
miRNAs are small, non-coding RNAs that bind target mRNAs leading to their degradation or disruption of their translation [1]. Because regulation of specific gene transcripts by miRNAs is essential to many developmental processes, it is not surprising that dysfunctional miRNA signaling contributes to diseases that arise from defective cellular proliferation and differentiation including acute myeloid leukemia (AML). AML is a highly heterogeneous disease characterized by failure of myeloid precursor cells to undergo terminal differentiation from proliferating precursor cells into mature blood cells. In addition to genetic and genomic aberrations, including chromosome translocations and inversions, gene deletions and mutations, changes in post-transcriptional regulation by miRNAs in AML have been reported. These miRNAs have functional relevance and some are sufficient to cause a cascade of downstream effects in malignant AML transformation in animal models, indicating the potential of miRNA-based therapeutic strategies. However, aberrant miRNAs are extensively associated with genetic and genomic alterations seen in AML. A comprehensive understanding of the full network of oncogenic events and downstream signaling pathways related to altered miRNAs could reveal new therapeutic strategies to modulate these miRNAs in AML.
The role of miRNAs in pathogenesis of AML
Recent studies have linked disruptions of miRNAs involved in gene regulation to AML pathogenesis. For example, miR-155 is upregulated in bone marrow of AML patients with mutations in nucleophosmin (NPM1) and FLT3-ITD [2, 3]. Overexpression of miR-155 in human hematopoietic stem cells increased proliferation of myeloid progenitors and blocked differentiation into erythrocyes and megakaryocytes. Overexpression of miR-155 in murine lymphocyte precursors induced polyclonal lymphocytosis but also led to high-grade lymphocytic leukemia [4, 5]. Src homology-2 domain containing inositol 5-phosphatase 1 (SHIP1), a negative regulator of immune cell signaling, is directly bound and inhibited by miR-155 [4, 6]. Mice lacking SHIP1 or overexpressing miR-155 exhibit similar myelo-proliferative disease characteristics, including increased granulocyte-monocyte cells and reduced B-lymphocytes [7]. Interestingly, loss of function SHIP1 mutations have been found in a small number of AML patients and have been linked to oncogenesis [8]. miR-155 also targets CEBPB [4], which is critical in granulopoiesis [9], suggesting that aberrant miR-155 signaling could deregulate both SHIP1 and CEBPB in AML.
The miR-125 family exists as three homologues (miR-125a, b, and c) all on different chromosomes. While studies of miR-125a suggested it has a tumor suppressor role in AML, miR-125b is considered as an oncomiRNA. miR-125a expression in cytogenetically normal AML (CN-AML) was most decreased in favorable and intermediate prognostic populations and associated with decreased survival [10]. In contrast, miR-125b is highly expressed in hematopoietic stem cells and when ectopically overexpressed leads to development of leukemia. The leukemic subtype developed is dependent on the level of miR-125b. Over-expression of miR-125b can block the terminal differentiation of HL60 and NB4 AML cell lines in vitro [11, 12]. miR-125b exists as two paralogs, miR-125b1 and miR-125b2, which contain the same seed region but are located on different chromosomes and are therefore independently regulated. MiR-125b-1 is strongly up-regulated in AML blasts, in particular those from patients with the t(2:11)(p21:q23) translocation, which shows a 90-fold increase [11-13]. In a study of CN-AML patients 60-year or older, miR-125b2 was one of the most overexpressed miRNAs of the 32 differentially expressed miRNAs examined [14]. miR-125b targets the pro-apoptotic Bak1 and Bmf transcripts and also negatively regulates p53 and many other genes in the p53 pathway, providing one mechanism for miR-125b’s role as an oncomiR in AML. In addition, NOTCH1 is also a target of miR-125b. While NOTCH activation plays oncogenic roles in acute T-lymphoblastic leukemia (T-ALL), NOTCH is suppressive for AML cell growth depending on cell conditions [15]. Romero et al. recently observed that C/EBPα binds directly to the promoter of miR-125b, increasing its transcription rate and subsequently reducing NOTCH1 levels in AML [16].
miR-100 is another oncomiR in AML with high expression in primary AML blasts [17]. The up-regulation of miR-100 represses its target RB serine phosphatase (RBSP3) [18], which is a phosphatase-like tumor suppressor, frequently mutated in human hematopoietic cell lines. RBSP3 can influence the phosphorylation status of pRB and then the release of E2F1 in controlling cell cycle. In vitro studies revealed that the mechanism underlying miR-100 in arresting human granulocyte and monocyte differentiation and promoting cell survival was through the RBSP3-pRB-E2F1 pathway [18].
Conversely, several miRNAs have been characterized as tumor suppressors in AML. Wang X, et al [19] saw that miR-29a and miR-142-3p were significantly increased in peripheral blood mononuclear cells (PBMNCs) and bone marrow (BM) white blood cells from AML patients. Increased miR-29a or miR-142-3p leads to increased differentiation into granulocytes and monocytes, while reduction of either miR-29a or miR-142-3p suppressed myeloid differentiation in leukemia cell models. Furthermore, co-transfection of both miR-29a and miR-142-3p inhibited both their common target, cyclin T2 (CCNT2), and their individual target genes cyclin-dependent kinase 6 (CDK6) and TGF-β activated kinase 1/MAP3K7 binding protein 2 (TAB2), respectively, leading to a larger synergistic reduction in myeloid differentiation [19-21]. miR-29a and miR-142-3p promote monocytopoiesis by suppressing CCNT2, which reduces pRb protein levels and cell proliferation. CDK6 interferes with Runx1 binding to DNA and C/EBPα in immature proliferating cells blocking their myeloid differentiation. Removal of CDK6 inhibition selectively activates Runx proteins, promoting terminal cell differentiation. miR-142-3p inhibits TAB2 expression and therefore an increase in miR-142-3p pushes monocytic precursors to differentiate into macrophages rather than osteoclasts. More recently, Gong et al. [22] reported that all the miR-29 family members, miR-29a, -29b and -29c, were reduced in PBMNCs and bone marrow CD34+ cells from AML patients. Reintroducing each miR-29 member into AML BM blasts was able to partially correct abnormal cell proliferation and apoptosis repression and myeloid differentiation arrest. Akt2 was also identified to be a target of the three miR-29 members that was significantly increased in the AML blasts. In myeloid leukemogenesis, overexpressed c-Myc inhibits miR-29 family expression, resulting in increased Akt2 and CCND2 protein expression in AML [20, 22-24].
A most recent study that profiled differentially expressed miRNAs from AML patients’ granulocytes versus healthy subjects identified two significantly under-expressed miRNAs miR-26a-5p and miR-23b-3p [25]. These two miRNAs have a common target the PrxIII gene. The accumulation of PrxIII caused by decreased miR-26a-5p and miR-23b-3p led to a considerable decrease in reactive oxygen species (ROS) in primary AML granulocyte samples and transfected cells [25]. Emerging evidence suggests that the ROS signal plays a critical role in regulating the balance between self-renewal and differentiation of hematopoietic stem cells (HSCs) [26]. The decreased ROS levels might push HSCs toward differentiation into myeloid lineage fates in hematopoietic systems, providing one mechanism for miR-26a-5p and miR-23b-3p’s role as tumor suppressors.
miR-223 was previously characterized as a negative regulator of AML pathogenesis [27]. In support of this, the miR-223 mutant mouse was found to exhibit granulocytosis and hemizygous loss of the miR-223 gene has been identified in AML patients [28]. Further studies identified that transcription factor myocyte-specific enhancer factor 2C (MEF2C) gene is one of the targets of miR-223 in mediating its anti-proliferative effects in granulopoiesis, while miR-223 transcription is activated by RUNX1/RUNX1T1-induced chromatin remodeling. Recently, Maria et al, using the miR-223-/Y and miR-223-/- mice, found that loss of miR-223 alone results in a modest expansion of myeloid progenitors, but does not induce myeloproliferative disorder or alter HSC long-term repopulating and self-renewal capacity [29]. These results suggest that miR-223 loss is not sufficient to cause AML but it may be a contributing factor in leukemogenesis that requires further study.
In addition to these miRNAs for which comprehensive studies have identified roles in AML pathogenesis and mechanism of actions, several new AML miRNAs have been highlighted in recent publications. For example, miR-9 (miR-9-5p) and miR-9* (miR-9-3p) are highly conserved miRNAs produced from a single precursor. Overexpression of miR-9 has been shown to enhance transformation of murine hematopoietic progenitor cells by MLL-AF9 [30]. Also, miR-9 targeting of the LIN28B/Let-7/HMGA2 axis induces monocytic differentiation in KASUMI-1 cells [31]. High expressions of both miR-9 and 9* (miR-9/9*) were detected in most cases from a cohort of 647 primary AML patients. Their expression levels varied among different AML subtypes with the highest expression in MLL-related leukemias harboring 11q23 abnormalities and in normal karyotype AML cases with mutations in NPM1 [32]. Ectopic expression of miR-9 or miR-9* blocked neutrophil development in the myeloid 32D cell line and in mouse primary lineage-negative bone marrow cells by inhibiting ETS-related gene (ERG) [32].
Studying AML caused by toxic DNA interstrand crosslinks (ICLs), Alemdehy et al [33] found that miR-139-3p and miR-199a-3p had opposite effects on hematopoiesis. In the study, both miR-139-3p and miR-199a-3p increased with age in myeloid progenitors from the nucleotide excision repair gene (Ercc1)-deficient mice. Ectopic expression of miR-139-3p inhibited myeloid progenitor proliferation of myeloid progenitors, increased miR-199a-3p enhanced proliferation of progenitors and accelerated the AML phenotype. Although this study directly supports the role of miR-199a-3p as an onco-miRNA, it also indicates that an auto-regulatory negative feedback from the elevated miR-139-3p as a suppressor is involved in the defective hematopoietic function in ICL-caused AML.
miR-638 is frequently down-regulated in various solid tumors, and it represses BaP-induced carcinogenesis by targeting breast cancer 1 (BRCA1) [34]. Lin et al recently saw that miR-638 was reduced in primary AML samples vs cells undergoing normal hematopoiesis [35]. Overexpression of miR-638 inhibited proliferation and promoted differentiation, while inhibition of miR-638 promoted proliferation and reduced differentiation. CDK2 has been identified as an miR-638 target and CDK2 overexpression rescued the miR-638- repressed colony formation of HL-60 cells. Considering that CDK2 is also commonly down-regulated during granulopoiesis, the miR-638/CDK2 axis may serve as a marker for prognosis or treatment response. However, miR-638 overexpression was not sufficient to overcome the failure of leukemic cells to differentiate. Rather, it reduced the threshold for leukemic cells to undergo forced myeloid differentiation. Thus, further studies in exploring the miR-638 regulatory network is necessary for fully clarifying the contribution of miR-638 to myeloid leukemia [35].
Accumulating evidence indicates that the miR-181 family plays important roles in AML pathogenesis [36]. Expression of all miR-181 family members was reduced in adult AML patients (M1-M3 subtypes), suggesting all function as tumor suppressors. miR-181a promotes a proliferative state by inhibiting terminal differentiation of both cultured HL-60 cells and CD34+ hematopoietic stem/progenitor cells (HSPCs). The in vivo expression of miR-181 partially reversed the lack of myeloid differentiation in AML patients and in the mice with CD34+ HSPCs from AML patients [37]
miRNAs were associated with AML stem cells
It has been demonstrated that, in normal hematopoiesis, some miRNAs were involved in progenitor lineage commitment[38] and controlling HSC [39-41] by coordinate repression of multiple targets [42]. For example, HSC self-renewal can be governed by miR-125a/b, miR-29a, and miR-126 [40, 43-45]. Recent studies have revealed important roles for miRNAs in leukemic stem cells (LSCs), linked to long-term self-renewal and transient cell-cycle quiescence or even dormancy [46]. LSCs are linked to treatment failure, recurrence, and chemotherapy resistant disease in patients with AML [47-49]. On the other hand, LSC frequency at the time of diagnosis and after treatment are prognostic indicators of poor AML outcomes [48, 50, 51]. Elucidation of the pattern of miRNA expression could help to elucidate the molecular mechanism of early hematopoietic differentiation and the pathogenic mechanisms underlying AML [52], risk categories, and prognosis [2, 53, 54].
Several LSC-associated miRNAs are linked to the development of leukemia. For example, miR-17-92 polycistron maintained LSC in Mixed-lineage leukemia (MLL) models [55]. Inhibiting miR-196 or miR-21 reduced LSCs in an experimental model of human MLL [56], miR-126 reduction reduced AML growth, and overexpression of some miRNAs induced murine leukemic transformation [44, 57]. Recent evidence indicates that LSCs-released microvesicles (LMVs) can regulate the malignance of AML cells, overexpression of tumor suppressive miR34a is able to interrupt this process, which indicates that modulating miR34a could offer a new approach for the management of AML [58]. In the other reports, overexpression of miR-29a in normal hematopoietic cells caused a myeloproliferative disorder that progressed to AML [23], and overexpression of miR-125b led to leukemia [11].
Furthermore, miR-126 was found to restrain cell cycle progression, prevent differentiation, and increase self-renewal of primary LSC in vivo, and the results demonstrate that miRNAs preserve LSC quiescence and promote chemotherapy resistance [59]. One study reported that targeting miR-126 in leukemic cells could reduce cell growth by inducing apoptosis [60]. Lechman and colleagues reported that miRNAs were involved in human LSC function [59], and another recent study revealed that the higher levels of miR-126 in older AML patients correlated with poor overall survival [61]. Thus, miRNA expression, including miR-126 in LSCs, impacts on the clinical outcome of AML patients. Furthermore, overexpression of miR-126 increased primitive quiescence in AML cells and reduced their differentiation into AML blasts. Knockdown of miR-126 promoted led primitive AML cells to exit their quiescent stem-like state into a more committed population of progenitors with decreased capacity for self-renewal. As to the mechanism of its cell cycle regulation, miR-126 controls the PI3K-AKT-mTOR pathway [40], a gatekeeper of LSC G0-G1 cycle control [62]. AML cells with increased miR-126 are more resistant to standard chemotherapy resulting in their enrichment during treatment. MiR-126 knockdown leads to increased proliferation of HSCs but impaired maintenance of LSCs, while its overexpression promotes LSC self-renewal and inhibits expansion of HSCs [40, 59]. Many studies have profiled miRNA expression in primary AML cells [2, 3, 53, 63, 64]. Reduced miR-126 induced AML cell apoptosis, but enhanced expansion of HSCs in normal BM. The role of miR-126 in LSC function appears to depend on age and AML subtype [61]. miRNAs appear to provide excellent LSC therapeutic targets due to their role in transformation of normal myeloid progenitors/stem cells into AML LSCs, but targeting LSCs without harming normal HSCs remains a significant hurdle [65]. Because the above-mentioned studies indicate that high miR-126 expression correlates with ‘stemness’ and that miR-126 is a viable target for eliminating the LSC in AML, the therapeutic feasibility of targeting miR-126 in LSCs is a subject of interest. Targeting the single miR-126 in LSCs reduced their colony formation and eliminated all leukemic cells, which suggests miR-126 as a target to specifically eradicate LSCs [60, 61]. miR-126 inhibition reduces LSC activity and chemotherapy resistance, which when used in conjunction with conventional leukemic therapies may significantly improve patient outcomes [66]. These observations suggest that selectively targeting LSCs by inhibiting miR-126, while simultaneously promoting the recovery of normal HSCs is a potential new therapy for AML (Figure 1).
There are many more miRNAs have been shown in publication that may be involved in the pathogenesis of AML (Table 1), suggesting incredible enthusiasm for research in this area in recent years. However, these studies share few similarities and there are several technical considerations to help understand the results. For example, applying multiple methods in parallel to detect a miRNA or the regulation of a miRNA is more appropriate based on the existence of related miRNAs with a high degree in sequence homology. Oftentimes, the regulatory changes in miRNA levels are small and might get lost in the biological noise when using a small number of samples. Using in vitro systems to study miRNA phenotypes might be different from what is happening in vivo. Lastly, efficacy of over-expression or anti-miR tools should be validated using downstream target readout to show the endogenous interaction between the miRNA and the targets.
Table 1. miRNAs that are involved in the pathogenesis of AML
Subtype |
Genetic subtype |
Up-regulated miRNA |
Down-regulated miRNA |
References |
AML |
(15; 17) |
miR-127, miR-134, miR-299-5p, miR-323, miR-376a, miR-382, miR-29b-3p, miR-224, miR-368, miR-181a-5p |
miR-17-5p, miR-20a, miR-126, miR-126*, miRNA-181a-3p, miR-126-5p |
|
AML |
t (8; 21) |
miR-27a, miR-126, miR-150, miR -223, miR-29b-3p |
miR-126-5p |
|
AML |
t(11q23)/MLL |
miR-326, miR-219, miR-194, miR-99b, miR-328, miR-196b, miR-29b-3p |
miR-34b, miR-15a, miR-29a, miR-29c, miR-372, miR-30a, miR-29b, miR-30e, miR-196a, let-7f, miR-102, miR-33, miR-299, miR-193, miR-126-5p |
|
CBF-AML |
t (8; 21) or inv (16) |
miR-126, miR-126*miR-29b-3p |
miR-126-5p |
|
APL |
14q32 |
miR-136-5p, miR-376a-3p, miR-29b-3p |
miR-126-5p |
|
AML |
t (9; 11) (p22; q23) |
- |
miR-126-5p |
[102] |
AML |
t(6;11)(q27;q23) |
miR-21, miR-26a, miR-26b, miR-29b-3p |
miR-126-5p |
|
AML |
three body (+8) |
miR-24a, miR-30d, miR-29b-3p |
miR-126-5p |
|
AML |
CBFB-MYH11 |
miR-377-3p, miR-495-3p, miR-29b-3p |
miR-126-5p |
|
AML |
RUNX1/RUNX1T1 |
miR-4516, miR-4739 |
- |
[104] |
CN-AML |
NPM1/HOX(+) |
miR-10a, miR-10b, miR-196a-1 |
- |
[112] |
AML (no APL) |
14q32 |
- |
miR-136-5p, miR-654-3p, miR-381-3p, miR -376a-3p, miR-377-3p, miR-376c-3p, miR-495-3p |
[104] |
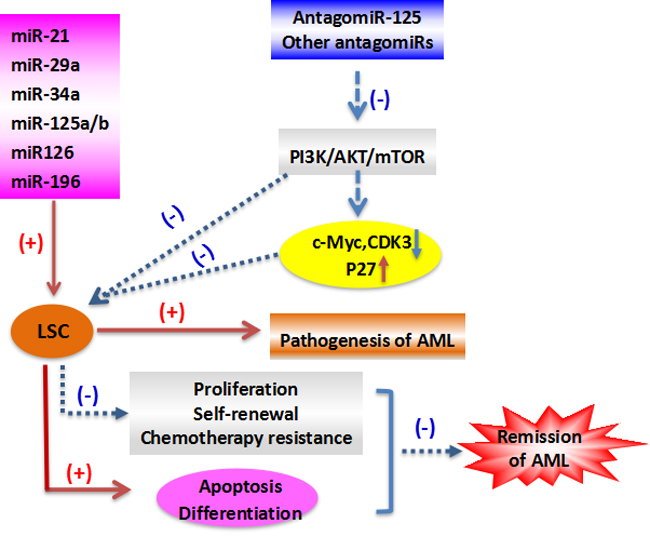
Figure 1: The role of LSC in the pathogenesis and remission of AML.
miRNA regulatory signaling pathways in AML
The associations of miRNAs with critical mutations and abnormal expressions of individual genes in AML have been nicely reviewed before [27]. It’s well recognized that miRNAs do not function through a single gene target and the combined regulation of many different genes determines the functionality of a miRNA. Recent progress sheds more lights on the systemic level miRNAs regulatory signaling networks in AML.
Pediatric AML patients with overexpressed granulocyte stimulating factor receptor class IV (G-CSFRIV), a major regulator of granulopoiesis [67], have been associated with defective differentiation and relapse risk. Zhang et al [68] identified that elevated miR-155 expression and reduced levels of miR-155 target genes (PU.1, GFI-1 and TP53INP1) were associated with a sustained Stat5 activation in G-CSF-stimulated HSPCs isolated from AML patients with G-CSFRIV overexpression. In addition, these HSPCs secreted more chemokine (C-C motif) ligand 2 (CCL2), a strong chemotactic factor for monocytes and macrophages, and the CCL2 levels were correlated with Stat5 activation and high miR-155 expression. miR-155 was found to indirectly regulate CCL2 expression [69] and CCL2 deficiency was shown to impair the secretion of G-CSF [70]. These observations clearly indicate the leukemogenic role of the G-CSF/G-CSFRIV−Stat5−miR-155−CCL2−G-CSF positive feedback loop in AML, and also pointed out the necessity in developing personalized effective anti-leukemia treatments for patients with increased G-CSFRIV, who are more susceptible to G-CSF-induced upregulation of miR-155 and subsequent de novo leukemogenicity or relapse (Figure 2).
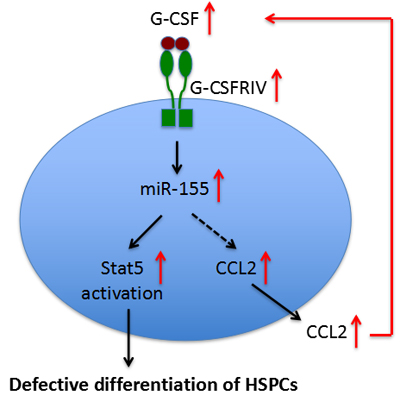
Figure 2: A positive-feedback mechanism involving miR-155 in defective differentiation of HSPCs in AML.
The two most common AML chromosome rearrangements generate the RUNX1-MTG8 (AML1-ETO) and CBFB-MYH11 fusion proteins. Both fusions are dominant negative on RUNX1 function. miR-223, one RUNX1 target, is critical for the establishment of granulocyte and monocyte lineages [71, 72]. The miR-222/221 gene cluster, also a RUNX1 target, regulates the kitproto-oncogene protein (KIT) receptor by targeting its 3’-UTR [73]. In (CBF)-AML samples with reduced miR-221 and miR-222 there is a concomitant up-regulation of KIT and KIT-induced proliferation [74]. KIT up-regulation was reported in 60-80% of all AML, including also non-CBF-AML. Fischer et al identified that miR-17, miR-18a, miR-20a and miR-93 all function as the CBF-AML fusion proteins in negative regulating their target RUNX1 and the RUNX1-miR-221-KIT axis [74]. miR-17 up-regulation is associated with the M5 subtype AML, which is frequently characterized by KIT up-regulation. miR-18a, miR-20a and miR-93 are frequently upregulated in distinct subtypes of non-CBF-AML [74]. Furthermore, this study found that increased KIT could delay myeloid differentiation. In the presence of factors that impair RUNX1 function/level this delay may act synergistically with deregulated of RUNX1-targets (e.g. miR-223) involved in differentiation of myeloid precursors. These data indicate that these miRNAs are linked to aberrant regulation of the network of RUNX1-miRNAs interactions underlying proliferation and myeloid differentiation in AML (Figure 3).
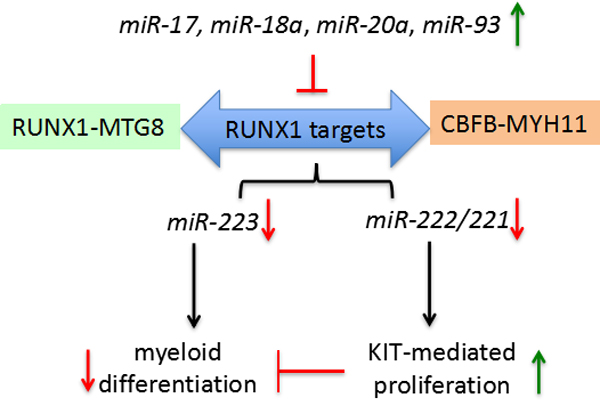
Figure 3: miRNAs (miR-17, miR-18a, miR-20a, miR-93) negatively regulate RUNX1 targets, including miR-223 and miR-222/221, in blocking myeloid differentiation by increasing KIT expression and enabling KIT-mediated proliferation.
Both gain- and loss-of-function in vivo studies of miR-126 in mouse models demonstrated that either enforced expression or knockout of miR-126 substantially promoted development of t(8;21) AML in mice [75]. miR-126 overexpression in mice more drastically reduced long-term survival and increased progression of leukemia stem/initiating cells (LSCs/LICs) through the AML1-ETO9a pathway than the miR-126 knockout. However, miR-126 knockout leukemia cells were significantly more responsive to standard chemotherapy. miR-126 overexpression leads to a gene expression profile similar to LSCs/LICs and/or primitive hematopoietic stem/progenitor cells by targeting ERRFI1 and SPRED1. miR-126 knockout, on the other hand, yields a gene expression profile more similar to that seen in more differentiated hematopoietic progenitor cells presumably by inducing FZD7 expression. Together, these findings show that miR-126 plays a dual role in leukemia, and uncovers a new layer of miRNA regulation in cancer (Figure 4).
MLL-AF9 (alias KMT2A-MLLT3), a gene fusion product of chromosomal translocation t(9;11)(p22;q23), causes acute leukemia in mice [76]. It is the most common fusion gene found in infant cases of AML and correlates with monoblastic AML. 41 genes and 21 miRNAs with MLL-AF9-dependent expression were identified in endogenous MLL-AF9 knockdown THP1 cells, derived from an AML patient [77, 78], including mediators of MLL-AF9 leukemogenic effects. The miRNA-target genes were validated and gene ontology analysis implicated the up-regulated miRNAs’ targets in cell cycle progression and the downregulated miRNAs’ targets in cell cycle checkpoints, indicating that MLL-AF9 via miRNAs promotes cell cycle progression and inhibits checkpoints. Stem cell maintenance and development and the stress response were among the processes predicted to be affected by miRNAs with MLL-AF9 expression. Of these, 5 miRNAs (miR-137, miR-214-3p, miR-301a-3p, miR-330-3p and miR-383-5p) affect all four categories, linking them strongly to leukemia pathogenesis and highlighting their potential as therapeutic targets. MLL-AF9 may cause leukemogenic effects by regulating both gene and miRNA expression. miRNA target genes were enriched in gene products related to the cell cycle, Wnt signaling, cell adhesion, myeloid differentiation, and the cellular stress response. These processes are likely directly regulated by MLL-AF9 through downstream miRNA expression.
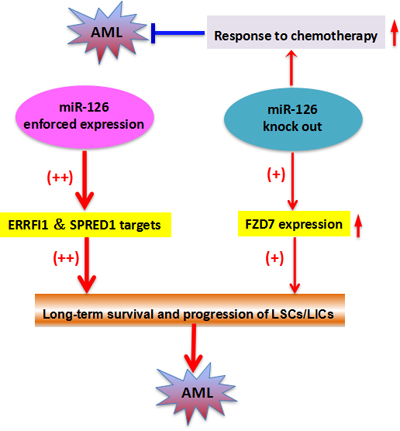
Figure 4: The two-faceted roles of miR126 in AML.
miRNAs as potential therapeutic targets for AML
miRNAs hold great promise as potential cancer therapeutics but safe and specific delivery to the tumor sites remains the principle hurdle to their clinical implementation [79]. For the heterogeneous AMLs, it is difficult to optimize targeted therapies for each case. Instead, finding common downstream miRNA targets of the fusion gene related to AML might yield novel therapeutic options. Using miRNA-based therapeutics whole molecular signaling network, which act synergistically leading to a disease such as AML, can be targeted.
Immunotherapy has become an increasingly appealing therapeutic strategy for cancer patients. Blocking inhibitory immune checkpoint molecules (such as PD-1/PD-L1) enhance the immune response to tumors. An inverse correlation between PD-L1 and miR-34a expression has been observed in AML samples [80]. Over-expression of miR-34a in AML cell lines reduced PD-L1 mRNA and cell surface expression of PD-L1 protein. Further studies identified that the PD-L1 mRNA is a direct target of miR-34a, and PD-L1 specific T cell apoptosis could be reduced following miR-34a transfection, suggesting the potential of miR-34a mimics in cancer immunotherapy. Furthermore, a positive feedback between PD-L1 expression and AKT (also known as PKB, Protein Kinase B) activation was observed in the AML cell lines [80], and the activation of AKT-mTOR pathway augmented immune escape by driving expression of PD-L1 [81]. Increased AKT-mTOR pathway activity promotes survival of leukemia stem cells and early committed leukemic precursors and its inhibition provides a potential therapeutic approach. Studies demonstrated that miR34a over-expression profoundly decreased AKT phosphorylation level in multiple cancer types [82, 83], indicating the promising efficacy of miR-34a mimics in inhibiting both PD-L1 and AKT activity as an AML immunotherapy. Activation of PI3K/Akt/mTOR is activated in more than 60 % of AML patients and is associated with decreased overall survival [84]. In AML cell lines with AML1/ETO fusion protein, the miR-193a is down-regulated, and treatment of a leukemia mouse model with synthetic miR-193a results in significant tumor regression and reduction of AML1/ETO, CCND1, MDM2 as well as concomitant up-regulation of phosphatase and tensin homolog (PTEN) [85]. The miR29 family members miR29a, -29b and -29c function as tumor suppressors in AML, regulating cell proliferation and apoptosis by the inhibiting AKT2 and CCND2, and show therapeutic potential for AML [22].
Inhibition of the ErbB pathway provides another potential new therapeutic target for leukemia [86, 87]. In AML NB4 cells ectopically expressing miR-125a ErbB pathway was significantly activated. In AML, ErbB receptors ErbB-1 and ErbB-3 were the main mediators of miR-125a, and the phosphorylation of the downstream effectors AKT and MAPK played key roles in driving proliferation and survival of the AML blasts. These findings indicate the potential of miR-125a as a new therapeutic target for miR-125a-low AML [10]. Furthermore, analysis of the upstream region of miR-125a and bisulfite sequencing revealed that miR-125a is suppressed by methylation in AML. The standard chemotherapy agent for AML, decitabine, also a de-methylating agent could significantly restore the expression of miR-125a through suppressing the global methylation in AML cell lines. It was known that elevated expression of ecotropic viral integration site 1 (Evi1) gene is associated with unfavorable prognosis in AML. In all, 5-10% of AML patients show Evi1 up-regulation. While Evi1-low patients showed >80% overall survival at 5 years, Evi1-high patients showed <60%. A recent study showed that miR-133 binding to Evi1 increases drug sensitivity specifically in Evi1 expressing leukemic cells, suggesting that miR-133 may be a promising therapeutic target for the Evi1 dys-regulated leukemia with poor prognosis [88]. Similarly, reduced expression of the miR-9 target Hes1 has been to be an indicator of poor prognosis for AML [89]. Coincidentally, high expression of miRNA-9 was identified in the leukemic progenitor cells (LPs) from CD34+ adult CN-AMLs [90]. Knockdown of miR-9 in a mouse leukemia model suppressed AML cell proliferation, decreased leukemic cell counts in blood and bone marrow, reduced splenomegaly, and increased survival times, indicating that miR-9 is a potential target for treatment of AML [90].
Su et al [37] demonstrated that miR-181 inhibition is a potential new treatment strategy for AML. miR-181 inhibits differentiation of AML cells into granulocytes and macrophages by down-regulating their direct targets PRKCD, CTDSPL, and CAMKK1 and then affecting the PRKCD-P38-C/EBPα pathway and reducing pRB phosphorylation. Knockdown of miR-181 in cultured bone marrow blasts from AML patients partially reversed blockage of myeloid differentiation. In AML CD34+ HSPC xenograft mice, inhibition of miR-181 increased differentiation of myeloid progenitors, reduced engraftment and infiltration of leukemic HSPCs into bone marrow and spleen, and ameliorated symptoms of leukemia. These findings suggest that miR-181 is a potential target for AML therapy. However, the temporal expression of miR-181b is also critical in determining the chemo-sensitivity in AML [91]. Human multidrug-resistant leukemia cells and relapsed/refractory AML patients have significantly lower levels of miR-181b. Leukemia cells overexpressing miR-181b, on the other hand, are more sensitive to cytotoxic chemotherapeutic agents and prone to drug-induced apoptosis. One of the underlying mechanisms is that miR-181b binds to the 3’-untranslated regions of HMGB1 and Mcl-1 and inhibits their expression. Direct suppression of HMGB1 sensitizes multidrug-resistant leukemia cells to chemotherapy and induces apoptotic cell death. These results demonstrate critical but complex roles of miR-181 in AML, and more importantly, the temporal changes of miRNA expression and function during AML progression highlight a rigorous evaluation of miRNA-based therapy in AML. A comprehensive list of potential miRNA targets for AML therapy is summarized in Table 2.
Table 2: Potential miRNAs as treatment targets for AML
miRNA |
target genes or pathways |
References |
miR-141 |
PI3K/Akt/mTOR |
[113] |
miR-125a |
ErbB pathway |
[10] |
miR-125b |
Mcl-1 |
|
miR-22-3p, let-7e-5p |
PLK1 |
[114] |
miR-34a |
PD-L1 |
[80] |
miR-638 |
CDK2 |
[34] |
miR-181a, b and c |
PRKCD, CTDSPL and CAMKK1 |
|
miR-191-5p, miR-142-3p |
PPP2R2A |
|
miR-181b |
MDR |
[36] |
miR-21, miR-196b |
HOX |
[98] |
miR-29a/b/c |
Dnmts |
The prognostic value of miRNAs in AML
Both single miRNAs and panel of miRNAs have potential prognostic value complementing information gained from cytogenetics, gene mutations, and altered gene expression. Chen and his colleagues reported that reduced miR-124-1 expression is commonly found in AML patients, most frequently in those with t(15;17). Patients with reduced miR-124-1 expression tended to have slightly longer overall survival and relapse-free survival than those without reduced miR-124-1, suggesting that miR-124-1 down-regulation predicts favorable survival in AML [92]. Liu et al reported that the down-regulation of circulating miR-328 in AML patients correlates with poor clinical outcome, and may provide a diagnostic and prognostic biomarker [93]. Eisfeld et al reported miR-3151 as an independent prognostic factor for outcome in older CN-AML patients [94]. In 179 CN-AML patients over 60 years of age high miR-3151 expression was associated with shorter disease-free and overall survival; high expressing patients also had a lower CR rate compared with low expressers. In other work, patients with increased miR-3151 had shorter overall and leukemia-free survival and higher cumulative incidence of relapse [95]. Moreover, multivariate analysis demonstrated that the combined observation of miR-3151 and its host gene BAALC improved this prognostic stratification. It was reported that patients with low levels of both miR-3151 and BAALC had better outcomes than patients with increased levels of either marker [95]. The down-regulation of miR-181 was associated with leukemia invasiveness, and miR-181 has been well studied to be a prognostic predictor of AML [6, 36, 96, 97]. It’s shown that the expression of miR-181a and miR-181b was positively associated with good clinical outcome in molecular high-risk CN-AML and inversely associated with the risk of an event, such as failure to achieve complete remission, relapse, and even death.
In a study of intermediate-risk cytogenetic AML (IR-AML), increased miR-196b or miR-644 were linked to shorter overall survival times, while reduced miR-135a and miR-409-3p were linked increased risk of relapse [98]. miR-135a, miR-196b, miR-409-3p, and mir-644 were identified as prognostic markers for IR-AML, while miR-122, miR-133b, miR-148a, and miR-409-3p were found to be be valuable in prognosis of AML and linked to adverse outcomes for older CN-AML patients [8]. Lin et al reported that the high let-7a-2-3p and low miR-188-5p expression could be potentially used as favorably prognostic biomarkers independently in CN-AML patients [9]. Recently, a study reported that the high serum miRNA-335 predicts poor outcomes and aggressive tumor progression in pediatric AML and may provide a prognostic indicator [99]. In another study, it was reported that miR-212 is significantly associated with increased overall, event-free, and relapse-free survival. Moreover, it was found that the prognostic significance and the prevalence of high miR-212 did not correlate with specific cytogenetic subtypes of AML, indicating that miR-212 may improve the current prognostic risk stratification of mixed AML including normal karyotype AML and AML with cytogenetic and molecular abnormalities [100].
Recently, there are many other miRNAs were demonstrated to correlate with clinical outcomes of de novo adult AML patients and pediatric AML patients (Table 3). However, each study reported unique miRNAs and each miRNAs showed significant prognostic values in certain AML populations. These may further testify the complex and heterogeneous property of AMLs, and the lack of uniformity among all the studies could be explained by the heterogeneity in mutational profiles among AML patient cohorts. Taken together, miRNAs as prognostication of AML subtypes or subgroups warrants further study.
Table 3: miRNAs that implicate clinical prognosis in AML
Prognosis status |
Up-regulated miRNA |
Down-regulated miRNA |
References |
Good prognosis |
miR-191, miR-199a, miR-30a, miR-30b, miR-30c, miR-181a, miR-181b, let-7a-2-3p |
miR-124-1, miR-125, miR-126 |
|
Bad prognosis |
miR-124, miR-128-1, miR-194, miR-219-5p, miR-220a, miR-320, miR-196b, miR-644, miR-3151, miR-146, miR-133b, miR-148a, miR-122, miR-409-3p, miR-126-5p/3p, miR-24, miR-331, miR-378 |
miR-150, miR-342, miR-135a, miR-409-3p, miR-96, miR-188-5p |
Concluding Remarks
miRNAs have emerged as major players in gene regulation underlying various aspects of AMLs. miRNAs and their regulatory signaling pathways must be placed alongside traditional protein-coding oncogenes and tumor suppressors if we aim to achieve a more thorough understanding of the complex mechanisms of malignant AML transformation. We are aware that a systems biological understanding of the miRNA regulatory networks is still superficial and limited. With in-depth studies, further clarifying the expression, function and regulatory mechanism of miRNAs will provide promising strategies for AML treatment.
Acknowledgments
This work is supported by the National Natural Science Foundation of China (81300426, 30771103, 81172792, 81573467, 81373670), the project for Shandong Medical and Health Science and Technology Plan project (2013WS0365), the Natural Science Foundation of Shandong Province of China (ZR2015YL028, 2015ZRC03102), the “twelfth five year” National Science and Technology Support program (2013BAI07B02). Shandong key project for Transformation of Results with Independent Innovation (2014CGZH1313, 2013ZHZX2A0405), and the Project for Laureate of Taishan Scholar (No. ts201511075). The Innovation Project of Shandong Academy of Medical Sciences. We greatly appreciate the assistance of Huiyuan Zhang in the Department of Immunology of the University of Texas MD Anderson Cancer Center for the revision of this review.
Conflicts of interests
The authors declare that they have no financial competing interests.
Authors’ Contributions
XL and GJ conceived the outline of the review and revised the manuscript. QL and BW drafted and revised the manuscript. All authors read and approved the final content.
References
1. Kim VN. Small RNAs: classification, biogenesis, and function. Molecules and cells. 2005; 19(1):1-15.
2. Garzon R, Volinia S, Liu CG, Fernandez-Cymering C, Palumbo T, Pichiorri F, Fabbri M, Coombes K, Alder H, Nakamura T, Flomenberg N, Marcucci G, Calin GA, Kornblau SM, Kantarjian H, Bloomfield CD, et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood. 2008; 111(6):3183-3189.
3. Jongen-Lavrencic M, Sun SM, Dijkstra MK, Valk PJ and Lowenberg B. MicroRNA expression profiling in relation to the genetic heterogeneity of acute myeloid leukemia. Blood. 2008; 111(10):5078-5085.
4. Fuster O, Llop M, Dolz S, Garcia P, Such E, Ibanez M, Luna I, Gomez I, Lopez M, Cervera J, Montesinos P, Moscardo F, Cordon L, Solves P, de Juan I, Palanca S, et al. Adverse prognostic value of MYBL2 overexpression and association with microRNA-30 family in acute myeloid leukemia patients. Leukemia research. 2013; 37(12):1690-1696.
5. Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W and Tuschl T. Identification of tissue-specific microRNAs from mouse. Current biology. 2002; 12(9):735-739.
6. Marcucci G, Radmacher MD, Maharry K, Mrozek K, Ruppert AS, Paschka P, Vukosavljevic T, Whitman SP, Baldus CD, Langer C, Liu CG, Carroll AJ, Powell BL, Garzon R, Croce CM, Kolitz JE, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. The New England journal of medicine. 2008; 358(18):1919-1928.
7. Zhao J, Lu Q, Zhu J, Fu J and Chen YX. Prognostic value of miR-96 in patients with acute myeloid leukemia. Diagnostic pathology. 2014; 9:76.
8. Niederwieser C, Kohlschmidt J, Volinia S, Whitman SP, Metzeler KH, Eisfeld AK, Maharry K, Yan P, Frankhouser D, Becker H, Schwind S, Carroll AJ, Nicolet D, Mendler JH, Curfman JP, Wu YZ, et al. Prognostic and biologic significance of DNMT3B expression in older patients with cytogenetically normal primary acute myeloid leukemia. Leukemia. 2015; 29(3):567-575.
9. Jinlong S, Lin F, Yonghui L, Li Y and Weidong W. Identification of let-7a-2-3p or/and miR-188-5p as prognostic biomarkers in cytogenetically normal acute myeloid leukemia. PloS one. 2015; 10(2):e0118099.
10. Ufkin ML, Peterson S, Yang X, Driscoll H, Duarte C and Sathyanarayana P. miR-125a regulates cell cycle, proliferation, and apoptosis by targeting the ErbB pathway in acute myeloid leukemia. Leukemia research. 2014; 38(3):402-410.
11. Bousquet M, Harris MH, Zhou B and Lodish HF. MicroRNA miR-125b causes leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(50):21558-21563.
12. Shaham L, Binder V, Gefen N, Borkhardt A and Izraeli S. MiR-125 in normal and malignant hematopoiesis. Leukemia. 2012; 26(9):2011-2018.
13. Huang K, Dong B, Wang Y, Tian T and Zhang B. MicroRNA-519 enhances HL60 human acute myeloid leukemia cell line proliferation by reducing the expression level of RNA-binding protein human antigen R. Molecular medicine reports. 2015; 12(6):7830-7836.
14. Whitman SP, Maharry K, Radmacher MD, Becker H, Mrozek K, Margeson D, Holland KB, Wu YZ, Schwind S, Metzeler KH, Wen J, Baer MR, Powell BL, Carter TH, Kolitz JE, Wetzler M, et al. FLT3 internal tandem duplication associates with adverse outcome and gene- and microRNA-expression signatures in patients 60 years of age or older with primary cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Blood. 2010; 116(18):3622-3626.
15. Tohda S. NOTCH signaling roles in acute myeloid leukemia cell growth and interaction with other stemness-related signals. Anticancer Res. 2014; 34(11):6259-6264.
16. Vargas Romero P, Cialfi S, Palermo R, De Blasio C, Checquolo S, Bellavia D, Chiaretti S, Foa R, Amadori A, Gulino A, Zardo G, Talora C and Screpanti I. The deregulated expression of miR-125b in acute myeloid leukemia is dependent on the transcription factor C/EBPalpha. Leukemia. 2015; 29(12):2442-2445.
17. Bai J, Guo A, Hong Z and Kuai W. Upregulation of microRNA-100 predicts poor prognosis in patients with pediatric acute myeloid leukemia. Onco Targets Ther. 2012; 5:213-219.
18. Zheng YS, Zhang H, Zhang XJ, Feng DD, Luo XQ, Zeng CW, Lin KY, Zhou H, Qu LH, Zhang P and Chen YQ. MiR-100 regulates cell differentiation and survival by targeting RBSP3, a phosphatase-like tumor suppressor in acute myeloid leukemia. Oncogene. 2012; 31(1):80-92.
19. Wang F, Wang XS, Yang GH, Zhai PF, Xiao Z, Xia LY, Chen LR, Wang Y, Wang XZ, Bi LX, Liu N, Yu Y, Gao D, Huang BT, Wang J, Zhou DB, et al. miR-29a and miR-142-3p downregulation and diagnostic implication in human acute myeloid leukemia. Molecular biology reports. 2012; 39(3):2713-2722.
20. Wang XS, Gong JN, Yu J, Wang F, Zhang XH, Yin XL, Tan ZQ, Luo ZM, Yang GH, Shen C and Zhang JW. MicroRNA-29a and microRNA-142-3p are regulators of myeloid differentiation and acute myeloid leukemia. Blood. 2012; 119(21):4992-5004.
21. Dahlhaus M, Roolf C, Ruck S, Lange S, Freund M and Junghanss C. Expression and prognostic significance of hsa-miR-142-3p in acute leukemias. Neoplasma. 2013; 60(4):432-438.
22. Gong JN, Yu J, Lin HS, Zhang XH, Yin XL, Xiao Z, Wang F, Wang XS, Su R, Shen C, Zhao HL, Ma YN and Zhang JW. The role, mechanism and potentially therapeutic application of microRNA-29 family in acute myeloid leukemia. Cell Death Differ. 2014; 21(1):100-112.
23. Han YC, Park CY, Bhagat G, Zhang J, Wang Y, Fan JB, Liu M, Zou Y, Weissman IL and Gu H. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. The Journal of experimental medicine. 2010; 207(3):475-489.
24. Teichler S, Illmer T, Roemhild J, Ovcharenko D, Stiewe T and Neubauer A. MicroRNA29a regulates the expression of the nuclear oncogene Ski. Blood. 2011; 118(7):1899-1902.
25. Jiang W, Min J, Sui X, Qian Y, Liu Y, Liu Z, Zhou H, Li X and Gong Y. MicroRNA-26a-5p and microRNA-23b-3p up-regulate peroxiredoxin III in acute myeloid leukemia. Leukemia & lymphoma. 2015; 56(2):460-471.
26. Ito K and Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014; 15(4):243-256.
27. Marcucci G, Mrozek K, Radmacher MD, Garzon R and Bloomfield CD. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood. 2011; 117(4):1121-1129.
28. Ramsingh G, Jacoby MA, Shao J, De Jesus Pizzaro RE, Shen D, Trissal M, Getz AH, Ley TJ, Walter MJ and Link DC. Acquired copy number alterations of miRNA genes in acute myeloid leukemia are uncommon. Blood. 2013; 122(15):e44-51.
29. Trissal MC, DeMoya RA, Schmidt AP and Link DC. MicroRNA-223 regulates granulopoiesis but is not required for HSC maintenance in mice. PloS one. 2015; 10(3):e0119304.
30. Chen P, Price C, Li Z, Li Y, Cao D, Wiley A, He C, Gurbuxani S, Kunjamma RB, Huang H, Jiang X, Arnovitz S, Xu M, Hong GM, Elkahloun AG, Neilly MB, et al. miR-9 is an essential oncogenic microRNA specifically overexpressed in mixed lineage leukemia-rearranged leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110(28):11511-11516.
31. Emmrich S, Katsman-Kuipers JE, Henke K, Khatib ME, Jammal R, Engeland F, Dasci F, Zwaan CM, den Boer ML, Verboon L, Stary J, Baruchel A, de Haas V, Danen-van Oorschot AA, Fornerod M, Pieters R, et al. miR-9 is a tumor suppressor in pediatric AML with t(8;21). Leukemia. 2014; 28(5):1022-1032.
32. Nowek K, Sun SM, Bullinger L, Bindels EM, Exalto C, Dijkstra MK, van Lom K, Dohner H, Erkeland SJ, Lowenberg B and Jongen-Lavrencic M. Aberrant expression of miR-9/9* in myeloid progenitors inhibits neutrophil differentiation by post-transcriptional regulation of ERG. Leukemia. 2016; 30(1):229-237.
33. Alemdehy MF, Haanstra JR, de Looper HW, van Strien PM, Verhagen-Oldenampsen J, Caljouw Y, Sanders MA, Hoogenboezem R, de Ru AH, Janssen GM, Smetsers SE, Bierings MB, van Veelen PA, von Lindern M, Touw IP and Erkeland SJ. ICL-induced miR139-3p and miR199a-3p have opposite roles in hematopoietic cell expansion and leukemic transformation. Blood. 2015; 125(25):3937-3948.
34. Li D, Wang Q, Liu C, Duan H, Zeng X, Zhang B, Li X, Zhao J, Tang S, Li Z, Xing X, Yang P, Chen L, Zeng J, Zhu X, Zhang S, et al. Aberrant expression of miR-638 contributes to benzo(a)pyrene-induced human cell transformation. Toxicological sciences. 2012; 125(2):382-391.
35. Lin Y, Li D, Liang Q, Liu S, Zuo X, Li L, Sun X, Li W, Guo M and Huang Z. miR-638 regulates differentiation and proliferation in leukemic cells by targeting cyclin-dependent kinase 2. The Journal of biological chemistry. 2015; 290(3):1818-1828.
36. Weng H, Lal K, Yang FF and Chen J. The pathological role and prognostic impact of miR-181 in acute myeloid leukemia. Cancer genetics. 2015; 208(5):225-229.
37. Su R, Lin HS, Zhang XH, Yin XL, Ning HM, Liu B, Zhai PF, Gong JN, Shen C, Song L, Chen J, Wang F, Zhao HL, Ma YN, Yu J and Zhang JW. MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene. 2015; 34(25):3226-3239.
38. Undi RB, Kandi R and Gutti RK. MicroRNAs as Haematopoiesis Regulators. Advances in hematology. 2013; 2013:695754.
39. Hu W, Dooley J, Chung SS, Chandramohan D, Cimmino L, Mukherjee S, Mason CE, de Strooper B, Liston A and Park CY. miR-29a maintains mouse hematopoietic stem cell self-renewal by regulating Dnmt3a. Blood. 2015; 125(14):2206-2216.
40. Lechman ER, Gentner B, van Galen P, Giustacchini A, Saini M, Boccalatte FE, Hiramatsu H, Restuccia U, Bachi A, Voisin V, Bader GD, Dick JE and Naldini L. Attenuation of miR-126 activity expands HSC in vivo without exhaustion. Cell stem cell. 2012; 11(6):799-811.
41. Mehta A, Zhao JL, Sinha N, Marinov GK, Mann M, Kowalczyk MS, Galimidi RP, Du X, Erikci E, Regev A, Chowdhury K and Baltimore D. The MicroRNA-132 and MicroRNA-212 Cluster Regulates Hematopoietic Stem Cell Maintenance and Survival with Age by Buffering FOXO3 Expression. Immunity. 2015; 42(6):1021-1032.
42. Ebert MS and Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012; 149(3):515-524.
43. Ooi AG, Sahoo D, Adorno M, Wang Y, Weissman IL and Park CY. MicroRNA-125b expands hematopoietic stem cells and enriches for the lymphoid-balanced and lymphoid-biased subsets. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(50):21505-21510.
44. O’Connell RM, Chaudhuri AA, Rao DS, Gibson WS, Balazs AB and Baltimore D. MicroRNAs enriched in hematopoietic stem cells differentially regulate long-term hematopoietic output. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(32):14235-14240.
45. Guo S, Lu J, Schlanger R, Zhang H, Wang JY, Fox MC, Purton LE, Fleming HH, Cobb B, Merkenschlager M, Golub TR and Scadden DT. MicroRNA miR-125a controls hematopoietic stem cell number. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(32):14229-14234.
46. Kreso A and Dick JE. Evolution of the cancer stem cell model. Cell stem cell. 2014; 14(3):275-291.
47. Holtz M, Forman SJ and Bhatia R. Growth factor stimulation reduces residual quiescent chronic myelogenous leukemia progenitors remaining after imatinib treatment. Cancer research. 2007; 67(3):1113-1120.
48. van Rhenen A, Feller N, Kelder A, Westra AH, Rombouts E, Zweegman S, van der Pol MA, Waisfisz Q, Ossenkoppele GJ and Schuurhuis GJ. High stem cell frequency in acute myeloid leukemia at diagnosis predicts high minimal residual disease and poor survival. Clinical cancer research. 2005; 11(18):6520-6527.
49. Costello RT, Mallet F, Gaugler B, Sainty D, Arnoulet C, Gastaut JA and Olive D. Human acute myeloid leukemia CD34+/CD38- progenitor cells have decreased sensitivity to chemotherapy and Fas-induced apoptosis, reduced immunogenicity, and impaired dendritic cell transformation capacities. Cancer research. 2000; 60(16):4403-4411.
50. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, Canty AJ, Danska JS, Bohlander SK, Buske C, Minden MD, Golub TR, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nature medicine. 2011; 17(9):1086-1093.
51. Terwijn M, Kelder A, Snel AN, Rutten AP, Scholten WJ, Oussoren YJ, Van De Loosdrecht AA, Zweegman S, Ossenkoppele GJ and Schuurhuis GJ. Minimal residual disease detection defined as the malignant fraction of the total primitive stem cell compartment offers additional prognostic information in acute myeloid leukaemia. International journal of laboratory hematology. 2012; 34(4):432-441.
52. Testa U and Pelosi E. MicroRNAs expressed in hematopoietic stem/progenitor cells are deregulated in acute myeloid leukemias. Leukemia & lymphoma. 2015; 56(5):1466-1474.
53. Li Z, Lu J, Sun M, Mi S, Zhang H, Luo RT, Chen P, Wang Y, Yan M, Qian Z, Neilly MB, Jin J, Zhang Y, Bohlander SK, Zhang DE, Larson RA, et al. Distinct microRNA expression profiles in acute myeloid leukemia with common translocations. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105(40):15535-15540.
54. Marcucci G, Radmacher MD, Mrozek K and Bloomfield CD. MicroRNA expression in acute myeloid leukemia. Current hematologic malignancy reports. 2009; 4(2):83-88.
55. Wong P, Iwasaki M, Somervaille TC, Ficara F, Carico C, Arnold C, Chen CZ and Cleary ML. The miR-17-92 microRNA polycistron regulates MLL leukemia stem cell potential by modulating p21 expression. Cancer research. 2010; 70(9):3833-3842.
56. Velu CS, Chaubey A, Phelan JD, Horman SR, Wunderlich M, Guzman ML, Jegga AG, Zeleznik-Le NJ, Chen J, Mulloy JC, Cancelas JA, Jordan CT, Aronow BJ, Marcucci G, Bhat B, Gebelein B, et al. Therapeutic antagonists of microRNAs deplete leukemia-initiating cell activity. The Journal of clinical investigation. 2014; 124(1):222-236.
57. Song SJ, Ito K, Ala U, Kats L, Webster K, Sun SM, Jongen-Lavrencic M, Manova-Todorova K, Teruya-Feldstein J, Avigan DE, Delwel R and Pandolfi PP. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell stem cell. 2013; 13(1):87-101.
58. Wang Y, Cheng Q, Liu J and Dong M. Leukemia Stem Cell-Released Microvesicles Promote the Survival and Migration of Myeloid Leukemia Cells and These Effects Can Be Inhibited by MicroRNA34a Overexpression. Stem cells international. 2016; 2016:9313425.
59. Lechman ER, Gentner B, Ng SW, Schoof EM, van Galen P, Kennedy JA, Nucera S, Ciceri F, Kaufmann KB, Takayama N, Dobson SM, Trotman-Grant A, Krivdova G, Elzinga J, Mitchell A, Nilsson B, et al. miR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer cell. 2016; 29(4):602-606.
60. de Leeuw DC, Denkers F, Olthof MC, Rutten AP, Pouwels W, Schuurhuis GJ, Ossenkoppele GJ and Smit L. Attenuation of microRNA-126 expression that drives CD34+38- stem/progenitor cells in acute myeloid leukemia leads to tumor eradication. Cancer research. 2014; 74(7):2094-2105.
61. Dorrance AM, Neviani P, Ferenchak GJ, Huang X, Nicolet D, Maharry KS, Ozer HG, Hoellarbauer P, Khalife J, Hill EB, Yadav M, Bolon BN, Lee RJ, Lee LJ, Croce CM, Garzon R, et al. Targeting leukemia stem cells in vivo with antagomiR-126 nanoparticles in acute myeloid leukemia. Leukemia. 2015; 29(11):2143-2153.
62. Miyata Y, Liu Y, Jankovic V, Sashida G, Lee JM, Shieh JH, Naoe T, Moore M and Nimer SD. Cyclin C regulates human hematopoietic stem/progenitor cell quiescence. Stem cells. 2010; 28(2):308-317.
63. Cammarata G, Augugliaro L, Salemi D, Agueli C, La Rosa M, Dagnino L, Civiletto G, Messana F, Marfia A, Bica MG, Cascio L, Floridia PM, Mineo AM, Russo M, Fabbiano F and Santoro A. Differential expression of specific microRNA and their targets in acute myeloid leukemia. American journal of hematology. 2010; 85(5):331-339.
64. Isken F, Steffen B, Merk S, Dugas M, Markus B, Tidow N, Zuhlsdorf M, Illmer T, Thiede C, Berdel WE, Serve H and Muller-Tidow C. Identification of acute myeloid leukaemia associated microRNA expression patterns. British journal of haematology. 2008; 140(2):153-161.
65. Trumpp A, Essers M and Wilson A. Awakening dormant haematopoietic stem cells. Nature reviews Immunology. 2010; 10(3):201-209.
66. Raffel S and Trumpp A. miR-126 Drives Quiescence and Self-Renewal in Leukemic Stem Cells. Cancer cell. 2016; 29(2):133-135.
67. Ehlers S, Herbst C, Zimmermann M, Scharn N, Germeshausen M, von Neuhoff N, Zwaan CM, Reinhardt K, Hollink IH, Klusmann JH, Lehrnbecher T, Roettgers S, Stary J, Dworzak M, Welte K, Creutzig U, et al. Granulocyte colony-stimulating factor (G-CSF) treatment of childhood acute myeloid leukemias that overexpress the differentiation-defective G-CSF receptor isoform IV is associated with a higher incidence of relapse. Journal of clinical oncology. 2010; 28(15):2591-2597.
68. Zhang H, Goudeva L, Immenschuh S, Schambach A, Skokowa J, Eiz-Vesper B, Blasczyk R and Figueiredo C. miR-155 is associated with the leukemogenic potential of the class IV granulocyte colony-stimulating factor receptor in CD34(+) progenitor cells. Molecular medicine. 2014; 20:736-746.
69. Nazari-Jahantigh M, Wei Y, Noels H, Akhtar S, Zhou Z, Koenen RR, Heyll K, Gremse F, Kiessling F, Grommes J, Weber C and Schober A. MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in macrophages. The Journal of clinical investigation. 2012; 122(11):4190-4202.
70. Strecker JK, Minnerup J, Gess B, Ringelstein EB, Schabitz WR and Schilling M. Monocyte chemoattractant protein-1-deficiency impairs the expression of IL-6, IL-1beta and G-CSF after transient focal ischemia in mice. PloS one. 2011; 6(10):e25863.
71. Ismail N, Wang Y, Dakhlallah D, Moldovan L, Agarwal K, Batte K, Shah P, Wisler J, Eubank TD, Tridandapani S, Paulaitis ME, Piper MG and Marsh CB. Macrophage microvesicles induce macrophage differentiation and miR-223 transfer. Blood. 2013; 121(6):984-995.
72. Rodriguez-Ubreva J, Ciudad L, van Oevelen C, Parra M, Graf T and Ballestar E. C/EBPa-mediated activation of microRNAs 34a and 223 inhibits Lef1 expression to achieve efficient reprogramming into macrophages. Mol Cell Biol. 2014; 34(6):1145-1157.
73. Felli N, Fontana L, Pelosi E, Botta R, Bonci D, Facchiano F, Liuzzi F, Lulli V, Morsilli O, Santoro S, Valtieri M, Calin GA, Liu CG, Sorrentino A, Croce CM and Peschle C. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(50):18081-18086.
74. Fischer J, Rossetti S, Datta A, Eng K, Beghini A and Sacchi N. miR-17 deregulates a core RUNX1-miRNA mechanism of CBF acute myeloid leukemia. Molecular cancer. 2015; 14:7.
75. Li Z, Chen P, Su R, Li Y, Hu C, Wang Y, Arnovitz S, He M, Gurbuxani S, Zuo Z, Elkahloun AG, Li S, Weng H, Huang H, Neilly MB, Wang S, et al. Overexpression and knockout of miR-126 both promote leukemogenesis. Blood. 2015; 126(17):2005-2015.
76. Chen W, Kumar AR, Hudson WA, Li Q, Wu B, Staggs RA, Lund EA, Sam TN and Kersey JH. Malignant transformation initiated by Mll-AF9: gene dosage and critical target cells. Cancer cell. 2008; 13(5):432-440.
77. Fleischmann KK, Pagel P, Schmid I and Roscher AA. RNAi-mediated silencing of MLL-AF9 reveals leukemia-associated downstream targets and processes. Molecular cancer. 2014; 13:27.
78. Fleischmann KK, Pagel P, von Frowein J, Magg T, Roscher AA and Schmid I. The leukemogenic fusion gene MLL-AF9 alters microRNA expression pattern and inhibits monoblastic differentiation via miR-511 repression. Journal of experimental & clinical cancer research. 2016; 35(1):9.
79. Naidu S, Magee P and Garofalo M. MiRNA-based therapeutic intervention of cancer. J Hematol Oncol. 2015; 8:68.
80. Wang X, Li J, Dong K, Lin F, Long M, Ouyang Y, Wei J, Chen X, Weng Y, He T and Zhang H. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cellular signalling. 2015; 27(3):443-452.
81. Lastwika KJ, Wilson W, 3rd, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, Liu LN, Gills JJ and Dennis PA. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer research. 2016; 76(2):227-238.
82. Rathod SS, Rani SB, Khan M, Muzumdar D and Shiras A. Tumor suppressive miRNA-34a suppresses cell proliferation and tumor growth of glioma stem cells by targeting Akt and Wnt signaling pathways. FEBS Open Bio. 2014; 4:485-495.
83. Cao W, Yang W, Fan R, Li H, Jiang J, Geng M, Jin Y and Wu Y. miR-34a regulates cisplatin-induce gastric cancer cell death by modulating PI3K/AKT/survivin pathway. Tumour Biol. 2014; 35(2):1287-1295.
84. Tamburini J, Elie C, Bardet V, Chapuis N, Park S, Broet P, Cornillet-Lefebvre P, Lioure B, Ugo V, Blanchet O, Ifrah N, Witz F, Dreyfus F, Mayeux P, Lacombe C and Bouscary D. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood. 2007; 110(3):1025-1028.
85. Li Y, Gao L, Luo X, Wang L, Gao X, Wang W, Sun J, Dou L, Li J, Xu C, Wang L, Zhou M, Jiang M, Zhou J, Caligiuri MA, Nervi C, et al. Epigenetic silencing of microRNA-193a contributes to leukemogenesis in t(8;21) acute myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood. 2013; 121(3):499-509.
86. Irwin ME, Nelson LD, Santiago-O’Farrill JM, Knouse PD, Miller CP, Palla SL, Siwak DR, Mills GB, Estrov Z, Li S, Kornblau SM, Hughes DP and Chandra J. Small molecule ErbB inhibitors decrease proliferative signaling and promote apoptosis in philadelphia chromosome-positive acute lymphoblastic leukemia. PloS one. 2013; 8(8):e70608.
87. Nordigarden A, Zetterblad J, Trinks C, Green H, Eliasson P, Druid P, Lotfi K, Ronnstrand L, Walz TM and Jonsson JI. Irreversible pan-ERBB inhibitor canertinib elicits anti-leukaemic effects and induces the regression of FLT3-ITD transformed cells in mice. Br J Haematol. 2011; 155(2):198-208.
88. Yamamoto H, Lu J, Oba S, Kawamata T, Yoshimi A, Kurosaki N, Yokoyama K, Matsushita H, Kurokawa M, Tojo A, Ando K, Morishita K, Katagiri K and Kotani A. miR-133 regulates Evi1 expression in AML cells as a potential therapeutic target. Scientific reports. 2016; 6:19204.
89. Tian C, Tang Y, Wang T, Yu Y, Wang X, Wang Y and Zhang Y. HES1 is an independent prognostic factor for acute myeloid leukemia. Onco Targets Ther. 2015; 8:899-904.
90. Tian C, You MJ, Yu Y, Zhu L, Zheng G and Zhang Y. MicroRNA-9 promotes proliferation of leukemia cells in adult CD34-positive acute myeloid leukemia with normal karyotype by downregulation of Hes1. Tumour biology. 2015.
91. Lu F, Zhang J, Ji M, Li P, Du Y, Wang H, Zang S, Ma D, Sun X and Ji C. miR-181b increases drug sensitivity in acute myeloid leukemia via targeting HMGB1 and Mcl-1. International journal of oncology. 2014; 45(1):383-392.
92. Chen XX, Lin J, Qian J, Qian W, Yang J, Ma JC, Deng ZQ, Xie D, An C, Tang CY and Qian Z. Dysregulation of miR-124-1 predicts favorable prognosis in acute myeloid leukemia. Clinical biochemistry. 2014; 47(1-2):63-66.
93. Liu L, Chen R, Zhang Y, Fan W, Xiao F and Yan X. Low expression of circulating microRNA-328 is associated with poor prognosis in patients with acute myeloid leukemia. Diagnostic pathology. 2015; 10:109.
94. Eisfeld AK, Marcucci G, Maharry K, Schwind S, Radmacher MD, Nicolet D, Becker H, Mrozek K, Whitman SP, Metzeler KH, Mendler JH, Wu YZ, Liyanarachchi S, Patel R, Baer MR, Powell BL, et al. miR-3151 interplays with its host gene BAALC and independently affects outcome of patients with cytogenetically normal acute myeloid leukemia. Blood. 2012; 120(2):249-258.
95. Diaz-Beya M, Brunet S, Nomdedeu J, Cordeiro A, Tormo M, Escoda L, Ribera JM, Arnan M, Heras I, Gallardo D, Bargay J, Queipo de Llano MP, Salamero O, Marti JM, Sampol A, Pedro C, et al. The expression level of BAALC-associated microRNA miR-3151 is an independent prognostic factor in younger patients with cytogenetic intermediate-risk acute myeloid leukemia. Blood cancer journal. 2015; 5:e352.
96. Li Z, Huang H, Li Y, Jiang X, Chen P, Arnovitz S, Radmacher MD, Maharry K, Elkahloun A, Yang X, He C, He M, Zhang Z, Dohner K, Neilly MB, Price C, et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood. 2012; 119(10):2314-2324.
97. Schwind S, Maharry K, Radmacher MD, Mrozek K, Holland KB, Margeson D, Whitman SP, Hickey C, Becker H, Metzeler KH, Paschka P, Baldus CD, Liu S, Garzon R, Powell BL, Kolitz JE, et al. Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. Journal of clinical oncology. 2010; 28(36):5257-5264.
98. Diaz-Beya M, Brunet S, Nomdedeu J, Tejero R, Diaz T, Pratcorona M, Tormo M, Ribera JM, Escoda L, Duarte R, Gallardo D, Heras I, Queipo de Llano MP, Bargay J, Monzo M, Sierra J, et al. MicroRNA expression at diagnosis adds relevant prognostic information to molecular categorization in patients with intermediate-risk cytogenetic acute myeloid leukemia. Leukemia. 2014; 28(4):804-812.
99. Lin X, Wang Z, Zhang R and Feng W. High serum microRNA-335 level predicts aggressive tumor progression and unfavorable prognosis in pediatric acute myeloid leukemia. Clinical & translational oncology. 2015; 17(5):358-364.
100. Sun SM, Rockova V, Bullinger L, Dijkstra MK, Dohner H, Lowenberg B and Jongen-Lavrencic M. The prognostic relevance of miR-212 expression with survival in cytogenetically and molecularly heterogeneous AML. Leukemia. 2013; 27(1):100-106.
101. Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP and Burge CB. Prediction of mammalian microRNA targets. Cell. 2003; 115(7):787-798.
102. Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R and Olson EN. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Developmental cell. 2008; 15(2):261-271.
103. Cattaneo C, Panzali A, Passi A, Borlenghi E, Lamorgese C, Petulla M, Re A, Caimi L and Rossi G. Serum posaconazole levels during acute myeloid leukaemia induction therapy: correlations with breakthrough invasive fungal infections. Mycoses. 2015; 58(6):362-367.
104. Cattaneo M, Pelosi E, Castelli G, Cerio AM, D’Angio A, Porretti L, Rebulla P, Pavesi L, Russo G, Giordano A, Turri J, Cicconi L, Lo-Coco F, Testa U and Biunno I. A miRNA Signature in Human Cord Blood Stem and Progenitor Cells as Potential Biomarker of Specific Acute Myeloid Leukemia Subtypes. Journal of cellular physiology. 2015; 230(8):1770-1780.
105. Daschkey S, Rottgers S, Giri A, Bradtke J, Teigler-Schlegel A, Meister G, Borkhardt A and Landgraf P. MicroRNAs distinguish cytogenetic subgroups in pediatric AML and contribute to complex regulatory networks in AML-relevant pathways. PloS one. 2013; 8(2):e56334.
106. Foran JM. New prognostic markers in acute myeloid leukemia: perspective from the clinic. Hematology. 2010; 2010:47-55.
107. Marcucci G, Maharry K, Radmacher MD, Mrozek K, Vukosavljevic T, Paschka P, Whitman SP, Langer C, Baldus CD, Liu CG, Ruppert AS, Powell BL, Carroll AJ, Caligiuri MA, Kolitz JE, Larson RA, et al. Prognostic significance of, and gene and microRNA expression signatures associated with, CEBPA mutations in cytogenetically normal acute myeloid leukemia with high-risk molecular features: a Cancer and Leukemia Group B Study. Journal of clinical oncology. 2008; 26(31):5078-5087.
108. Bernot KM, Nemer JS, Santhanam R, Liu S, Zorko NA, Whitman SP, Dickerson KE, Zhang M, Yang X, McConnell KK, Ahmed EH, Munoz MR, Siebenaler RF, Marcucci GG, Mundy-Bosse BL, Brook DL, et al. Eradicating acute myeloid leukemia in a Mll(PTD/wt):Flt3(ITD/wt) murine model: a path to novel therapeutic approaches for human disease. Blood. 2013; 122(23):3778-3783.
109. Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT, Jiang J, Schmittgen TD and Patel T. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006; 130(7):2113-2129.
110. Alcalay M, Tiacci E, Bergomas R, Bigerna B, Venturini E, Minardi SP, Meani N, Diverio D, Bernard L, Tizzoni L, Volorio S, Luzi L, Colombo E, Lo Coco F, Mecucci C, Falini B, et al. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood. 2005; 106(3):899-902.
111. Hackanson B, Bennett KL, Brena RM, Jiang J, Claus R, Chen SS, Blagitko-Dorfs N, Maharry K, Whitman SP, Schmittgen TD, Lubbert M, Marcucci G, Bloomfield CD and Plass C. Epigenetic modification of CCAAT/enhancer binding protein alpha expression in acute myeloid leukemia. Cancer research. 2008; 68(9):3142-3151.
112. Mrozek K, Marcucci G, Paschka P, Whitman SP and Bloomfield CD. Clinical relevance of mutations and gene-expression changes in adult acute myeloid leukemia with normal cytogenetics: are we ready for a prognostically prioritized molecular classification? Blood. 2007; 109(2):431-448.
113. Bertacchini J, Heidari N, Mediani L, Capitani S, Shahjahani M, Ahmadzadeh A and Saki N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cellular and molecular life sciences. 2015; 72(12):2337-2347.
114. Fontemaggi G, Bellissimo T, Donzelli S, Iosue I, Benassi B, Bellotti G, Blandino G and Fazi F. Identification of post-transcriptional regulatory networks during myeloblast-to-monocyte differentiation transition. RNA biology. 2015; 12(7):690-700.
115. Ruvolo PP, Ruvolo VR, Jacamo R, Burks JK, Zeng Z, Duvvuri SR, Zhou L, Qiu Y, Coombes KR, Zhang N, Yoo SY, Pan R, Hail N, Jr., Konopleva M, Calin G, Kornblau SM, et al. The protein phosphatase 2A regulatory subunit B55alpha is a modulator of signaling and microRNA expression in acute myeloid leukemia cells. Biochimica et biophysica acta. 2014; 1843(9):1969-1977.
116. Zhao JL, Rao DS, Boldin MP, Taganov KD, O’Connell RM and Baltimore D. NF-kappaB dysregulation in microRNA-146a-deficient mice drives the development of myeloid malignancies. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108(22):9184-9189.
117. Organista-Nava J, Gomez-Gomez Y, Illades-Aguiar B, Del Carmen Alarcon-Romero L, Saavedra-Herrera MV, Rivera-Ramirez AB, Garzon-Barrientos VH and Leyva-Vazquez MA. High miR-24 expression is associated with risk of relapse and poor survival in acute leukemia. Oncology reports. 2015; 33(4):1639-1649.
118. Butrym A, Rybka J, Baczynska D, Tukiendorf A, Kuliczkowski K and Mazur G. Expression of microRNA-331 can be used as a predictor for response to therapy and survival in acute myeloid leukemia patients. Biomarkers in medicine. 2015; 9(5):453-460.
119. Feng M, Li Z, Aau M, Wong CH, Yang X and Yu Q. Myc/miR-378/TOB2/cyclin D1 functional module regulates oncogenic transformation. Oncogene. 2011; 30(19):2242-2251.
120. Lee DY, Deng Z, Wang CH and Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104(51):20350-20355.
121. Fayyad-Kazan H, Bitar N, Najar M, Lewalle P, Fayyad-Kazan M, Badran R, Hamade E, Daher A, Hussein N, ElDirani R, Berri F, Vanhamme L, Burny A, Martiat P, Rouas R and Badran B. Circulating miR-150 and miR-342 in plasma are novel potential biomarkers for acute myeloid leukemia. Journal of translational medicine. 2013; 11:31.