were originally created to examine the possible role of gastrin in regulating islet differentiation. The INS-GAS transgenic mouse contains two exons of the human gastrin gene, which encode progastrin precursor under control of insulin promoter [26]. INS-GAS mice have also been used to investigate the effect of gastrin on gastric cancer development [27]. INS-GAS mice showed increased maximal gastric acid secretion and increased parietal cell number in early stage, but gradually changed into hypochlorhydria and decreased parietal cell number later (five months later). On the age of 20 months, INS-GAS mice exhibited metaplasia, dysplasia and gastric cancer. H felis infection could accelerate (≤8 months) these lesions [28]. The tumorigenesis of INS-GAS mice revealed differences in strain and gender. The mice with FVB/N background is more susceptible to gastric cancer, while C57/BL6 background mice is not susceptible to gastric cancer, and only develop hyperplasia and low-grade dysplasia [29]. In addition, male mouse is more susceptible to gastric cancer compared to female mouse [30]. INS-GAS mouse with helicobacter infection has been widely used as a model of intestinal type gastric cancer. Scientists use this mouse model to study the change of gene expression induced by H pylori infection at the high gastrin status and found a group of differential expressed genes [31]. Lofgren compared INS-GAS mice raised in germ-free (GF) and SPF with intestinal flora conditions and found that intestinal flora promoted gastric lesions [32]. Lertpiriyapong compared INS-GAS mice raised in germ-free, altered Schaedler’s flora and intestinal flora conditions, and then infected by H Pylori. They found that mice at altered Schaedler’s flora and intestinal flora conditions could develop severer gastric lesions. They believe that colonization efficiency of commensals is important [33]. Whary infect INS-GAS mice with Heligmosomoidespolygyrus and H Pylori, and found that coinfection of Heligmosomoidespolygyrus with H Pylori could decrease H Pylori -induced atrophy and dysplasia [34].
Gastrin knockout mice
Gastrin knockout mice (C57BL/6 strain, GAS-/-) were originally generated by Koh and Friis separately through gene targeting [35, 36]. Mutant mice were viable and fertile, but secretion of gastric acid was abolished, leading to a marked change in gastric architecture, with decreased parietal cell number and enterochromaffin-like (ECL) cells, and increased mucous neck cells. There were no differences in the proliferation labeling index of the stomach between gastrin-deficient mice and wild type littermates. Some researchers found that GAS-/-mice of 129/Sv strain kept in nonbarrier rooms led to bacterial overgrowth and elevated numbers of parietal cell, G cell and inflammation [37]. At the age of 12 months, these mice developed stomach tumors [38]. GAS-/-mice are proper for studying biological functions of gastrin-regulated gene (Tff1 and Tff2) in maintenance and repair of gastric mucosa [39, 40]. INS-GAS transgenic mice and GAS knockout mice are often used together in one research to study the role of gastrin in gastric carcinogenesis, but the results were not always concordant. The different results may be due to the different backgrounds of mice strain and different location on stomach, for example the corpus or antrum. It seemed that gastrin promote cancer development in corpus but not antrum [29].
Tff1 knockout mice
Trefoil factor 1 (TFF1, formerly known as pS2) is a tumor suppressor gene that encodes a peptide belonging to the trefoil factor family. TFF1 expression is frequently lost in gastric carcinomas [41]. As early as in 1996, Lefebvre and colleagues generated Tff1 knockout (Tff1-/-) mice to explore biological function of this gene [42]. Homozygous mutant mice of Tff1 (Tff1-/-) develop antropyloric adenoma, and even multifocal carcinomas, consistent with increased inflammatory scores [43]. They identified NF-κB to be a network hub that is activated in Tff1 knockout mice through mRNA microarray analysis on antral samples from Tff1-/- mice and wild type mice. Another group reported MNU-driven tumorigenesis on Tff1+/- mice. They found increased antral proliferation and progenitor cell number at age of 18 weeks, and increased malignant tumors in heterozygous mice (Tff1+/-), compared to wild type mice. In addition, they found that mRNA expression of Tff1 was almost lost in heterozygous mice (Tff1+/-) [44]. The Tff1+/- mice are applied for studies on gene heterozygocity and transcript regulation.
IL-1β transgenic mice
Polymorphisms of interleukin-1beta (IL-1β) involved in enhancing production of IL-1β are associated with an increase risk for both hypochlorhydria induced by H. pylori and gastric cancer [45]. Tu et al. generated IL-1β transgenic mice by using an H/K-ATPase/hIL-β transgenic mouse that express human IL-1β specifically in stomach. The IL-1β transgenic mice spontaneously develop chronic gastritis, metaplasia and high-grade dysplasia/carcinoma. In the setting of H felis infection, these mice show accelerated development of gastric inflammation and carcinoma compared to control mice [46]. In human beings, IL-1β synergizes with H pylori and increases the risk of gastric cancer. Moreover, overexpression of IL-1β in transgenic mice could recruit the accumulation of myeloid-derived suppressor cells (MDSCs) through NF-κB signal pathway. MDSCs are important for carcinogenesis in the early stage of gastric carcinogenesis. IL-1β transgenic mouse is widely used for testing the efficacy of anti- IL-1β therapies in cancer prevention and function of MDSCs in tumor microenvironment [47]. IL-1β knockout mice were also created and treated with H pylori. These mice exhibited decreased recruitment of macrophages and neutrophils by H pylori infection and reduced activation of NF-kB [48].
K-ras transgenic/knockin mice
K-ras gene mutations have been found in about ~6% or ~18% of diffuse type or intestinal type of gastric cancer, respectively [49]. The effect of RAS protein is complex with either positive or negative effects on cell growth, differentiation and death [50]. Brembeck and coworkers created K-ras transgenic mice under control of cytokeratin19 (K19) promoter [51]. K19 was expressed in an epithelial-specific pattern, restricted to ductal epithelial cells in the pancreas, surface colonocytes, small intestinal villi and gastric isthmus cells. Despite the findings in pancreas, K-ras transgenic mice exhibited concomitant parietal cell decrease and mucous neck cell hyperplasia (3-6 months age) [52]. The CK19CreERT; LSL-KrasG12D mouse is a kind of K-ras mutation (G12D) mouse, which showed metaplasia, foveolar hyperplasia, reduced presence of parietal cells and a deeper proliferative zone in the fundus of stomach at the age of 4-6 months after tamoxifen administration [53]. Matkar and colleagues found that systemic activation of K-ras leads to rapid changes in gastric cellular homeostasis and causes hyperplasia of the forestomach and the glandular stomach, depletion of parietal cells, accompanied by upregulation of inflammatory response factor (COX-2), stem cell marker (Dcamkl1, CD44) and activated MAPK pathway. Therefore, systemic K-rasG12D activated mice function as a tool for studying the early molecular events of gastric carcinogenesis [54].
Apc and Wnt1 transgenic mice
Adenomatous polyposis coli (APC) is a key tumor suppressor gene that acts as an antagonist of Wnt signaling pathway by maintaining cytoplasmic levels of β-catenin [55]. About 88% patients with familial adenomatous polyposis caused by APC germline mutations develop to fundic gland polyps, while fundic glandular polyp may transform to adenocarcinoma [56]. However, APC gene mutation is frequently found in gastric adenomas, but rarely in gastric cancer. For instance, one Apc mutation (Apc1638) led to gastric dysplasia and polyposis in antrum and pyloric junction [57, 58]. A transgenic mouse carrying Apc gene mutations infected by H felis developed less gastritis, less epithelial proliferation and inflammation, compared to wild type mice. It means the immune and inflammatory response of Apc gene mutations were not serious [59]. To explore Wnt pathway on gastric carcinogenesis, Oshima and coworkers constructed K19-Wnt1 transgenic mice, which express Wnt1 in gastric mucosa. They crossed K19-Wnt1 mice with K19-C2mE transgenic mice to investigate the role of Wnt and PGE2 on gastric carcinogenesis [60]. The phenotype of K19-C2mE mice is overexpressed COX-2 and microsomal prostaglandin E synthase-1 (mPGES-1) in gastric epithelium, resulted in increased metaplasia, hyperplasia and tumors in the glandular stomach with heavy macrophage infiltrations [61]. In addition, Akaboshi and coworkers used Hmga1-knockin mice crossed with K19-Wnt/C2mE mice and proved that Hmga1 is involved in gastric carcinogenesis via Wnt/β-catenin pathway [62].
p53 knockout mice
Mutations of p53 gene constitute one of the most frequent molecular events in human cancers. As we know, p53 knockout mice are highly susceptible to spontaneous tumorigenesis at age of 6 months [63, 64]. Since homozygous knockout animals could not be maintained long term, most of experiments of p53 knockout mice are based on heterozygous knockout mice (p53+/-) rather than homozygous knockout mice (p53-/-). The incidence of invasive adenocarcinomas in the stomach of p53+/-mice was significantly higher than that in WT mice [65]. Infection of H felis for both WT and p53+/-mice was disclosed active chronic inflammation and marked mucosal hyperplasia at the age of 6 months [66]. There is a synergistic action between infection with H felis and p53 deficiency in the accumulation of mutations in stomach [67]. However, another research revealed that H pylori (24w after infection) did not result in significant difference on the level of gastric epithelial apoptosis and proliferation between p53+/- mice and WT mice [68]. In order to explore the synergistic effect of two or more different genes, Shimada and coworkers created double conditional knockout (DCKO) mice by crossing Atp4b-Cre mice with Cdh1fl/fl and p53fl/f lmice, to examine the synergistic effect of E-cadherin loss and p53 loss on stomach. The DCKO mice exhibited phenotypes of loss of cell polarity for parietal cells and proton pump-negative atypical foci, ultimately progressed to intramucosal cancer (9 months) and invasive cancer (12 months) [69]. Moreover, Park and coworkers crossed Pdx-1-Cre mice with Smad4fl/fl p53fl/flCdh1fl/+ and found that E-cadherin loss and Smad4 loss cooperate with p53 loss could promote the development of gastric cancer [17, 18].
Klf4 knockout mice
The Kruppel-like factor 4 (KLF4) is a zinc finger transcription factor that regulate numerous biological processes including proliferation, differentiation, development and apoptosis. KLF4 expression is found primarily in post mitotic phase and terminal-differentiated epithelial cells such as skin, lungs, and gastrointestinal tract [70,71]. Loss of KLF4 expression was significantly associated with poor survival in gastric cancer [72]. Li and coworkers created Klf4 conditional knockout mouse models by crossing Villin-cre transgenic mice and Klf4fl/fl mice [13]. Villin is an actin-bundling protein located in the apical brush border of absorptive epithelium of intestine, as well as gastric progenitor cell (GPC). Villin positive GPCs are quiescent in unstimulated stomach with multilineage potential. By inflammation stimulation, these cells could undergo symmetric or asymmetric division and gradually replace pyloric glands [73]. The antrum mucosa cells revealed disturbed Klf4 expression in Klf4 knockout mice. At the age of 35 to 50 weeks, these Villin-cre-Klf4 knockout mice developed preneoplasia in antrum, and 29% of them progressed to gastric cancer at 80 weeks. Chemical reagent MNU could accelerate tumor formation at 35 to 50 weeks of age.
Histological features appeared on mouse stomach
In order to evaluate experimental effects, researchers need to observe gastric histology on different stages by necropsy. The adult mouse′s stomach is located in the left cranial part of the abdominal cavity. The forestomach forms the left half, the glandular stomach forms the right half of the stomach. The wall has three layers: the mucosa with submucosa, the muscularis, and the serosa. In the forestomach, the mucosa is lined by stratified squamous epithelium covered by cornified tissue layer. It borders on that of the glandular stomach by a mucosal fold called the limiting ridge. The mucosa of the glandular stomach is lined by single columnar epithelium forming deep foveolae. The main part of the glandular stomach is fundic glands containing surface mucous cells in the pit zone, granular eosinophilic parietal cells (oxyntic cells) secreting hydrochloric acid and mucin-secreting non-chief cells lining the neck, and basophilic chief cells (zymogenic cells) producing prepepsin and located at the base [74, 75].
(Figure 3) indicates the dissecting method of mouse stomach. The first incision is cut along the greater curvature from esophagus through proximal duodenum. After opening the stomach, the stomach is laid flat on a cutting board. Two or three linear strips are cut from the lesser curvature including squamocolumnar junction, corpus, antrum and pyloru and proximal duodenum. In general, stomach tissues are collected and fixed in 4% formalin in PBS overnight, and processed for standard paraffin histology. Slides are stained by hematoxylin-eosin reagent or mucin stain and scored for pathology on a scale of 0 to 4. Regarding to H pylori-associated gastric histology, there are six parameters for histological evaluation, such as inflammation, epithelial defect, oxyntic atrophy, hyperplasia, pseudopyloric metaplasia, and dysplasia or tumor. Sometimes, some parameters are not appeared on chemistry-induced lesions. Here, we present several crucial histological evaluation parameters: corpus and antral inflammation; corpus glandular atrophy; mucus metaplasia; dysplasia and carcinoma. Among them, mucus metaplasia and dysplasia belong to precancerous lesions. The corpus and antral inflammation is defined by sub-mucosal and mucosal presence of polymorphous nuclear and mononuclear cells, which is scored according to the extent of inflammatory cells. Infiltration: 0, no inflammatory cells; 1, inflammatory cells infiltration of the submucosa with or without infiltration at the very base of the mucosa; 2, inflammatory cells infiltration of the submucosa and the bottom half of the mucosa; 3, inflammatory cells infiltration of the submucosa and greater than 50% of the mucosa; 4, transmural infiltration of inflammatory cells. Corpus glandular atrophy is defined as loss of parietal and zymogenic cells. Corpus glandular atrophy is scored based on the estimated percentage of parietal cell and chief cell loss within the corpus: 0, no visible parietal cell and chief cell loss; 1, 25% parietal cell loss and 50% chief cell loss; 2, 50% parietal cell loss and greater than 75% chief cell loss; 3, 75% parietal cell loss and 100% chief cell loss; 4, greater than 75% parietal cell loss and no chief cells. Mucus metaplasia is defined as the ectopic presence of Alcian blue stained acidic mucin associated with the acquisition of an elongated antral glandular structure within corpus glands. Mucus metaplasia is scored based on the percentage of the corpus mucosa showing replacement of oxyntic glands with elongated Alcian-blue stained glands reminiscent of antral mucosa: 0, no visible mucus metaplasia; 1, small foci were present; 2, up to one-third of the corpus was affected; 3, two thirds of the corpus were affected; 4, greater than two thirds of the corpus were affected. Dysplasia is defined as disturbed or haphazard glandular arrangement, loss of vertical orientation, back-to-back associations without intervening stroma, branching and infolding glands. At the cellular level, dysplastic features include altered nuclear size, hyperpleomorphism, poorly defined cell junctions, loss of nuclear polarity, and hyperchromatin with increased nuclear-cytoplasmic ratio [76]. Dysplasia is scored based on the following criteria: 0, no visible dysplasia; 1, appearing aberrant crypt foci including distortion of normal columnar orientation, increased diameter, asymmetrical cell piling, and back-to-back forms. 2, there is glandular infolding, branching, and more advanced cellular atypia such as increased nuclear-cytoplasmic ratio. 3, cellular distortion with haphazard arrangements, the lesion developed to carcinoma in situ. 4, highly dysplastic glands invade into the submucosa or beyond, such as deeper layers, vessels and lymphatics [77]. Table 2 summarized the names of genetically engineered mouse models and observed lesions.
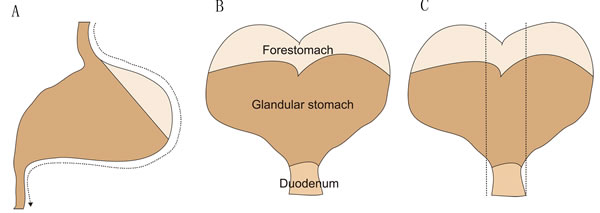
Figure 3: The schematic figure of dissecting method for mouse stomach. A. The first incision is cut along the greater curvature from esophagus through proximal duodenum .B. After opening the stomach, the stomach is laid flat on a cutting board. C. Two or three linear strips are cut from the lesser curvature including squamocolumnar junction, corpus, antrum and pyloru and proximal duodenum.
Table 2: Genetically engineered mouse models and observed lesions*
Model name |
Time\Lesion |
Metaplasia |
Hyperplasia |
Dysplasia |
Carcinoma |
Invasion |
Metastasis |
Cancer type |
References |
Tff1-/- |
12 months |
ND |
Y |
Y |
Y |
Y |
N |
/ |
|
gp130757F/F |
3 months |
Y |
Y |
Y |
Y |
Y |
N |
Intestinal |
|
Cdx2 transgenic |
12 weeks |
Y |
Y |
N |
Y |
N |
N |
Intestinal |
|
INS-GAS |
20 months |
Y |
ND |
Y |
Y |
Y |
N |
Intestinal |
|
ACT-GAS |
20 months |
ND |
Y |
ND |
Y |
N |
N |
N |
|
Gastrin-/- |
12 months |
Y |
Y |
Y |
Y |
N |
N |
N |
|
Atp4a-/- |
12 months |
Y |
Y |
N |
N |
N |
N |
N |
|
NHE2-/- |
3 months |
ND |
Y |
N |
N |
N |
N |
N |
[97] |
NHE4-/- |
9 weeks |
N |
N |
N |
N |
N |
N |
N |
[98] |
Kvlqt1-/- |
3 months |
ND |
Y |
N |
N |
N |
N |
N |
[99] |
H2R-/- |
16 weeks |
N |
Y |
N |
N |
N |
N |
N |
|
HDC-/- |
12 months |
Y |
Y |
N |
N |
N |
N |
N |
[103] |
IQGAP1-/- |
15 months |
ND |
Y |
Y |
N |
N |
N |
N |
[104] |
Tgf β1-/- |
20 days |
Y |
Y |
N |
N |
N |
N |
N |
[105] |
Smad4-/- |
9 months |
ND |
Y |
Y |
Y |
Y |
N |
/ |
|
Runx3-/- |
8 months |
ND |
Y |
Y |
N |
N |
N |
N |
|
Apc +/- |
20 weeks |
ND |
Y |
Y |
Y |
Y |
N |
/ |
[111] |
MTH1-/- |
18 months |
ND |
Y |
Y |
Y |
N |
N |
Intestinal |
[112] |
K19-C2mE transgenic |
45 weeks |
Y |
Y |
Y |
Y |
N |
N |
/ |
|
Tsp-/- |
3 weeks |
Y |
Y |
N |
N |
N |
N |
N |
[114] |
Tgf α transgenic |
6 weeks |
ND |
Y |
Y |
N |
N |
N |
/ |
|
AhR transgenic |
3 months |
Y |
Y |
Y |
Y |
Y |
N |
Intestinal |
|
Klf4 -/- |
35 weeks |
ND |
Y |
Y |
Y |
N |
N |
/ |
|
p27-/- |
60 weeks |
Y |
Y |
Y |
Y |
N |
N |
Intestinal |
[121] |
Car9-/- |
4 weeks |
ND |
Y |
N |
N |
N |
N |
N |
|
CEA SV40 transgenic |
37 days |
ND |
ND |
Y |
Y |
Y |
N |
Intestinal |
|
H+/K+-ATPase β subunit SV40 transgenic |
12 months |
N |
N |
Y |
Y |
N |
N |
/ |
|
H+/K+-ATPase β subunit-/- |
20 months |
ND |
ND |
Y |
Y |
Y |
N |
/ |
[128] |
Shh-/- |
18.5 day embryo |
Y |
Y |
N |
N |
N |
N |
/ |
[129] |
Occludin-/- |
10 months |
N |
N |
Y |
N |
N |
N |
/ |
[130] |
ClC2-/- Cdh1-/-p53-/- |
9 weeks 6 months |
N Y |
N Y |
Y Y |
N Y |
N Y |
N Y |
/ Diffuse |
[131] |
*Y, Yes; N, No; ND, Not detectable
Summary
Mice are important experimental model for human gastric cancer study. Along with the findings of multiple mutations of crucial functional genes by whole genome sequencing, the biological functions of multiple genes should be verified on cell models as well as on genetically engineered mice model. Scientists could make conditional knockout mice by Cre mice that express CRE protein specifically in stomach. Scientists could make transgenic mice via specific promoter, such as K19 (epithelial cell specific) and H-K-ATPase (parietal cell specific). At present, there are no perfect promoters for gastric carcinogenesis study. New technologies such as CRISPR-Cas system are under study. Regarding to animal experiment, combination of chemical carcinogen and biological carcinogen could accelerate the tumorigenesis. In near future, more and more mouse models will be created. Although development of a new mouse model is time-consuming, it is valuable for gene function study. However, we must understand that there are some differences such as immune system or their anatomic organs between mouse model and human being. The histological scoring criteria are helpful to improve comparison of results between different laboratories. The right evaluation will assist scientific investigators and medical professionals in understanding and objectively scoring disease progression in mouse models.
Acknowledgments
This project was partially supported by the Chinese National Key Program (2016YFC1303200, 2012AA02A504, 2012AA02A203), the National Natural Science Foundation of China (81172329 and 81372644) and the Innovation Foundation of Translational Medicine of Shanghai Jiao Tong University School of Medicine (15ZH1002 and 15ZH4001).
CONFLICTs OF INTEREST
The authors declare no conflicts of interest.
References
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. International journal of cancer Journal international du cancer. 2015; 136:E359-386.
2. Mouse Genome Sequencing C, Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002; 420:520-562.
3. Doyle A, McGarry MP, Lee NA and Lee JJ. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic research. 2012; 21:327-349.
4. Swank RT, Novak EK, McGarry MP, Rusiniak ME and Feng L. Mouse models of Hermansky Pudlak syndrome: a review. Pigment cell research / sponsored by the European Society for Pigment Cell Research and the International Pigment Cell Society. 1998; 11:60-80.
5. Bosma GC, Custer RP and Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983; 301:527-530.
6. Russell WL, Kelly EM, Hunsicker PR, Bangham JW, Maddux SC and Phipps EL. Specific-locus test shows ethylnitrosourea to be the most potent mutagen in the mouse. Proceedings of the National Academy of Sciences of the United States of America. 1979; 76:5818-5819.
7. Onoyama I, Suzuki A, Matsumoto A, Tomita K, Katagiri H, Oike Y, Nakayama K and Nakayama KI. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. The Journal of clinical investigation. 2011; 121:342-354.
8. Xiao C, Ogle SA, Schumacher MA, Orr-Asman MA, Miller ML, Lertkowit N, Varro A, Hollande F and Zavros Y. Loss of parietal cell expression of Sonic hedgehog induces hypergastrinemia and hyperproliferation of surface mucous cells. Gastroenterology. 2010; 138:550-561, 561 e551-558.
9. Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV and van der Oost J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016; 353:aad5147.
10. Shibata W, Takaishi S, Muthupalani S, Pritchard DM, Whary MT, Rogers AB, Fox JG, Betz KS, Kaestner KH, Karin M and Wang TC. Conditional deletion of IkappaB-kinase-beta accelerates helicobacter-dependent gastric apoptosis, proliferation, and preneoplasia. Gastroenterology. 2010; 138:1022-1034 e1021-1010.
11. Maloum F, Allaire JM, Gagne-Sansfacon J, Roy E, Belleville K, Sarret P, Morisset J, Carrier JC, Mishina Y, Kaestner KH and Perreault N. Epithelial BMP signaling is required for proper specification of epithelial cell lineages and gastric endocrine cells. American journal of physiology Gastrointestinal and liver physiology. 2011; 300:G1065-1079.
12. Garrison WD, Battle MA, Yang C, Kaestner KH, Sladek FM and Duncan SA. Hepatocyte nuclear factor 4alpha is essential for embryonic development of the mouse colon. Gastroenterology. 2006; 130:1207-1220.
13. Li Q, Jia Z, Wang L, Kong X, Li Q, Guo K, Tan D, Le X, Wei D, Huang S, Mishra L and Xie K. Disruption of Klf4 in villin-positive gastric progenitor cells promotes formation and progression of tumors of the antrum in mice. Gastroenterology. 2012; 142:531-542.
14. Katz JP, Perreault N, Goldstein BG, Actman L, McNally SR, Silberg DG, Furth EE and Kaestner KH. Loss of Klf4 in mice causes altered proliferation and differentiation and precancerous changes in the adult stomach. Gastroenterology. 2005; 128:935-945.
15. Zhao Z, Sun Y, Hou N, Teng Y, Wang Y and Yang X. Capn8 promoter directs the expression of Cre recombinase in gastric pit cells of transgenic mice. Genesis. 2009; 47:674-679.
16. Larsson LI, Madsen OD, Serup P, Jonsson J and Edlund H. Pancreatic-duodenal homeobox 1 -role in gastric endocrine patterning. Mechanisms of development. 1996; 60:175-184.
17. Park JW, Park DM, Choi BK, Kwon BS, Seong JK, Green JE, Kim DY and Kim HK. Establishment and characterization of metastatic gastric cancer cell lines from murine gastric adenocarcinoma lacking Smad4, p53, and E-cadherin. Molecular carcinogenesis. 2015; 54:1521-1527.
18. Park JW, Jang SH, Park DM, Lim NJ, Deng C, Kim DY, Green JE and Kim HK. Cooperativity of E-cadherin and Smad4 loss to promote diffuse-type gastric adenocarcinoma and metastasis. Molecular cancer research. 2014; 12:1088-1099.
19. Sundaresan S, Kang AJ, Hayes MM, Choi EK and Merchant JL. Deletion of Men1 and somatostatin induces hypergastrinemia and gastric carcinoids. Gut. 2016.
20. Cacioppo JA, Koo Y, Lin PC, Gal A and Ko C. Generation and characterization of an endothelin-2 iCre mouse. Genesis. 2015; 53:245-256.
21. Thiem S, Eissmann MF, Elzer J, Jonas A, Putoczki TL, Poh A, Nguyen P, Preaudet A, Flanagan D, Vincan E, Waring P, Buchert M, Jarnicki A and Ernst M. Stomach-Specific Activation of Oncogenic KRAS and STAT3-Dependent Inflammation Cooperatively Promote Gastric Tumorigenesis in a Preclinical Model. Cancer research. 2016; 76:2277-2287.
22. Syder AJ, Karam SM, Mills JC, Ippolito JE, Ansari HR, Farook V and Gordon JI. A transgenic mouse model of metastatic carcinoma involving transdifferentiation of a gastric epithelial lineage progenitor to a neuroendocrine phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:4471-4476.
23. Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, van den Brink S, Korving J, et al. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell stem cell. 2010; 6:25-36.
24. Quante M, Marrache F, Goldenring JR and Wang TC. TFF2 mRNA transcript expression marks a gland progenitor cell of the gastric oxyntic mucosa. Gastroenterology. 2010; 139:2018-2027 e2012.
25. Engevik AC, Feng R, Yang L and Zavros Y. The acid-secreting parietal cell as an endocrine source of Sonic Hedgehog during gastric repair. Endocrinology. 2013; 154:4627-4639.
26. Wang TC, Bonner-Weir S, Oates PS, Chulak M, Simon B, Merlino GT, Schmidt EV and Brand SJ. Pancreatic gastrin stimulates islet differentiation of transforming growth factor alpha-induced ductular precursor cells. The Journal of clinical investigation. 1993; 92:1349-1356.
27. Wang TC, Koh TJ, Varro A, Cahill RJ, Dangler CA, Fox JG and Dockray GJ. Processing and proliferative effects of human progastrin in transgenic mice. The Journal of clinical investigation. 1996; 98:1918-1929.
28. Wang TC, Dangler CA, Chen D, Goldenring JR, Koh T, Raychowdhury R, Coffey RJ, Ito S, Varro A, Dockray GJ and Fox JG. Synergistic interaction between hypergastrinemia and Helicobacter infection in a mouse model of gastric cancer. Gastroenterology. 2000; 118:36-47.
29. Takaishi S, Tu S, Dubeykovskaya ZA, Whary MT, Muthupalani S, Rickman BH, Rogers AB, Lertkowit N, Varro A, Fox JG and Wang TC. Gastrin is an essential cofactor for helicobacter-associated gastric corpus carcinogenesis in C57BL/6 mice. The American journal of pathology. 2009; 175:365-375.
30. Fox JG, Rogers AB, Ihrig M, Taylor NS, Whary MT, Dockray G, Varro A and Wang TC. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer research. 2003; 63:942-950.
31. Takaishi S and Wang TC. Gene expression profiling in a mouse model of Helicobacter-induced gastric cancer. Cancer science. 2007; 98:284-293.
32. Lofgren JL, Whary MT, Ge Z, Muthupalani S, Taylor NS, Mobley M, Potter A, Varro A, Eibach D, Suerbaum S, Wang TC and Fox JG. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011; 140:210-220.
33. Lertpiriyapong K, Whary MT, Muthupalani S, Lofgren JL, Gamazon ER, Feng Y, Ge Z, Wang TC and Fox JG. Gastric colonisation with a restricted commensal microbiota replicates the promotion of neoplastic lesions by diverse intestinal microbiota in the Helicobacter pylori INS-GAS mouse model of gastric carcinogenesis. Gut. 2014; 63:54-63.
34. Whary MT, Muthupalani S, Ge Z, Feng Y, Lofgren J, Shi HN, Taylor NS, Correa P, Versalovic J, Wang TC and Fox JG. Helminth co-infection in Helicobacter pylori infected INS-GAS mice attenuates gastric premalignant lesions of epithelial dysplasia and glandular atrophy and preserves colonization resistance of the stomach to lower bowel microbiota. Microbes and infection / Institut Pasteur. 2014; 16:345-355.
35. Friis-Hansen L, Sundler F, Li Y, Gillespie PJ, Saunders TL, Greenson JK, Owyang C, Rehfeld JF and Samuelson LC. Impaired gastric acid secretion in gastrin-deficient mice. The American journal of physiology. 1998; 274:G561-568.
36. Koh TJ, Goldenring JR, Ito S, Mashimo H, Kopin AS, Varro A, Dockray GJ and Wang TC. Gastrin deficiency results in altered gastric differentiation and decreased colonic proliferation in mice. Gastroenterology. 1997; 113:1015-1025.
37. Zavros Y, Rieder G, Ferguson A, Samuelson LC and Merchant JL. Genetic or chemical hypochlorhydria is associated with inflammation that modulates parietal and G-cell populations in mice. Gastroenterology. 2002; 122:119-133.
38. Zavros Y, Eaton KA, Kang W, Rathinavelu S, Katukuri V, Kao JY, Samuelson LC and Merchant JL. Chronic gastritis in the hypochlorhydric gastrin-deficient mouse progresses to adenocarcinoma. Oncogene. 2005; 24:2354-2366.
39. Khan ZE, Wang TC, Cui G, Chi AL and Dimaline R. Transcriptional regulation of the human trefoil factor, TFF1, by gastrin. Gastroenterology. 2003; 125:510-521.
40. Tu S, Chi AL, Lim S, Cui G, Dubeykovskaya Z, Ai W, Fleming JV, Takaishi S and Wang TC. Gastrin regulates the TFF2 promoter through gastrin-responsive cis-acting elements and multiple signaling pathways. American journal of physiology Gastrointestinal and liver physiology. 2007; 292:G1726-1737.
41. Beckler AD, Roche JK, Harper JC, Petroni G, Frierson HF, Jr., Moskaluk CA, El-Rifai W and Powell SM. Decreased abundance of trefoil factor 1 transcript in the majority of gastric carcinomas. Cancer. 2003; 98:2184-2191.
42. Lefebvre O, Chenard MP, Masson R, Linares J, Dierich A, LeMeur M, Wendling C, Tomasetto C, Chambon P and Rio MC. Gastric mucosa abnormalities and tumorigenesis in mice lacking the pS2 trefoil protein. Science. 1996; 274:259-262.
43. Soutto M, Belkhiri A, Piazuelo MB, Schneider BG, Peng D, Jiang A, Washington MK, Kokoye Y, Crowe SE, Zaika A, Correa P, Peek RM, Jr. and El-Rifai W. Loss of TFF1 is associated with activation of NF-kappaB-mediated inflammation and gastric neoplasia in mice and humans. The Journal of clinical investigation. 2011; 121:1753-1767.
44. Tomita H, Takaishi S, Menheniott TR, Yang X, Shibata W, Jin G, Betz KS, Kawakami K, Minamoto T, Tomasetto C, Rio MC, Lerkowit N, Varro A, Giraud AS and Wang TC. Inhibition of gastric carcinogenesis by the hormone gastrin is mediated by suppression of TFF1 epigenetic silencing. Gastroenterology. 2011; 140:879-891.
45. El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF, Jr. and Rabkin CS. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature. 2001; 412:99.
46. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG and Wang TC. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer cell. 2008; 14:408-419.
47. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, Baik GH, Shibata W, Diprete B, Betz KS, Friedman R, Varro A, Tycko B and Wang TC. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer cell. 2011; 19:257-272.
48. Shigematsu Y, Niwa T, Rehnberg E, Toyoda T, Yoshida S, Mori A, Wakabayashi M, Iwakura Y, Ichinose M, Kim YJ and Ushijima T. Interleukin-1beta induced by Helicobacter pylori infection enhances mouse gastric carcinogenesis. Cancer letters. 2013; 340:141-147.
49. Ushijima T and Sasako M. Focus on gastric cancer. Cancer cell. 2004; 5:121-125.
50. Frame S and Balmain A. Integration of positive and negative growth signals during ras pathway activation in vivo. Current opinion in genetics & development. 2000; 10:106-113.
51. Brembeck FH, Moffett J, Wang TC and Rustgi AK. The keratin 19 promoter is potent for cell-specific targeting of genes in transgenic mice. Gastroenterology. 2001; 120:1720-1728.
52. Brembeck FH, Schreiber FS, Deramaudt TB, Craig L, Rhoades B, Swain G, Grippo P, Stoffers DA, Silberg DG and Rustgi AK. The mutant K-ras oncogene causes pancreatic periductal lymphocytic infiltration and gastric mucous neck cell hyperplasia in transgenic mice. Cancer research. 2003; 63:2005-2009.
53. Ray KC, Bell KM, Yan J, Gu G, Chung CH, Washington MK and Means AL. Epithelial tissues have varying degrees of susceptibility to Kras(G12D)-initiated tumorigenesis in a mouse model. PloS one. 2011; 6:e16786.
54. Matkar SS, Durham A, Brice A, Wang TC, Rustgi AK and Hua X. Systemic activation of K-ras rapidly induces gastric hyperplasia and metaplasia in mice. American journal of cancer research. 2011; 1:432-445.
55. Minde DP, Anvarian Z, Rudiger SG and Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Molecular cancer. 2011; 10:101.
56. Oliveira C, Pinheiro H, Figueiredo J, Seruca R and Carneiro F. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. The Lancet Oncology. 2015; 16:e60-70.
57. Maesawa C, Tamura G, Suzuki Y, Ogasawara S, Sakata K, Kashiwaba M and Satodate R. The sequential accumulation of genetic alterations characteristic of the colorectal adenoma-carcinoma sequence does not occur between gastric adenoma and adenocarcinoma. The Journal of pathology. 1995; 176:249-258.
58. Lee JH, Abraham SC, Kim HS, Nam JH, Choi C, Lee MC, Park CS, Juhng SW, Rashid A, Hamilton SR and Wu TT. Inverse relationship between APC gene mutation in gastric adenomas and development of adenocarcinoma. The American journal of pathology. 2002; 161:611-618.
59. Fox JG, Dangler CA, Whary MT, Edelman W, Kucherlapati R and Wang TC. Mice carrying a truncated Apc gene have diminished gastric epithelial proliferation, gastric inflammation, and humoral immunity in response to Helicobacter felis infection. Cancer research. 1997; 57:3972-3978.
60. Oshima H, Matsunaga A, Fujimura T, Tsukamoto T, Taketo MM and Oshima M. Carcinogenesis in mouse stomach by simultaneous activation of the Wnt signaling and prostaglandin E2 pathway. Gastroenterology. 2006; 131:1086-1095.
61. Oshima H, Oshima M, Inaba K and Taketo MM. Hyperplastic gastric tumors induced by activated macrophages in COX-2/mPGES-1 transgenic mice. The EMBO journal. 2004; 23:1669-1678.
62. Akaboshi S, Watanabe S, Hino Y, Sekita Y, Xi Y, Araki K, Yamamura K, Oshima M, Ito T, Baba H and Nakao M. HMGA1 is induced by Wnt/beta-catenin pathway and maintains cell proliferation in gastric cancer. The American journal of pathology. 2009; 175:1675-1685.
63. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS and Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356:215-221.
64. Donehower LA, Harvey M, Vogel H, McArthur MJ, Montgomery CA, Jr., Park SH, Thompson T, Ford RJ and Bradley A. Effects of genetic background on tumorigenesis in p53-deficient mice. Molecular carcinogenesis. 1995; 14:16-22.
65. Ohgaki H, Fukuda M, Tohma Y, Huang H, Stoica G, Tatematsu M and Donehower LA. Effect of intragastric application of N-methylnitrosourea in p53 knockout mice. Molecular carcinogenesis. 2000; 28:97-101.
66. Fox JG, Li X, Cahill RJ, Andrutis K, Rustgi AK, Odze R and Wang TC. Hypertrophic gastropathy in Helicobacter felis-infected wild-type C57BL/6 mice and p53 hemizygous transgenic mice. Gastroenterology. 1996; 110:155-166.
67. Jenks PJ, Jeremy AH, Robinson PA, Walker MM and Crabtree JE. Long-term infection with Helicobacter felis and inactivation of the tumour suppressor gene p53 cumulatively enhance the gastric mutation frequency in Big Blue transgenic mice. The Journal of pathology. 2003; 201:596-602.
68. Suzuki H, Miyazawa M, Kai A, Suzuki M, Suematsu M, Miura S and Ishii H. No difference in the level of gastric mucosal cell apoptosis and proliferation in Helicobacter pylori-colonized p53 heterozygous knockout mice. Alimentary pharmacology & therapeutics. 2002; 16 Suppl 2:158-166.
69. Shimada S, Mimata A, Sekine M, Mogushi K, Akiyama Y, Fukamachi H, Jonkers J, Tanaka H, Eishi Y and Yuasa Y. Synergistic tumour suppressor activity of E-cadherin and p53 in a conditional mouse model for metastatic diffuse-type gastric cancer. Gut. 2012; 61:344-353.
70. Shields JM, Christy RJ and Yang VW. Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. The Journal of biological chemistry. 1996; 271:20009-20017.
71. McConnell BB, Ghaleb AM, Nandan MO and Yang VW. The diverse functions of Kruppel-like factors 4 and 5 in epithelial biology and pathobiology. BioEssays. 2007; 29:549-557.
72. Wei D, Gong W, Kanai M, Schlunk C, Wang L, Yao JC, Wu TT, Huang S and Xie K. Drastic down-regulation of Kruppel-like factor 4 expression is critical in human gastric cancer development and progression. Cancer research. 2005; 65:2746-2754.
73. Qiao XT, Ziel JW, McKimpson W, Madison BB, Todisco A, Merchant JL, Samuelson LC and Gumucio DL. Prospective identification of a multilineage progenitor in murine stomach epithelium. Gastroenterology. 2007; 133:1989-1998.
74. Krueger S, Roessner A and Kuester D. Murine models of H. pylori-induced gastritis and gastric adenocarcinoma. Pathology, research and practice. 2011; 207:599-607.
75. Rogers AB. Histologic scoring of gastritis and gastric cancer in mouse models. Methods in molecular biology. 2012; 921:189-203.
76. Menheniott TR, O’Connor L, Chionh YT, Dabritz J, Scurr M, Rollo BN, Ng GZ, Jacobs S, Catubig A, Kurklu B, Mercer S, Minamoto T, Ong DE, et al. Loss of gastrokine-2 drives premalignant gastric inflammation and tumor progression. The Journal of clinical investigation. 2016; 126:1383-1400.
77. Kwon HJ, Won YS, Nam KT, Yoon YD, Jee H, Yoon WK, Nam KH, Kang JS, Han SU, Choi IP, Kim DY and Kim HC. Vitamin D(3) upregulated protein 1 deficiency promotes N-methyl-N-nitrosourea and Helicobacter pylori-induced gastric carcinogenesis in mice. Gut. 2012; 61:53-63.
78. Tomasetto C and Rio MC. Pleiotropic effects of Trefoil Factor 1 deficiency. Cellular and molecular life sciences. 2005; 62:2916-2920.
79. Johnson AH, Frierson HF, Zaika A, Powell SM, Roche J, Crowe S, Moskaluk CA and El-Rifai W. Expression of tight-junction protein claudin-7 is an early event in gastric tumorigenesis. The American journal of pathology. 2005; 167:577-584.
80. Soutto M, Romero-Gallo J, Krishna U, Piazuelo MB, Washington MK, Belkhiri A, Peek RM, Jr. and El-Rifai W. Loss of TFF1 promotes Helicobacter pylori-induced beta-catenin activation and gastric tumorigenesis. Oncotarget. 2015; 6:17911-17922. doi: 10.18632/oncotarget.3772.
81. Soutto M, Chen Z, Katsha AM, Romero-Gallo J, Krishna US, Piazuelo MB, Washington MK, Peek RM, Jr., Belkhiri A and El-Rifai WM. Trefoil factor 1 expression suppresses Helicobacter pylori-induced inflammation in gastric carcinogenesis. Cancer. 2015; 121:4348-4358.
82. Tebbutt NC, Giraud AS, Inglese M, Jenkins B, Waring P, Clay FJ, Malki S, Alderman BM, Grail D, Hollande F, Heath JK and Ernst M. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nature medicine. 2002; 8:1089-1097.
83. Jones GW, McLeod L, Kennedy CL, Bozinovski S, Najdovska M and Jenkins BJ. Imbalanced gp130 signalling in ApoE-deficient mice protects against atherosclerosis. Atherosclerosis. 2015; 238:321-328.
84. Mutoh H, Hakamata Y, Sato K, Eda A, Yanaka I, Honda S, Osawa H, Kaneko Y and Sugano K. Conversion of gastric mucosa to intestinal metaplasia in Cdx2-expressing transgenic mice. Biochemical and biophysical research communications. 2002; 294:470-479.
85. Mutoh H, Sakurai S, Satoh K, Tamada K, Kita H, Osawa H, Tomiyama T, Sato Y, Yamamoto H, Isoda N, Yoshida T, Ido K and Sugano K. Development of gastric carcinoma from intestinal metaplasia in Cdx2-transgenic mice. Cancer research. 2004; 64:7740-7747.
86. Mutoh H, Sakurai S, Satoh K, Osawa H, Hakamata Y, Takeuchi T and Sugano K. Cdx1 induced intestinal metaplasia in the transgenic mouse stomach: comparative study with Cdx2 transgenic mice. Gut. 2004; 53:1416-1423.
87. Sakamoto H, Asahara T, Chonan O, Yuki N, Mutoh H, Hayashi S, Yamamoto H and Sugano K. Comparative analysis of gastrointestinal microbiota between normal and caudal-related homeobox 2 (cdx2) transgenic mice. Intestinal research. 2015; 13:39-49.
88. El-Zaatari M, Tobias A, Grabowska AM, Kumari R, Scotting PJ, Kaye P, Atherton J, Clarke PA, Powe DG and Watson SA. De-regulation of the sonic hedgehog pathway in the InsGas mouse model of gastric carcinogenesis. British journal of cancer. 2007; 96:1855-1861.
89. Burns M, Muthupalani S, Ge Z, Wang TC, Bakthavatchalu V, Cunningham C, Ennis K, Georgieff M and Fox JG. Helicobacter pylori Infection Induces Anemia, Depletes Serum Iron Storage, and Alters Local Iron-Related and Adult Brain Gene Expression in Male INS-GAS Mice. PloS one. 2015; 10:e0142630.
90. Fox JG, Wang TC, Rogers AB, Poutahidis T, Ge Z, Taylor N, Dangler CA, Israel DA, Krishna U, Gaus K and Peek RM, Jr. Host and microbial constituents influence Helicobacter pylori-induced cancer in a murine model of hypergastrinemia. Gastroenterology. 2003; 124:1879-1890.
91 Kanda N, Seno H, Kawada M, Sawabu T, Uenoyoma Y, Nakajima T, Konda Y, Fukui H, Takeuchi T and Chiba T. Involvement of cyclooxygenase-2 in gastric mucosal hypertrophy in gastrin transgenic mice. American journal of physiology Gastrointestinal and liver physiology. 2006; 290:G519-527.
92. Konda Y, Kamimura H, Yokota H, Hayashi N, Sugano K and Takeuchi T. Gastrin stimulates the growth of gastric pit with less-differentiated features. The American journal of physiology. 1999; 277:G773-784.
93. Todisco A, Mao M, Keeley TM, Ye W, Samuelson LC and Eaton KA. Regulation of gastric epithelial cell homeostasis by gastrin and bone morphogenetic protein signaling. Physiological reports. 2015; 3: e12501.
94. Kang W, Rathinavelu S, Samuelson LC and Merchant JL. Interferon gamma induction of gastric mucous neck cell hypertrophy. Laboratory investigation; a journal of technical methods and pathology. 2005; 85:702-715.
95. Judd LM, Andringa A, Rubio CA, Spicer Z, Shull GE and Miller ML. Gastric achlorhydria in H/K-ATPase-deficient (Atp4a(-/-)) mice causes severe hyperplasia, mucocystic metaplasia and upregulation of growth factors. Journal of gastroenterology and hepatology. 2005; 20:1266-1278.
96. Spicer Z, Miller ML, Andringa A, Riddle TM, Duffy JJ, Doetschman T and Shull GE. Stomachs of mice lacking the gastric H,K-ATPase alpha -subunit have achlorhydria, abnormal parietal cells, and ciliated metaplasia. The Journal of biological chemistry. 2000; 275:21555-21565.
97. Schultheis PJ, Clarke LL, Meneton P, Harline M, Boivin GP, Stemmermann G, Duffy JJ, Doetschman T, Miller ML and Shull GE. Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. The Journal of clinical investigation. 1998; 101:1243-1253.
98. Gawenis LR, Greeb JM, Prasad V, Grisham C, Sanford LP, Doetschman T, Andringa A, Miller ML and Shull GE. Impaired gastric acid secretion in mice with a targeted disruption of the NHE4 Na+/H+ exchanger. The Journal of biological chemistry. 2005; 280:12781-12789.
99. Lee MP, Ravenel JD, Hu RJ, Lustig LR, Tomaselli G, Berger RD, Brandenburg SA, Litzi TJ, Bunton TE, Limb C, Francis H, Gorelikow M, Gu H, et al. Targeted disruption of the Kvlqt1 gene causes deafness and gastric hyperplasia in mice. The Journal of clinical investigation. 2000; 106:1447-1455.
100. Kobayashi T, Tonai S, Ishihara Y, Koga R, Okabe S and Watanabe T. Abnormal functional and morphological regulation of the gastric mucosa in histamine H2 receptor-deficient mice. The Journal of clinical investigation. 2000; 105:1741-1749.
101. Ogawa T, Maeda K, Tonai S, Kobayashi T, Watanabe T and Okabe S. Utilization of knockout mice to examine the potential role of gastric histamine H2-receptors in Menetrier’s disease. Journal of pharmacological sciences. 2003; 91:61-70.
102. Fukuhara S, Matsuzaki J, Tsugawa H, Masaoka T, Miyoshi S, Mori H, Fukushima Y, Yasui M, Kanai T and Suzuki H. Mucosal expression of aquaporin-4 in the stomach of histamine type 2 receptor knockout mice and Helicobacter pylori-infected mice. Journal of gastroenterology and hepatology. 2014; 29 Suppl 4:53-59.
103. Nozaki K, Weis V, Wang TC, Falus A and Goldenring JR. Altered gastric chief cell lineage differentiation in histamine-deficient mice. American journal of physiology Gastrointestinal and liver physiology. 2009; 296:G1211-1220.
104. Li S, Wang Q, Chakladar A, Bronson RT and Bernards A. Gastric hyperplasia in mice lacking the putative Cdc42 effector IQGAP1. Molecular and cellular biology. 2000; 20:697-701.
105. Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP and Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998; 93:1159-1170.
106. Kim BG, Li C, Qiao W, Mamura M, Kasprzak B, Anver M, Wolfraim L, Hong S, Mushinski E, Potter M, Kim SJ, Fu XY, Deng C and Letterio JJ. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature. 2006; 441:1015-1019.
107. Redman RS, Katuri V, Tang Y, Dillner A, Mishra B and Mishra L. Orofacial and gastrointestinal hyperplasia and neoplasia in smad4+/- and elf+/-/smad4+/- mutant mice. Journal of oral pathology & medicine. 2005; 34:23-29.
108. Takeda H, Rust AG, Ward JM, Yew CC, Jenkins NA and Copeland NG. Sleeping Beauty transposon mutagenesis identifies genes that cooperate with mutant Smad4 in gastric cancer development. Proceedings of the National Academy of Sciences of the United States of America. 2016; 113: e2057-65.
109. Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ, Yamamoto H, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002; 109:113-124.
110. Brenner O, Levanon D, Negreanu V, Golubkov O, Fainaru O, Woolf E and Groner Y. Loss of Runx3 function in leukocytes is associated with spontaneously developed colitis and gastric mucosal hyperplasia. Proceedings of the National Academy of Sciences of the United States of America. 2004; 101:16016-16021.
111. Tomita H, Yamada Y, Oyama T, Hata K, Hirose Y, Hara A, Kunisada T, Sugiyama Y, Adachi Y, Linhart H and Mori H. Development of gastric tumors in Apc(Min/+) mice by the activation of the beta-catenin/Tcf signaling pathway. Cancer research. 2007; 67:4079-4087.
112. Tsuzuki T, Egashira A, Igarashi H, Iwakuma T, Nakatsuru Y, Tominaga Y, Kawate H, Nakao K, Nakamura K, Ide F, Kura S, Nakabeppu Y, Katsuki M, Ishikawa T and Sekiguchi M. Spontaneous tumorigenesis in mice defective in the MTH1 gene encoding 8-oxo-dGTPase. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:11456-11461.
113. Oshima M, Oshima H, Matsunaga A and Taketo MM. Hyperplastic gastric tumors with spasmolytic polypeptide-expressing metaplasia caused by tumor necrosis factor-alpha-dependent inflammation in cyclooxygenase-2/microsomal prostaglandin E synthase-1 transgenic mice. Cancer research. 2005; 65:9147-9151.
114. Ludlow A, Yee KO, Lipman R, Bronson R, Weinreb P, Huang X, Sheppard D and Lawler J. Characterization of integrin beta6 and thrombospondin-1 double-null mice. Journal of cellular and molecular medicine. 2005; 9:421-437.
115. Dempsey PJ, Goldenring JR, Soroka CJ, Modlin IM, McClure RW, Lind CD, Ahlquist DA, Pittelkow MR, Lee DC, Sandgren EP and et al. Possible role of transforming growth factor alpha in the pathogenesis of Menetrier’s disease: supportive evidence form humans and transgenic mice. Gastroenterology. 1992; 103:1950-1963.
116. Sharp R, Babyatsky MW, Takagi H, Tagerud S, Wang TC, Bockman DE, Brand SJ and Merlino G. Transforming growth factor alpha disrupts the normal program of cellular differentiation in the gastric mucosa of transgenic mice. Development. 1995; 121:149-161.
117. Takagi H, Fukusato T, Kawaharada U, Kuboyama S, Merlino G and Tsutsumi Y. Histochemical analysis of hyperplastic stomach of TGF-alpha transgenic mice. Digestive diseases and sciences. 1997; 42:91-98.
118. Nomura S, Settle SH, Leys CM, Means AL, Peek RM, Jr., Leach SD, Wright CV, Coffey RJ and Goldenring JR. Evidence for repatterning of the gastric fundic epithelium associated with Menetrier’s disease and TGFalpha overexpression. Gastroenterology. 2005; 128:1292-1305.
119. Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, Hanberg A and Poellinger L. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proceedings of the National Academy of Sciences of the United States of America. 2002; 99:9990-9995.
120. Kuznetsov NV, Andersson P, Gradin K, Stein P, Dieckmann A, Pettersson S, Hanberg A and Poellinger L. The dioxin/aryl hydrocarbon receptor mediates downregulation of osteopontin gene expression in a mouse model of gastric tumourigenesis. Oncogene. 2005; 24:3216-3222.
121. Kuzushita N, Rogers AB, Monti NA, Whary MT, Park MJ, Aswad BI, Shirin H, Koff A, Eguchi H and Moss SF. p27kip1 deficiency confers susceptibility to gastric carcinogenesis in Helicobacter pylori-infected mice. Gastroenterology. 2005; 129:1544-1556.
122. Gut MO, Parkkila S, Vernerova Z, Rohde E, Zavada J, Hocker M, Pastorek J, Karttunen T, Gibadulinova A, Zavadova Z, Knobeloch KP, Wiedenmann B, Svoboda J, Horak I and Pastorekova S. Gastric hyperplasia in mice with targeted disruption of the carbonic anhydrase gene Car9. Gastroenterology. 2002; 123:1889-1903.
123. Leppilampi M, Karttunen TJ, Kivela J, Gut MO, Pastorekova S, Pastorek J and Parkkila S. Gastric pit cell hyperplasia and glandular atrophy in carbonic anhydrase IX knockout mice: studies on two strains C57/BL6 and BALB/C. Transgenic research. 2005; 14:655-663.
124. Thompson J, Epting T, Schwarzkopf G, Singhofen A, Eades-Perner AM, van Der Putten H and Zimmermann W. A transgenic mouse line that develops early-onset invasive gastric carcinoma provides a model for carcinoembryonic antigen-targeted tumor therapy. International journal of cancer Journal international du cancer. 2000; 86:863-869.
125. Nockel J, van den Engel NK, Winter H, Hatz RA, Zimmermann W and Kammerer R. Characterization of gastric adenocarcinoma cell lines established from CEA424/SV40 T antigen-transgenic mice with or without a human CEA transgene. BMC cancer. 2006; 6:57.
126. Li Q, Karam SM and Gordon JI. Simian virus 40 T antigen-induced amplification of pre-parietal cells in transgenic mice. Effects on other gastric epithelial cell lineages and evidence for a p53-independent apoptotic mechanism that operates in a committed progenitor. The Journal of biological chemistry. 1995; 270:15777-15788.
127. Stewart LA, van Driel IR and Gleeson PA. Perturbation of gastric mucosa in mice expressing the temperature-sensitive mutant of SV40 large T antigen. Potential for establishment of an immortalised parietal cell line. European journal of cell biology. 2002; 81:281-293.
128. Bakkelund KE, Waldum HL, Nordrum IS, Hauso O and Fossmark R. Long-term gastric changes in achlorhydric H(+)/K(+)-ATPase beta subunit deficient mice. Scandinavian journal of gastroenterology. 2010; 45:1042-1047.
129. Ramalho-Santos M, Melton DA and McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000; 127:2763-2772.
130. Saitou M, Furuse M, Sasaki H, Schulzke JD, Fromm M, Takano H, Noda T and Tsukita S. Complex phenotype of mice lacking occludin, a component of tight junction strands. Molecular biology of the cell. 2000; 11:4131-4142.
131. Nighot MP, Nighot PK, Ma TY, Malinowska DH, Shull GE, Cuppoletti J and Blikslager AT. Genetic Ablation of the ClC-2 Cl- Channel Disrupts Mouse Gastric Parietal Cell Acid Secretion. PloS one. 2015; 10:e0138174.