
Introduction
Lung cancer is the most common malignancy but curative therapy for metastatic disease is limited. Although tyrosine kinase inhibitor (TKI) therapy achieves a higher objective response rate and longer progression free survival in cancers with epidermal growth factor receptor (EGFR) mutations or anaplastic lymphoma kinase (ALK)-rearrangement, relapse is the rule after 10-14 months due to drug resistance [1-5]. Increasing evidences have shown in many cancers, enhanced tumorigenicity resides in a tumor cell population that exhibits stem cell-like properties such as self-renewal, differentiation, cell mobility and toxicity resistance, designated as cancer stem cells (CSC) or tumor initiating cells (TIC) [6, 7]. Inhibition of morphogenesis regulatory pathways such the Hedgehog, Notch and Wnt/β-catenin pathways can retard tumor transplantability, further supporting TIC-targeting could be useful for lung cancer control [8-11]. However, a deeper understanding of the cell targets and maintenance mechanisms is essential for further therapy development.
Lung cancer cells expressing various molecules such as CD133, CD166, ALDH, CXCR4, GLDC, etc. have been shown to demonstrate TIC phenotypic characteristics [12-15]. On the other hand, to identify TIC with higher specificity, more stringent selection strategies such as the adoption of co-expressed markers and verification in larger numbers of cell lines including patient-derived samples are advocated. We have previously demonstrated lung cancer cells with high CD44 expression were enriched for stem cell-like properties [16]. Moreover, CD44 is expressed in breast, colon and gastric cancer stem cells [17-19]. Aldehyde dehydrogenase 1 (ALDH) is expressed in murine embryonic lungs and has been reported to select for human lung TIC [15, 20, 21]. The specificity of combined ALDH/CD44 expression as lung TIC marker compared to either marker alone is not known. In this study, we have used a range of established and patient-derived lung cancer cell lines (PDCL) to show cells with high ALDH and CD44 co-expression (ALDHhiCD44hi) possessed in vitro and in vivo TIC properties with enhanced tumorigenicity and drug resistance compared to the low-expressing (ALDHloCD44lo) compartment or un-selected cells. Simultaneous ALDH inhibition and CD44 depletion as well as pharmacologic inhibition of Hedgehog or Notch attenuated TIC characteristics. In clinical lung cancers, recurrence-free survival was longer for patients with low abundance ALDH/CD44-coexpressing cells (p = 0.053). Our data demonstrated lung TIC are enhanced through ALDH and CD44 co-regulating pathways. Further investigation of the ALDHhiCD44hi population would enable a better understanding of TIC regulation and facilitate development of therapeutic strategies for long term lung cancer control.
Results
ALDHhiCD44hi population displayed in vitro TIC properties
The ALDH/CD44 co-expression profiles of 11 lung cancer cell lines including PDCL and drug-induced resistant cells were analyzed by flow cytometry. In 6 cell lines, ALDH/CD44 co-expressing cells (ALDH+/CD44+) comprised the smallest subset with ALDH/CD44 non-expressing cells (ALDH-/CD44-) forming the largest population (Supplementary Table S1). Subsequently, the top/bottom 1 to 5% of cells showing highest/lowest expression of the markers (ALDHhiCD44hi, ALDHhiCD44lo, ALDHloCD44hi and ALDHloCD44lo) were freshly isolated from H1650 and HCC827 cell lines for further in vitro tests. In the spheroid formation assay, the ALDHhiCD44hi populations generated more abundant and larger spheroid bodies than the other 3 subsets (Figure 1A). In the cell invasion assay, they demonstrated the highest percentage of invading cells while the ALDHloCD44lo subset showed the lowest (Figure 1B). In vitro differentiation in normal culture conditions showed only the ALDHhiCD44hi subset was able to differentiate into all 4 cell populations with similar distribution profile as the parental cell line while compositions of the other 3 subsets remained largely unchanged from their fresh, post-sorting profiles (Figure 1C).
The ALDHhiCD44hi population showed expression profiles that were characteristic of TIC. They had significantly higher expression of the pluripotency genes NANOG, POU5F1 and SOX2 at both the mRNA and protein levels compared to ALDHloCD44lo and unsorted populations (Figure 1D to F). They also showed higher mRNA expression of the epithelial to mesenchymal transition (EMT) transcription factors ZEB1 and SNAIL2, the mesenchymal gene VIM, the DNA double strand break repair gene RAD51, the TGFβ/IL6 axis gene IL6ST, and a lower expression level of the cell adhesion molecule CDH1 (Figure 1D & E).
ALDHhiCD44hi showed G2/M shift compared to ALDHloCD44lo subset in cell cycle analysis
Cell cycle analysis showed the ALDHhiCD44hi subset of H1650 had a significantly higher proportion in G2/M phase (14.57 ± 3.23%) compared to ALDHloCD44lo (3.74 ± 0.59%, p < 0.05) and unsorted controls (5.81 ± 0.23%, p < 0.01) while cells in G0/1 phase were less abundant (48.42 ± 4.48%) than the other populations (ALDHloCD44lo, 71.84 ± 2.58%, p < 0.01; unsorted, 72.06 ± 9.98%, p < 0.05) (Figure 1G). Cell growth kinetics analysis showed a higher growth rate of ALDHhiCD44hi than unsorted and ALDHloCD44lo cells (Figure 1H). Correspondingly, the mRNA expression of the G2/M phase cell cycle genes CCNB1 and CCNB2 were significantly higher in ALDHhiCD44hi than either ALDHloCD44lo or unsorted cells (Figure 1I).
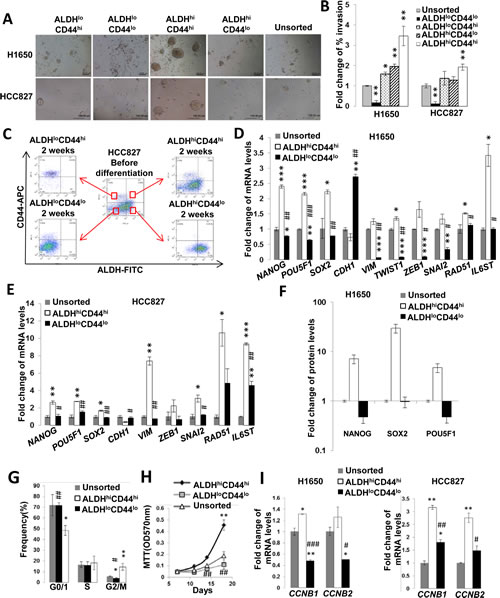
Figure 1: ALDHhiCD44hi lung cancer cells showed in vitro TIC characteristics. A, Spheroid formation assay. FACS-isolated lung cancer cell populations with differential ALDH/CD44 expressions and unsorted cell controls were kept in serum-free non-adherent plates for 21 days. B, Matrigel invasion assay. The proportions of invading cells from respective cell subsets were normalized to the unsorted control. C, In vitro differentiation assay. The 4 freshly isolated populations were separately cultured in adhesive plates containing normal medium for 2 weeks. Cells were then freshly harvested and re-analyzed by flow cytometry for ALDH/CD44 expression profile. The central profile represented parental unsorted cells and profiles of the subsets were as labeled. D and E, Normalized mRNA expressions of pluripotency, EMT and other genes by QPCR. F, Pluripotency proteins expression analyzed by flow cytometry. Results were normalized to unsorted control. G, Cell cycle analysis. Freshly isolated ALDHhiCD44hi and ALDHloCD44lo populations of H1650 were stained with propidium iodide and analyzed by flow cytometry for DNA content. H, Cell proliferation assay. Respective subsets of freshly isolated H1650 cells were analyzed by MTT. I, Expression of CCNB1 and CCNB2 analyzed by QPCR.*, p < 0.05; **, p < 0.01; ***, p < 0.001, compared with unsorted; #, p < 0.05; ##, p < 0.01; ###, p < 0.001, compared with ALDHhiCD44hi. All data represent the mean ± SD of triplicate experiments.
ALDHhiCD44hi subset showed the highest tumorigenicity in immunosuppressed mice
To test for in vivo tumorigenicity, the 4 freshly harvested ALDH/CD44 populations and unsorted controls were transplanted subcutaneously into SCID mice according to their assigned locations (Figure 2A & B) from 3 established cell lines and 4 PDCLs. As shown in Table 1, the ALDHhiCD44hi population of all analyzed cell lines demonstrated higher graft rates, shorter latency and larger volumes of xenografts compared to the same dose of ALDHloCD44hi, ALDHhiCD44lo or ALDHloCD44lo cells (Figure 2C & D). As few as 500 ALDHhiCD44hi cells of HCC827 or H1650 but not other populations were able to initiate tumors at this low seeding dose (Supplementary Table S2). Interestingly, the HKULC3 cell line, which did not form xenografts from 108 unsorted cells transplanted in nude mice for 6 months in our previous study, was able to initiate tumor in SCID mice from 4,000 ALDHhiCD44hi cells in 3 months ([22] , Table 1).
Table 1: Serial transplantation of ALDH/CD44 subsets from different NSCLC cell lines
Cell line and subset |
1st generation |
2nd generation |
3rd generation |
||||
Tumor incidence |
Latency (days) |
Tumor incidence |
Latency (days) |
Tumor incidence |
Latency (days) |
||
H1650 |
2,500 cells injection |
2,500 cells injection |
2,500 cells injection |
||||
Unsorted |
3/6 |
69.6 ± 5.6 |
0/5 |
N/A |
ND |
ND |
|
ALDHloCD44lo |
2/6 |
75.0 ± 4.0 |
1/6 |
89 |
1/6 |
90 |
|
ALDHloCD44hi |
3/6 |
72.7 ± 6.7 |
4/6 |
64.0 ± 10.8 |
3/5 |
35.3 ± 1.0 |
|
ALDHhiCD44lo |
4/6 |
43.5 ± 2.5 |
5/6 |
53.4 ± 3.8 |
4/5 |
50.8 ± 1.3 |
|
ALDHhiCD44hi |
6/6 |
41.0 ± 5.0 |
6/6 |
25.8 ± 2.0 |
6/6 |
19.0 ± 1.8 |
|
HCC827 |
2,500 cells injection |
2,500 cells injection |
2,500 cells injection |
||||
Unsorted |
2/3 |
42.7 ± 2.9 |
3/4 |
60.0 ± 11.1 |
0/4 |
N/A |
|
ALDHloCD44lo |
1/3 |
59 |
2/4 |
65.5 ± 4.9 |
0/4 |
N/A |
|
ALDHloCD44hi |
2/3 |
41.0 ± 2.0 |
3/3 |
63.0 ± 6.9 |
1/4 |
43 |
|
ALDHhiCD44lo |
2/3 |
41.0 ± 7.5 |
4/4 |
53.3 ± 9.5 |
2/4 |
44.5 ± 2.1 |
|
ALDHhiCD44hi |
3/3 |
29.0 ± 1.0 |
4/4 |
22.0 ± 1.6 |
4/4 |
20.2 ± 1.8 |
|
PDCL #24 |
30,000 cells injection |
2,500 cells injection |
2,500 cells injection |
||||
Unsorted |
3/3 |
22.0 ± 1.2 |
1/3 |
44.0 |
1/3 |
50.0 |
|
ALDHloCD44lo |
2/3 |
33.5 ± 2.0 |
1/3 |
44.0 |
0/3 |
N/A |
|
ALDHloCD44hi |
3/3 |
22.0 ± 1.2 |
3/3 |
34.0 ± 1.0 |
2/3 |
45.2 ± 3.7 |
|
ALDHhiCD44lo |
N/A |
N/A |
0/2 |
N/A |
N/A |
N/A |
|
ALDHhiCD44hi |
3/3 |
8.7 ± 1.5 |
3/3 |
20.7 ± 2.1 |
3/3 |
18.7 ± 3.2 |
|
PDCL #2 |
8,000 cells injection |
8,000 cells injection |
ND |
||||
Unsorted |
0/3 |
N/A |
0/3 |
N/A |
ND |
ND |
|
ALDHloCD44lo |
0/3 |
N/A |
0/3 |
N/A |
ND |
ND |
|
ALDHloCD44hi |
0/3 |
N/A |
0/3 |
N/A |
ND |
ND |
|
ALDHhivCD44lo |
0/3 |
N/A |
0/3 |
N/A |
ND |
ND |
|
ALDHhiCD44hi |
2/3 |
150.0 ± 42.4 |
3/3 |
39.0 ± 1.7 |
ND |
ND |
|
PDCL #10 |
1,500 cells injection |
1,200 cells injection |
600 cells injection |
||||
Unsorted |
0/3 |
N/A |
0/3 |
N/A |
ND |
ND |
|
ALDHloCD44lo |
0/3 |
N/A |
0/3 |
N/A |
ND |
ND |
|
ALDHloCD44hi |
3/3 |
20.0 ± 5.0 |
0/3 |
N/A |
0/3 |
N/A |
|
ALDHhiCD44lo |
0/3 |
N/A |
0/3 |
N/A |
0/3 |
N/A |
|
ALDHhiCD44hi |
3/3 |
9.0 ± 2.0 |
3/3 |
12.0 ± 3.0 |
3/3 |
52.1 ± 8.5 |
|
PDCL #18 |
10,000 cells injection |
ND |
ND |
||||
Unsorted |
0/2 |
N/A |
ND |
ND |
ND |
ND |
|
ALDHloCD44lo |
0/2 |
N/A |
ND |
ND |
ND |
ND |
|
ALDHloCD44hi |
1/2 |
35.0 |
ND |
ND |
ND |
ND |
|
ALDHhiCD44lo |
0/2 |
N/A |
ND |
ND |
ND |
ND |
|
ALDHhiCD44hi |
2/2 |
17.0 ± 4.0 |
ND |
ND |
ND |
ND |
|
HKULC3 |
4,000 cells injection |
4,000 cells injection |
ND |
||||
Unsorted |
0/3 |
N/A |
ND |
ND |
ND |
ND |
|
ALDHloCD44lo |
0/3 |
N/A |
ND |
ND |
ND |
ND |
|
ALDHloCD44hi |
0/3 |
N/A |
ND |
ND |
ND |
ND |
|
ALDHhiCD44lo |
0/3 |
N/A |
ND |
ND |
ND |
ND |
|
ALDHhiCD44hi |
3/3 |
92.3 ± 4.0 |
3/3 |
76.3 ± 6.9 |
ND |
ND |
|
N/A, tumor dimension not available; ND, not done
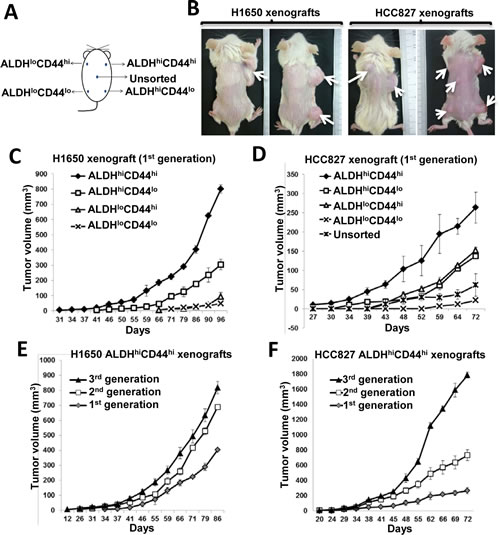
Figure 2: ALDHhiCD44hi population showed TIC properties in vivo. A, Schematic diagram showing transplantation sites of respective ALDH/CD44 populations in SCID mice. B, Representative photographs of H1650 or HCC827 xenografts derived from respective ALDH/CD44 populations after 3 months tumor development. C and D, Tumor growth curves of 1st generation xenografts derived from 2,500 cells of respective ALDH/CD44 populations. E and F, tumor growth curves of 3 serially transplanted generations of xenografts derived from 2,500 ALDHhiCD44hi cells of respective cell lines. Data represent the mean ± SD of tumor volumes at different time points of different groups, there are 6 mice in each group.
Serial transplantation showed ALDHhiCD44hi–derived tumors had increasing induction frequencies, reduced latency and increased growth rates in successive generations compared to the first-generation transplants (Table 1, Figure 2E & F). In contrast, the tumor frequencies of ALDHloCD44hi, ALDHhiCD44lo, ALDHloCD44lo or unsorted cells decreased in the second and tertiary generations (Table 1).
ALDHhiCD44hi population showed in vitro and in vivo resistance to anti-cancer drugs
The response of ALDH/CD44 subsets to both targeted drugs and cytotoxic chemotherapy were compared using in vitro and in vivo models. Gefitinib treatment of the TKI-sensitive HCC827 (EGFR exon19 deletion) led to significant elevation of ALDH+CD44+ and ALDH+CD44- subsets (Figure 3A). In cells induced by chronic exposure to increasing gefitinib dosage (HCC827-GR), progressive resistance was associated with stepwise increase in ALDH+CD44+ proportions (Figure 3B). This population was also higher in HCC827 induced for cisplatin-resistance (HCC827-CR) (Figure 3C). Cell viability to gefitinib treatment was significantly higher for ALDHhiCD44hi compared to unsorted HCC827 cells (Figure 3D). Conversely, the apoptotic fractions after 24 hrs cisplatin treatment were significantly higher in ALDHloCD44lo (p < 0.05) and unsorted H1650 cells (TKI-resistant, cisplatin-sensitive) (p < 0.01) while no effect was observed in the ALDHhiCD44hi population (Figure 3E). Amongst 5 TKI-treated mice bearing HCC827 xenografts, only ALDHhiCD44hi-derived tumors persisted after 4-5 weeks intraperitoneal gefitinib while no residual tumor was detected histologically at other injection sites (Figure 3F & 3G). No reduction in tumor size was observed in mice receiving control treatment.
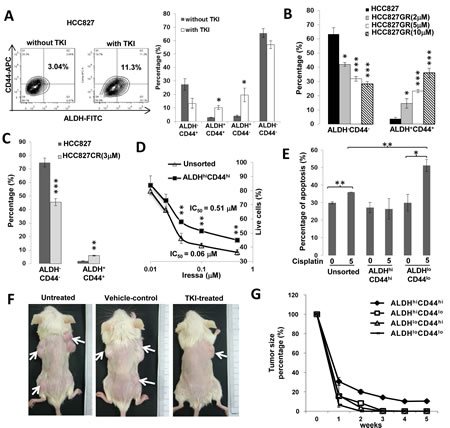
Figure 3: ALDHhiCD44hi cells showed drug resistance in vitro and in vivo. A, Effects of 30 nM gefitinib treatment for 24 hr on ALDH/CD44 subsets of HCC827 analyzed by flow cytometry. B and C, Proportions of ALDH+CD44+ and ALDH-CD44- cells in drug-resistant HCC827 cells. Resistance was induced by chronic exposure to increasing gefitinib doses (HCC827-GR) or to cisplatin (HCC827-CR). D, In vitro cell viability response. Freshly isolated ALDHhiCD44hi and unsorted cells of HCC827 were treated with a range of gefitinib doses for 24 hr and cell viability was assayed by MTT. E, Apoptosis response. Freshly isolated respective populations and unsorted H1650 were treated with 5 µM cisplatin for 24 hr and apoptosis fractions were assayed by Annexin V/PI staining. F, In vivo xenograft response after TKI treatment. Mice bearing HCC827 xenografts from respective ALDH/CD44 subsets as depicted in Figure 2A were given intraperitoneal gefitinib 5 times per week for 5 weeks. G, In vivo tumor response curve. Tumor volumes of respective HCC827 xenografts in TKI-treated (n = 5) or control-treated (n = 3) mice were measured twice weekly. Data represent mean ± SD of tumor volume normalized to pre-treatment size in gefitinib-treated mice. *, p < 0.05; **, p < 0.01; ***, p < 0.001, compared with control treatment or unsorted cells.
Hedgehog and Notch pathway inhibition suppressed ALDHhiCD44hi and spheroid formation
To investigate whether morphogenesis pathways were involved in ALDHhiCD44hi subset maintenance, the effects of pharmacological inhibition of Hedgehog and Notch pathways were analyzed. The expression of sonic Hedgehog (SMO, GLI1) and Notch (NOTCH1, NOTCH3, HEY1) pathway genes were significantly higher in ALDHhiCD44hi compared to ALDHloCD44lo populations in both H1650 and HCC827 (Figure 4A & B). Cyclopamine treatment led to mRNA suppression of the Hedgehog transcription factor GLI1 by >50% while RO4929097 suppressed the Notch transcription factor HES1 by 30%, respectively (Figure 4C). These treatments resulted in reduction of the ALDH+CD44+ in both cell lines as well as inhibited spheroid formation, indicating their involvement in ALDHhiCD44hi subset maintenance (Figure 4D to 4F).
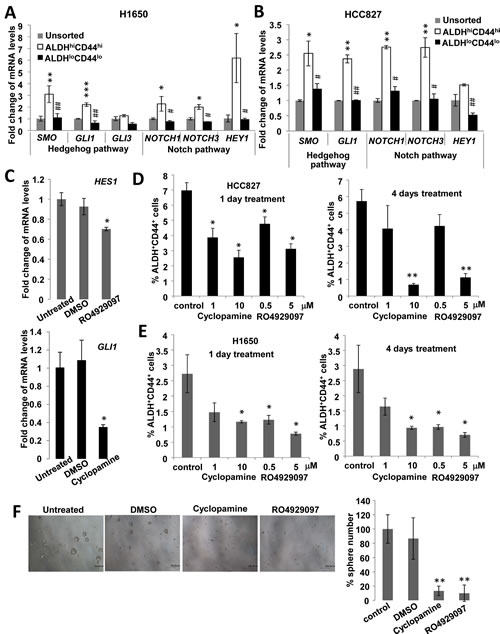
Figure 4: Hedgehog and Notch signaling were involved in ALDHhiCD44hi TIC maintenance. A and B, mRNA levels of Hedgehog and Notch signaling genes by QPCR in sorted cells from H1650 and HCC827. C, mRNA levels of HES1 or GLI1 by QPCR in HCC827 after Notch inhibition by RO4929097 or Hedgehog inhibition by cyclopamine. D and E, Proportions of ALDH+CD44+ by flow cytometry in HCC827 and H1650 after pathway inhibition for 1 or 4 days. F, Spheroid formation assay of HCC827 cells treated with inhibitors. *, p < 0.05; **, p < 0.01; ***, p < 0.001, compared with unsorted or control; #, p < 0.05; ##, p < 0.01, compared with ALDHhiCD44hi. Data represent mean ± SD of triplicate experiments.
CD44 knockdown and ALDH inhibition sensitized tumor cells to in vitro anti-cancer drugs
To investigate the role of ALDH and CD44 in TIC maintenance, ALDH enzymatic activity was completely inhibited by DEAB and CD44 was down-regulated with 80-90% reduction in mRNA and protein expression in multiple cancer cell lines (Figure 5A & B). These treatments led to a variable reduction of developmental regulatory genes expression by 30-50% including POU5F1, NANOG, SOX2 and BMI1 (Figure 5A & B). It also resulted in significant sensitization to cisplatin (H1299, H1650, PDCL #24, p < 0.05) and gefitinib treatment ((HCC827-GR, p < 0.01; H1650-GR, p < 0.001) with reduced cell viability (Figure 5C).
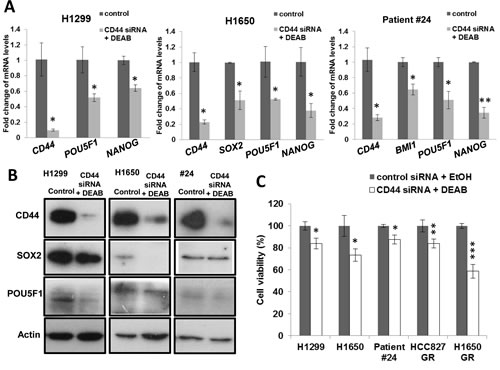
Figure 5: CD44 knockdown and ALDH inhibition reduced pluripotency gene expression and sensitized cells to drug treatment. A and B, mRNA and protein expressions of CD44 and pluripotency genes. CD44 was depleted by siRNA and ALDH inhibited by DEAB (100 µM) in H1299, H1650 and PDCL #24, control siRNA and Ethanol were used as control. Histograms represent mean ± SD of mRNA measured by QPCR in triplicates and protein expression was analyzed by western blot. C, Cell viability assay. CD44 and ALDH-inhibited or control cells were treated respectively with cisplatin (H1650, 10 µM; H1299 & PDCL #24, 15 µM) or gefitinib (HCC827 GR, 10 µM; H1650 GR, 20 µM) for 24 hrs. *, p < 0.05; **, p < 0.01; ***, p < 0.001, compared with control. Data represent mean ± SD of triplicate experiments.
Lung cancer patients with lower abundance of tumor cells co-expressing ALDH and CD44 had longer recurrence-free survival
Immunohistochemical analysis of 193 resected lung cancers of mixed histological types showed ALDH was expressed in 82 (42.5%) and CD44 in 119 (61.7%) cases, respectively (Supplementary Table S4). ALDH and CD44 expression were both associated with squamous cell carcinomas (χ2 test, p = 0.035 and p = 0.005, respectively). No association of either marker with tumor differentiation, pathological stage, patient gender or smoking history was found. There was no correlation between cases that showed ALDH and CD44 expression. Clinicopathological variables such as gender, smoking history, age, pathological stage, tumor type, differentiation, and ALDH or CD44 single marker expression were included in Cox regression multivariate analysis for recurrence-free survival (RFS), and results showed the male gender (HR 2.89, 95% CI 1.48-5.63, p = 0.002) and advanced pathological stage (HR 1.96, 95% CI 1.50-2.56, p < 0.001) were independent prognostic factors associated with shorter RFS.
In 43 (22.3%) tumors, neither ALDH nor CD44 was expressed. In 47 (24.4%) cases, ALDH/CD44 co-expressing cells were detected, including 27 (57.4%) showing only a few scattered co-expressing cells (low abundance) and 20 (42.6%) cases that showed large aggregates or confluent sheets of such tumor cells (high abundance) (Figure 6A to D). In this group, survival analysis by log rank test showed there was no association between RFS and ALDH or CD44 single marker expression (Figure 6E & F). However, the low abundance ALDH/CD44 co-expressing group had longer RFS than the high abundance group (p = 0.053) (Figure 6G). The effect on overall survival did not reach statistical significance (p = 0.141).
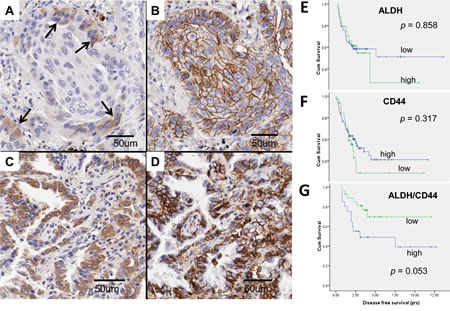
Figure 6: ALDH and CD44 expression in clinical lung cancers. A to D, ALDH/CD44 co-expression patterns were variable in different tumors. A and B, Cytoplasmic ALDH expression (A) was observed in only a few tumor cells (→) at the invasion front of this squamous cell carcinoma while membranous CD44 expression (B) was present in all tumor cells in the same cluster. Co-localizing cells showing both ALDH and CD44 expression were few and thus graded as low abundance. C and D, This adenocarcinoma showed co-expression of ALDH (C) and CD44 (D) in almost all tumor cells and was thus graded as high abundance. E to G, Kaplan Meier survival curves comparing recurrence-free survival (RFS) in different tumor groups. For single marker analysis of ALDH (E) or CD44 (F), no significant difference in RFS was observed between tumor groups showing low or high abundance. For ALDH/CD44 dual marker analysis (G), RFS was shorter for patients with high compared to low abundance ALDH/CD44-coexpressing TIC (p = 0.053).
Discussion
Cellular heterogeneity encompassing a TIC population that carries enhanced tumorigenic potentials has been demonstrated in many cancers but the mechanisms maintaining such populations remain unclear. To study the regulatory mechanisms, the distinction of TIC from non-TIC is required but opinions differ on the most appropriate method of TIC identification. Some investigators employ the cell spheroid as an in vitro functional TIC indicator but the TIC purity, particularly of large spheroids, has seldom been documented and its applicability in mechanistic studies is unclear. On the other hand, the employment of cell surface markers allows the in vitro isolation of a particular cell population but no single marker has been found to be universally applicable for all cancers and the specific scenario being investigated needs to be noted. In this study, we have employed a series of well-validated in vitro, in vivo and expression assays to demonstrate the association of TIC properties with the ALDHhiCD44hi phenotype in a panel of established lung cancer cell lines as well as PDCL. Transplantation studies using multiple cell lines and PDCL consistently showed tumors derived from ALDHhi/CD44hi cells had the highest tumor induction and growth rate, lowest seeding dose, and shortest latency in immunodeficient mice that could be serially passaged for at least 3 generations. Early passage PDCL which are generally believed to be less adapted to in vitro growth conditions could be serially transplanted through the ALDHhiCD44hi subset but much less readily through the other 3 populations. Compared to CD44 or ALDH single marker, tumorigenicity was enhanced in cells selected by ALDHhiCD44hi combined marker ([16] and Supplementary Table S3). Moreover, the ALDH+CD44+ population and cell spheres formation were inhibited by pharmacological disruption of developmental programs such as the sonic Hedgehog and Notch pathways. The results of these comprehensive assays showed under de novo experimental conditions, tumor cells showing the highest expression of ALDHhiCD44hi combined markers demonstrated enhanced TIC properties. This marker combination has also been shown to associate with TIC potentials in head and neck as well as breast cancers [23, 24].
Interestingly, results of in vitro differentiation studies were consistent with the expected identity of ALDHhiCD44hi population as TIC as it yielded 4 progenies distributed in proportions resembling the parental cell line while the others remained largely as the original phenotype lacking differentiation capacity. However, in the xenografts, tumors derived from all populations including ALDHloCD44lo showed similar ALDH/CD44 profiles (data not shown). This could be due to phenotypic plasticity with conversion of non-TIC into TIC under tumor microenvironment stimulation. Experimentally, the TGFβ/IL6 axis has been proposed to be involved in CSC maintenance. We found that exogenous TGFβ stimulation of HCC827 induced a significant increase of ALDH+CD44+ cells from 3.63% to 9.38% (p < 0.05) (Supplementary Figure S1). TGFβ is shown to be involved in CD44 regulation in breast CSC maintenance while interleukin 6 (IL6) can promote growth and survival of glioma stem cells [25, 26]. These data not only caution against a purely in vitro approach in studying TIC regulation but also indicate disrupting stroma-cancer interactions is important in anti-TIC therapy.
One of the major implications of the CSC theory is that chemotherapy or target therapy effects are mainly incident on the non-TIC thus compromising long term cancer control [27-29]. Our in vitro and in vivo results argue in favor of this hypothesis. The IC50 of gefitinib was almost 8.5 fold higher for TIC than unsorted cells of HCC827, while cisplatin produced higher apoptotic rate in non-TIC than TIC of H1650 (Figure 3). Only subcutaneous xenografts derived from ALDHhiCD44hi progenies persisted after intraperitoneal gefitinib injection. The mechanism mediating the differential drug resistance is likely to be multifactorial. In both animal and human studies, ALDH mediates detoxification and cyto-protection against multiple agents such as aldehydes, alkylating drugs, oxidative stress, etc. On the other hand, through the retinoid signaling pathway, it is involved in self-renewal and tissue differentiation [30, 31]. For CD44, binding to hyaluronan increases NANOG phosphorylation and nuclear translocation, subsequently leading to the upregulation and stabilization of multidrug-resistant protein 1 (MDR1) for drug export [32]. CD44 interaction with POU5F1-SOX2-NANOG signaling also plays a pivotal role in CSC maintenance from head and neck cancers [33]. Together, these date suggest ALDH and CD44 could mediate drug-resistance directly or through stem cell regulatory programs. Indeed, in our study, concomitant CD44 depletion and ALDH inhibition led to sensitization of resistant cells to gefitinib and cisplatin, as well as down-regulation of pluripotency genes NANOG, SOX2 and POU5F1.
Other potential mechanisms enhancing tumorigenicity and drug resistance of the ALDHhiCD44hi TIC include a higher G2/M fraction, elevated cyclin B1 and B2 expression and correspondingly, a higher population growth rate. This population also showed elevated expression of the double strand break DNA repair gene RAD51 which is associated with resistance to radiation, chemotherapeutic agents and tyrosine kinase inhibitor therapy [34-36]. Thus, enhanced DNA damage repair and more rapid cell replenishment could facilitate maintenance of the ALDHhiCD44hi population. Furthermore, TGFβ-dependent IL6 secretion has been shown to contribute to primary and acquired erlotinib resistance in lung cancer patients [37]. Our data showed ALDHhiCD44hi population had higher mRNA expression of the IL6 co-receptor IL6ST, suggesting TGFβ-induced resistance could reside mainly in TIC.
Expression profiling studies have shown enrichment of embryonic or tissue stem cell signatures is associated with increased risk of tumor metastasis, poor morphological differentiation and adverse patient outcomes, raising the possibility patient prognosis might be predicted by an appropriate TIC marker. Previous reports on the relevance of ALDH or CD44 single marker have yielded controversial information. For example, ALDH has been reported both as an adverse and a favorable prognostic indicator [21, 38]. For CD44, our previous study has shown a contradictory association with well-differentiated AD and a longer patient survival in lung adenocarcinomas [16]. In this study, there was no statistical evidence of global ALDH and CD44 co-regulated expressions. Notably, when analysis was limited to tumors containing ALDH/CD44-coexpressing cells, those with a higher abundance of TIC was observed to associate with a shorter recurrence free survival (p = 0.053). The finding suggested ALDH/CD44 co-expression could be a useful prognostic indicator and supported the relevance of these molecules as lung TIC marker. Some tumors lacked expression of either marker, suggesting other lung TIC indicators remain to be described.
We have shown comprehensive in vitro, in vivo data as well as clinical support that the ALDHhiCD44hi compartment carries TIC capabilities and is the cellular mediator of drug resistance in lung cancer. These findings are useful in future studies addressing TIC-related cancer biology and development of TIC-targeting therapy.
Materials and Methods
Cell lines
Established human NSCLC cell lines were obtained from ATCC. PDCL were raised from resected lung cancers or malignant effusions and only 1st to 5th passage cells were used for study. Gefitinib or cisplatin-resistant (-GR or -CR) cells were generated by chronic graded exposure of parental cells to increasing doses of the respective drugs. Cells were maintained in RPMI-1640 (Life Technologies Inc.) or ACL4 with 10% FBS [22].
Flow cytometry and fluorescence activated cell sorting (FACS)
ALDH activity was analyzed by the Aldefluor kit (Stem Cell Technologies) according to manufacturer’s instructions. Briefly, cells were suspended with activated Aldefluor substrate with or without diethylaminobenzaldehyde (DEAB) specific inhibitor as control at 37˚C for 30 minutes. CD44 expression was stained by anti-CD44-APC or anti-CD44-PE (BD Pharmingen) as previously described [16]. Cells were fixed and permeabilized (Invitrogen) before antibody incubation for NANOG (Cell signaling), POU5F1 (Chemicon) and SOX2 (Cell signaling) analyses. Corresponding isotype-matched immunoglobulins were used as controls (BD Pharmingen). Non-viable cells were identified by propidium iodide inclusion. Mouse cells were gated out by Pacific blue-labeled anti-mouse CD31 and lineage and H-2Kd staining (BioLegend). Flow cytometry was performed using FACSCanto II (BD Biosciences) and figures were produced using FlowJo (Tree star). Cells showing ALDH and CD44 co-expression compared to staining controls were designated as ALDH+CD44+, those showing no expression of either marker as ALDH-CD44-, and those showing either marker only as ALDH+CD44- or ALDH-CD44+, respectively. For comparative studies between different ALDH/CD44 populations, cells showing the top/bottom 1 to 5% expression levels were freshly isolated by FACS using BD Aria (BD Biosciences) and designated accordingly as ALDHhiCD44hi, ALDHhiCD44lo, ALDHloCD44hi and ALDHloCD44lo populations. Each batch of cells was re-analyzed after collection to ensure a purity of > 90%.
Sphere formation assay
One thousand freshly isolated cells were cultured in an ultra-low plate (Costar) with serum-free medium containing FGF, EGF and IGF for 21 days as previously described [16].
Cell invasion assay
Cell invasion assay was performed using transwell® (Costar) coated with matrigel (BD Pharmingen) according to manufacturer’s instructions.
Cell cycle analysis
Freshly sorted cells were fixed in cold 70% ethanol, incubated in PBS buffer containing 50 µg/mL propidium iodide, 100 µg/mL RNAse A and 0.05% Triton X-100 at 37˚C for 45 minutes and analyzed using flow cytometry. The proportion of cells in G0/G1, S and G2/M was calculated by FlowJo (Tree star).
Drug sensitivity and apoptosis assays
Drug sensitivity assay was performed by MTT test and apoptosis was quantified by Annexin V-FITC and PI staining as previously described [16].
Gene expression analysis
Gene mRNA expression was analyzed by quantitative RT-PCR (QPCR) (7900HT, Applied Biosystems) and SYBR green detection. Expressions of GAPDH and beta-2-microglobulin (B2M) were averaged and used as internal controls. Primers were listed in Supplementary Table S5.
In vivo tumorigenicity and TKI response
All animal experiments were approved and performed according to guidelines by the Animal Ethics Committee, the University of Hong Kong. Briefly, freshly sorted cells mixed with an equal volume of matrigel (BD Pharmingen) were injected subcutaneously at the back of 6-week severe combined immunodeficiency (SCID) mice. For serial transplantation, the previous generation xenografts derived from the respective ALDH/CD44 subsets were digested to obtain single cell suspensions, stained, subjected to FACS to isolate the parental subset and transplanted into subsequent recipients. To investigate the in vivo TKI response, mice bearing xenografts from HCC827 were subjected to intraperitoneal gefitinib (Selleck) injections at 50 mg/kg in 0.2 mL of 1% Tween 80 five times per week for 4-5 weeks [39, 40]. Gefitinib was dissolved in DMSO; a mixture of 1% Tween 80 with an equal volume of DMSO was used as vehicle control.
CD44 knockdown, ALDH, Hedgehog and Notch pathways inhibition
Small interfering RNA (siRNA) targeting human CD44 and non-targeting siRNA (Qiagen) were used for CD44 knockdown. Transfection was performed using Genemute (SignaGen Laboratories) according to the manufacturer’s instructions. DEAB (Sigma) was constituted in 100% ethanol and diluted in media to 100 µM. The ALDEFLUOR Assay was used in conjunction with flow cytometry to assess ALDH activity following DEAB treatment. Cyclopamine (Selleck) or RO4929097 (Selleck) was used for inhibition of Hedgehog or Notch pathway, respectively.
Immunohistochemistry (IHC) for ALDH and CD44 expression in clinical lung cancers
Formalin-fixed paraffin-embedded tumor tissues from 193 surgically resected primary lung carcinomas without neoadjuvent therapy were arrayed on tissue micro-array (TMA) with each case represented by 3 to 5 tumor cores. Tissues from fetal lung, reactive pulmonary lesions and a representative lung cancer were included in each TMA block for standardization. IHC for CD44 and ALDH were performed separately using anti-CD44 standard form (clone 156-3C11, 1:200 dilution, BiocareMedical) or anti-ALDH1A1 (1:1000 dilution, Abcam) as previously described [16]. Primary antibodies were replaced by SignalStain® Antibody Diluent (Cell Signaling Technology) in the negative controls. Expression levels were semi-quantitatively analyzed using an automated image capturing and analysis system (Aperio) [16]. Briefly, representative tumor areas in each tissue core excluding stroma, necrosis or inflammatory regions were gated on the scanned images. The staining intensities and cell abundance in each annotated area were computed using standard algorithms. Data from all tissue cores were integrated and a single score reflecting the abundance of ALDH or CD44-expressing cells, respectively, was assigned to each case. For ALDH/CD44-coexpresssion grading, only co-localized tumor cells with simultaneous ALDH and CD44 staining were counted. Tumor typing and pathological staging were according to W.H.O. criteria [41]. Patient outcomes were collated by the clinician in-charge.
Statistical methods
Differences between groups were analyzed by student’s t test for continuous variables. Correlation between clinicopathological variables and ALDH or CD44 expression were analyzed by χ2 test or Fisher Exact tests. Differences in patient survival were analyzed by log rank test and survival curves were drawn by Kaplan Meier method. Independent prognostic indicators for survival were analyzed by the Cox regression model. All statistical tests were analyzed by SPSS 18 for Windows (SPSS Inc., Chicago, IL). A 2-tailed p value of < 0.05 was considered as the threshold for statistical significance.
Acknowledgement
The study was supported by the Hong Kong Research Grants Council General Research Fund HKU 783011/M, and the University of Hong Kong research grant 201209176184. The authors would like to thank LKS Faculty of Medicine at The University of Hong Kong for the Faculty Core Facility.
Disclosure of Potential Conflicts of Interest
The authors disclose no potential conflicts of interest.
References
1. Pao W and Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010; 10(11):760-774.
2. Tam IY, Chung LP, Suen WS, Wang E, Wong MC, Ho KK, Lam WK, Chiu SW, Girard L, Minna JD, Gazdar AF and Wong MP. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006; 12(5):1647-1653.
3. Wong DW, Leung EL, So KK, Tam IY, Sihoe AD, Cheng LC, Ho KK, Au JS, Chung LP, Pik Wong M and University of Hong Kong Lung Cancer Study G. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009; 115(8):1723-1733.
4. Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, Jessop NA, Wain JC, Yeo AT, Benes C, Drew L, Saeh JC, Crosby K, Sequist LV, Iafrate AJ and Engelman JA. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Science translational medicine. 2012; 4(120):120ra117.
5. Dienstmann R, Martinez P and Felip E. Personalizing therapy with targeted agents in non-small cell lung cancer. Oncotarget. 2011; 2(3):165-177.
6. Visvader JE and Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012; 10(6):717-728.
7. Magee JA, Piskounova E and Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012; 21(3):283-296.
8. Takebe N, Harris PJ, Warren RQ and Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nature reviews Clinical oncology. 2011; 8(2):97-106.
9. Garcia Campelo MR, Alonso Curbera G, Aparicio Gallego G, Grande Pulido E and Anton Aparicio LM. Stem cell and lung cancer development: blaming the Wnt, Hh and Notch signalling pathway. Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico. 2011; 13(2):77-83.
10. Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP and Wicha MS. Notch Pathway Activity Identifies Cells with Cancer Stem Cell-like Properties and Correlates with Worse Survival in Lung Adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013; 19(8):1972-1980.
11. Pacheco-Pinedo EC and Morrisey EE. Wnt and Kras signaling-dark siblings in lung cancer. Oncotarget. 2011; 2(7):569-574.
12. Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, Bhakoo KK, Jayapal SR, Nichane M, Yu Q, Ahmed DA, Tan C, et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012; 148(1-2):259-272.
13. Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S, Roz E, Caserini R, Lo Vullo S, Camerini T, Mariani L, Delia D, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106(38):16281-16286.
14. Jung MJ, Rho JK, Kim YM, Jung JE, Jin YB, Ko YG, Lee JS, Lee SJ, Lee JC and Park MJ. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene. 2013; 32(2):209-221.
15. Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, Honorio S, Xie Y, Scaglioni PP, DiMaio JM, Gazdar AF, Shay JW, et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer research. 2010; 70(23):9937-9948.
16. Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, Fink LM, Ma Y and Wong MP. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PloS one. 2010; 5(11):e14062.
17. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ and Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100(7):3983-3988.
18. Kimura Y, Goi T, Nakazawa T, Hirono Y, Katayama K, Urano T and Yamaguchi A. CD44variant exon 9 plays an important role in colon cancer initiating cells. Oncotarget. 2013; 4(5):785-791.
19. Takaishi S, Okumura T, Tu S, Wang SS, Shibata W, Vigneshwaran R, Gordon SA, Shimada Y and Wang TC. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem cells (Dayton, Ohio). 2009; 27(5):1006-1020.
20. Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, Wang de Y, Lim B, Chow VT, Crum CP, Xian W and McKeon F. Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell. 2011; 147(3):525-538.
21. Jiang F, Qiu Q, Khanna A, Todd NW, Deepak J, Xing L, Wang H, Liu Z, Su Y, Stass SA and Katz RL. Aldehyde dehydrogenase 1 is a tumor stem cell-associated marker in lung cancer. Mol Cancer Res. 2009; 7(3):330-338.
22. Lam DC, Girard L, Suen WS, Chung LP, Tin VP, Lam WK, Minna JD and Wong MP. Establishment and expression profiling of new lung cancer cell lines from Chinese smokers and lifetime never-smokers. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2006; 1(9):932-942.
23. Croker AK, Goodale D, Chu J, Postenka C, Hedley BD, Hess DA and Allan AL. High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. Journal of cellular and molecular medicine. 2009; 13(8B):2236-2252.
24. Krishnamurthy S, Dong Z, Vodopyanov D, Imai A, Helman JI, Prince ME, Wicha MS and Nor JE. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer research. 2010; 70(23):9969-9978.
25. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J and Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133(4):704-715.
26. Wang H, Lathia JD, Wu Q, Wang J, Li Z, Heddleston JM, Eyler CE, Elderbroom J, Gallagher J, Schuschu J, MacSwords J, Cao Y, McLendon RE, Wang XF, Hjelmeland AB and Rich JN. Targeting interleukin 6 signaling suppresses glioma stem cell survival and tumor growth. Stem cells (Dayton, Ohio). 2009; 27(10):2393-2404.
27. Dean M, Fojo T and Bates S. Tumour stem cells and drug resistance. Nature reviews Cancer. 2005; 5(4):275-284.
28. Singh A and Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010; 29(34):4741-4751.
29. Cufi S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, Martin-Castillo B and Menendez JA. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget. 2012; 3(4):395-398.
30. Sladek NE. Human aldehyde dehydrogenases: potential pathological, pharmacological, and toxicological impact. J Biochem Mol Toxicol. 2003; 17(1):7-23.
31. Ma I and Allan AL. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2011; 7(2):292-306.
32. Bourguignon LY, Spevak CC, Wong G, Xia W and Gilad E. Hyaluronan-CD44 interaction with protein kinase C(epsilon) promotes oncogenic signaling by the stem cell marker Nanog and the Production of microRNA-21, leading to down-regulation of the tumor suppressor protein PDCD4, anti-apoptosis, and chemotherapy resistance in breast tumor cells. The Journal of biological chemistry. 2009; 284(39):26533-26546.
33. Bourguignon LY, Wong G, Earle C and Chen L. Hyaluronan-CD44v3 interaction with Oct4-Sox2-Nanog promotes miR-302 expression leading to self-renewal, clonal formation, and cisplatin resistance in cancer stem cells from head and neck squamous cell carcinoma. The Journal of biological chemistry. 2012; 287(39):32800-32824.
34. Bello VE, Aloyz RS, Christodoulopoulos G and Panasci LC. Homologous recombinational repair vis-a-vis chlorambucil resistance in chronic lymphocytic leukemia. Biochemical pharmacology. 2002; 63(9):1585-1588.
35. Slupianek A, Hoser G, Majsterek I, Bronisz A, Malecki M, Blasiak J, Fishel R and Skorski T. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Mol Cell Biol. 2002; 22(12):4189-4201.
36. Ko JC, Ciou SC, Jhan JY, Cheng CM, Su YJ, Chuang SM, Lin ST, Chang CC and Lin YW. Roles of MKK1/2-ERK1/2 and phosphoinositide 3-kinase-AKT signaling pathways in erlotinib-induced Rad51 suppression and cytotoxicity in human non-small cell lung cancer cells. Mol Cancer Res. 2009; 7(8):1378-1389.
37. Yao Z, Fenoglio S, Gao DC, Camiolo M, Stiles B, Lindsted T, Schlederer M, Johns C, Altorki N, Mittal V, Kenner L and Sordella R. TGF-beta IL-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(35):15535-15540.
38. Dimou A, Neumeister V, Agarwal S, Anagnostou V, Syrigos K and Rimm DL. Measurement of aldehyde dehydrogenase 1 expression defines a group with better prognosis in patients with non-small cell lung cancer. The American journal of pathology. 2012; 181(4):1436-1442.
39. Yan Y, Lu Y, Wang M, Vikis H, Yao R, Wang Y, Lubet RA and You M. Effect of an epidermal growth factor receptor inhibitor in mouse models of lung cancer. Mol Cancer Res. 2006; 4(12):971-981.
40. Helfrich BA, Raben D, Varella-Garcia M, Gustafson D, Chan DC, Bemis L, Coldren C, Baron A, Zeng C, Franklin WA, Hirsch FR, Gazdar A, Minna J and Bunn PA, Jr. Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006; 12(23):7117-7125.
41. Travis W, Brambilla, E., Muller-Hermelink, H. and Harris, C. World Health Organization Classification of Tumors: Pathology and Genetics of tumors of the Lung, Pleura, Thymus and Heart. 2004.