
INTRODUCTION
Elevated mitogen-activated protein kinase (MAPK) signaling is common in adult and pediatric gliomas. Among the gene alterations that promote increased MAPK signaling is a single nucleotide substitution in BRAF kinase exon 15, which results in expression of the constitutively-active BRAFV600E mutant kinase [1]. For pediatric glioma, mutation screening studies have revealed that BRAFV600E frequencies are highest in grade II and III pleomorphic xanthoastrocytoma (PXA) (60-66%) [2]. BRAFV600E has also been detected in 18-50% of ganglioglioma [3, 4], 5-33% of grade II-IV malignant astrocytoma [4–7], and 9% of pilocytic astrocytoma [4, 5]. We previously reported that five out of seven (71%) of BRAFV600E mutant pediatric grade II-IV astrocytoma have homozygous deletion of CDKN2A, which encodes the p16 tumor suppressor [6]. A subsequent study reported that the BRAFV600E mutation and CDKN2A deletion are more frequent in pediatric low-grade glioma that transform to high-grade malignancy than in non-transforming tumors [8]. BRAFV600E is less common in adult glioma, in which MAPK signaling is most often stimulated by gene amplification and mutation of upstream receptor tyrosine kinases [9, 10]. Cumulatively, the results of mutation screening studies indicate that BRAFV600E is important to multiple pediatric glioma types, and suggest that this oncogenic alteration cooperates with CDKN2A deletion to promote neoplastic transformation and tumor malignant progression.
In a previous study, we modeled the combined effects of BRAFV600E expression and CDKN2A deficiency by crossing the cre-conditional BrafCA [11] and hGFAP-cre [12] mice to mice lacking Ink4a-Arf [13], a locus that contains the murine homolog of CDKN2A. Triple transgenic mice expressed BrafV600E in Gfap+ cells under control of the endogenous Braf promoter, and lacked Cdkn2a expression [14]. These mice died prior to developing tumors but cells isolated from the ganglionic eminence of BrafCA Ink4a-Arf knock-out mice and infected with adenovirus expressing cre recombinase (Ad-cre) in culture, became tumorigenic upon intracranial injection into SCID mice. We also observed intracranial tumor formation by inducing BrafV600E expression and Cdkn2a deficiency through injection of Ad-cre into the subventricular zone (SVZ) of the lateral ventricle of BrafCA mice bred with a cre-conditional knock-out allele of Ink4a-Arf (Ink4a-ArfLoxP/LoxP (fl/fl)) [14].
Results from the use of BrafV600E Ink4a-Arf knock-out murine allografts and BRAFV600E + CDKN2A-deficient human glioma xenografts demonstrated the anti-tumor activity of PLX4720 [14, 15], a tool compound of the FDA-approved BRAFV600E-inhibitor vemurafenib. These studies helped motivate an active clinical trial for assessing vemurafenib in treating children with recurrent BRAFV600E glioma (ClinicalTrials.gov Identifier NCT01748149). There are early indications that this personalized approach benefits some patients with BRAFV600E positive ganglioglioma [16, 17], recurrent PXA [18] and recurrent glioblastoma [19]. Moreover, patients with relapsed or refractory high-grade and low-grade BRAFV600E glioma have shown radiographic response to treatment with BRAFV600E inhibitor dabrafenib in a phase 1 clinical trial. In some cases, however, tumors showed progression despite dabrafenib treatment, suggesting that some glioma have inherent, primary resistance to BRAFV600E targeted therapy [20]. The observation of progressive tumor growth during treatment is consistent with our more recent preclinical studies that showed no significant impact on survival rates from PLX4720 monotherapy when treating mice with distinct BRAFV600E mutant and CDKN2A deficient tumors models (intracranial xenografts from pilocytic astrocytoma [21] and glioblastoma [22]).
Here, we present results from the characterization and therapeutic testing of a newly developed BrafV600E-expressing Cdkn2a deficient glioma model, the first to involve the use of BrafV600E glioma cells in a syngeneic, immunocompetent host. Our study examines the relative anti-tumor activity of BRAFV600E vs. MEK targeted monotherapy, and of combination therapy using the same inhibitors. Compared with the effects of either inhibitor alone, combination therapy significantly decreased Ki67 positivity, reduced bioluminescence signaling, and conferred the most substantial survival benefit to animal subjects with lentivirus-luciferase modified, BrafV600E expressing Ink4a-Arf knock-out murine allografts. Our results demonstrate the utility of this model for testing small molecule inhibitors, and should as well, prove useful for testing therapies for modulating immune response against BRAFV600E mutant glioma.
RESULTS
BrafV600E + Ink4a-Arf deficient 2341luc cells produce intracranial tumors in FVB/N mice with features characteristic of high-grade glioma
To establish a tumor-derived glioma cell line carrying the BrafV600E mutation and deficient for Cdkn2a, we injected adenovirus expressing cre recombinase (Ad-cre) into the corpus callosum of ten week-old, cre-conditional, FVB/N BrafCA/+ Ink4a/ArfLoxP/LoxP (fl/fl)transgenic mice (Figure 1A). Mice consistently developed tumors by eight weeks post-injection (Figure 1B). Tumor isolated from one such animal (animal number 2341) was used in preparing an explant culture that was subsequently expanded and modified with firefly luciferase-encoding lentivirus (2341luc; Figure 1A). PCR analysis of DNA extracted from these cells showed that the BrafV600E transgene was expressed (Figure 1C). Deletion of Ink4a-Arf, which encodes p16Ink4a and p19Arf, was confirmed by quantitative PCR for p16Ink4a (Figure 1D).
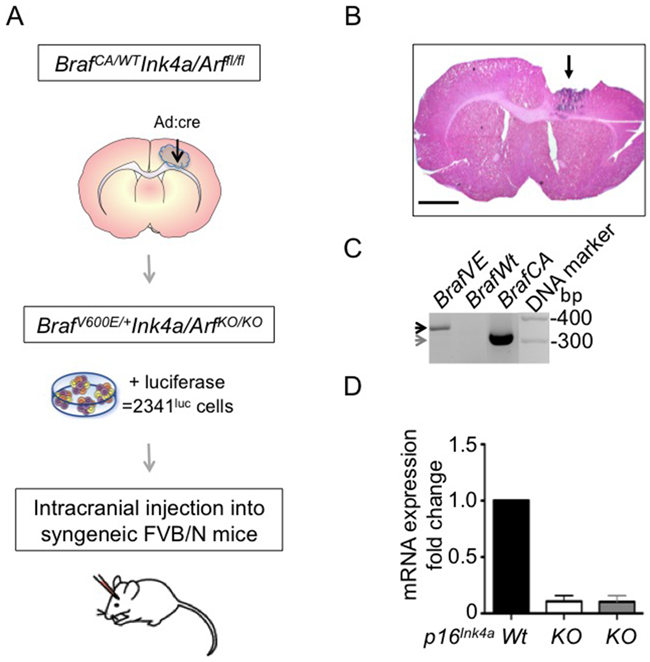
Figure 1: BrafV600E mutant Ink4a-Arf deleted tumor-derived cells form syngeneic grafts. A. Schematic of 2341luc model development. Cells were isolated from a tumor developed in an 18-week-old BrafCA/+ Ink4a/ArfLoxP/LoxP (fl/fl) mouse (animal number 2341) that had received adenovirus-cre (Ad:cre) virus injection in the corpus callosum at ten weeks of age. Tumor cells were subsequently modified with lentivirus expressing luciferase (2341luc), for injection into syngeneic FVB/N mice. B. Hematoxylin and Eosin staining of a tumor developed by injection of Ad:cre as described in A. Arrow points to the location of the tumor and parts of the tumor were excised for cell culturing. Scale bar is 1000 μm. C. PCR detection of mutant BrafV600E and BrafCA alleles. Specific primers were used to distinguish between the 308 bp band for cre-conditional BrafCA (grey arrow) and the 335 bp band for BrafV600E (black arrow). Panel depicts 2341luc cells, which express BrafV600E (BrafVE), control subventricular zone-derived cells with the wild type Braf (BrafWt) or the BrafCA (BrafCA) allele. D. QPCR detection of p16Ink4a. Reduced expression of p16Ink4a mRNA was detected by real-time PCR in 2341luc cells (KO; white bar), and subventricular zone-derived neurosphere cells homozygous deleted for Ink4a-Arf (KO; grey bar). The mRNA expression levels are normalized to expression levels in subventricular zone-derived wild type (Wt) cells, used as control.
To determine whether the 2341 cell line has acquired additional genetic changes in selected oncogenes, and in particular during modification with luciferase lentivirus, we analyzed 2341 cells at initial passage, and following lentiviral modification and expansion. Targeted sequencing of known oncogenic driver genes, including Akt3, Kras, Map2K1, Pik3ca, Pik3r2, Pten, H3f3a and H1h3a showed no alteration from wild-type sequence for these genes. This result supports earlier observations that expression of BrafV600E and deletion of Cdkn2a are sufficient for neoplastic transformation (Supplementary Figure S1) [14].
Serial bioluminescence imaging (BLI) of 2341luc cells, following intracranial injection of 30,000 cells in immunodeficient, Nod scid gamma (NSG) mice, as well as in immune competent, syngeneic FVB/N mice, revealed progressive increase in luminescence in all injected mice. The increase in luminescence is more rapid in the NSG animals compared with the FVB/N animals (Figure 2A). Median symptom-free survival of mice for NSG and FVB/N mice was 45 and 58.5 days, respectively (p=0.01; Figure 2B).
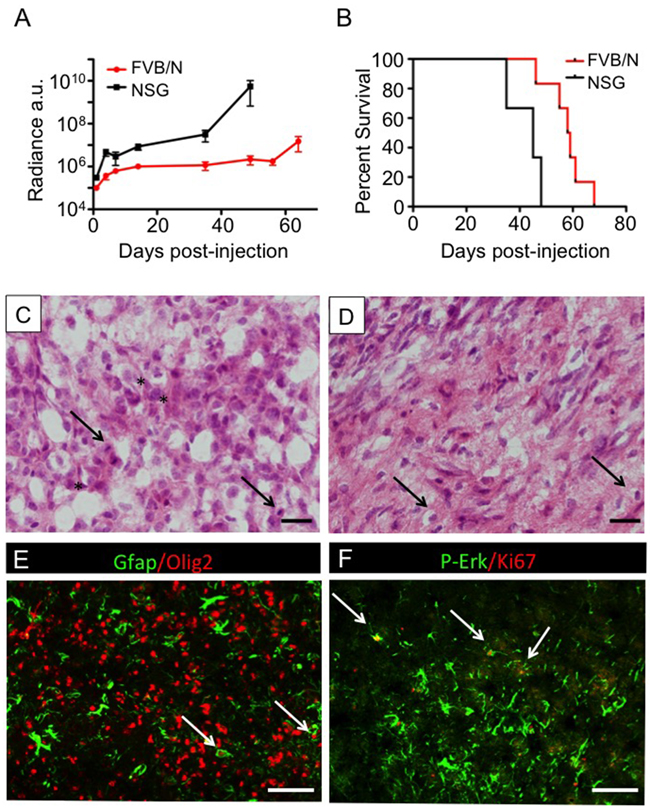
Figure 2: 2341luc syngeneic grafts showed characteristics of high-grade glioma. A. Bioluminescence imaging (BLI) growth plots of intracranial tumors established from 2341luc cells in syngeneic, immunecompetent FVB/N and immunecompromised Nod scid gamma (NSG) mice, respectively. Values are displayed in arbitrary units (a.u.). The linear plots of log base 10 values indicated that exponential increase of bioluminescence was achieved by all tumors, though the rate of increase was slower in FVB/N mice. Standard deviations are indicated. B. Kaplan-Meier survival plots for 12 NSG and 10 FVB/N mice, respectively, following intracranial injection of 30,000 2341luc cells per animal. The median survival post-injection is 58.5 days for FVB/N mice and 45 days for NSG mice (*=p=0.010). C, D. H&E-stained tumor tissues showed histopathological features of high-grade glioma, including mitotic figures (C; arrows), pleomorphic and hyperchromatic nuclei (C; asterisks) and invading tumor cells (D; arrows). E, F. Immunofluorescence of 2341luc syngeneic grafts for astrocyte marker Gfap (green), oligodendrocyte lineage and glioma cell marker Olig2 (red) (E) phospho-Erk (p-Erk; green) and proliferation antigen Ki67 (red), is indicative of a high-grade astrocytic tumor (F) White arrows point to double-positive cells. Scale bars are 20 μm in C, D and 200 μm in E, F.
H&E staining of sections from resected brains showed tumor that was highly cellular, infiltrative, and pleomorphic (Figures 2C and 2D). Tumors expressed the astrocyte marker Gfap, oligodendrocyte lineage and glioma marker Olig2, proliferative marker Ki67, and phosphorylated extracellular kinase (phospho-Erk), indicative of active MAPK pathway signaling (Figures 2E and 2F).
BrafV600E-expressing Ink4a-Arf knock-out 2341luc tumor cells become unresponsive to BRAFV600E-targeted inhibition
We compared the effects of treating 2341luc cells, in vitro, with vemurafenib, which continues to see extensive use in treating BRAFV600E mutant cancer, as well as with a second FDA-approved inhibitor of BRAFV600E, dabrafenib (Db; GSK2118436). Results from inhibitor treatment indicated IC50 values of 11.92 nM for Db and 25.61 nM for vemurafenib (Figure 3A). A human BRAF wild-type pediatric glioma cell line (SF188) exhibited no significant response to Db up to concentrations as high as 0.5 µM. Vemurafenib concentrations as high as 10 µM only reduced cell viability by 40% (Supplementary Figure S2). The latter result suggests off-target effects of vemurafenib that have been reported by others [27].
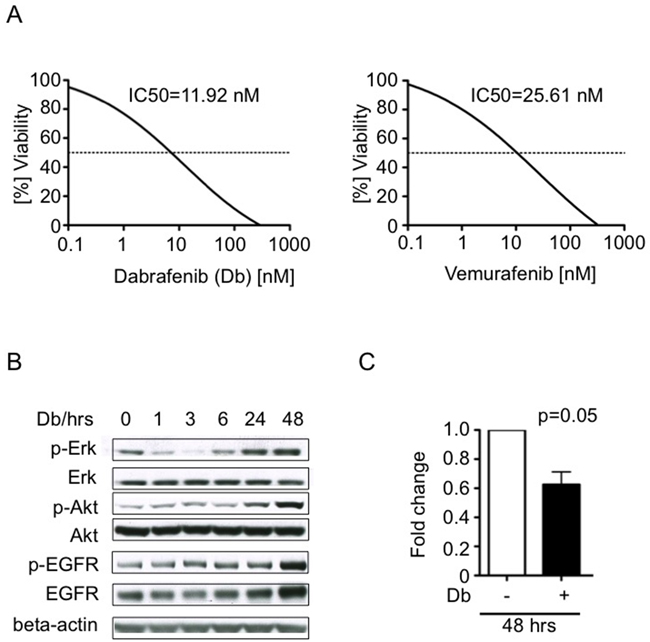
Figure 3: Tumor cell response to BRAFV600E inhibitors dabrafenib and vemurafenib in vitro. A. Dose-response-curves showing in vitro viability effects of increasing concentrations of dabrafenib (Db; left panel) and vemurafenib (right panel) on 2341luc cells (n= 3). B. EGFR, Akt and Erk activities increase in 2341luc cells during treatment with BRAFV600E inhibitor Db. Cells were treated with Db at IC50 concentration for up to 48 hours (hrs) and protein was extracted following a 20 min stimulation with EGF (25 ng/ml), and analyzed by immunoblotting for phospho-Erk (p-Erk), total Erk (Erk), phospho-Akt (p-Akt), total Akt (Akt), phospho-EGFR (p-EGFR) and total EGFR (EGFR). After an initial decrease, p-ERK signal increased from six to 48 hrs, p-Akt signal increased from 24 to 48 hrs and p-EGFR as well as EGFR increased at 48 hrs, beyond phosphorylation levels detected before treatment was started. Beta-actin was detected as loading control. C. Short-term Db treatment inhibits 2341luc cell proliferation. Cells were treated with IC50 concentrations of Db for 48 hrs, incubated with Hoechst dye, and analyzed for DNA content (HO>2), indicative of actively proliferating cells, by flow cytometry. The graph shows fold change in proliferative cells (p=0.05).
We further investigated the 2341luc cell response to BRAFV600E inhibitor Db by examining phospho-Erk (p-Erk) levels, a commonly used surrogate of MAPK pathway activity. Immunoblot results showed p-Erk levels decreasing over the first six hours of exposure to dabrafenib, with increases evident beyond 24 hours, and that ultimately exceeded the phosphorylation levels of untreated cells (Figure 3B). Based on our previous work [21] as well as the published work of others [28–31] (Supplementary Figure S3), we analyzed EGFR and PI3k-Akt activity in Db-treated 2341luc cells, as potential mediators of MAPK signaling rebound. Immunoblot results showed increased phospho-Akt (p-Akt) from 24 to 48 hours, and increased phospho-EGFR (p-EGFR) as well as total EGFR at 48 hours (Figure 3B).
Db treatment was next examined for anti-proliferative activity. For this, cells were treated for 48 hours at Db IC50 concentrations, and then analyzed for DNA content by flow cytometry. Db treated cell samples showed significantly reduced DNA content relative to untreated cells, indicative of reduced proliferation (Figure 3C).
Combination therapy is more effective than trametinib or dabrafenib monotherapy in preventing MAPK activation and inhibiting tumor cell proliferation
Clinical results using Db in combination with the MEK inhibitor trametinib (Tr) to treat melanoma showed improved progression-free survival, compared to either inhibitor used as monotherapy [32]. To evaluate the potential of this combination therapy for treating BRAFV600E high-grade glioma, we first compared single vs. dual agent effects on 2341luc cell cultures. After determining the IC50 value for Tr (IC50 = 30.5 nM) in 2341luc cells (Supplementary Figure S4), we found this concentration to be effective at decreasing p-Erk levels in EGF stimulated cells (25 ng/ml). This suppressive effect was further augmented by combining Tr with Db (Figure 4A). Flow cytometry for p-Erk using 2341luc cells, treated for five days with IC50 concentrations of both agents, showed that Tr but not Db significantly decreased p-Erk-positive cell frequency, with combination treatment resulting in further decrease (p<0.001 for Tr versus Db + Tr; Figure 4B).
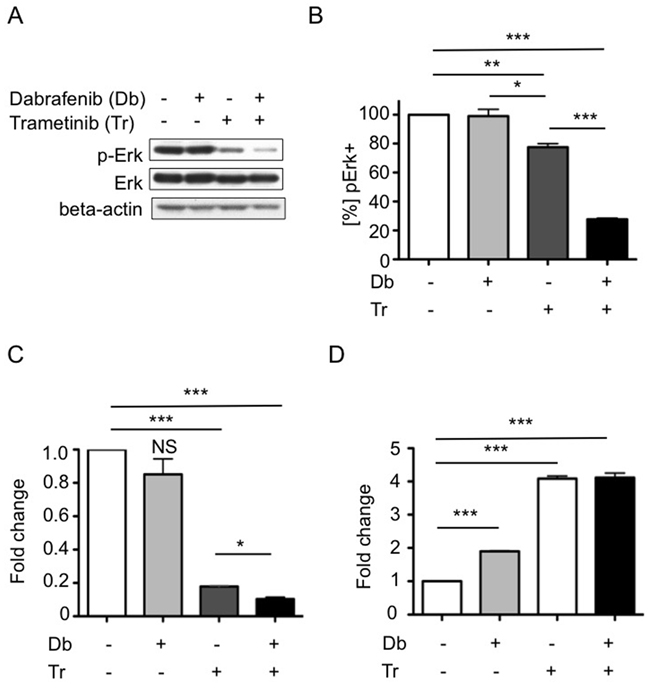
Figure 4: Combined BRAFV600E and MEK inhibition effects on MAPK pathway activity, tumor cell growth and apoptosis. A, B. Combined BRAFV600E and MEK inhibition inhibits p-Erk levels more effectivey than MEK inhibition alone. (A) 2341luc cells treated with IC50 doses of dabrafenib (Db), trametinib (Tr) or a combination of both (Db+Tr) for four days and stimulated with EGF (25 ng/ml) for 20 min before protein extraction were analyzed by immunoblotting for p-Erk and total Erk (Erk). Beta-actin was detected as loading control. P-ERK was suppressed by Tr and further suppressed by combination treatment. (B) Flow cytometric quantification of p-Erk levels in 2341luc cells treated with Db, Tr or a combination of both (Db+Tr) for five days. Db did not alter p-Erk levels. Tr significantly decreased p-Erk-positive cell frequency (**=p=0.0060 for control versus Tr) and combined Db and Tr treatment resulting in further decrease (***=p<0.0001 for Tr versus Db + Tr and for control versus Db + Tr; *=p=0.0412 for Db versus Tr). Graph shows percent p-ERK-positive ([%] pERK+) cells normalized to control-treated cells = 100%. C. Combination treatment reduces 2341luc cell cycling more effectively than single agent treatment. Cells were treated with IC50 concentrations of Db, Tr or a combination of both (Db+Tr) for four days, incubated with Hoechst dye, and analyzed for DNA content (HO>2) by flow cytometry. Graph shows fold change in the frequency of cells with a DNA content of HO>2. Tr but not Db decreased proliferation significantly (***=p=0.0001 for Tr versus control) and this effect was enhanced by combination treatment (***=p<0.0001 for Db+Tr versus control; *=p=0.0149 for Db+Tr versus Tr) (n=3). Graph depicts the fold change of frequency of proliferative cells normalized to control-treated cells = 1. D. Single agent and combination treatment increases apoptosis. Cells were treated as in C. and frequency of annexin V-positive cells was determined as surrogate for apoptotic cells. Single drug Db and Tr treatment and, more effectively, a combination of both significantly increased apoptosis (***=p<0.0001 for Db versus control and Tr versus control; ***=p=0.0002 for Db+Tr versus control). Graph depicts fold change of annexin V-positive cell frequency normalized to control-treated cells = 1.
Single and dual agent treatments were also examined for anti-proliferative and pro-apoptotic activities. Cells treated for 4 days with IC50 concentrations of Db, Tr or a combination, were analyzed by flow cytometry for DNA content of HO>2, which is indicative for proliferative cells. Db had no significant effect on proliferation. In contrast, Tr significantly decreased proliferation (p=0.0001 for Tr versus control) and, as above, this effect was enhanced by adding Db (p=0.0149 for Db + Tr versus Tr; Figure 4C). In parallel, cells were analyzed for apoptosis by Annexin V staining, with results showing that all treatments significantly increased apoptotic cell fractions, with the most substantial increases observed for Tr only and combination treatments (Figure 4D).
Combination therapy effectively prevents MAPK activation, proliferation and increases apoptosis in orthotopic tumors
We next examined the effects of treating FVB/N mice, bearing syngeneic, orthotopic 2341luc tumors, using Db and Tr mono- and combination therapies. Brains with tumor, resected at the end of 14-days treatment, were stained for p-Erk (Figures 5A and 5B). Single agent Db increased p-Erk-positive (p-Erk+) tumor cell frequency (p=0.075), while Tr monotherapy had insignificant effect, relative to control (p=0.31). Consistent with the in vitro data, Tr in combination with Db significantly decreased p-Erk+ cell frequency (p=0.004 for Db+Tr versus control). The combination effect was significant in comparison with each monotherapy as well (p=0.0007 for Db+Tr versus Tr; p<0.0001 for Db+Tr versus Db; Figure 5B). We compared differences in p-Akt between treated groups at two timepoints during treatment. We observed an increase of p-Akt levels after three days of Db treatment (Supplementary Figure S5), consistent with the increase within 48 hrs of Db treatment in vitro (Figure 3B). P-Akt levels increased further within 14 days of Db treatment, in association with adaptive resistance. Combined Db and Tr therapy prevented the increase of p-Akt levels (Supplementary Figures 5A and 5B).
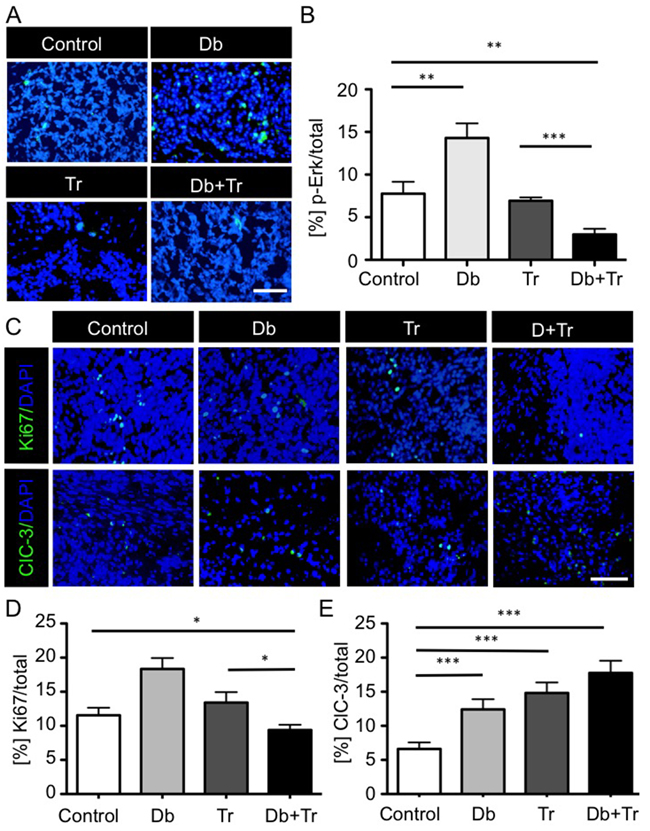
Figure 5: BRAFV600E and MEK inhibitor mono- and combination therapy effects on MAPK pathway activity, proliferation and apoptosis in syngeneic 2341luc grafts. A, B. Combined BRAFV600E and MEK inhibition inhibits p-Erk levels more effectively than MEK inhibition alone. A. Immunofluorescence for phospho-Erk (p-Erk) in 2341luc syngeneic grafts treated with dimethylsulfoxide (Control), dabrafenib (Db), trametinib (Tr), or a combination of both (Db+Tr), for 14 days starting at day 30 post-implantation (n=8/group). P-Erk staining is in green. DNA is stained with DAPI and is in blue. Scale bars are 20 μm. B. Quantification of p-Erk+ tumor cells in 2341luc syngeneic grafts for each treatment group. P-Erk+ cell frequency was determined using Image J. Db monotherapy increased p-Erk+ cell frequency (**=p=0.0075 for Db versus control). Tr monotherapy decreased pErk+ cell frequency, compared with Db monotherapy (**p<0.0023 for Tr versus Db), but not with control (p=0.31). Tr in combination with Db significantly decreased p-Erk + cell frequency (**=p=0.004 for Db+Tr versus control; ***=p=0.0007 for Db+Tr versus Tr; ***=p<0.0001 for Db+Tr versus Db). C–E. Effects of 14-days consecutive inhibitor treatment on proliferation and apoptosis. C. Immunofluorescence images of Ki67 (top panels) and cleaved caspase-3 (ClC-3; bottom panels) stained tumor tissue from 2341luc syngeneic grafts at day 14 of treatment (n=8/group). Ki67 and ClC-3 are in green. DNA was stained with DAPI and is shown in blue. Scale bar is 20 μm. D, E. Quantification of Ki67+ and ClC-3+ cells in tumors from each treatment group. D. Db or Tr monotherapy did not significantly change Ki67+ Db or Tr monotherapy did not significantly change Ki67 positivity. Combination treatment significantly decreased Ki67+ cell frequency (*=p=0.0339 for Db+Tr versus control; *=p=0.017 for Db+Tr versus Tr). E. Each treatment significantly increased apoptosis compared with control (***=p=0.0008 for Db versus control; ***=p<0.0001 for Db+Tr versus control; ***=p<0.0001 for Db+Tr versus Tr).
We also examined treatment effects on tumor cell proliferation and apoptosis by staining for Ki67 and cleaved caspase-3, respectively. Whereas Db monotherapy significantly increased Ki67 positivity and Tr monotherapy showed no significant effect in relation to control, combination therapy significantly decreased Ki67 positive cells (p=0.0339 for Db+Tr versus control; p=0.017 for Db+Tr versus Tr; Figures 5C and 5D). As treatment effects on apoptosis were concerned, all therapies significantly increased cleaved caspase-3 positivity, with the most substantial increase observed for combined inhibitor treatment (Figures 5C and 5E).
Combination therapy effectively inhibits tumor cell growth and extends survival
Effects of treatment on lentivirus-luciferase modified, syngeneic, orthotopic 2341luc tumors were assessed by serial bioluminescence imaging (BLI) of mice treated with mono- or combination therapy. The results showed that Db and Tr monotherapy reduced BLI for three and seven days, respectively, following treatment initiation. Beyond these time-points, BLI values increased, despite continuously administrating inhibitor daily. In contrast, combination therapy inhibited BLI over the entire period of treatment (14 days; Figure 6A). Consistent with the changes in BLI, Db monotherapy provided no survival benefit (median survival control = 46 days versus median survival Db = 42 days: p = 0.253). Tr monotherapy increased survival significantly in relation to control group mice (median survival= 51 days; p=0.044 for Tr versus control), and combination therapy further extended survival (median survival = 70 days; p=0.049 for Db + Tr versus control; Figure 6B).
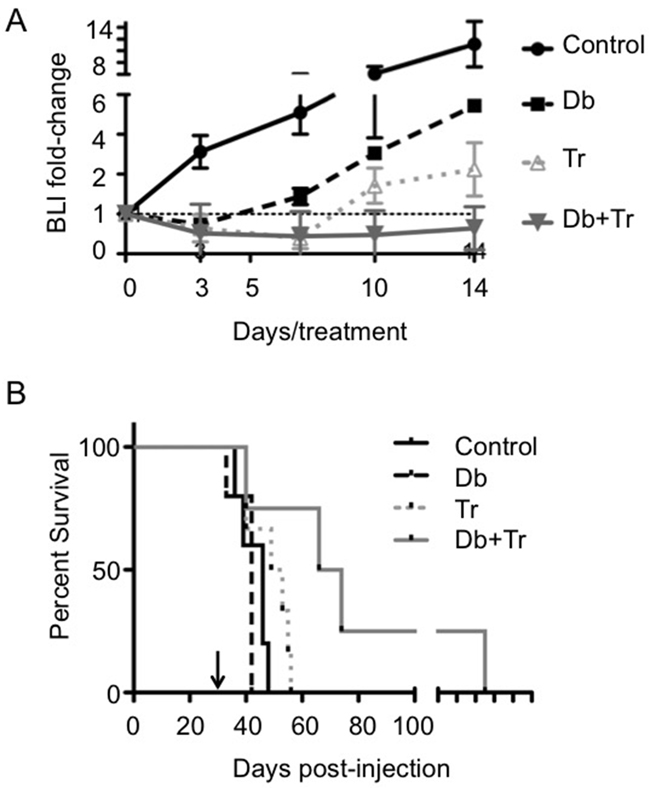
Figure 6: BRAFV600E and MEK inhibitor mono- and combination therapy effects on bioluminescence intensity and survival in syngeneic BrafV600E expressing Ink4a-Arf knock-out grafts. A. Dabrafenib (Db) and trametinib (Tr) combination therapy but not monotherapy shows durable reduction of bioluminescence intensity (BLI). Plots for BLI of 2341luc syngeneic grafts over 14 day treatments with Db, Tr, or a combination of both (Db+Tr) (n=8/group). Treatment was initiated at day 30 post-implantation of 2341luc and BLI was measured on treatment days 3, 7, 10, and 14, and expressed as normalized BLI (fold-change from the start of treatment). B. Db and Tr combination therapy but not monotherapy significantly extends survival. Graph shows Kaplan-Meier survival plot of mice with 2341luc syngeneic grafts treated with Db or Tr monotherapy or a combination of both, until they developed neurologic symptoms. Db monotherapy (median survival = 42 days) failed to extend survival while Tr monotherapy significantly increased survival (median survival Tr = 51 days; median survival control = 46 days; p=0.044 for Tr versus control). The combination treatment was most effective in extending survival (median survival = 70 days; p=0.049 for Db+Tr versus control).
DISCUSSION
The BRAFT1799A point mutation, which results in the expression of constitutively active BRAFV600E kinase, occurs in many cancers, including gliomas. Array CGH and exome sequencing of a population of matched pediatric low-grade glioma (PLGG) and secondary high-grade glioma (sHGG) identified BRAFV600E mutation and CDKN2A deletion as the most recurrent alterations in sHGG, at 39% and 57% respectively [8]. This study determined that BRAFV600E identifies a clinically distinct subset of patients whose tumors are more likely to undergo malignant progression [8].
Earlier, we assessed whether BRAF activation alone would be sufficient for tumor formation, by inducing expression of BrafV600E under control of its endogenous promoter in Gfap+ cells, by breeding BrafCA/+ hemizygous and hGFAP-cre transgenic mice [14]. We found that mice only survived for up to three weeks of age without any evidence of tumor formation. We concluded that BrafV600E expression during development is associated with lethality unrelated to glioma formation, and that BrafV600E expression alone is insufficient to induce gliomagenesis during the first month of age. To test whether BrafV600E cooperates with Cdkn2a (Ink4a-Arf) deletion to induce glioma formation, we injected Ad-cre near the SVZ of adult BrafCA/+ and BrafCA/+ Ink4a-arffl/fl mice, respectively. Combined expression of BrafV600E and homozygous deletion of Ink4a-Arf, but not expression of BrafV600E alone, induced gliomagenesis. These data corroborated that BrafV600E expression cooperates with homozygous inactivation of Cdkn2a to generate tumors [14]. In the current study, we obtained tumors by injecting Ad-cre into the corpus callosum of BrafCA/+ Ink4a-arffl/fl mice, which were genetically identical to the mice used in the earlier study (Figures 1A and 1B). From one such tumor (#2341), we have developed a syngeneic and orthotopic BrafV600E mutant and Cdkn2a deficient engraftment model. Our current findings shows that non-SVZ cells expressing the BrafV600E mutant and lacking Cdkn2a are capable to give rise to transplantable tumors.
Established human BrafV600E GBM lines are capable of generating xenografts and are typically kept in serum-conditions. Prolonged maintenance of cell cultures in the presence of serum significantly alters transcriptional and genomic cell profiles, in comparison with their parental tumor. In contrast, serum-free conditions, such as the ones used to maintain the 2341 cells, preserve better the expression profiles of the parental tumors [33, 34]. The 2341luc experimental glioma model thus provides a tool complementary to the already existing human BRAFV600E cell lines.
Histopathology and immunohistochemical analyses of 2341luc syngeneic grafts identified high-grade astrocytoma-like features and tumor cell phosphorylation of Erk, a surrogate for MAPK pathway activation. Tumors also co-expressed astrocyte and neural stem cell marker Gfap and Olig2, which is rare in normal brain cells, and indicates aberrant cell fate specification typical of tumor cells (Figures 2C and 2F) [35].
We found that vemurafenib and Db had IC50 values in 2341luc cells in the nanomolar range (Figure 4A), which is consistent with IC50 values of Db in some human melanoma cells [36]. With Db being more specific for BrafV600E than vemurafenib in 2341luc cells (Supplementary Figure S2) and its use in clinical trials for BRAFV600E mutant glioma [20], we focused subsequent analyses on effects of Db on 2341 cells and grafts. In total, our data support transient 2341 cell response to Db, as indicated by effects on MAPK pathway activity and cell proliferation (Figures 4B and 4C). MAPK pathway activity is restored, and in addition Akt and EGFR activities are heightened beyond six hours of Db treatment, as indicated by phosphoprotein immunoblot analyses of Db-treated cells (Figure 3B). Moreover, mice carrying 2341 grafts showed elevated p-Akt levels after three days of Db treatment, when compared with control and combination treatment (Supplementary Figure S5). The transient effect of Db on p-Erk levels and subsequent increase in p-Akt suggests the ability of 2341 cells to adapt to BRAFV600E inhibition in a manner similar to that seen in melanoma, where mutant BRAF inhibition provides strong selective pressure for upregulation of MAPK and PI3k-Pten-Akt pathways through epigenetic and genetic mechanisms [37].
Moreover, primary resistance or adaptive tumor response to BRAF inhibition in melanoma include upregulation of receptor tyrosine kinases [30]. A point of distinction between glioma and melanoma adaptation to BRAFV600E inhibition involves EGFR. We have previously shown that human BRAFV600E glioma but not melanoma cell lines express high levels of EGFR. Moreover, BRAFV600E inhibition of glioma cells with the vemurafenib tool compound PLX4720 releases a negative feedback loop that is placed on EGFR signaling by BRAFV600E [21]. This relationship has also been observed in BRAFV600E colorectal cancer [38] and the increase in pEGFR as well as total EGFR levels in Db-treated 2341 cells (Figure 3B), suggests a similar feedback mechanism in these mouse glioma cells (Supplementary Figure S3). Emerging clinical data and our preclinical study argue for a similar mechanism in glioma where BRAF mutation does not always appear to be predictive for responsiveness to BRAF inhibitor therapy.
The occurrence of acquired resistance to BRAFV600E inhibition in the treatment of various types of cancer [39–42] has motivated efforts to develop combination therapies to prevent and/or delay tumor adaptation. BRAFV600E + MEK inhibitor combination treatment of melanoma patients with BRAFV600E mutant tumors have shown this to be an effective strategy, as indicated by the survival benefit to this patient population [32]. These studies have motivated FDA-approval of Tr, a potent mitogen-activated protein kinase-1 (MEK)-1/2 inhibitor, for use in combination with Db as first-line therapy for metastatic melanoma. A further reason to combine BRAF and MEK inhibitors in patients is to decrease the development of secondary RAS-driven cancers, such as squamous cell carcinomas [43] and chronic lymphoblastic leukemia [44], that have been reported in patients inhibitor treated with BRAF monotherapy. These cancers develop due to the paradoxical activation of MAPK signaling in BRAF wild-type cells with upstream activation of RAS when treated with BRAF inhibitors. This MAPK activation is significantly impeded through the addition of MEK blockade [45].
Here, our results show that the BRAFV600E inhibitor Db has modest effect on intracranial tumor growth (Figures 6A and 6B), with rapid and substantial increases in MAPK and Akt pathway activities (Figures 5A and 5B; Supplementary Figure S5). Using this regiment, Db reportedly showed remarkable efficacy in reducing tumor size in the brains of patients with brain metastasis [41] and an intact blood-brain-barrier only partially prevents Db from entering the brain [24].
Presumably, as a result of MAPK activity rebound and rapidly increasing Akt activity, however, Db monotherapy had virtually no effect on animal subject survival (Figure 6B). Tr decreased 2341 MAPK pathway activity and cell proliferation while increasing apoptosis in vitro (Figures 4A-4D), and this translated to a small but significant survival benefit for Tr-treated animals bearing orthotopic 2341 tumors (Figure 6B). Nonetheless, our results shown that 2341 grafts adapt to Tr monotherapy, whereas combination treatment delayed acquired resistance (Figure 6A). Our data are consistent with those obtained by studying human BRAFV600E mutant glioma cell lines, which point to re-activation of MAPK pathway and increased AKT and EGFR signaling activity as a mechanisms of adaptation and resistance to BRAFV600E inhibition (Supplementary Figure S3) [21, 46]. These similarities suggest that our mouse model has clinical relevance. Future studies will examine the effects of extended combination therapy, including the responses of cancer stem cell subpopulations.
The 2341luc model exhibits multiple features of a useful preclinical engraftment model, as discussed in Oh et al [47], and include in vivo recapitulation of the diagnostic histopathology features of a human high-grade astrocytoma, in vitro tumor cell sustainability and amenability to genetic manipulation, engraftment consistency, and reproducible as well as predictable growth characteristics. As a result of 2341 cells being syngeneic to immunocompetent FVB/N mice, we anticipate our model as facilitating investigation of immunotherapy + small molecule inhibitor treatment of BRAFV600E gliomas.
MATERIALS AND METHODS
Isolation of BrafV600E Ink4a-Arf deleted tumor cells and cell culturing
Mouse breeding and procedures were conducted under an approved Animal Study Protocol according to UCSF Animal Care and Use Committee guidelines. Adenovirus-cre (Vector-BioLabs) was injected intracerebral using the following coordinates 1 mm anterior, 1 mm lateral and 2 mm deep to target the corpus callosum of BrafCA/+ Ink4a/ArfLoxP/LoxP (fl/fl)young adult FVB/N mice to induce BrafV600E expression under control of the endogenous Braf promoter [11], and deletion of p16Ink4a and p19Arf. At eight weeks post-injection, we sacrificed mice because they exhibited neurologic symptoms and harvested tumor tissue and dissociated it with the Neural Dissociation Kit (Miltenyi Biotec Inc., San Diego) according to manufacturer’s instructions. Primary cells were passaged using Accutase (Innovative Cell technologies) in non-adherent conditions at 37°C under 5% CO2 in Neurobasal-A media (1x B27-A, 1x N2, pen/strep, L-glutamine, 5 ng/ml EGF, 5 ng/ml bFGF2 and 5 ng/ml PDGF).
Genotyping, PCR and targeted sequencing
Genomic PCR to distinguish between BrafCA and BrafV600E, was conducted as described [11]. Isolation of genomic DNA from cells was performed using 0.5 mg/ml Proteinase K for one hour followed by precipitation with 0.1 M sodium acetate and ethanol extraction. Primers were 5’-tgagtatttttgtggcaactgc (forward) and 5’-ctctgctgggaaagcggc (reverse) for Braf. Semi-quantitative PCR for p16Ink4a was conducted as described elsewhere [14].
Generation of the 2341luc mouse model, bioluminescence imaging and preclinical treatment
2341 cells were infected with lentivirus expressing luciferase (Genecopoeia LPP-FLUC-Lv105-025) and infected cells were selected in the presence of 0.5 µg/mL puromycin. Adding 500 µg/mL luciferin to 1 x 106 cells and measuring bioluminescence verified luciferase expression. For orthotopic tumor growth 6-week old Nod scid gamma (NSG; Jackson Lab) and FVB/N mice (Simonsen Laboratories, Gilroy, CA) were implanted with 30,000 cells/animal and for preclinical testing FVB/N mice were injected with up to 4 x 105 2341luc cells/mouse as previously described [23]. The protocol was modified by suspending cells in 3 µl Matrigel (Corning Life Sciences). Using a 10 μl Hamilton gas-tight syringe and a stereotactic device (Stoelting; Wood Dale, IL), cells were implanted and the needle was withdrawn 120 seconds after injection. The burr hole was plugged with sterile bone wax and skin closed with Vicryl Rapide (Ethicon). For preclinical testing, at 30 days post-implantation, mice were randomly assigned to treatment, which consisted of vehicle control (DMSO), dabrafenib at 2.5 mg/kg daily by intraperitoneal injections [24], trametinib treatment at 1 mg/kg daily by oral gavage [25] or a combination of both for up to 14 days.
Bioluminescence imaging (BLI) was conducted twice weekly and signal intensity was quantified as the sum of detected photons per second within the region of interest using the Living Image software package and expressed as normalized BLI (fold-change from the start of treatment or original radiance). For survival studies, all animals were kept on treatment until reaching the major endpoint, which was animal neurological symptom free survival. BLI was measured using a Xenogen IVIS Spectrum Imaging System.
Alamar Blue cell viability assay and agent treatment
Viability assays were conducted as previously described [22] with the modification that 1000 cells per well were plated and Alamar Blue was added at 24 hours and fluorescence was measured 48 hours after plating. To assess dose-inhibition curves and IC50 values, we used concentrations between 0.1 nM and 0.5 µM of vemurafenib, dabrafenib, or trametinib (Selleckchem). Each drug was tested alone or in combination in three replicates in three independent experiments. DMSO-treated cell values were considered as 100% viability. The IC50 concentration was used to treat cells for four or five days at 48 hours intervals with dabrafenib, trametinib, or both before flow cytometry and immunoblotting.
Flow cytometry
Treated 2341 cells were fixed in 2% paraformaldehyde, permeabilized in 100% ice-cold methanol and stained with rabbit p-ERK antibody (Cell Signaling #4377S) and donkey anti-rabbit IgG-647 (Jackson Labs #711-136-152). Cell cycle analyses were conducted by treating cells with Hoechst 33342 at a final concentration of 2 ng/ml and DNA content (2n versus 4 n) was determined as described [26]. Analyses were conducted using the FACS ARIA II (BD Biosciences). Annexin V staining was performed on unfixed cells using the FITC Apoptosis detection Kit according to manufacturer instructions (BD Cell Analysis).
Immunoblotting
Treated 2341 cells were stimulated with EGF (20ng/ml) for 20 min prior to protein extraction which was performed using cell lysis buffer (Cell Signaling) supplemented with proteinase and phosSTOP phosphatase inhibitor cocktail (Roche). Proteins were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes, which were then probed with primary antibodies, followed by horseradish peroxidase-conjugated secondary antibody, and visualized by ECL (GE Healthcare). Antibodies specific for p-EGFR (Tyr1068), p-Akt (Ser473), total Akt, p-ERK, and beta-actin were from Cell Signaling Technology. Total EGFR and total ERK antibodies were from Santa Cruz Biotechnology, Inc.
Immunofluorescence (IF)
Tissue sections from 4% paraformaldehyde (PFA)-perfused mouse brains were cut and stained as 10 μm sections. Sections were post-fixed with 4% PFA for 10 minutes, washed and blocked with PBS-0.1% triton-5% normal donkey serum. All sections were incubated with primary antibody and secondary antibody, as well as streptavidin conjugate as described [22].
The following antibodies and streptavidin conjugates were used: Rabbit anti-pERK (Cell Signaling Technology, dilution 1:200), rabbit anti-p-Akt (Ser473, D9E, Cell Signaling Technology, Inc, dilution 1:200), rabbit anti-Gfap (DAKO, dilution 1:400), goat anti-Olig2 (R&D, dilution 1:500), goat anti-Ki67 (Santa Cruz, dilution 1:200), rabbit anti-Ki67 (Cell Signaling Technology, dilution 1:200) and rabbit anti-cleaved caspase-3 (Cell Signaling Technology, dilution 1:400). The secondary antibodies used were donkey IgG-fluorescence conjugates (Jackson ImmunoResearch, dilution 1:400). Images were obtained using a Zeiss AxioImager M1 microscope with Zen 2 software (Carl Zeiss microcopy 2011). Images were also acquired at the 6D high throughput epifluorescent system (Nikon Imaging Center at UCSF) The presence of tumors was confirmed by DAPI staining and H&E staining of consecutive sections.
Statistics and analysis
Statistical analyses were conducted as indicated in the figure legends using GraphPad Prism 5.0 and Microsoft Excel. For determination of the IC50s in vitro drug efficacy studies non-linear regression analyses were utilized. A two-tailed paired t-test was conducted to analyze for significant differences between two treatment groups in flow cytometry experiments assessing DNA content and annexin V. An unpaired t-test was conducted to analyze differences between two treatment groups in flow cytometry experiments detecting p-ERK. One-way ANOVA was used to analyze differences between three or more groups. For Kaplan–Meier survival rate analyses, the number of surviving mice in the three groups of animals were recorded daily after 2341luc implantation. The data were subjected to Log-rank test in order to determine if significant differences existed in survival between the experimental groups. For statistical tests *=p<0.05, **=p<0.01, ***=p<0.001.
ACKNOWLEDGMENTS
The authors would like to thank David Deng, Sista Sugiarto, Hannah Y. Collins and Patrizia Hanecker for technical assistance, Helena Oft for editing, Sandra Gomez-Lopez for the schematic in Figure 1, and Martin Oft and Derek Wainwright for critical discussion.
CONFLICT OF INTEREST
All authors declare no conflict of interest.
GRANT SUPPORT
National Institute of Health/National Cancer Institute (CA16476-04 to C.K.P); National Institute of Neurological Disorders and Stroke (NS080619 to C.K.P and C.D.J); Childhood Brain Tumor Foundation (to C.K.P.); the Loglio Research Program (to C.K.P), the Research Allocation Program Investigator Grant CTSI-UCSF (to C.K.P) and California Institute of Regenerative Medicine (TB1-01190 to I.D.M).
REFERENCES
1. Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R, Cancer Genome P. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004; 116: 855–67.
2. Dias-Santagata D, Lam Q, Vernovsky K, Vena N, Lennerz JK, Borger DR, Batchelor TT, Ligon KL, Iafrate AJ, Ligon AH, Louis DN, Santagata S. BRAF V600E mutations are common in pleomorphic xanthoastrocytoma: diagnostic and therapeutic implications. PLoS One [Internet]. 2011/04/12 ed. 2011; 6: e17948. doi: 10.1371/journal.pone.0017948.
3. Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, Storm PB, Biegel JA. Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol [Internet]. 2010 [cited 2016 Apr 12]; 12: 621–30. doi: 10.1093/neuonc/noq007.
4. Schindler G, Capper D, Meyer J, Janzarik W, Omran H, Herold-Mende C, Schmieder K, Wesseling P, Mawrin C, Hasselblatt M, Louis DN, Korshunov A, Pfister S, et al. Analysis of BRAF V600E mutation in 1,320 nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol [Internet]. 2011/01/29 ed. 2011; 121: 397–405. doi: 10.1007/s00401-011-0802-6.
5. Eisenhardt AE, Olbrich H, Röring M, Janzarik W, Anh TN Van, Cin H, Remke M, Witt H, Korshunov A, Pfister SM, Omran H, Brummer T. Functional characterization of a BRAF insertion mutant associated with pilocytic astrocytoma. Int J cancer [Internet]. 2011 [cited 2016 Apr 12]; 129: 2297–303. doi: 10.1002/ijc.25893.
6. Schiffman JD, Hodgson JG, VandenBerg SR, Flaherty P, Polley MY, Yu M, Fisher PG, Rowitch DH, Ford JM, Berger MS, Ji H, Gutmann DH, James CD. Oncogenic BRAF mutation with CDKN2A inactivation is characteristic of a subset of pediatric malignant astrocytomas. Cancer Res [Internet]. 2010/01/14 ed. 2010; 70: 512–9. doi: 0008-5472.CAN-09-1851 [pii]10.1158/0008-5472.CAN-09-1851.
7. Nicolaides TP, Li H, Solomon DA, Hariono S, Hashizume R, Barkovich K, Baker SJ, Paugh BS, Jones C, Forshew T, Hindley GF, Hodgson JG, Kim JS, et al. Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res. 2011; 17: 7595–604. doi: 10.1158/1078-0432.CCR-11-1456.
8. Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, Stavropoulos J, Alon N, Pole JD, Ray PN, Navickiene V, Mangerel J, Remke M, et al. BRAF mutation and CDKN2A deletion define a clinically distinct subgroup of childhood secondary high-grade glioma. J Clin Oncol [Internet]. 2015 [cited 2016 Apr 12]; 33: 1015–22. doi: 10.1200/JCO.2014.58.3922.
9. Basto D, Trovisco V, Lopes JM, Martins A, Pardal F, Soares P, Reis RM. Mutation analysis of B-RAF gene in human gliomas. Acta Neuropathol [Internet]. 2005/03/26 ed. 2005; 109: 207–10. doi: 10.1007/s00401-004-0936-x.
10. Knobbe CB, Reifenberger J, Reifenberger G. Mutation analysis of the Ras pathway genes NRAS, HRAS, KRAS and BRAF in glioblastomas. Acta Neuropathol [Internet]. 2004/11/02 ed. 2004; 108: 467–70. doi: 10.1007/s00401-004-0929-9.
11. Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev [Internet]. 2007/02/15 ed. 2007; 21: 379–84. doi: gad.1516407 [pii]10.1101/gad.1516407.
12. Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci [Internet]. 1994 [cited 2016 Apr 13]; 14: 1030–7. Available from http://www.ncbi.nlm.nih.gov/pubmed/8120611.
13. Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, Redston MS, DePinho RA. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev [Internet]. 2003; 17: 3112–26. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14681207.
14. Huillard E, Hashizume R, Phillips JJ, Griveau a., Ihrie R a., Aoki Y, Nicolaides T, Perry a., Waldman T, McMahon M, Weiss W a., Petritsch C, James CD, et al. Cooperative interactions of BRAFV600E kinase and CDKN2A locus deficiency in pediatric malignant astrocytoma as a basis for rational therapy. Proc Natl Acad Sci. 2012; 109: 8710–5. doi: 10.1073/pnas.1117255109.
15. Nicolaides T, Li H, Solomon D, Hariono S, Hashizume R, Barkovich K, Baker SJ, Paugh BS, Jones C, Forshew T, Hindley G, Hodgson JG, Kim JS, et al. Targeted Therapy for BRAFV600E Malignant Astrocytoma. Clin Cancer Res [Internet]. 2011/11/01 ed. 2011; doi: 1078-0432.CCR-11-1456 [pii]10.1158/1078-0432.CCR-11-1456.
16. Rush S, Foreman N, Liu A. Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol [Internet]. 2013 [cited 2016 Apr 13]; 31: e159–60. doi: 10.1200/JCO.2012.44.1568.
17. del Bufalo F, Carai A, Figà-Talamanca L, Pettorini B, Mallucci C, Giangaspero F, Antonelli M, Badiali M, Moi L, Bianco G, Cacchione A, Locatelli F, Ferretti E, et al. Response of recurrent BRAFV600E mutated ganglioglioma to Vemurafenib as single agent. J Transl Med [Internet]. 2014 [cited 2016 Apr 13]; 12: 356. doi: 10.1186/s12967-014-0356-1.
18. Chamberlain MC. Salvage therapy with BRAF inhibitors for recurrent pleomorphic xanthoastrocytoma: a retrospective case series. J Neurooncol [Internet]. 2013 [cited 2016 Apr 13]; 114: 237–40. doi: 10.1007/s11060-013-1176-5.
19. Robinson GW, Orr BA, Gajjar A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer [Internet]. 2014 [cited 2015 Sep 17]; 14: 258. doi: 10.1186/1471-2407-14-258.
20. Kieran M, Hargrave D, Cohen K, Aerts I, Dunkel I, Hummel T, Jimenez I, Pearson A, Pratilas C, Whitlock J, Bouffet E, Shen W, Broniscer A, et al. Phase 1 study of dabrafenib in pediatric patients (pts) with relapsed or refractory BRAF V600E high- and low-grade gliomas (HGG, LGG), Langerhans cell histiocytosis (LCH), and other solid tumors (OST). J Clin O. 2015.
21. Yao T-W, Zhang J, Prados M, Weiss WA, James CD, Nicolaides T. EGFR blockade prevents glioma escape from BRAFV600E targeted therapy. Oncotarget [Internet]. 2015 [cited 2016 May 21]; 6: 21993–2005. doi: 10.18632/oncotarget.4014.
22. Lerner RG, Grossauer S, Kadkhodaei B, Meyers I, Sidorov M, Koeck K, Hashizume R, Ozawa T, Phillips JJ, Berger MS, Nicolaides T, James CD, Petritsch CK. Targeting a Plk1-Controlled Polarity Checkpoint in Therapy-Resistant Glioblastoma-Propagating Cells. Cancer Res [Internet]. 2015 [cited 2015 Dec 27]; 75: 5355–66. doi: 10.1158/0008-5472.CAN-14-3689.
23. Sugiarto S, Persson AI, Munoz EG, Waldhuber M, Lamagna C, Andor N, Hanecker P, Ayers-Ringler J, Phillips J, Siu J, Lim DA, Vandenberg S, Stallcup W, et al. Asymmetry-Defective Oligodendrocyte Progenitors Are Glioma Precursors. Cancer Cell. 2011; 20: 328–40. doi: 10.1016/j.ccr.2011.08.011.
24. Mittapalli RK, Vaidhyanathan S, Dudek AZ, Elmquist WF. Mechanisms limiting distribution of the threonine-protein kinase B-RaF(V600E) inhibitor dabrafenib to the brain: implications for the treatment of melanoma brain metastases. J Pharmacol Exp Ther [Internet]. 2013 [cited 2016 Apr 27]; 344: 655–64. doi: 10.1124/jpet.112.201475.
25. Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, Yang J, Seestaller-Wehr L, Zhang S-Y, Hopson C, Tsvetkov L, Jing J, Zhang S, et al. The BRAF and MEK Inhibitors Dabrafenib and Trametinib: Effects on Immune Function and in Combination with Immunomodulatory Antibodies Targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res [Internet]. 2015 [cited 2016 Apr 27]; 21: 1639–51. doi: 10.1158/1078-0432.CCR-14-2339.
26. Daynac M, Morizur L, Kortulewski T, Gauthier LR, Ruat M, Mouthon M-A, Boussin FD. Cell Sorting of Neural Stem and Progenitor Cells from the Adult Mouse Subventricular Zone and Live-imaging of their Cell Cycle Dynamics. J Vis Exp [Internet]. 2015 [cited 2016 Apr 28]; doi: 10.3791/53247.
27. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature [Internet]. 2010/09/09 ed. 2010; 467: 596–9. doi: nature09454 [pii]10.1038/nature09454.
28. Shi H, Moriceau G, Kong X, Lee M-K, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, Sosman JA, Kefford RF, Long G V, et al. Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nat Commun [Internet]. 2012 [cited 2016 Apr 13]; 3: 724. doi: 10.1038/ncomms1727.
29. Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, Meyerson M, Garraway LA. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol [Internet]. 2011/03/09 ed. 2011; 29: 3085–96. doi: JCO.2010.33.2312 [pii]10.1200/JCO.2010.33.2312.
30. Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee M-K, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature [Internet]. 2010 [cited 2013 Aug 22]; 468: 973–7. doi: 10.1038/nature09626.
31. Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, Kelley MC, Kefford RF, Chmielowski B, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov [Internet]. 2014 [cited 2016 Apr 14]; 4: 80–93. doi: 10.1158/2159-8290.CD-13-0642.
32. Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov L V, Hassel JC, Rutkowski P, Mohr P, Dummer R, Trefzer U, Larkin JM, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012; 367: 107–14. doi: 10.1056/NEJMoa1203421.
33. Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, Park JK, Fine HA. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell [Internet]. 2006; 9: 391–403. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=16697959.
34. Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ, Rosenblum M, Mikkelsen T, Zenklusen JC, Fine HA. Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol Cancer Res [Internet]. 2008 [cited 2016 Jul 17]; 6: 21–30. doi: 10.1158/1541-7786.MCR-07-0280.
35. Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A [Internet]. 2003; 100: 15178–83. Available from http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=14645703.
36. King AJ, Arnone MR, Bleam MR, Moss KG, Yang J, Fedorowicz KE, Smitheman KN, Erhardt JA, Hughes-Earle A, Kane-Carson LS, Sinnamon RH, Qi H, Rheault TR, et al. Dabrafenib; Preclinical Characterization, Increased Efficacy when Combined with Trametinib, while BRAF/MEK Tool Combination Reduced Skin Lesions. Smalley K, editor. PLoS One [Internet]. Public Library of Science; 2013 [cited 2016 Jul 17]; 8: e67583. doi: 10.1371/journal.pone.0067583.
37. Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, Sosman JA, Ribas A, Lo RS. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell [Internet]. 2015 [cited 2015 Sep 10]; 162: 1271–85. doi: 10.1016/j.cell.2015.07.061.
38. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature [Internet]. 2012 [cited 2016 Feb 21]; 483: 100–3. doi: 10.1038/nature10868.
39. Hauschild A, Grob JJ, Demidov L V, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller Jr. WH, Kaempgen E, Martin-Algarra S, Karaszewska B, Mauch C, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012; 380: 358–65. doi: 10.1016/S0140-6736(12)60868-X.
40. Sloot S, Fedorenko I V, Smalley KS, Gibney GT. Long-term effects of BRAF inhibitors in melanoma treatment: friend or foe? Expert Opin Pharmacother. 2014; 15: 589–92. doi: 10.1517/14656566.2014.881471.
41. Falchook GS, Long G V, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O’Day SJ, Blackman SC, Curtis CM, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet [Internet]. 2012 [cited 2013 Aug 22]; 379: 1893–901. doi: 10.1016/S0140-6736(12)60398-5.
42. Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C, Aranda E, Nordlinger B, Cisar L, et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol [Internet]. 2010 [cited 2016 Mar 11]; 28: 466–74. doi: 10.1200/JCO.2009.23.3452.
43. Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis-Filho JS, Kong X, Koya RC, Flaherty KT, Chapman PB, Kim MJ, Hayward R, et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med [Internet]. 2012 [cited 2016 Apr 29]; 366: 207–15. doi: 10.1056/NEJMoa1105358.
44. Callahan MK, Wolchok JD. Clinical Activity, Toxicity, Biomarkers, and Future Development of CTLA-4 Checkpoint Antagonists. Semin Oncol [Internet]. 2015 [cited 2015 Sep 17]; 42: 573–86. doi: 10.1053/j.seminoncol.2015.05.008.
45. Sanlorenzo M, Choudhry A, Vujic I, Posch C, Chong K, Johnston K, Meier M, Osella-Abate S, Quaglino P, Daud A, Algazi A, Rappersberger K, Ortiz-Urda S. Comparative profile of cutaneous adverse events: BRAF/MEK inhibitor combination therapy versus BRAF monotherapy in melanoma. J Am Acad Dermatol [Internet]. 2014 [cited 2016 May 20]; 71: 1102–9.e1. doi: 10.1016/j.jaad.2014.09.002.
46. Olow A, Mueller S, Yang X, Hashizume R, Meyerowitz J, Weiss WA, Resnick AC, Waanders AJ, Stalpers LJA, Berger MS, Gupta N, James CD, Petritsch CK, et al. BRAF status in personalizing treatment approaches for pediatric gliomas. Clin Cancer Res [Internet]. 2016 [cited 2016 Jun 27]; doi: 10.1158/1078-0432.CCR-15-1101.
47. Oh T, Fakurnejad S, Sayegh ET, Clark AJ, Ivan ME, Sun MZ, Safaee M, Bloch O, James CD, Parsa AT. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014; 12: 107. doi: 10.1186/1479-5876-12-107.