
INTRODUCTION
The most hotly debated and yet unresolved issue in the area of employing calorie restriction (CR) mimetics (CRMs) as anti-cancer agents with mammalian target of rapamycin (mTOR)-inhibiting activity is the relative effects of nature versus nurture, i.e., the relative contributions of systemic factors versus cancer cell-autonomous effects [1-3]. The comparison of energy-balance regimens, such as CR, and pharmacological interventions with well-established mTOR inhibitors has revealed that rapamycin conspicuously mimics CR in its ability to decrease the mammary tumor burden in obese animals [4-6]. However, constitutively active mTOR has been found to fully ablate the beneficial effects of CR on mammary tumor growth [7]. Similarly, tumors with constitutively active phosphatidylinositol-3 kinase (PI3K) have been found to be refractory to the anti-cancer activity of dietary restriction (DR) [8, 9]. Because the binding of insulin to its receptors activates the PI3K/AKT pathway, which is known to stimulate mTOR activity, Kalaany and Sabatini’s observations that tumor cells bearing constitutively activated PI3K mutations are proliferative in vitro in the absence of insulin or IGF-1 and form DR-resistant tumors in vivo [8] clearly suggest that cancer cell-autonomous alterations (e.g., activating mutations of PI3K) may ultimately determine the response of cancer cells to CR, DR, or CRMs.
One mTOR-inhibiting drug with great promise as an anti-cancer CRM is metformin [10-16], a biguanide drug that is commonly used to treat type 2 diabetes. Epidemiological studies have consistently suggested that type 2 diabetic patients treated with metformin have a lower risk of developing and dying from cancer than do diabetic patients receiving sulfonylurea, insulin, or other therapies [17, 18]. Because insulin resistance and consequent hyperinsulinemia can promote carcinogenesis directly through the insulin receptor or indirectly by increasing the levels of insulin-like growth factors or promoting persistently elevated plasma glucose, the association between metformin consumption and the reduced risk of cancer among type 2 diabetic patients may be explained simply by the metformin-driven improvement of insulin levels and blood glucose [19-22]. Indeed, the serum insulin- and IGF-1-lowering effects of metformin have been thought to explain why the administration of metformin suppresses tumor development or growth in multiple experimental models, including colon, hematopoietic, and mammary cancer models. However, decreases in hyperglycemia and hyperinsulinemia are not always correlated with the anti-cancer efficacy of metformin, as can be observed in non-diabetic mouse models. Thus, the anti-tumoral action of metformin is not precluded in PTEN+/- [23, 24], HER2 [25, 26], and APCmin/+ [27] mouse tumor models, in which insulin-related markers are not significantly attenuated by treatment with metformin. The notion that metformin suppresses cancer growth through pathways other than insulin/IGF-1-dependent, indirect drug action has recently been supported by experiments in liver IGF-1-deficient (LID) mice, which had naturally decreased lung tumor multiplicity and burden compared with wild-type mice [28, 29]. In this model, metformin further decreased tobacco carcinogen (NNK)-induced lung tumorigenesis without affecting IGF-1 levels. The authors demonstrated that metformin can act through IGF-1-independent mechanisms by promoting the systemic inhibition of circulating growth factors and local receptor tyrosine kinase signaling in cancer tissues [28]. Although these findings suggest an insulin-independent, direct anti-tumoral activity for metformin, it remains unknown whether cancer cell-autonomous alterations that are known to drive insulin/IGF-1-independent, DR-resistant tumors preclude the anti-cancer activity of metformin.
We recently proposed that CR/DR-unresponsive tumors with activating mutations in the PIK3CA oncogene should be monitored for their responsiveness to clinically relevant concentrations of metformin. We now present evidence that human cancer xenotumors harboring the insulin-unresponsive PIK3CA-activating mutation H1047R remain sensitive to metformin. Our data confirm that metformin cannot be considered a bona fide CRM, at least in terms of its anti-cancer activity. Given the high number of PIK3CA mutations in human cancer, this finding could have a significant impact on the design of metformin-based therapies that aim to influence both the early stages of tumor formation and progression and cancer recurrence in advanced tumors.
RESULTS
PIK3CA H1047R-mutated, DR-resistant MCF10DCIS.com xenotumors are highly sensitive to intraperitoneally (i.p.) administered metformin. To model the oncogenic PIK3CA-driven early stages of human cancer, we employed the clonal breast cancer cell line MCF10DCIS.com, which was derived from a xenograft originating from premalignant MCF10AT cells that were injected into SCID mice. The MCF10DCIS.com cell line has been shown to have a missense mutation, H1047R, in the kinase domain of PI3K. This gain-of-function mutation is one of the “hot-spot” mutations in the catalytic domain p110α of the PI3KCA gene, and it generates the most potently oncogenic PI3K that is found with high frequency in various cancers. More importantly for this study, MCF10DCIS.com cells have been shown to proliferate regardless of the presence or absence of insulin in vitro and form tumors that are refractory to DR in vivo [8].
Two different metformin treatment protocols were tested (n=5 mice per group). In the first protocol, mice were exposed to control (water) or metformin (250 mg kg-1) ad libitum 1 week prior to tumor cell inoculation. The water was changed twice weekly and provided continuously for a total of 56 days. In the second protocol, metformin was administered by i.p. injection, and mice were treated daily with 200 mg kg-1 metformin beginning 1 week before cell inoculation. This i.p. schedule with metformin was continued for a total of 56 days. Metformin was well tolerated in both treatment regimens, and it did not significantly affect the weight (Supplementary Fig. 1) or the diet consumption profiles (data not shown) of the mice throughout the course of treatment.
Efficacy of oral metformin in the DR-resistant MCF10DCIS.com xenograft model. Fig. 1A shows the rate of tumor growth in the three treatment groups, with the data plotted as the mean tumor volume in each group over time. Compared with the animals in the vehicle-treated group (mean xenografted MCF10DCIS.com tumor volume of 1794±273 mm3), the animals that received 8 weeks of treatment with oral metformin exhibited slightly decelerated tumor growth, with a final mean tumor volume of 1186±293 mm3 (Fig. 1A). However, none of the differences between the untreated controls and the oral metformin-treated xenotumors over time reached statistical significance. This finding was better reflected by the percent MCF10DCIS.com tumor growth inhibition, which was calculated as follows: 1-treated/control volume ratio (1-T/C). The inhibitory effect of oral metformin was modest, reaching a maximum of 43% at 4 weeks after cell inoculation and decreasing toward the end of the treatment (approximately 30-35%).
Efficacy of i.p. metformin in the DR-resistant MCF10DCIS.com xenograft model. We investigated whether the i.p. dosing schedule with metformin was more effective in preventing the growth of MCF10DCIS.com xenotumors and found that i.p. metformin was highly effective. Compared with the mean xenograft tumor volume (approximately 1800 mm3) in the untreated control animals, daily i.p. treatment with metformin resulted in a dramatic reduction in mean tumor volume to 280±171 mm3 (Fig. 1A). Notably, the anti-tumor activity of i.p. metformin at only 7 days after cell inoculation (49%) was already greater than the maximum activity achieved at any time with oral metformin. Moreover, the inhibitory effect of i.p metformin increased in a time-dependent manner, reaching a maximum of 84% at 42 days after cell inoculation.
Intraperitoneally administered metformin significantly decreases mitotic activity in MCF10DCIS.com cancer tissues. We analyzed MCF10DCIS.com tumor xenografts to investigate the potential proapoptotic and/or anti-proliferative effects of metformin; these effects may have played a role in the overall anti-tumor efficacy of the drug. No significant differences were observed in the number of apoptotic cells between control tumors and the tumors from the two therapeutic regimens (i.e., mice treated with oral metformin and mice treated with i.p. metformin). We then compared the mitotic counts in hematoxylin and eosin (H&E)-stained paraffin-embedded sections according to the Scarff-Bloom-Richardson histoprognostic grading system, as modified by Elston and Ellis (Fig. 1B). For the first protocol, microscopic analysis of the H&E staining of MCF10DCIS.com tumors showed a moderate decrease (21%) in average mitotic counts in the oral metformin group (26±8) compared with the untreated control (33±1), but this difference did not reach statistical significance (p=0.5). For the second protocol, however, the quantification of average mitotic counts revealed a statistically significant (p=0.01) 42% decrease in the mitotic activity index in the i.p. metformin group (19±1) compared with the control group.
The microscopic appearance of MCF10DCIS.com carcinomas changed slightly after metformin treatment. Histological examination confirmed that the untreated MCF10DCIS.com tumors consisted of cells that appeared cytologically malignant, with high-grade nuclei, pleomorphism, and abundant central necrosis (i.e., Grade 3 invasive ductal carcinomas; Fig. 1C, panels a and a’). Anatomopathological examination of MCF10DCIS.com breast xenotumors in animals treated with oral metformin revealed that the cancer tissues remained poorly differentiated, despite visual evidence of a reduction in tumor size (Fig. 1C, panels b and b’). Interestingly, in the very small MCF10DCIS.com tumors that developed in the presence of daily i.p. metformin, a significant trend of decreasing cellularity, accompanied by an increase in the amount of extracellular connective tissue matrix, was observed (Fig. 1C, panels c and c’).
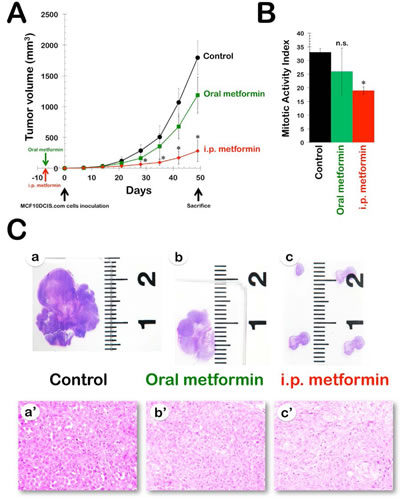
Figure 1: Efficacy of oral and i.p. metformin in the DR-resistant MCF10DCIS.com xenograft model. A. Shown are the mean tumor volumes (±SD) of MCF10DCIS.com xenograft-bearing nude mice following oral (ad libitum access to water containing 250 mg kg-1 metformin) and i.p. (daily i.p. injections of 200 mg kg-1 metformin) administration of metformin for 8 weeks. Tumor growth rates were significantly different between the control and the i.p. metformin groups (* Student’s t-test P<0.01). B. The bar graph (mean ± SD) shows the quantification of the mitotic activity in the xenografts. The number of mitosis per high power field was quantified by light microscopy in at least ten high power fields per tumor in all xenografts (n=2 per group, three groups; * Student’s t-test P<0.01 versus control group). C. Metformin-treated MCF10DCIS.com xenotumors have reduced tumor growth and altered histological features. a-c. Sections from xenografts were stained with H&E and taken at low magnification. a’-c’. Histopathological comparison among MCF10DCIS.com xenografts (evaluation was performed under 400X objective magnification)
Insulin-independent growth of PIK3CA H1047R-mutated tumor cells is inhibited by metformin in vitro. Because the constitutive activation of the PI3K pathway drives MCF10DCIS.com cells to form DR-resistant tumors in vivo and to grow in an insulin-independent manner in vitro, we determined whether exogenous supplementation with metformin significantly affects tumor cell unresponsiveness to insulin in vitro. DR-resistant MCF10DCIS.com cells grew similarly in culture in an insulin-independent fashion, i.e., insulin failed to cause a dose-dependent increase in cell number. Metformin did not act as an insulin sensitizer for insulin-resistant PIK3CA H1047R-mutated MCF10DCIS.com cells; rather, it acted as a growth inhibitor in a dose-dependent manner (Fig. 2).
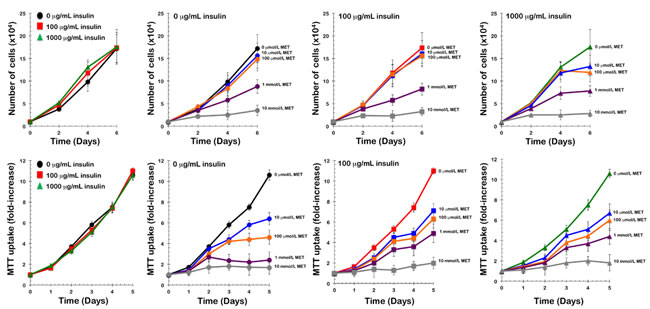
Figure 2: Metformin inhibits insulin-independent growth of PIK3CA-mutated tumor cells in vitro. A. Top. Proliferation curves of MCF10DCIS.com cells cultured in the presence of increasing concentrations of insulin and/or metformin. MCF10DCIS.com cells were plated in 24-well plates at a density of 5,000 cells/well and cultured in 0.1% horse serum in the absence or presence of insulin (0, 100 and 1,000 ng/mL), metformin (1 µmol/L, 10 µmol/L, 100 µmol/L, 1 mmol/L, and 10 mmol/L), or a combination of insulin and metformin as specified. The data presented are the means of number cells × 104/well (±SD) from one representative experiment made in triplicate and obtained after 0, 2, 4, and 6 days. Bottom. MTT uptake curves of MCF10DCIS.com cells cultured in the presence of increasing concentrations of insulin and/or metformin. MCF10DCIS.com cells were plated in 96-well plates at a density of ∼2,000 cells/well and cultured in 0.1% horse serum in the absence or presence of insulin (0, 100 and 1,000 ng/mL), metformin (1 µmol/L, 10 µmol/L, 100 µmol/L, 1 mmol/L, and 10 mmol/L), or a combination of insulin and metformin as specified. The data presented are the means ±SD of fold-increases in OD570.
Isogenic conversion of a PIK3CA-wild-type tumor into a PIK3CA H1047R-mutated tumor promotes sensitization to the anti-cancer effects of metformin. To model PIK3CA-activating mutations in the late stages of human cancer, we used two types of cancer cells derived from the highly metastatic SW48 colorectal cancer cell line. These cell lines were isogenic, except that one line (SW48-WT) carries a wild-type allele of the PIK3CA oncogene, and the other (SW48-Mut) carries a constitutively active mutant allele (H1047R). In xenograft studies, the subcutaneous injection of SW48-WT or SW48-Mut cells into nude mice yielded tumors of approximately equal volume within 27 days (Fig. 3, left panel). The administration of an oral regimen of metformin to xenografted mice beginning 1 week before tumor cell injection failed to significantly alter tumor size in SW48-WT xenotumors over time. In contrast, SW48-Mut cells formed metformin-sensitive tumors that showed significant decreases in tumor volume under the metformin feeding condition. Compared with the mean xenograft tumor volume (1138±188 mm3) in the untreated control animals, oral treatment with metformin resulted in a notable reduction in the mean tumor volume, to 572±134 mm3 (Fig. 3, right panel). Indeed, the inhibitory effect of oral metformin against SW48-Mut xenotumors increased in a time-dependent manner, reaching a maximum of approximately 50% at 21 days after cell inoculation.
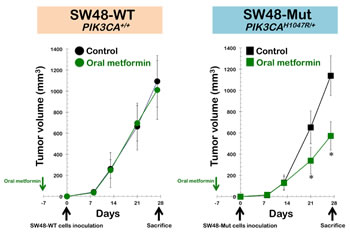
Figure 3. Isogenic conversion of a PIK3CA-wild-type tumor into a PIK3CA H1047R-mutated tumor promotes sensitization to the anti-cancer effects of metformin. Shown are the mean tumor volumes (±SD) of SW48-WT (left panel) and SW48-Mut (right panel) xenograft-bearing nude mice following oral (ad libitum access to water containing 250 mg kg-1 metformin) administration of metformin for ∼5 weeks. Tumor growth rates were significantly different between the control and the oral metformin groups in SW48-Mut xenografts (* Student’s t-test P<0.01).
DISCUSSION
The metabolism of tumors deviates significantly from that of corresponding normal cells and tissues [30-37]. Accordingly, the therapeutic use of DR/CR-mimetic agents to exploit the differential susceptibility of malignant versus normal cells to achieve energy metabolism inhibition is attracting great interest, and promising results have been obtained from cultured cells, animal models, and human trials. Although drugs targeting the cancer metabolic phenotype are expected to be “magic bullets” because many cancers show similar metabolic characteristics, it is important to note that molecular markers that can distinguish between CRM-sensitive and CRM-resistant tumors are lacking.
When searching for a cellular mechanism to explain the effect of DR on tumors, Kalaany and Sabatini found that DR reduces the activity of insulin-mediated signaling [8]. This signaling involves the activation of cell-surface receptors by insulin and the triggering of a downstream signaling cascade in the cell. This cascade is mediated by the enzyme PI3K and its suppression by DR enhances cell death and reduces tumor size in several, but not all, types of tumor cells. Cancer cell lines with mutations that cause the constitutive activation of PI3K (or loss-of-function mutations in PTEN, which normally antagonizes PI3K) are fully refractory to the tumor growth-inhibitory effects of DR. These findings are consistent with the effects of DR on the growth of early tumors due to both systemic changes in the host (e.g., circulating levels of insulin or IGF-1) and signaling events that are intrinsic to the tumor cells (e.g., those involving AMPK, mTOR, or SIRT1, all of which interact with the PI3K pathway at multiple levels). In this scenario, the pivotal involvement of the insulin-mediated pathway in determining the ability of DR to induce anti-tumor effects supports the hypothesis that drugs that ameliorate insulin resistance in type 2 diabetes might be beneficial in preventing cancer, even in non-diabetic patients. The same model, however, can be used to predict which tumors would be vulnerable to such DR-mimicking pharmacological treatments based on their mutation profiles, particularly in the genes encoding PI3K and PTEN. Because it is now evident that cancers of the liver, colon, and breast are the most likely to harbor PIK3CA mutations [38-44], with average mutational frequencies (across reported studies) of 36%, 26%, and 25%, respectively, a lack of responsiveness of PI3K-mutated carcinomas to putative DR mimetics such as metformin could have a substantial impact on the ever-growing number of randomized trials that are underway to evaluate whether and how metformin influences both the early stages of tumor formation and progression and cancer recurrence in advanced tumors [45-50].
Here, for the first time, we reveal that PIK3CA H1047R-mutated, DR-resistant MCF10DCIS.com xenotumors are highly sensitive (>80% tumor growth inhibition) to the daily i.p. administration of metformin. Because the oral dosing schedule of metformin only modestly affected tumor growth (approximately 40% of tumor growth inhibition), it is tempting to suggest that i.p. injections of metformin provoke peak plasma concentrations that are higher than the levels achieved with oral administration [29]. Pharmacokinetic analyses are currently underway to confirm whether the i.p. administration of metformin produces higher plasma levels of metformin, which might cause more significant anti-tumor effects. Nevertheless, there was a correlation between the metformin-stimulated decrease in proliferation factor mitotic activity index (MAI) in orally or intraperitoneally treated tumors and the overall efficacy of the route of metformin administration in preventing tumor growth in DR-resistant MCF10DCIS.com xenotumors. Because the significant decrease in the number of mitotic figures in tumors treated with i.p. metformin compared with untreated controls was not coupled with a change in the number of apoptotic cells, these findings suggested that the antitumor activity of i.p. metformin against MCF10DCIS.com breast cancer tumor growth mainly involved the inhibition of cell proliferation. Moreover, tumors treated with i.p. metformin showed a relatively substantial increase in stromal connective tissue that was not supportive of tumor growth [51], as this increase was accompanied by a decrease in mitotic figure numbers and reduced cellularity in the small amount of tumor tissue that remained at the end of the treatment. Proliferation factor MAI is the strongest prognostic factor in early breast cancer [52-54], a loss of cellularity has been shown to correlate with better prognosis and clinical outcome [55-57], and, ideally, a combination of residual tumor size and changes in tumor cellularity is useful in documenting treatment response and outcome. For these reasons, the response of PIK3CA H1047R-mutated breast cancer xenotumors to the anti-cancer effects elicited by the i.p. administration of metformin might be predictive of more favorable patient outcomes in a clinical setting. It would be interesting to investigate whether longer i.p. metformin regimens could still suppress PIK3CA H1047R-mutated tumors or whether tumors recur after some interval. Future studies should elucidate whether responsive tumors harboring PI3K-activating mutations will become resistant to the beneficial effects of metformin with time, as most tumors acquire resistance to chemotherapy.
At advanced tumor stages, signaling pathways other than the PI3K pathway have been suggested to play key roles in mediating the anti-cancer effects of DR. Hence, DR may no longer modulate signaling in advanced tumors once these tumors activate growth and survival pathways in a growth factor-independent fashion. This study is the first to reveal that, remarkably, the isogenic conversion of a PIK3CA-wild-type tumor into a PIK3CA H1047R-mutated tumor is sufficient to promote sensitization to the anti-cancer activity of metformin in an in vivo model of metastatic colon cancer. Considering the emerging role of PIK3CA mutation status in the treatment selection process [58-60], these findings might have a significant impact on the design of future trials evaluating the potential of combining metformin with targeted therapy. The hyperactivation of PI3K signaling downstream of the tyrosine kinase receptor HER2, either through loss-of-function PTEN mutations or dominant activating mutations in the catalytic subunit of PI3K, has been shown to significantly impair the efficacy of HER2-targeted drugs such as trastuzumab [61-68]. Accordingly, PI3K inhibitors, mTOR inhibitors, and dual PI3K/mTOR inhibitors are being evaluated as effective therapies for overcoming trastuzumab resistance. In this regard, it is interesting that the JIMT-1 breast cancer cell line, a model of intrinsic resistance to trastuzumab that harbors an activating mutation in the PIK3CA gene and low expression of PTEN [69], was notably responsive to metformin in vitro and in vivo [70, 71]. Moreover, when trastuzumab was combined with metformin to treat PIK3CA-mutated HER2+ breast carcinomas, tumor volume decreased sharply, thus suggesting that metformin is sufficient to overcome in vivo primary resistance to trastuzumab in these tumors [71]. Because of the accumulating evidence that PIK3CA mutation/PTEN expression status predicts colon carcinoma response to the EGFR inhibitor cetuximab [72-74], we are currently investigating whether the combination of cetuximab with metformin could be an effective therapeutic option in PIK3CA-mutated colon cancer.
After the TP53 suppressor gene, PIK3CA is one of the most frequently mutated (gain-of-function) genes in several human carcinomas. A landmark study by Buzzai et al. [75] used the paired isogenic colon cancer cell lines HCT116 p53+/+ and HCT116 p53-/- to test systemic treatment with metformin (250 mg kg-1 administered by daily i.p. injections). Using the clonal breast cancer cell line MCF10DCIS.com, which harbors the gain-of-function H1047R hot-spot mutation in the catalytic domain of the PI3KCA gene and has been shown to form DR-refractory xenotumors [8], and taking advantage of the isogenic conversion of a wild-type tumor into a PIK3CA H1047R-mutated tumor using the highly metastatic SW48 colorectal cancer cell line, we have shown that metformin can no longer be considered a bona fide DR mimetic, at least in terms of anti-cancer activity. In particular, tumors harboring the insulin-unresponsive, DR-resistant, PIK3CA-activating mutation H1047R remain sensitive to the anti-tumoral effects of the drug. Given the high prevalence of PIK3CA mutations in human carcinomas, and considering the emerging role of PIK3CA mutation status in the treatment selection process, these findings may have a significant impact on the design of future trials evaluating the potential of combining metformin with targeted therapy.
METHODS
Cell lines.
MCF10DCIS.COM cells were purchased from Asterand, Inc. (Detroit, MI, USA). X-MAN™ isogenic cell lines were obtained from Horizon Discovery Ltd (http://www.horizondiscovery.com). The X-MAN™ isogenic cell line SW48 PI3Kα (H1047R/+), heterozygous knock-in of PIK3CA kinase domain activating mutation, was used in this study (HD103-005). The parental cell line, SW48 PI3K (+/+) was also used. Cells were maintained according to the supplier recommendations.
Tumor xenograft study.
Mice were randomly divided into groups of five mice each and orally or intraperitoneally treated as described in the Results section. To produce xenografts, approximately 5 × 106 cells were subcutaneously injected into the dorsal flanks of female athymic nude mice (4-5 weeks old, 23-25 g; Harlan Laboratories). In both treatment regimens, body weight and diet consumption was determined weekly after dosing; tumor size was measured daily with electronic calipers; and tumor volume was calculated using the following formula: volume (mm3) = length × width2 × 0.5. The experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Institut d’Investigació Biomèdica de Bellvitge (IDIBELL; Animal Use Protocol #6302 authorized by the Animal Experimental Commission of the Catalan Government, Barcelona, Spain). After 56 days of metformin treatment, the mice were euthanized by cervical dislocation. The tumors were excised and weighed, and one portion of the tissue was fixed in buffered formalin, whereas the remaining portion was stored at -80°C for further analysis. Tumor volumes were compared using Student’s t-test. At the end of the tumorigenesis studies, plasma was isolated by centrifugation of blood samples, and the levels of circulating insulin were measured using the Rat/Mouse Insulin ELISA Kit (Millipore, Billerica, MA).
Histology.
At the appropriate time points, animals were euthanized, and tumors were removed and cut in half. One half was fixed in 10% buffered formalin, and the other was snap frozen. To assess apoptosis and mitosis in the tumor tissues, sections from formalin-fixed, paraffin-embedded xenograft tissues were stained with H&E.
MAI.
The mitotic figures observable following the H&E staining protocol were defined according to van Diest et al. [52], with some modifications, as follows: a) the absence of nuclear membrane signifying the end of prophase, and b) the presence of condensed chromosomes that were clustered together (beginning metaphase), arranged in a plane (metaphase or anaphase), or in separate clusters (telophase) all counted as one mitotic figure. Hyperchromatic nuclei, fragmented chromatin, and apoptotic nuclei were ignored. All H&E-stained sections were examined carefully (magnification, ×400), and the section showing the highest proliferation was selected for an assessment of mitotic activity. Starting from the subjectively most mitotically active area of the tumor and moving between consecutive fields, approximately 10 consecutive high-power fields (HPFs) were counted with an Olympus BH-2 microscope. No attempts were made to maximize counting by selecting those fields with a greater number of mitotic figures. Mitotic counts were performed without the knowledge of the treatment group-based immunohistochemical staining. MAI activities were compared using a two-tailed, two-sample, equal-variance Student’s t-test.
Proliferation assay.
On day 0, cell lines were seeded in the appropriate media in 24-well plates at a density of 5,000 cells/well, and all plates were incubated overnight. On day 1, the assay plates for each cell line were washed twice with regular medium in the absence of serum, and the medium was replaced with medium supplemented with 0.1% serum alone, 0.1% serum and one of two different concentrations of insulin (100 and 1,000 ng/mL), 0.1% serum and one of seven different concentrations of metformin (10 nmol/L, 100 nmol/L, 1 µmol/L, 10 µmol/L, 100 µmol/L, 1 mmol/L, and 10 mmol/L), or a combination of insulin and metformin, as specified. Three wells per media condition were included in each plate. An additional plate for each cell line was used as a baseline day 0 measurement of cell number without addition of the assay media. Cell numbers were counted at days 0, 2, 4, and 6 using a Coulter Counter (Coulter Electronics, Inc.).
Cell viability assays.
The effect of metformin on cell viability in the absence or presence of insulin was determined using a standard colorimetric 3,4,5-dimethylthiazol-2-yl-2,5-diphenyl-tetrazolium bromide (MTT) reduction assay. For each treatment, the percent cell viability was calculated using the following equation: (OD570 of the treated sample/OD570 of the untreated sample) × 100.
Acknowledgments.
This work was financially supported by the Instituto de Salud Carlos III (Ministerio de Sanidad y Consumo, Fondo de Investigación Sanitaria (FIS), Spain, grants EC10-125, CP05-00090, PI06-0778 and RD06-0020-0028), the Fundación Científica de la Asociación Española Contra el Cáncer (AECC, Spain), and the Ministerio de Ciencia e Innovación (SAF2009-11579 and SAF2012-38914, Plan Nacional de I+D+i, MICINN, Spain). Sílvia Cufí received a research fellowship (BES-2010-032066, Formación de Personal Investigador, FPI) from the Ministerio de Ciencia e Innovación (MICINN, Spain).
Collaborators (METTEN-01 Investigators):
Maria Luque Cabal (Medical Oncology Service, Hospital Central de Asturias, Oviedo, Spain), César A. Rodríguez-Sánchez (Medical Oncology Service, Hospital Universitario de Salamanca, Salamanca, Spain), Jose Luis Alonso Romero (Department of Medical Oncology, Hospital Universitario Virgen de la Arrixaca, Murcia, Spain), Ricardo Cubedo Cervera (Department of Medical Oncology, Hospital Puerta de Hierro, Madrid, Spain), Jose Manuel López-Vega (Medical Oncology Department, Hospital Universitario Marqués de Valdecilla, Santander, Spain), Noelia Martínez Jáñez (Medical Oncology Department, Hospital Universitario Ramón y Cajal, Madrid, Spain), Kepa Amillano Parraga (Medical Oncology Service, Hospital Universitario San Joan de Reus, Reus, Spain).
References
1. Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN. Calories and carcinogenesis: lessons learned from 30 years of calorie restriction research. Carcinogenesis. 2010; 31:83-89.
2. Hursting SD, Berger NA. Energy balance, host-related factors, and cancer progression. J Clin Oncol. 2010; 28:4058-4065.
3. Hursting SD, Dunlap SM, Ford NA, Hursting MJ, Lashinger LM. Calorie restriction and cancer prevention: a mechanistic perspective. Cancer & Metabolism. 2013; 1:10.
4. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460:392-395.
5. Lashinger LM, Malone LM, Brown GW, Daniels EA, Goldberg JA, Otto G, Fischer SM, Hursting SD: Rapamycin partially mimics the anticancer effects of calorie restriction in a murine model of pancreatic cancer. Cancer Prev Res. 2011, 4:1041-1051.
6. De Angel RE, Conti CJ, Wheatley KE, Brenner AJ, Otto G, Degraffenried LA, Hursting SD. The enhancing effects of obesity on mammary tumor growth and Akt/mTOR pathway activation persist after weight loss and are reversed by RAD001. Mol Carcinog. 2013;52:446-458.
7. Nogueira LM, Dunlap SM, Ford NA, Hursting SD. Calorie restriction and rapamycin inhibit MMTV-Wnt-1 mammary tumor growth in a mouse model of postmenopausal obesity. Endocr Relat Cancer. 2012; 19:57-68.
8. Kalaany NY, Sabatini DM. Tumours with PI3K activation are resistant to dietary restriction. Nature. 2009; 458:725-731.
9. Brunet A. Cancer: When restriction is good. Nature. 2009; 458:713-714.
10. Martin-Castillo B, Vazquez-Martin A, Oliveras-Ferraros C, Menendez JA. Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle. 2010; 9:1057-1064.
11. Vazquez-Martin A, Oliveras-Ferraros C, Cufí S, Martin-Castillo B, Menendez JA. Metformin and energy metabolism in breast cancer: from insulin physiology to tumour-initiating stem cells. Curr Mol Med. 2010;10:674-691.
12. Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY). 2010; 2:760-774.
13. Menendez JA, Cufí S, Oliveras-Ferraros C, Vellon L, Joven J, Vazquez-Martin A. Gerosuppressant metformin: less is more. Aging (Albany NY). 2011; 3:348-362.
14. Del Barco S, Vazquez-Martin A, Cufí S, Oliveras-Ferraros C, Bosch-Barrera J, Joven J, Martin-Castillo B, Menendez JA. Metformin: multi-faceted protection against cancer. Oncotarget. 2011; 2:896-917.
15. Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer Discov. 2012, 2:778-790.
16. Dowling RJ, Niraula S, Stambolic V, Goodwin PJ: Metformin in cancer: translational challenges. J Mol Endocrinol. 2012; 48:R31–R43.
17. Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonammi B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res. 2010; 3:1451-1461.
18. Currie CJ, Poole CD, Gale EA. The influence of glucose-lowering therapies on cancer risk in type 2 diabetics. Diabetologia. 2009, 52:1766-1777.
19. Goodwin PJ, Stambolic V. Obesity and insulin resistance in breast cancer--chemoprevention strategies with a focus on metformin. Breast. 2011; 20 Suppl 3:S31-S35.
20. Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012; 12:159-169.
21. Cohen DH, LeRoith D. Obesity, type 2 diabetes, and cancer: the insulin and IGF connection. Endocr Relat Cancer. 2012; 19:F27-F45.
22. Onitilo AA, Engel JM, Glurich I, Stankowski RV, Williams GM, Doi SA. Diabetes and cancer II: role of diabetes medications and influence of shared risk factors. Cancer Causes Control. 2012; 23:991-1008.
23. Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008; 412:211-221.
24. Rosilio C, Lounnas N, Nebout M, Imbert V, Hagenbeek T, Spits H, Asnafi V, Pontier-Bres R, Reverso J, Michiels JF, Ben Sahra I, Bost F, Peyron JF. The metabolic perturbators metformin, phenformin and AICAR interfere with the growth and survival of murine PTEN-deficient T cell lymphomas and human T-ALL/T-LL cancer cells. Cancer Lett. 2013; 336:114-126.
25. Anisimov VN, Egormin PA, Bershtein LM, Zabezhinskii MA, Piskunova TS, Popovich IG, Semenchenko AV. Metformin decelerates aging and development of mammary tumors in HER-2/neu transgenic mice. Bull Exp Biol Med. 2005; 139:721-723.
26. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005; 40:685-693.
27. Tomimoto A, Endo H, Sugiyama M, Fujisawa T, Hosono K, Takahashi H, Nakajima N, Nagashima Y, Wada K, Nakagama H, Nakajima A. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 2008; 99:2136-2141.
28. Quinn BJ, Dallos M, Kitagawa H, Kunnumakkara AB, Memmott RM, Hollander MC, Gills JJ, Dennis PA. Inhibition of Lung Tumorigenesis by Metformin Is Associated with Decreased Plasma IGF-I and Diminished Receptor Tyrosine Kinase Signaling. Cancer Prev Res (Phila). 2013 Jul 25. [Epub ahead of print]
29. Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen--induced lung tumorigenesis. Cancer Prev Res (Phila). 2010; 3:1066-1076.
30. Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 2009; 23:537-548.
31. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029-1033.
32. Cuperlovic-Culf M, Culf AS, Touaibia M, Lefort N. Targeting the latest hallmark of cancer: another attempt at ‘magic bullet’ drugs targeting cancers’ metabolic phenotype. Future Oncol. 2012; 8:1315-1330.
33. Cantor JR, Sabatini DM. Cancer cell metabolism: one hallmark, many faces. Cancer Discov. 2012; 2:881-898.
34. Soga T. Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. 2013;104:275-81
35. Butler EB, Zhao Y, Muñoz-Pinedo C, Lu J, Tan M. Stalling the engine of resistance: targeting cancer metabolism to overcome therapeutic resistance. Cancer Res. 2013; 73:2709-2717.
36. Menendez JA, Joven J, Cufí S, Corominas-Faja B, Oliveras-Ferraros C, Cuyàs E, Martin-Castillo B, López-Bonet E, Alarcón T, Vazquez-Martin A.The Warburg effect version 2.0: metabolic reprogramming of cancer stem cells. Cell Cycle. 2013; 12:1166-1179.
37. Menendez JA, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle. 2012; 11:2782-2792.
38. Karakas B, Bachman KE, Park BH. Mutation of the PIK3CA oncogene in human cancers. Br J Cancer. 2006; 94:455-459.
39. Zhao L, Vogt PK. Class I PI3K in oncogenic cellular transformation. Oncogene. 2008; 27:5486-5496.
40. Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011; 13:224.
41. Braccini L, Ciraolo E, Martini M, Pirali T, Germena G, Rolfo K, Hirsch E. PI3K keeps the balance between metabolism and cancer. Adv Biol Regul. 2012; 52:389-405.
42. Janku F, Wheler JJ, Naing A, Stepanek VM, Falchook GS, Fu S, Garrido-Laguna I, Tsimberidou AM, Piha-Paul SA, Moulder SL, Lee JJ, Luthra R, Hong DS, Kurzrock R.PIK3CA mutations in advanced cancers: characteristics and outcomes. Oncotarget. 2012; 3:1566-1575.
43. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Bäsecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, Tafuri A, Cocco L, Evangelisti C, Chiarini F, Martelli AM. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012; 3:954-987.
44. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, Mazzarino MC, Fagone P, Nicoletti F, Bäsecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, Tafuri A, Chiarini F, Evangelisti C, Cocco L, Martelli AM. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget. 2012; 3:1068-1111.
45. Martin-Castillo B, Dorca J, Vazquez-Martin A, Oliveras-Ferraros C, Lopez-Bonet E, Garcia M, Del Barco S, Menendez JA. Incorporating the antidiabetic drug metformin in HER2-positive breast cancer treated with neo-adjuvant chemotherapy and trastuzumab: an ongoing clinical-translational research experience at the Catalan Institute of Oncology. Ann Oncol. 2010; 21:187-189.
46. Gonzalez-Angulo AM, Meric-Bernstam F. Metformin: a therapeutic opportunity in breast cancer. Clin Cancer Res. 2010; 16:1695-1700.
47. Jalving M, Gietema JA, Lefrandt JD, de Jong S, Reyners AK, Gans RO, de Vries EG. Metformin: taking away the candy for cancer? Eur J Cancer. 2010; 46:2369-2380
48. Goodwin PJ, Stambolic V, Lemieux J, Chen BE, Parulekar WR, Gelmon KA, Hershman DL, Hobday TJ, Ligibel JA, Mayer IA, Pritchard KI, Whelan TJ, Rastogi P, Shepherd LE. Evaluation of metformin in early breast cancer: a modification of the traditional paradigm for clinical testing of anti-cancer agents. Breast Cancer Res Treat. 2011; 126:215-220.
49. Belda-Iniesta C, Pernía O, Simó R. Metformin: a new option in cancer treatment. Clin Transl Oncol. 2011; 13:363-367.
50. Bost F, Sahra IB, Le Marchand-Brustel Y, Tanti JF. Metformin and cancer therapy. Curr Opin Oncol. 2012; 24:103-108.
51. Appleyard MV, Murray KE, Coates PJ, Wullschleger S, Bray SE, Kernohan NM, Fleming S, Alessi DR, Thompson AM. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br J Cancer. 2012; 106:1117-1122.
52. van Diest PJ, van der Wall E, Baak JP. Prognostic value of proliferation in invasive breast cancer: a review. J Clin Pathol. 2004; 57:675-681.
53. Skaland I, Janssen EA, Gudlaugsson E, Klos J, Kjellevold KH, Søiland H, Baak JP. Phosphohistone H3 expression has much stronger prognostic value than classical prognosticators in invasive lymph node-negative breast cancer patients less than 55 years of age. Mod Pathol. 2007; 20:1307-1315.
54. Baak JP, Gudlaugsson E, Skaland I, Guo LH, Klos J, Lende TH, Søiland H, Janssen EA, Zur Hausen A. Proliferation is the strongest prognosticator in node-negative breast cancer: significance, error sources, alternatives and comparison with molecular prognostic markers. Breast Cancer Res Treat. 2009; 115:241-254.
55. Ogston KN, Miller ID, Payne S, Hutcheon AW, Sarkar TK, Smith I, Schofield A, Heys SD. A new histological grading system to assess response of breast cancers to primary chemotherapy: prognostic significance and survival. Breast. 2003; 12:320-327.
56. Sharkey FE, Addington SL, Fowler LJ, Page CP, Cruz AB. Effects of preoperative chemotherapy on the morphology of resectable breast carcinoma. Mod Pathol. 1996; 9:893-900.
57. Bhargava R, Dabbs DJ, Beriwal S, Yildiz IA, Badve P, Soran A, Johnson RR, Brufsky AM, Lembersky BC, McGuire KP, Ahrendt GM. Semiquantitative hormone receptor level influences response to trastuzumab-containing neoadjuvant chemotherapy in HER2-positive breast cancer. Mod Pathol. 2011; 24:367-374.
58. Gonzalez-Angulo AM, Ferrer-Lozano J, Stemke-Hale K, Sahin A, Liu S, Barrera JA, Burgues O, Lluch AM, Chen H, Hortobagyi GN, Mills GB, Meric-Bernstam F. PI3K pathway mutations and PTEN levels in primary and metastatic breast cancer. Mol Cancer Ther. 2011; 10:1093-1101.
59. Loi S, Michiels S, Baselga J, Bartlett JM, Singhal SK, Sabine VS, Sims AH, Sahmoud T, Dixon JM, Piccart MJ, Sotiriou C. PIK3CA genotype and a PIK3CA mutation-related gene signature and response to everolimus and letrozole in estrogen receptor positive breast cancer. PLoS One. 2013; 8:e53292.
60. Ludovini V, Bianconi F, Pistola L, Pistola V, Chiari R, Colella R, Bellezza G, Tofanetti FR, Siggillino A, Baldelli E, Flacco A, Giuffrida D, Sidoni A, Crinò L. Optimization of patient selection for EGFR-TKIs in advanced non-small cell lung cancer by combined analysis of KRAS, PIK3CA, MET, and non-sensitizing EGFR mutations. Cancer Chemother Pharmacol. 2012; 69:1289-1299.
61. Berns K, Horlings HM, Hennessy BT, Madiredjo M, Hijmans EM, Beelen K, Linn SC, Gonzalez-Angulo AM, Stemke-Hale K, Hauptmann M, Beijersbergen RL, Mills GB, van de Vijver MJ, Bernards R. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell. 2007; 12:395-402.
62. Serra V, Markman B, Scaltriti M, Eichhorn PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S, Maira M, Garcia-Echeverria C, Parra JL, Arribas J, Baselga J. NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K signaling and inhibits the growth of cancer cells with activating PI3K mutations. Cancer Res. 2008; 68:8022-8030.
63. Eichhorn PJ, Gili M, Scaltriti M, Serra V, Guzman M, Nijkamp W, Beijersbergen RL, Valero V, Seoane J, Bernards R, Baselga J. Phosphatidylinositol 3-kinase hyperactivation results in lapatinib resistance that is reversed by the mTOR/phosphatidylinositol 3-kinase inhibitor NVP-BEZ235. Cancer Res. 2008; 68:9221-9230.
64. Hynes NE, Dey JH. PI3K inhibition overcomes trastuzumab resistance: blockade of ErbB2/ErbB3 is not always enough. Cancer Cell. 2009; 15:353-355.
65. van der Heijden MS, Bernards R. Inhibition of the PI3K pathway: hope we can believe in? Clin Cancer Res. 2010; 16:3094-3099.
66. Razis E, Bobos M, Kotoula V, Eleftheraki AG, Kalofonos HP, Pavlakis K, Papakostas P, Aravantinos G, Rigakos G, Efstratiou I, Petraki K, Bafaloukos D, Kostopoulos I, Pectasides D, Kalogeras KT, Skarlos D, Fountzilas G. Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Res Treat. 2011; 128:447-456.
67. Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis C, King TA. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012; 18:6784-6791.
68. Cizkova M, Dujaric ME, Lehmann-Che J, Scott V, Tembo O, Asselain B, Pierga JY, Marty M, de Cremoux P, Spyratos F, Bieche I. Outcome impact of PIK3CA mutations in HER2-positive breast cancer patients treated with trastuzumab. Br J Cancer. 2013; 108:1807-1809.
69. Köninki K, Barok M, Tanner M, Staff S, Pitkänen J, Hemmilä P, Ilvesaro J, Isola J. Multiple molecular mechanisms underlying trastuzumab and lapatinib resistance in JIMT-1 breast cancer cells. Cancer Lett. 2010; 294:211-219.
70. Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B, Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast Cancer Res Treat. 2011; 126:355-364.
71. Cufi S, Corominas-Faja B, Vazquez-Martin A, Oliveras-Ferraros C, Dorca J, Bosch-Barrera J, Martin-Castillo B, Menendez JA. Metformin-induced preferential killing of breast cancer initiating CD44+CD24-/low cells is sufficient to overcome primary resistance to trastuzumab in HER2+ human breast cancer xenografts. Oncotarget. 2012; 3:395-398.
72. De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V, Papamichael D, Laurent-Puig P, Penault-Llorca F, Rougier P, Vincenzi B, Santini D, Tonini G, Cappuzzo F, Frattini M, Molinari F, Saletti P, De Dosso S, Martini M, Bardelli A, Siena S, Sartore-Bianchi A, Tabernero J, Macarulla T, Di Fiore F, Gangloff AO, Ciardiello F, Pfeiffer P, Qvortrup C, Hansen TP, Van Cutsem E, Piessevaux H, Lambrechts D, Delorenzi M, Tejpar S. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol. 2010; 11:753-762.
73. Sood A, McClain D, Maitra R, Basu-Mallick A, Seetharam R, Kaubisch A, Rajdev L, Mariadason JM, Tanaka K, Goel S. PTEN gene expression and mutations in the PIK3CA gene as predictors of clinical benefit to anti-epidermal growth factor receptor antibody therapy in patients with KRAS wild-type metastatic colorectal cancer. Clin Colorectal Cancer. 2012; 11:143-150.
74. Tian S, Simon I, Moreno V, Roepman P, Tabernero J, Snel M, van’t Veer L, Salazar R, Bernards R, Capella G. A combined oncogenic pathway signature of BRAF, KRAS and PI3KCA mutation improves colorectal cancer classification and cetuximab treatment prediction. Gut. 2013; 62:540-549.
75. Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007; 67:6745-6752.