
INTRODUCTION
Lung cancer is the main cause of cancer-related human death in Korea and worldwide [1–3]. The development of molecular targeted anticancer agents has resulted in better therapeutic responses in some types of lung cancer, including non-small cell lung cancer (NSCLC) with mutant epidermal growth factor receptor (EGFR) [4]. Nevertheless, drug resistance reduces the effectiveness of these targeted anticancer drugs [5], and the 5-year survival of lung cancer remains less than 20% [2]. In this regard, cancer chemoprevention, an approach to prevent, retard or reverse the carcinogenic process using dietary or synthetic chemicals [6, 7], can be an effective strategy for controlling lung cancer. Lung carcinogenesis is a multi-step, multi-path, and multi-focal process [6]. Hence, chemopreventive agents are anticipated to control each step of carcinogenesis to prevent normal cells from carcinogen-mediated genetic or epigenetic alterations and to suppress clonal expansion, proliferation, invasion, and metastasis of malignant cells. Numerous clinical trials investigating the effectiveness of lung cancer chemoprevention have been conducted using several natural or synthetic chemicals, including aspirin, β-carotene, vitamin E, and selenium [7]. However, the chemoprevention trials for lung cancer to date have shown negative or somewhat harmful effects; currently, no agents have been identified to be effective for the prevention of lung cancer. These findings suggest the necessity to discover novel strategies to control lung cancer.
Tobacco smoking (TS) is responsible for the majority of NSCLC. Therefore, understanding the signaling changes involved in tobacco carcinogen-mediated lung carcinogenesis is important to identify appropriate targets for lung cancer chemoprevention. Epidermal growth factor receptor (EGFR) is a plausible target [8]; however, EGFR mutations are primarily found in lung cancer in never-smokers [9]. KRAS is frequently mutated in smoking-related lung cancer [9]; however, the failure of the development of anti-Ras inhibitors [10] has dampened the enthusiasm for targeting KRAS. Hence, the development of novel targets involved in TS-mediated lung carcinogenesis is an appropriate strategy for lung cancer chemoprevention.
In the present study, we investigated the mechanisms of tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced lung tumor formation. We found that NNK triggered insulin-like growth factor 1 receptor (IGF-1R) phosphorylation via β-adrenergic receptor (β-AR)-mediated increases in IGF2 transcription. Treatment with β-AR antagonists suppressed NNK-induced IGF-1R phosphorylation, significantly inhibiting NNK-stimulated lung epithelial cell transformation and murine lung tumorigenesis. These results suggest that suppression of IGF-1R activation by blockade of β-AR can be an effective approach to prevent smoking-induced lung cancer.
RESULTS
NNK increased IGF-1R phosphorylation in human lung epithelial cells
Our previous study showed that IGF-1R is activated in human high-grade dysplasia tissues and in NNK-exposed murine lung tissues [11]. Recently, we found that NNK stimulates IGF-1R activation in lung epithelial cells, promoting tumor formation (manuscript submitted for publication). We showed that NNK induced a rapid IGF-1R activation in primary cultured normal human lung epithelial (HBE) cells derived from large airways, in various immortalized, normal HBE cell lines, including BEAS-2B, and in premalignant HBE cell line carrying loss of p53 expression (HBE/p53i). In the current study, we confirmed that IGF-1R is activated in a sustained manner during chronic exposure to NNK. HBE/p53i and BEAS-2B cells displayed time-dependent increases in IGF-1R phosphorylation with a maximal phosphorylation at 24 h after stimulation with NNK (Figure 1A). NNK also induced a dose-dependent upregulation of IGF-1R phosphorylation in BEAS-2B cells (Figure 1B). The NNK treatment stimulated the IGF-1R signaling cascade as shown by the increased phosphorylation of protein kinase B (Akt) (Figure 1C). Immunofluorescence staining further revealed increases in IGF-1R phosphorylation in HBE/p53i and BEAS-2B cells in response to the NNK treatment for 24 h (Figure 1D). Because IGF-1R and IR are structurally similar to each other (84% homology in the cytoplasmic β subunit, which contains intrinsic tyrosine kinase activity) [12], commercially available antibodies detecting phosphorylated IGF-1R cross-react with phosphorylated IR. Hence, we confirmed that NNK induced IGF-1R phosphorylation by utilizing HCC-15 non-small cell lung cancer cells that lack IR expression (Figure 1E).
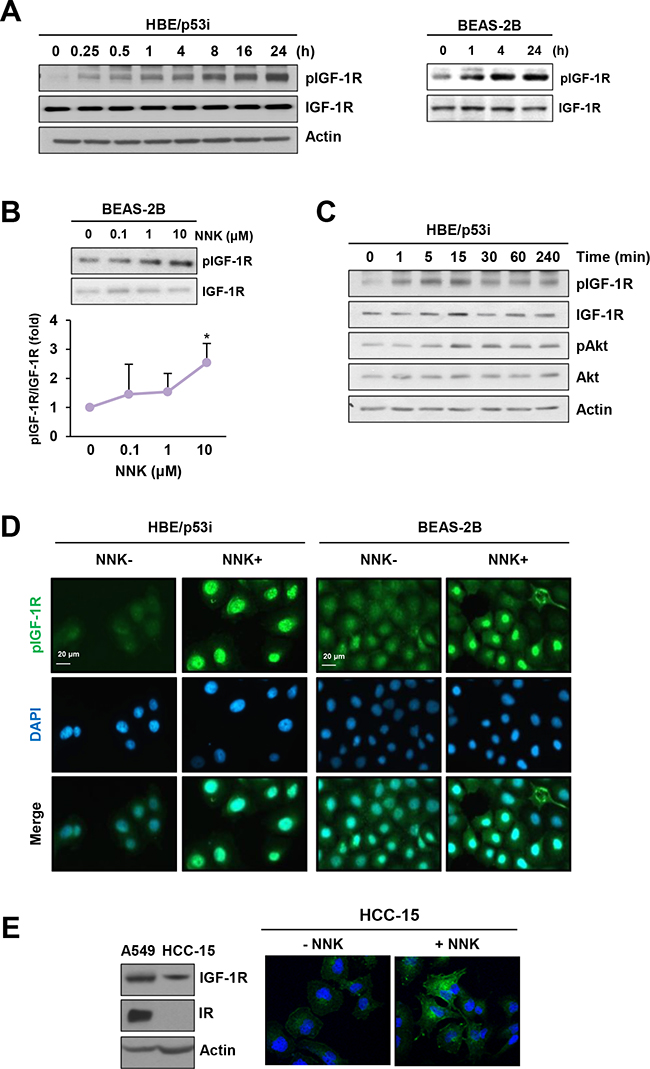
Figure 1: NNK induces IGF-1R phosphorylation. A. Immunoblot analysis demonstrating a time-dependent increase in IGF-1R phosphorylation by treatment with NNK (10 μM) in HBE/p53i and BEAS-2B cells. B. Immunoblot analysis demonstrating a dose-dependent increase in IGF-1R phosphorylation by treatment of BEAS-2B cells with NNK (10 μM) for 24 h (n = 3 per group). Densitometric analysis of total and phosphorylated IGF-1R blots (normalized to actin) was performed using the Image J software. Data are presented as the mean ± SD. The statistical significance of difference was determined by Student’s t-test (*: P < 0.05). C. Immunoblot analysis demonstrating a time-dependent increase in the phosphorylation of IGF-1R and Akt by treatment with NNK (10 μM) in HBE/p53i cells D. Immunofluorescence staining for the detection of NNK-induced IGF-1R phosphorylation. Cells were stimulated with NNK (10 μM) for 24 h. E. Up-regulation of IGF-1R phosphorylation in HCC-15 cells as determined by fluorescence microscopy.
Association of β-adrenergic receptor with NNK-induced IGF-1R phosphorylation in lung epithelial cells
We investigated the mechanism underlying NNK-induced IGF-1R phosphorylation. Because NNK has been shown to bind β-adrenergic receptor (β-AR) [13], we investigated the possible involvement of β-AR in NNK-induced IGF-1R phosphorylation. We first assessed whether β-AR agonists, including isoproterenol (nonselective), dobutamine (selective to β1-AR), and metaproterenol (selective to β2-AR), induces IGF-1R activation in BEAS-2B cells. Isoproterenol induced a dose-dependent increase in IGF-1R phosphorylation, and activation of IGF-1R was evident with isoproterenol doses as low as 5 μM (Figure 2A). In addition, treatment with isoproterenol, dobutamine, and metaproterenol for 24 h displayed a substantial IGF-1R phosphorylation in BEAS-2B cells (Figure 2B).
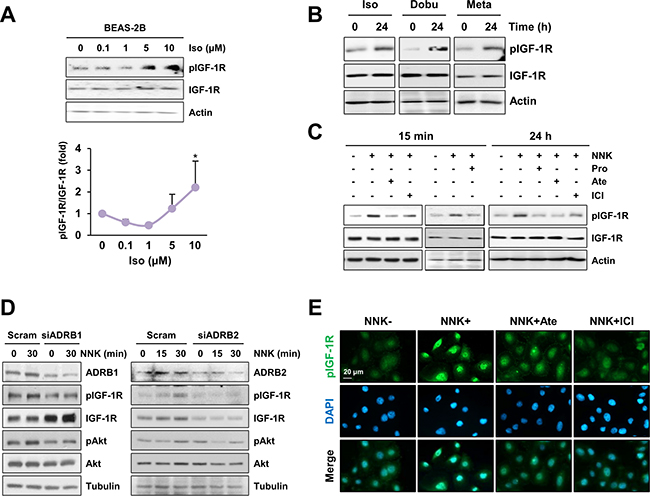
Figure 2: β-AR is involved in NNK-mediated IGF-1R phosphorylation. A. Immunoblot analysis demonstrating a dose-dependent increase in IGF-1R phosphorylation by treatment with NNK in BEAS-2B cells (n = 3 per group). Densitometric analysis of total and phosphorylated IGF-1R blots (normalized to actin) was performed using the Image J software. Data are presented as the mean ± SD. The statistical significance of difference was determined by Student’s t-test (*: P < 0.05; ***: P < 0.001). Iso: isoproterenol. B. Immunoblot analysis demonstrating the activation of IGF-1R by stimulation with β-AR agonists (10 μM) in BEAS-2B cells. Iso: isoproterenol; Dobu: dobutamine; Meta: metaproterenol. C. Inhibition of NNK-induced IGF-1R phosphorylation by treatment with β-AR antagonists (10 μM) in BEAS-2B cells was analyzed by Western blot analysis. Pro: propranolol; Ate: atenolol; ICI: ICI-118,551. Cells were stimulated with NNK for 15 min (left) or 24 h (right) in the presence or absence of indicated inhibitors. In case of a short-term NNK stimulation, cells were pretreated with inhibitors for 3 h. D. Immunoblot analysis evaluating the suppression of IGF-1R phosphorylation by silencing β1- or β2-AR expression in HBE/p53i cells. E. Immunofluorescence staining to detect the modulation of IGF-1R phosphorylation by blockade of β-AR in HBE/p53i cells. Cells were treated with NNK (10 μM) in the absence or presence of atenolol (Ate; 10 μM) or ICI-118,551 (ICI; 10 μM) for 15 min.
To assess the involvement of β-AR in NNK-induced IGF-1R phosphorylation, we tested the effects of the blockade of β-AR on NNK-induced IGF-1R phosphorylation. Treatment with propranolol (a nonselective β-AR antagonist), atenolol (a β1-AR antagonist) or ICI-118,551 (a β2-AR antagonist) effectively suppressed IGF-1R phosphorylation during NNK exposure in BEAS-2B cells (Figure 2C). Moreover, HBE/p53i cells transfected with either β1-AR or β2-AR siRNAs exhibited markedly decreased phosphorylation of IGF-1R and Akt upon NNK exposure compared with those transfected with control scrambled siRNA (Figure 2D). Immunofluorescence staining also confirmed that NNK induced a substantial increase in IGF-1R phosphorylation, which was attenuated by pretreatment with ICI-118,551 or atenolol (Figure 2E). Together, these results indicate that β-AR is associated with NNK-induced IGF-1R phosphorylation.
We next investigated the role of downstream signaling pathways of β-AR in NNK-induced IGF-1R phosphorylation. β-AR is a G protein-coupled receptor (GPCR) that consists of Gαs, Gβ, and Gγ subunits [14]. Activated β-AR results in the dissociation of Gβγ subunits from the Gα subunit; the Gαs subunit activates adenylyl cyclase and converts ATP to cyclic AMP (cAMP), mediating the activation of downstream effectors, including protein kinase A (PKA) and Epac [14]. The Gβγ subunits modulate the function of several effectors, including phospholipase C (PLC; PLCβ and PLCε), PI3Kγ, and calcium channels, and regulating calcium mobilization and signal transduction to promote cell proliferation and survival [15]. Accordingly, the effectors responsible for NNK-induced IGF-1R phosphorylation were determined using pharmacological inhibitors – i.e., H-89 (a PKA inhibitor), ESI-09 (an Epac inhibitor), U73122 (a PLC inhibitor), and gallein (a Gβγ subunit inhibitor). Treatment of HBE/p53i cells with H-89 or ESI-09 displayed no obvious effect on NNK-mediated IGF-1R phosphorylation (Figure 3A), whereas treatment with gallein (Figure 3B) or U73122 (Figure 3C) effectively decreased NNK-induced IGF-1R phosphorylation. Collectively, these results suggest that NNK-induced activation of the β-AR signaling pathway followed by Gβγ-mediated PLC activation is crucial for IGF-1R phosphorylation.
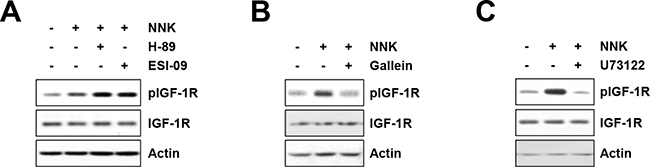
Figure 3: The Gβγ-mediated PLC activation mediates NNK-induced IGF-1R phosphorylation. A–C. Suppression of NNK (10 μM)-induced IGF-1R phosphorylation by blocking Gβγ subunit and PLC but not PKA and Epac in HBE/p53i cells, as determined by Western blot analysis. HBE/p53i cells were pretreated with H-89 (A; 10 μM), ESI-09 (A; 10 μM), gallein (B; 10 μM), and U73122 (C; 1 μM) for 3 h, and then were stimulated with NNK for 15 min. The level of total and phosphorylated IGF-1R was evaluated by Western blot analysis.
β-AR activation by NNK induced IGF2 transcription through STAT3 and NF-κB activation
We investigated the mechanisms underlying NNK-induced IGF-1R phosphorylation via the β-AR signaling pathway. We found that exposure to NNK stimulated the regulatory pathway of IGF2 secretion within 5 min, leading to a rapid activation of IGF-1R in lung epithelial cells (manuscript submitted for publication). In the current study, we further assessed whether sustained exposure to NNK modulated the mRNA expression levels of IGF2. The transcription of IGF-1R signaling components, including IGF1, IGF1R, and IGFBP3, remained unchanged in BEAS-2B cells after treatment with NNK for 24 h (Figure 4A, left panel). In contrast, BEAS-2B cells treated with NNK exhibited a significant up-regulation of IGF2 transcription (Figure 4A, left panel). A substantial increase in IGF2 transcription was observed as early as 6 h after NNK treatment, and the increase was statistically significant after 24 h of NNK exposure (Figure 4A, right panel). Stimulation with the β-AR agonist isoproterenol also increased IGF2 transcription (Figure 4B), and treatment with β-AR antagonists significantly suppressed NNK-induced increases in IGF2 mRNA expression (Figure 4C). These findings indicate that transcriptional up-regulation of IGF2 via β-AR is responsible at least in part for the NNK-induced IGF-1R phosphorylation in lung epithelial cells.
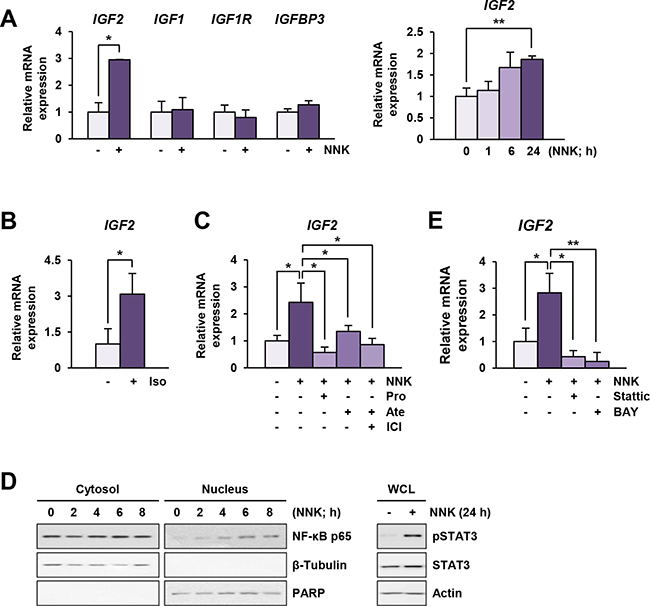
Figure 4: NNK induces an increase in IGF2 transcription via β-AR, STAT3 and NF-κB activation. A and B. Real-time PCR analyses demonstrating IGF2, but not IGF1, IGF1R, and IGFBP3, transcription by stimulation with NNK (10 μM) (A) or isoproterenol (Iso; 10 μM) (B) (n = 3 per group) in BEAS-2B cells. Data are presented as the mean ± SD. Statistical significance of difference was determined by Student’s t-test or one-way ANOVA (*: P < 0.05; **: P < 0.01). C. Suppression of NNK-induced IGF2 transcription by treatment with β-AR antagonists in BEAS-2B cells, as analyzed by real-time PCR (n = 3 per group). Data are presented as the mean ± SD. Statistical significance was determined by one-way ANOVA (*: P < 0.05). Cells were stimulated with NNK (10 μM) in the absence or presence of β-AR antagonists (10 μM) for 24 h. Pro: propranolol; Ate: atenolol; ICI: ICI-118,551. D. NNK-mediated phosphorylation of STAT3 and nuclear translocation of a NF-κB p65 subunit in BEAS-2B cells were analyzed by Western blot analysis. E. Suppression of NNK-induced IGF2 transcription by treatment with Stattic (2 μM) or BAY 11-7082 (BAY; 5 μM) in BEAS-2B cells was analyzed by real-time PCR (n = 3 per group). Cells were stimulated with NNK (10 μM) with or without inhibitors for 24 h. Data are presented as the mean ± SD. Statistical significance was determined by one-way ANOVA (*: P < 0.05; **: P < 0.01).
We investigated the mechanism underlying the NNK-mediated increase in IGF2 mRNA expression. Based on our recent findings and previous reports showing 1) direct binding of STAT3 to IGF2 P3 and P4 promoters [16], 2) NF-κB-mediated up-regulation of IGF2 transcription [17], and 3) activation of STAT3 and NF-κB by NNK [18], we examined the involvement of STAT3 and NF-κB in NNK-induced IGF2 transcription. Indeed, NNK increased the nuclear translocation of NF-κB p65 subunit and the level of phosphorylated STAT3 (pSTAT3) expression (Figure 4D). Moreover, treatment with the STAT3 inhibitor Stattic or the NF-κB inhibitor BAY 11-7082 significantly down-regulated NNK-stimulated IGF2 transcription (Figure 4E). These results suggest that activation of STAT3 and NF-κB is associated with NNK-induced IGF2 transcription.
Suppression of transformed phenotypes and in vivo tumor formation by blockade of β-AR
To investigate whether blockade of β-AR could inhibit the NNK-induced acquisition of transformed phenotypes in lung epithelial cells, we examined the effects of β-AR antagonists on NNK-mediated transformed phenotypes, including anchorage-dependent and -independent colony formation and foci formation resulting from single-cell survival, anchorage-independent growth, and loss of contact inhibition, respectively [19–21]. HBE cells exposed to NNK formed significantly more colonies than those of vehicle-treated control cells, and treatment with β-AR antagonists substantially suppressed the NNK-induced anchorage-dependent (Figure 5A) and anchorage-independent (Figure 5B) colony-forming abilities. The NNK-treated HBE cells also exhibited a significantly increased ability to form foci, and blockade of β-AR markedly suppressed the NNK-induced foci formation (Figure 5C). These results suggest that β-AR inhibition suppresses NNK-mediated HBE cell transformation.
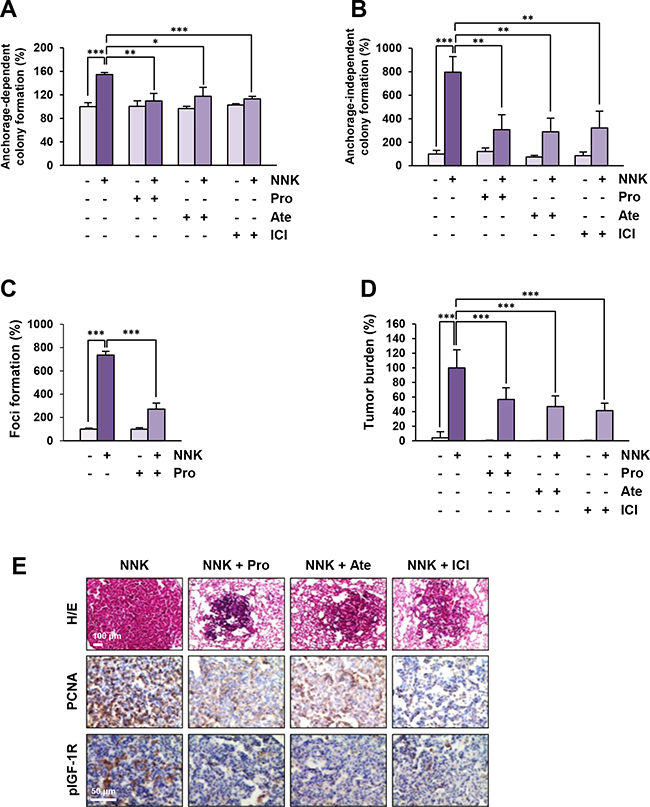
Figure 5: Blockade of β-AR suppresses NNK-induced cell transformation and murine lung tumor formation. A and B. Decreases in anchorage-dependent (A; n = 3 per group) and –independent (B; n = 4 per group) colony formation of BEAS-2B cells by treatment with β-AR antagonists. Data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA (*: P < 0.05; **: P < 0.01; ***: P < 0.001). C. Inhibition of NNK-induced foci formation of HBE/p53i cells by treatment with propranolol (n = 3 per group). Data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA (***: P < 0.001). D. Suppression of the NNK-mediated increase in tumor burden by blockade of β-AR (n = 7-14 per group). Data are presented as mean ± SD. Statistical significance was determined by one-way ANOVA (***: P < 0.001). E. Immunohistochemical analysis demonstrating the suppression of IGF-1R phosphorylation and cell proliferation (determined by PCNA expression) in a nodule of the lung from mice treated with NNK and β-AR antagonists. Pro: propranolol; Ate: atenolol; ICI: ICI-118,551.
Finally, we investigated whether blocking β-AR could inhibit NNK-induced lung tumor formation in vivo. Female A/J mice were treated by oral gavage with vehicle (PBS) or NNK, either alone or together with β-AR antagonists for 20 weeks. As shown in Figure 5D, mice treated with NNK alone displayed a profoundly increased tumor burden compared with the control group, and the tumor burden was significantly decreased by the administration of β-AR antagonists. As shown by the results from immunohistochemical analysis, IGF-1R phosphorylation and PCNA expression, a marker of cell proliferation [22], were markedly lower in the lungs of mice treated with β-AR antagonists than those from control mice (Figure 5E). Therefore, along with the in vitro results, these in vivo results suggest that blockade of β-AR effectively suppresses NNK-mediated lung tumor formation by inhibiting IGF2-mediated IGF-1R phosphorylation.
DISCUSSION
In this study, we demonstrated that 1) β-AR mediates NNK-induced IGF-1R activation through the up-regulation of IGF2 transcription; 2) the Gβγ subunits dissociated from β-AR upon NNK exposure stimulate IGF-1R through the PLC-mediated pathway; and 3) the inhibition of β-AR suppresses NNK-mediated malignant cell transformation and tumor formation in mice. These results suggest that inhibition of NNK-induced IGF2-IGF-1R activation by blocking β-AR could be a novel approach for the chemoprevention of smoking-associated lung cancer.
Despite extensive investigation to develop effective strategies for the prevention of lung cancer, smoking cessation is the only way to prevent the onset or progression of lung cancer [7]. The alteration of several genes is implicated in the carcinogenic process of lung cancer [7]; therefore, targeting these mutated genes or related signaling pathways is an efficacious approach for lung cancer prevention. Although EGFR and KRAS are frequently mutated in non-smoking- and smoking-related lung cancer, respectively, and thus can be considered targets for lung cancer prevention, their direct modulation by chemicals is not applicable for lung cancer prevention due to potential side effects and toxicity when applied as long-term treatment. Hence, considering that smoking remains the primary cause of lung cancer, targeting signal transduction that can be modulated by a smoking component is a plausible approach for lung cancer prevention.
The activation of receptor tyrosine kinases (RTKs) is strongly implicated in oncogenesis, cancer progression, and drug resistance [23]. Of these processes, IGF-1R signaling is critical for cell proliferation, survival, invasion, and angiogenesis [24]. Our previous study demonstrated the association of IGF-1R activation with lung carcinogenesis [11]. We found that IGF-1R signaling was activated in premalignant and malignant human lung tissues (high-grade dysplasia and metaplasia) and NNK-exposed lung epithelial cells and murine lung tissues [11]. These results suggest that IGF-1R may be an important target for the prevention of smoking-induced lung cancer formation. However, inhibition of IGF-1R directly is not applicable for prevention because of the potential side effects of small-molecule IGF-1R inhibitors, such as hyperglycemia due to cross-reactivity to the insulin receptor (IR) [25]. Hence, suppression of signaling networks responsible for aberrant IGF-1R signaling activation by smoking would be more suitable for further clinical utility; however, the underlying mechanisms have been poorly investigated. Therefore, the present study aimed to explore alternative means to control the IGF-1R pathway by investigating the mechanisms involved in NNK-induced IGF-1R activation.
We found that the β-AR pathway is activated by exposure to NNK, leading to IGF-1R activation. It has been proposed that various RTKs, including EGFR and IGF-1R, can be transactivated by GPCRs via several mechanisms, including ligand-dependent activation by metalloproteinase-dependent shedding of ligands and reactive oxygen species (ROS)- or Src-mediated ligand-independent activation [26]. RTK transactivation via direct association between RTKs and GPCRs through the formation of a complex with other non-receptor tyrosine kinases, adaptor proteins, and β-arrestin has also been proposed [27, 28]. Our results demonstrate that NNK treatment leads to increased IGF2 transcription via activating β-AR, STAT3, and NF-κB. Together with our recent observation on the association of the β-AR pathway with rapid IGF-1R phosphorylation via increased IGF2 secretion (manuscript submitted for publication), exposure to NNK is likely to stimulate IGF-1R phosphorylation via dual pathways through a rapid increase in IGF2 secretion followed by a transcriptional increase in IGF2 expression.
Our results demonstrate blockade of NNK-induced IGF-1R activation via genomic or pharmacological approaches that block β-AR, Gβγ, or PLC. Considering that Gβγ-mediated PLC and subsequent IP3R activation play important roles in the increase in intracellular calcium [29] and STAT3 and NF-κB are known to be modulated by a calcium/calmodulin-dependent protein kinase II [30, 31], Gβγ-mediated PLC activation couple the NNK-stimulated β-AR signaling pathway with IGF2 regulation, resulting in activation of the IGF-1R signaling pathway. These results appear to be novel and distinct from the previously reported IGF-1R transactivation. Most importantly, our data provide evidence that blockade of the β-AR signaling pathway effectively suppresses NNK-stimulated lung tumor formation in mice. These findings corroborate the in vitro studies, supporting the novel concept that blocking the β-AR signaling pathway is an alternative strategy to suppress aberrant IGF-1R activation and, thus, prevent lung cancer formation in smokers. Indeed, β-blockers (β1-AR antagonists) have been widely used for the treatment of hypertension, and the evaluation of their efficacy and safety has been thoroughly tested; therefore, the use of β-blockers can be an effective approach for cancer prevention. In agreement with this notion, β-AR has also emerged as an attractive target for adjuvant cancer therapy based on its intrinsic role as a receptor of stress-associated neurotransmitters – i.e., epinephrine and norepinephrine – and its downstream signaling pathways responsible for cell proliferation, survival, metastasis, and angiogenesis [32]. In addition, the inhibition of β-AR provides better therapeutic responses to conventional anticancer therapies in neuroblastoma, hemangioendothelioma, and angiosarcoma [33, 34]. Moreover, although controversial results have also been reported [35], the use of β-blockers may reduce the mortality of lung, breast, ovary, and prostate cancer [36–40]. Although the correlation between the use of β-blockers and the reduction of cancer risk remains questionable and needs to be further investigated [41, 42], our results provide preclinical evidence for using β-AR antagonists in prevention of smoking-associated lung cancer. In addition, previous studies have reported a possible positive correlation between hypertension and lung cancer in current smokers [43, 44]. Considering the traditional use of β-blockers as a therapy for hypertension, the relationship between smoking-related lung cancer and hypertension also needs to be examined.
In summary, the present study demonstrates that NNK activates IGF-1R through the β-AR-mediated activation of Gβγ-PLC and up-regulation of IGF2 transcription via activation of β-AR, STAT3, and NF-κB. Blockade of β-AR significantly reduced the acquisition of transformed phenotypes in lung epithelial cells and NNK-induced lung tumor formation in mice. These results suggest that blockade of β-AR may be efficacious for the prevention of smoking-related lung cancer. Further studies are warranted to investigate the effectiveness of blockade of β-AR in smoking-associated lung cancer chemoprevention using additional preclinical and clinical settings.
MATERIALS AND METHODS
Cell culture
Human bronchial epithelial (HBE) cells knocked down for p53 expression using RNA interference (HBE/p53i) [45] were kindly provided by Dr. John D. Minna (The University of Texas Southwestern Medical Center, Dallas, TX, USA). BEAS-2B cells were purchased from American Type Culture Collection (ATCC; Manassas, VA, USA). These cells were cultured in K-SFM (Invitrogen, Grand Island, NY, USA) supplemented with 5 ng/ml recombinant epidermal growth factor (EGF) and 50 μg/ml bovine pituitary extracts. A549 and HCC-15 cells, purchased from ATCC, were cultured in RPMI 1640 supplemented with 10% fetal bovine serum and antibiotics (all from Welgene, Daegu, Republic of Korea). Cells were maintained at 37°C with 5% CO2 in a humidified atmosphere.
Reagents
Dimethyl sulfoxide (DMSO), crystal violet, 3-(4,5-dimethyltrizaol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and other chemicals, unless otherwise indicated, were purchased from Sigma-Aldrich (St. Louis, MO, USA). 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) was purchased from Sigma or Toronto Research Chemicals (TRC; Toronto, ON, Canada). Isoproterenol, dobutamine, metaproterenol, propranolol, atenolol, ICI-118,551, gallein, and U73122 were purchased from Tocris Bioscience (Bristol, UK). ESI-09 was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Antibodies against phosphorylated IGF-1R (pIGF-1R) at Y1131 or Y1135/36, IGF-1R, phosphorylated STAT3 at Y705, STAT3, phosphorylated Akt at S473, Akt, β-tubulin, α/β-tubulin (tubulin), and PARP were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against actin, β1-AR, and β2-AR were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-proliferating cell nuclear antigen (PCNA) antibody was purchased from Abcam (Cambridge, MA, USA).
Anchorage-dependent colony formation assay
BEAS-2B cells were seeded into 6-well plates at a density of 300 cells/well and were incubated for 24 h. Cells were stimulated with 10 μM NNK in the presence or absence of inhibitors for 10 days. Colonies were stained with 0.01% crystal violet and were counted.
Anchorage-independent colony formation assay
Before seeding, the base agar (final agar concentration: 1%) was prepared in 24-well plates. BEAS-2B cells (final 2 x 103 cells/well) were mixed with sterile low-melting agar solution and immediately poured onto the base agar. NNK (10 μM) with or without inhibitors diluted in 0.5 ml of complete medium were added. Cells were incubated for 3 weeks, and the drug-containing medium was replaced every 3-4 days. Colonies were stained with the MTT solution (final 500 μg/ml), photographed, and counted.
Foci formation assay
HBE/p53i cells at 70-80% confluence in 6-well plates were treated with NNK (10 μM) with or without inhibitors for 2 weeks. The drug-containing media were replaced every 2-3 days. After incubation, foci were photographed and counted.
Preparation of cytosol/nuclear extracts
BEAS-2B cells were stimulated with NNK for the indicated time points. Cytosol fraction was prepared with hypotonic Buffer A [10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1 mM EDTA, protease inhibitor cocktail (Roche Applied Sciences, Indianapolis, IN, USA) and phosphatase inhibitor cocktail (Roche)]. After washing with Buffer A, pellets were suspended in hypertonic Buffer C [20 mM HEPES (pH 7.9), 1.5 mM MgCl2, 420 mM NaCl, 0.5 mM DTT, 0.2 mM EDTA, 25% glycerol, protease inhibitor cocktail, and phosphatase inhibitor cocktail]. After centrifugation, supernatants were collected and stored at -70°C. The cytosolic and nuclear localization of the NF-κB p65 subunit was determined by Western blot analysis as described below.
Western blot analysis
Cells were seeded into 6-well plates at a density of 3 × 105 cells/well. On the next day, culture media were replaced by supplement-free K-SFM (for HBE/p53i and BEAS-2B), and the cells were incubated for 1-2 days. When necessary, the cells were pretreated with inhibitors 3-6 h before stimulation and then stimulated with NNK (10 μM) or β-AR agonists (10 μM) for the indicated times. Cells were harvested with modified RIPA lysis buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1 mM EDTA, 0.25% sodium deoxycholate, 1% Triton X-100, 100 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μg/ml aprotinin, and 1 μg/ml leupeptin]. Equal amounts of proteins were resolved by 8% SDS-PAGE and then electrically transferred onto a PVDF membrane (ATTO Corp., Osaka, Japan). Membranes were blocked with blocking buffer [3% BSA in Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBST)] for 1 h at room temperature, and then were incubated with primary antibodies diluted in blocking buffer (1:1,000) overnight at 4°C. After washing several times with TBST, membranes were incubated with corresponding secondary antibodies diluted in 3% non-fat dry milk in TBST (1:5,000) for 1-2 h at room temperature. Membranes were washed several times with TBST and were visualized using an enhanced chemiluminescence (ECL) detection kit (Thermo Fisher Scientific, Grand Island, NY, USA). When necessary, densitometric analysis was performed using the Image J software (National Institutes of Health, Bethesda, MD, USA).
Transfection
For transient knockdown of β1- or β2-ARs, cells were transfected with scrambled or gene-specific targeting small interfering RNAs (siRNAs; purchased from Integrated DNA Technologies, Coralville, Iowa, USA) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Knockdown of these genes was confirmed by Western blot analysis.
Reverse transcription-polymerase chain reaction (RT-PCR)
RT-PCR and real-time PCR analyses were performed as described previously [46]. The primer sequences used for this study are described in Table 1.
Table 1: Primer sequences used for real-time PCR analysis
Gene |
Forward (5’-3’) |
Reverse (5’-3’) |
IGF1 |
ATGTATTGCG CACCCCTCAA |
GGGCACGG ACAGAGCG |
IGF2 |
CCGTGCTTCC GGACAACTT |
CTGCTTCCAG GTGTCATATTGC |
IGF1R |
TGAAAGTGACG TCCTGCATTTC |
GGTACCGGTG CCAGGTTATG |
IGFBP3 |
TCTGCGTCAA CGCTAGTGC |
GCTCTGAGACT CGTAGTCAACT |
ACTB |
GCGAGAAGAT GACCCAGATC |
GGATAGCAC AGCCTGGATAG |
Animal experiments
All of the animal experiments were performed using protocols approved by the Seoul National University Institutional Animal Care and Use Committee. Five-week-old A/J mice were randomly grouped and exposed to NNK (3 μmol; dissolved in sterile PBS) twice per week. After 2-4 weeks, several inhibitors (propranolol: 1 mg/kg; atenolol: 5 mg/kg; ICI-118,551: 0.5 mg/kg; drugs were dissolved in sterile PBS) were administered with or without NNK by oral gavage for an additional 20 weeks. The mice were euthanized, and tumor formation was evaluated and compared with that of the vehicle-treated control group. After gross evaluation of tumor formation, microscopic evaluation of formalin-fixed and paraffin-embedded lung tissue stained with hematoxylin and eosin (H&E) was performed to measure the mean tumor number (N) and volume (V) in a blinded fashion. The tumor volume was calculated using the following formula: V (mm3) = (long diameter × short diameter2)/2. The tumor burden was calculated using the following formula: mean tumor number (N) × mean tumor volume (V). The number and size of tumors were calculated in five sections uniformly distributed throughout each lung.
Immunofluorescence and immunohistochemistry
Immunofluorescence and immunohistochemical analyses to detect phosphorylated IGF-1R or PCNA expressions were performed as described previously [46].
Statistics
The data are presented as the mean ± SD. All in vitro experiments were independently performed at least twice, and a representative result is presented. The statistical significance was analyzed using a two-sided Student’s t-test or one-way analysis of variance (ANOVA) followed by Dunnett’s post-hoc test using GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA). P values less than 0.05 were considered statistically significant.
ACKNOWLEDGMENTS
This work was supported by grants from the National Research Foundation of Korea (NRF), the Ministry of Science, ICT & Future Planning (MSIP), Republic of Korea (No. and NRF-2016R1A3B1908631).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Jung KW, Won YJ, Kong HJ, Oh CM, Seo HG and Lee JS. Cancer statistics in Korea: incidence, mortality, survival and prevalence in 2010. Cancer Res Treat. 2013; 45:1-14.
2. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
3. Jemal A, Bray F, Center MM, Ferlay J, Ward E and Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011; 61:69-90.
4. Janku F, Stewart DJ and Kurzrock R. Targeted therapy in non-small-cell lung cancer--is it becoming a reality? Nature reviews Clinical oncology. 2010; 7:401-414.
5. Holohan C, Van Schaeybroeck S, Longley DB and Johnston PG. Cancer drug resistance: an evolving paradigm. Nature reviews Cancer. 2013; 13:714-726.
6. William WN, Jr., Heymach JV, Kim ES and Lippman SM. Molecular targets for cancer chemoprevention. Nature reviews Drug discovery. 2009; 8:213-225.
7. Keith RL and Miller YE. Lung cancer chemoprevention: current status and future prospects. Nature reviews Clinical oncology. 2013; 10:334-343.
8. Dannenberg AJ, Lippman SM, Mann JR, Subbaramaiah K and DuBois RN. Cyclooxygenase-2 and epidermal growth factor receptor: pharmacologic targets for chemoprevention. Journal of clinical oncology. 2005; 23:254-266.
9. Dogan S, Shen R, Ang DC, Johnson ML, D’Angelo SP, Paik PK, Brzostowski EB, Riely GJ, Kris MG, Zakowski MF and Ladanyi M. Molecular epidemiology of EGFR and KRAS mutations in 3,026 lung adenocarcinomas: higher susceptibility of women to smoking-related KRAS-mutant cancers. Clinical cancer research. 2012; 18:6169-6177.
10. Brunner TB, Hahn SM, Gupta AK, Muschel RJ, McKenna WG and Bernhard EJ. Farnesyltransferase inhibitors: an overview of the results of preclinical and clinical investigations. Cancer research. 2003; 63:5656-5668.
11. Kim WY, Jin Q, Oh SH, Kim ES, Yang YJ, Lee DH, Feng L, Behrens C, Prudkin L, Miller YE, Lee JJ, Lippman SM, Hong WK, Wistuba, II and Lee HY. Elevated epithelial insulin-like growth factor expression is a risk factor for lung cancer development. Cancer research. 2009; 69:7439-7448.
12. Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E, Jacobs S, Francke U, Ramachandran J, Fujita-Yamaguchi Y. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. The EMBO journal. 1986; 5:2503-2512.
13. Schuller HM, Tithof PK, Williams M and Plummer H, 3rd. The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer research. 1999; 59:4510-4515.
14. Lappano R and Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nature reviews Drug discovery. 2011; 10:47-60.
15. Smrcka AV. G protein betagamma subunits: central mediators of G protein-coupled receptor signaling. Cellular and molecular life sciences. 2008; 65:2191-2214.
16. Lee SC, Min HY, Jung HJ, Park KH, Hyun SY, Cho J, Woo JK, Kwon SJ, Lee HJ, Johnson FM and Lee HY. Essential role of insulin-like growth factor 2 in resistance to histone deacetylase inhibitors. Oncogene. 2016; 35:5515-5526.
17. Schmeisser MJ, Baumann B, Johannsen S, Vindedal GF, Jensen V, Hvalby OC, Sprengel R, Seither J, Maqbool A, Magnutzki A, Lattke M, Oswald F, Boeckers TM and Wirth T. IkappaB kinase/nuclear factor kappaB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. The Journal of neuroscience. 2012; 32:5688-5703.
18. Akopyan G and Bonavida B. Understanding tobacco smoke carcinogen NNK and lung tumorigenesis. Int J Oncol. 2006; 29:745-752.
19. Franken NA, Rodermond HM, Stap J, Haveman J and van Bree C. Clonogenic assay of cells in vitro. Nature protocols. 2006; 1:2315-2319.
20. Casto BC, Janosko N and DiPaolo JA. Development of a focus assay model for transformation of hamster cells in vitro by chemical carcinogens. Cancer research. 1977; 37:3508-3515.
21. Freedman VH and Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell. 1974; 3:355-359.
22. Kubben FJ, Peeters-Haesevoets A, Engels LG, Baeten CG, Schutte B, Arends JW, Stockbrugger RW and Blijham GH. Proliferating cell nuclear antigen (PCNA): a new marker to study human colonic cell proliferation. Gut. 1994; 35:530-535.
23. Gschwind A, Fischer OM and Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nature reviews Cancer. 2004; 4:361-370.
24. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nature reviews Cancer. 2008; 8:915-928.
25. Cohen DH and LeRoith D. Obesity, type 2 diabetes, and cancer: the insulin and IGF connection. Endocrine-related cancer. 2012; 19:F27-45.
26. Cattaneo F, Guerra G, Parisi M, De Marinis M, Tafuri D, Cinelli M and Ammendola R. Cell-surface receptors transactivation mediated by g protein-coupled receptors. Int J Mol Sci. 2014; 15:19700-19728.
27. Lin X, Li X, Jiang M, Chen L, Xu C, Zhang W, Zhao H, Sun B, Xu X, Nan F and Liu J. An activity-based probe reveals dynamic protein-protein interactions mediating IGF-1R transactivation by the GABA(B) receptor. The Biochemical journal. 2012; 443:627-634.
28. Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, Lefkowitz RJ and Luttrell LM. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. The Journal of biological chemistry. 2000; 275:9572-9580.
29. Bootman MD. Calcium signaling. Cold Spring Harb Perspect Biol. 2012; 4:a011171.
30. Cuschieri J, Bulger E, Garcia I, Jelacic S and Maier RV. Calcium/calmodulin-dependent kinase II is required for platelet-activating factor priming. Shock. 2005; 23:99-106.
31. Si J and Collins SJ. Activated Ca2+/calmodulin-dependent protein kinase IIgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer research. 2008; 68:3733-3742.
32. Cole SW and Sood AK. Molecular pathways: beta-adrenergic signaling in cancer. Clinical cancer research. 2012; 18:1201-1206.
33. Stiles JM, Amaya C, Rains S, Diaz D, Pham R, Battiste J, Modiano JF, Kokta V, Boucheron LE, Mitchell DC and Bryan BA. Targeting of beta adrenergic receptors results in therapeutic efficacy against models of hemangioendothelioma and angiosarcoma. PloS one. 2013; 8:e60021.
34. Pasquier E, Street J, Pouchy C, Carre M, Gifford AJ, Murray J, Norris MD, Trahair T, Andre N and Kavallaris M. beta-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. British journal of cancer. 2013; 108:2485-2494.
35. Cardwell CR, Coleman HG, Murray LJ, O’Sullivan JM and Powe DG. Beta-blocker usage and prostate cancer survival: a nested case-control study in the UK Clinical Practice Research Datalink cohort. Cancer epidemiology. 2014; 38:279-285.
36. Wang HM, Liao ZX, Komaki R, Welsh JW, O’Reilly MS, Chang JY, Zhuang Y, Levy LB, Lu C and Gomez DR. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Annals of oncology. 2013; 24:1312-1319.
37. Melhem-Bertrandt A, Chavez-MacGregor M, Lei XD, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD, Hortobagyi GN and Gonzalez-Angulo AM. Beta-Blocker Use Is Associated With Improved Relapse-Free Survival in Patients With Triple-Negative Breast Cancer. Journal of Clinical Oncology. 2011; 29:2645-2652.
38. Diaz ES, Karlan BY and Li AJ. Impact of beta blockers on epithelial ovarian cancer survival. Gynecologic oncology. 2012; 127:375-378.
39. Barron TI, Connolly RM, Sharp L, Bennett K and Visvanathan K. Beta Blockers and Breast Cancer Mortality: A Population-Based Study. Journal of Clinical Oncology. 2011; 29:2635-2644.
40. Grytli HH, Fagerland MW, Fossa SD and Tasken KA. Association Between Use of beta-Blockers and Prostate Cancer-Specific Survival: A Cohort Study of 3561 Prostate Cancer Patients with High-Risk or Metastatic Disease. European urology. 2014; 65:635-641.
41. Jansen L, Below J, Chang-Claude J, Brenner H and Hoffmeister M. Beta blocker use and colorectal cancer risk Population-based case-control study. Cancer. 2012; 118:3911-3919.
42. Monami M, Filippi L, Ungar A, Sgrilli F, Antenore A, Dicembrini I, Bagnoli P, Marchionni N, Rotella CM and Mannucci E. Further data on beta-blockers and cancer risk: observational study and meta-analysis of randomized clinical trials. Current medical research and opinion. 2013; 29:369-378.
43. Lindgren A, Pukkala E, Nissinen A and Tuomilehto J. Blood pressure, smoking, and the incidence of lung cancer in hypertensive men in North Karelia, Finland. American journal of epidemiology. 2003; 158:442-447.
44. Lee SY, Kim MT, Jee SH and Im JS. Does hypertension increase mortality risk from lung cancer? A prospective cohort study on smoking, hypertension and lung cancer risk among Korean men. Journal of hypertension. 2002; 20:617-622.
45. Sato M, Vaughan MB, Girard L, Peyton M, Lee W, Shames DS, Ramirez RD, Sunaga N, Gazdar AF, Shay JW and Minna JD. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer research. 2006; 66:2116-2128.
46. Jin Q, Lee HJ, Min HY, Smith JK, Hwang SJ, Whang YM, Kim WY, Kim YH and Lee HY. Transcriptional and posttranslational regulation of insulin-like growth factor binding protein-3 by Akt3. Carcinogenesis. 2014; 35:2232-2243.