
INTRODUCTION
Src-associated in mitosis of 68 kDa (Sam68), also known as KHDRBS1, is a KH-type RNA binding protein of the signal transduction and activation of RNA metabolism (STAR) family [1–3]. The expression of Sam68 is often elevated in human cancers, including breast, colorectal, esophageal, endometrial, cervical, renal, lung, bladder, ovarian, neural, and prostate cancers [4–15]. Furthermore, overexpression of Sam68 has been shown to correlate with poor survival prognosis in renal cell carcinoma, colorectal cancer, and non-small cell lung cancer patients [5, 9, 10]. In some cancers, high expression of Sam68 correlates with low expression of certain miRNAs shown to target Sam68, such as in the case of miR-203 and miR-204 observed in neuroblastoma and breast cancer, respectively [13, 16]. Collectively, these findings suggest that Sam68 has a positive role in tumor progression.
To further investigate the physiological roles of Sam68, a mouse model was generated by homologous recombination [17]. These whole body knockout mice are born with no visible deformities and live to old age [17]. Phenotypically, Sam68-null mice are lean and are protected against age-related bone loss [17, 18]. These mice also have significant motor coordination defects attributed to RNA metabolism dysregulation in neurons [19–22]. Interestingly, crossbreeding with spinal muscular atrophy (SMAΔ7) mice rescued the body weight and viability of SMAΔ7, suggesting that the loss of Sam68 could be a therapeutic avenue for some patients with spinal muscular atrophy [23]. Sam68-null mice do not develop spontaneous tumors, demonstrating that Sam68 is not a bona fide tumor suppressor like p53 [24, 25]. However, Sam68-haploinsufficiency delays the onset of MMTV-PyMT-driven mammary tumors and reduces dissemination of lung metastasis, implicating that Sam68 is required for mammary tumorigenesis [26].
As a prototypic member of the STAR proteins, Sam68 has signaling properties and functions downstream of Src family kinases to promote cell survival [26, 27]. In addition, Sam68 regulates cell cycle progression and apoptosis. Specifically, Sam68 modulates transcription and mRNA translation, and functions as a major regulator of pre-mRNA alternative splicing [2]. It has been demonstrated that Sam68 depletion by siRNA promotes cell cycle arrest, and decreases the proliferation of many cancer cell lines [4, 7, 8, 15]. In particular, Sam68 has been shown to enhance the inclusion of CD44 variable exon 5 and the splicing of cyclin D1b isoform [28, 29], both of which are known to promote cell proliferation. Depletion of Sam68 from breast cancer cell lines upregulates cyclin-dependent kinase inhibitors p21 and p27, while also reducing Akt phosphorylation [4]. Moreover, Sam68 contributes to the protection of prostate cancer cells from genotoxic agents [15]. Hence, these findings strongly suggest Sam68 as an RNA binding protein required for the proliferation of certain cancer cells.
Sam68 has also been shown to harbor tumor suppressive functions, but this activity is not well understood, nor is there any evidence of this tumor suppressive role in vivo. It has been proposed that the loss of Sam68 leads to NIH3T3 cell transformation; however, this was not rescued by Sam68, suggesting that additional events were responsible for transformation in this report [30]. Consistent with its tumor suppressor-like activities, the overexpression of Sam68 in murine fibroblasts induces cell cycle arrest and apoptosis [31]. Moreover, Sam68 expression favors alternative splicing of the pro-apoptotic Bcl-x(s) isoform, which further demonstrates its tumor suppressor-like activity [32, 33].
The development of mouse cancer models has greatly advanced our understanding of the mechanisms underlying tumor initiation, progression, metastasis, and acquired chemoresistance [34–36]. Specifically, the chemical carcinogen-based inducible tumor models are essential tools for investigating tumor development in a controlled manner [34, 35]. For instance, the azoxymethane (AOM)-inducible mouse model provides a reproducible system for studying spontaneous colon cancer, where tumors develop frequently in the distal part of the colon, and exhibit features similar to spontaneous colon cancer found in humans [37]. Additionally, genetically engineered mice, such as the p53-deficient mice [38], have been particularly valuable for the identification of molecular mechanisms and pathways affected by p53-deficiency, as the loss or mutation of TP53 occurs in approximately 50% of all cancers [25, 39]. Indeed, p53-deficient mice are predisposed to a range of spontaneous tumors, including lymphomas and sarcomas [40, 41]. Recently, we have identified Sam68 as a transcriptional co-activator of p53 [42], suggesting that Sam68 may also have tumor suppressor activities like p53.
In this study, we report the role of Sam68 in tumorigenesis using both p53-proficient and p53-deficient mouse models of cancer. We show that in p53 wild type mice, Sam68-deficiency enhanced AOM-induced colon tumorigenesis, whereas in p53-deficient mice, Sam68-haploinsufficiency delayed the onset of spontaneous tumors. Hence, we demonstrate for the first time that p53 status can influence the role of Sam68 during tumorigenesis.
RESULTS
Sam68-deficiency increases AOM-induced tumor burden and malignancy in p53 wild type mice
To investigate the role of Sam68 in tumor development where p53 is wild type, we chose a carcinogen-based mouse cancer model [34, 40]. Specifically, we used an azoxymethane (AOM)-inducible mouse model of colon carcinogenesis. AOM exerts colonotropic carcinogenicity in a non-specific manner by alkylating DNA to drive spontaneous tumor initiation [37]. Importantly, p53 is not generally mutated in AOM-induced lesions [43], allowing us to study the effect of Sam68 in this cancer model. Cohorts of fifteen p53+/+; Sam68+/+ and p53+/+; Sam68−/− mice, backcrossed 7 generations in the FVB background, were treated with AOM for 8 weeks and examined each week for 13 weeks. Three of the p53+/+; Sam68−/− mice were sacrificed early due to acute clinical features. In the remaining p53+/+; Sam68−/− mice, as well as the p53+/+; Sam68+/+ mice, aberrant growths were restricted to the colon with no macroscopically visible metastasis. The p53+/+; Sam68−/− mice had a statistically significant increase in colon weight (0.69 g ± 0.03 versus 0.43 g ± 0.03) because of the higher tumor burden (Figure 1A and 1B). Moreover, p53+/+;Sam68−/− mice displayed increased number of tumors per colon (49.83 ± 2.93 versus 32.07 ± 3.07; Figure 1C), and generally developed larger tumors (2–3 mm, ≥ 4 mm) compared to the p53+/+;Sam68+/+ mice (Figure 1D). Further examinations revealed that AOM-treated p53+/+; Sam68−/− mice had a statistically significant increase in the number of adenomas (12.67 ± 1.45 versus 5.5 ± 0.5) and carcinomas (8.67 ± 0.33 versus 6.5 ± 0.5) compared to p53+/+;Sam68+/+ mice (Figure 2). Concomitantly, p53+/+;Sam68−/− mice showed a marked increase in the cell proliferation marker Ki-67 compared to p53+/+;Sam68+/+ mice (45.77 ± 2.43 versus 23.07 ± 2 per crypt; Figure 3). Taken together, p53+/+;Sam68−/− mice exhibited a higher incidence of AOM-induced colon cancer, suggesting a crucial function for Sam68 in suppressing the malignant progression of these tumors.
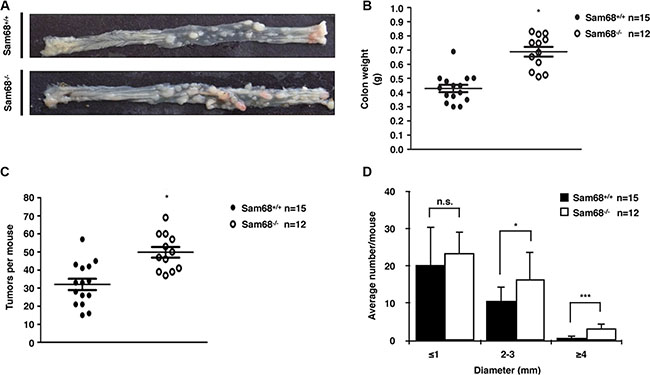
Figure 1: Sam68-deficent mice showed higher AOM-induced tumor burden and size. (A) Colons isolated from p53+/+;Sam68+/+ and p53+/+;Sam68−/− FVB mice after AOM treatment. (B) Weight of colons isolated from p53+/+;Sam68+/+ and p53+/+;Sam68−/− mice after AOM treatment. (C–D) Quantification of AOM-induced colon tumor numbers and average tumor size in p53+/+;Sam68+/+ and p53+/+;Sam68−/− mice. All error bars represent S.D. of the mean (*p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005).
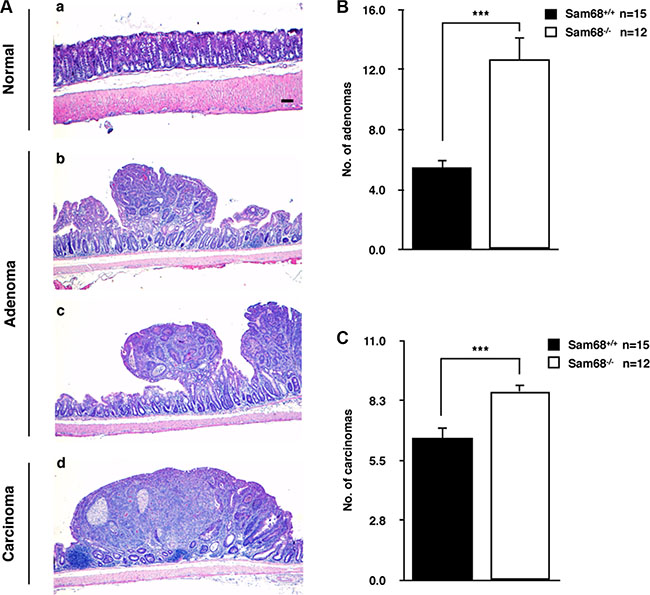
Figure 2: Sam68-deficient mice developed colon tumors with increased malignancy. (A) Representative H&E stains of untreated normal colon (a), adenoma from AOM-treated p53+/+;Sam68+/+ mice (b) and p53+/+;Sam68−/− mice (c), and carcinoma from AOM-treated p53+/+;Sam68−/− mice. Scale bar, 100μm. (B–C) Quantification of adenomas and carcinomas from colons of AOM-treated p53+/+;Sam68+/+ and p53+/+;Sam68−/− mice. All error bars represent S.D. of the mean (*p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005).
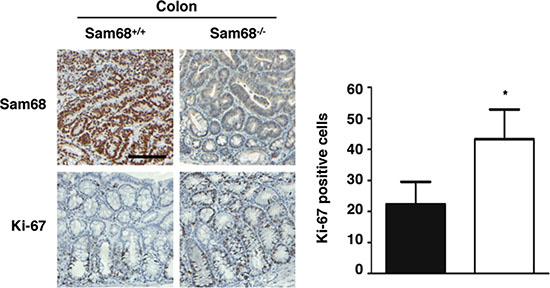
Figure 3: Sam68-deficient mouse colons contain more Ki-67-positive cells. Immunohistochemistry analysis of Sam68 and Ki-67 in colons isolated from AOM-treated p53+/+;Sam68+/+ and p53+/+;Sam68−/− mice. Scale bar, 100 μm. All error bars represent S.D. of the mean (*p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005).
Sam68-haploinsufficiency delayed tumor onset and prolonged survival in p53-deficient mice
We next examined whether the absence of Sam68 affects the onset of spontaneous tumors in p53-null mice [38, 40], given that we have recently reported Sam68 as a transcriptional co-activator of p53 [42]. We initially crossbred Sam68−/− (> 14 generations in C57BL/6 background) with p53−/− mice, but the breeding was complicated by the fact that Sam68 homozygous females have impaired fertility and males are infertile [26, 44]. For these reasons, only Sam68 heterozygotes were used for breeding, which are known to express lower levels of Sam68 than wild type mice [26]. Additionally, few p53−/− females were obtained, therefore the breeding was performed with p53−/−; Sam68+/− males and p53+/−;Sam68+/− females. Few p53−/−;Sam68−/− mice were obtained for unknown reasons but they did survive into adulthood. Thus, we compared tumor latency between p53−/−;Sam68+/+ and p53−/−;Sam68+/− mice. We observed that Sam68-haploinsufficiency significantly delayed tumor onset in p53-deficient mice from 140 days (p53−/−;Sam68+/+ mice, n = 21) to 195 days (p53−/−;Sam68+/− mice, n = 20, p < 0.0001; Figure 4). Like p53−/−;Sam68+/+ mice , p53−/−;Sam68+/− mice succumbed to mostly lymphomas and sarcomas [38]. Consistent with previous studies, none of the control p53+/+;Sam68−/− mice (n = 10) developed tumors in the 300 days of the experiment (Figure 4) [17]. Altogether, our findings suggest that Sam68 has pro-tumorigenic properties in the absence of p53.
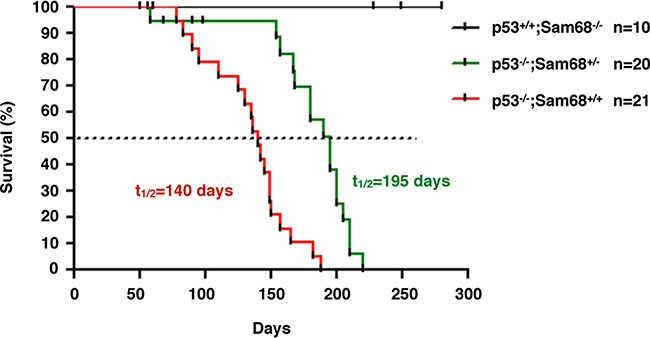
Figure 4: Sam68-haploinsufficiency delayed tumor onset in p53-deficient mice. Kaplan-Meier survival curve of tumor incidence in p53+/+; Sam68−/−, p53−/−; Sam68+/+, and p53−/−; Sam68+/− C57BL/6 mice. Tumor formation was monitored over a period of 10 months, and sacrificed at first clinical signs of discomfort (p < 0.0001).
DISCUSSION
In the present study, we examined the physiological role of Sam68 in tumor development using two different mouse cancer models. For Sam68-proficient and deficient mice in a p53 wild type FVB background, we implemented the azoxymethane (AOM)-inducible colon carcinogenesis model. We observed that Sam68-deficient (p53+/+;Sam68−/−) mice displayed higher tumor burden with increased malignancy than wild type controls (p53+/+;Sam68+/+), demonstrating that Sam68 has tumor suppressor-like activities in vivo. We also generated p53-null mice expressing Sam68 (p53−/−;Sam68+/+) or haploinsufficient dose of Sam68 (p53−/−;Sam68+/−). In this p53−/− C57BL/6 background, we observed that Sam68 haploinsufficiency caused significant delays in the onset of spontaneous tumors compared to Sam68 wild type mice. These findings show that in the absence of p53, Sam68 can exhibit pro-tumorigenic properties. Collectively, our results demonstrate that p53 seems to act as a switch to influence whether Sam68 slows or accelerates tumor development. Taken together, these findings provide insight into a previously unknown relation between Sam68 and the p53 tumor suppressor in tumorigenesis.
RNA binding proteins are becoming increasingly appreciated as fundamental players in human cancers [45, 46]. Like tumor suppressors, abnormal expression or mutations in RNA binding proteins have been shown to alter their function, and many are associated with a cancer phenotype [46]. We posit for the first time that Sam68-deficiency leads to increased tumorigenesis, where p53+/+;Sam68−/− mice displayed significantly higher AOM-induced tumor burden and malignancy compared to p53+/+;Sam68+/+ mice. Therefore, in this context, Sam68 behaves as a tumor suppressor. Indeed, Sam68 has been shown to be pro-apoptotic mainly by regulating Bcl-x(s) splicing [31–33]. Moreover, the fact that Sam68 functions as a co-activator of p53 is consistent with its tumor suppressor role [42]. Interestingly, it has been shown that p53 does not influence the rate of tumor initiation, but instead is required to prevent the malignant progression of tumors [47], which parallels the phenotype of the AOM mouse model. However, untreated Sam68−/− mice are not predisposed to spontaneous tumor development [17], similar to the phenotypes of p21−/− and p21−/−;Puma−/−;Noxa−/− mice [48, 49]. This perhaps explains why, as observed with the p21 gene, that no known mutations of Sam68 have been found in human tumors [50]. Furthermore, unlike p53, the function of Sam68 may be partially redundant with other STAR family RNA binding proteins, such as Sam68 paralogs SLM-1 and SLM-2 [51, 52].
Given the interplay between Sam68 and p53, it is almost expected that Sam68 would lose its tumor suppressive function when p53 is depleted. Indeed, studies in p53−/− mice showed that decreased Sam68 expression significantly delayed the onset of spontaneous tumors, indicating a pro-tumorigenic role for Sam68 in this mouse model. Fu et al. recently reported that Sam68 lessens colon tumor development in Apcmin716/+ mice via regulation of the PARP1-NF-κB-anti-apoptotic gene axis [53]. Furthermore, Sam68-deficient human colon cancer cells are sensitive to genotoxic stress-induced apoptosis [53].
Although it is uncertain how p53 loss or inactivation can contribute to the pro-tumorigenic function of Sam68, a large body of literature has elucidated molecular mechanisms by which Sam68 can support tumor growth. As a STAR family protein, Sam68 can link signal transduction with RNA processing to modulate cancer-relevant splicing events that enhance cell survival and proliferation [1–3]. For instance, Fyn-mediated tyrosine phosphorylation of Sam68 promotes splicing of the anti-apoptotic Bcl-x(L) isoform in prostate and pancreatic cancer cells [32, 54]. Additionally, Sam68 has been identified as a downstream target of the MAPK ERK1/2 pathway, where serine/threonine phosphorylation of Sam68 enhances tumorigenic CD44 variable exon 5 inclusion [28]. Similarly, activation of the MAPK pathway also stimulates Sam68-mediated splicing of cyclin D1b isoform to favor cell proliferation [29]. The involvement of Sam68 in signaling during tumorigenesis has also been observed previously in a MMTV-PyMT mouse model, where Sam68-haploinsufficiency delayed mammary tumorigenesis [26], consistent with the phenotype of our p53−/−;Sam68+/− mice. Of note, mammary tumor cells derived from the MMTV-PyMT mice are p53 wild type and are sensitive to p53 gene therapy, but the antitumor mechanisms remain elusive [55]. Furthermore, Sam68 may promote tumorigenesis by taking part in the transcription machinery. In prostate cancer cells, Sam68 functions as a transcriptional co-activator of the androgen receptor (AR) [14], a nuclear hormone receptor driving the onset and progression of prostate cancer [56]. Sam68 also modulates the promoter specificity of NF-κB to induce CD25 expression implicated in tumorigenesis [57]. Additionally, Sam68 is an essential component of the MLL oncogenic transcriptional complex by acting as a bridging molecule for MLL and PRMT1, and the knockdown of Sam68 suppressed MLL-mediated transformation [58]. In further support of its pro-tumorigenic properties, Sam68 overexpression may drive tumor progression by downregulating tumor suppressive miRNAs, including miR-203 and miR-204 [13, 16].
Sam68 overexpression, phosphorylation, and its cytoplasmic localization have been associated with a significant risk factor for poor prognosis [8, 9, 59]. It is also known that extensive post-translational modifications such as phosphorylation can influence its RNA binding activity [28, 32, 60]. For example, the elevated expression, mislocalization, and tyrosine phosphorylation of Sam68 all contribute to poor prognosis in breast cancer patients [4, 59]. Consistently, aberrant tyrosine phosphorylation and cytoplasmic localization of Sam68 has also been observed in various other cancer cells [8, 9, 26, 61, 62]. Together, the evidence suggests that aberrant Sam68 regulation, including sequestration of Sam68 from its nuclear role or inactivation by phosphorylation contributes to exacerbate tumorigenesis.
Although further studies are required to understand the mechanism underlying p53’s ability to switch the functional outcome of Sam68, we have illustrated the importance of defining the molecular contexts in which targeting RNA binding proteins may be beneficial for anticancer therapy. Much remains to be defined on how RNA binding proteins and their regulatory networks contribute to tumor initiation and progression.
MATERIALS AND METHODS
Mice breeding, AOM treatment, and colon tumor preparations
All animal procedures followed the Canadian Council on Animal Care guidelines and were approved by the McGill Animal Care Committee. Age-matched (10 to 12 weeks) p53+/+;Sam68+/+ and p53+/+; Sam68−/− mice in FVB background (cohorts of 15 in each group) were injected intraperitoneally once a week for 8 weeks with 10 mg/kg of body weight azoxymethane (AOM) (Sigma-Aldrich) dissolved in saline. Animals were systematically examined twice per week for the appearance of clinical symptoms. Animals showing signs of discomfort such as weight loss, rectal bleed, and prolapse were sacrificed. End points were defined as 13 weeks after the first injection. After sacrificing the mice, entire colons were removed, rinsed with PBS, opened longitudinally and fixed flat on strips of 4% paraformaldehyde-soaked Whatman filter paper. Colons were assessed in a blinded fashion under a stereo-dissecting microscope as previously described [63]. Tumors were measured using a clear transparency of 1 mm2 graph paper, and the total surface area was determined based on the total number of squares overlaying the tumor, as described [64].
The p53−/− mice (Catalog# 002101) were obtained from JAX laboratories. Both Sam68−/− and p53−/− mice were maintained on a C57BL/6 background and crossbred. Animals showing signs of discomfort such as weight loss were sacrificed.
Mouse genotyping
Total genomic DNA was extracted from mouse earpiece using AccuStart II Mouse Genotyping Kit (Quanta) following the manufacturer’s protocol. Genomic PCR was performed with the following primers: Sam68 (forward 1) 5′- GAT ATG ATG GAT GAT ATC TTG TCA G-3′; (forward 2) 5′- CTA AAG CGC ATG CTC CAG A-3′; (reverse) 5′- AAA TCC TAA CCC TCC TCA GTC A-3′; p53 (forward) 5′- ACA GCG TGG TGG TAC CTT AT-3′; (reverse 1) 5′-TAT ACT CAG AGC CGG CCT-3′; (reverse 2) 5′-CTA TCA GGA CAT AGC GTT GG-3′. PCR products were analyzed on 1.5% agarose gel stained with ethidium-bromide.
H&E staining and immunohistochemistry
Hematoxylin and eosin (H&E) or immunohistochemistry staining were performed on p53+/+; Sam68+/+ and p53+/+; Sam68−/− mice colons at the IRIC (Université de Montréal). Quantitative assessment was done as previously described [65].
Statistical analysis
Statistical analysis was performed using Student’s t test with the GraphPad software. A p-value of < 0.05 was considered to be statistically significant.
ACKNOWLDGMENTS
We thank Claire Turbide for expert technical assistance. We thank Zhenbao Yu and Gillian Vogel for helpful discussions and for critically reading the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
N. L. is supported by the CIHR/FRSQ training grant in cancer research of the McGill Integrated Cancer Research Training Program (FRN53888). The work was supported by a grant from the Canadian Institute of Health Research (MOP-123531) to S. R.
REFERENCES
1. Richard S. Reaching for the stars: Linking RNA binding proteins to diseases. Adv Exp Med Biol. 2010; 693:142–157.
2. Bielli P, Busa R, Paronetto MP, Sette C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocrine-related cancer. 2011; 18:R91–R102.
3. Frisone P, Pradella D, Di Matteo A, Belloni E, Ghigna C, Paronetto MP. SAM68: Signal Transduction and RNA Metabolism in Human Cancer. Biochemistry research international. 2015; 2015:528954.
4. Song L, Wang L, Li Y, Xiong H, Wu J, Li J, Li M. Sam68 up-regulation correlates with, and its down-regulation inhibits, proliferation and tumourigenicity of breast cancer cells. The Journal of pathology. 2010; 222:227–237.
5. Liao WT, Liu JL, Wang ZG, Cui YM, Shi L, Li TT, Zhao XH, Chen XT, Ding YQ, Song LB. High expression level and nuclear localization of Sam68 are associated with progression and poor prognosis in colorectal cancer. BMC Gastroenterol. 2013; 13:126–135.
6. Wang Y, Liang L, Zhang J, Li M, Zhu J, Gong C, Yang L, Zhu J, Chen L, Ni R. Sam68 promotes cellular proliferation and predicts poor prognosis in esophageal squamous cell carcinoma. Tumour Biol. 2015; 36:8735–8745.
7. Wang Q, Li Y, Zhou J, Liu J, Qin J, Xing F, Zhang J, Cheng J. Clinical significance of Sam68 expression in endometrial carcinoma. Tumour Biol. 2015; 36:4509–4518.
8. Li Z, Yu CP, Zhong Y, Liu TJ, Huang QD, Zhao XH, Huang H, Tu H, Jiang S, Zhang Y, Liu JH, Song LB. Sam68 expression and cytoplasmic localization is correlated with lymph node metastasis as well as prognosis in patients with early-stage cervical cancer. Ann Oncol. 2012; 23:638–646.
9. Zhang Z, Li J, Zheng H, Yu C, Chen J, Liu Z, M. L, Zeng M, Zhou F, Song L. Expression and cytoplasmic localization of SAM68 is a significant and independent prognostic marker for renal cell carcinoma. Cancer Epidemiol Biomarkers Prev. 2009; 18:2685–2693.
10. Zhang Z, Xu Y, Sun N, Zhang M, Xie J, Jiang Z. High Sam68 expression predicts poor prognosis in non-small cell lung cancer. Clin Transl Oncol. 2014; 16:886–891.
11. Zhang Z, Yu C, Li Y, Jiang L, Zhou F. Utility of SAM68 in the progression and prognosis for bladder cancer. BMC Cancer. 2015; 15:364.
12. Wang Y, Zhang W, Wang X, Wang D, Xie J, Tang C, Xi Q, Zhong J, Deng Y. Expression of Sam68 Correlates With Cell Proliferation and Survival in Epithelial Ovarian Cancer. Reprod Sci. 2016; pii:1933719116650757.
13. Zhao D, Tian Y, Li P, Wang L, Xiao A, Zhang M, Shi T. MicroRNA-203 inhibits the malignant progression of neuroblastoma by targeting Sam68. Mol Med Rep. 2015; 12:5554–5560.
14. Rajan P, Gaughan L, Dalgliesh C, El-Sherif A, Robson CN, Leung HY, Elliott DJ. The RNA-binding and adaptor protein Sam68 modulates signal-dependent splicing and transcriptional activity of the androgen receptor. The Journal of pathology. 2008; 215:67–77.
15. Busa R, Paronetto MP, Farini D, Pierantozzi E, Botti F, Angelini DF, Attisani F, Vespasiani G, Sette C. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene. 2007; 26:4372–4382.
16. Wang L, Tian H, Yuan J, Wu H, Wu J, Zhu X. CONSORT: Sam68 Is Directly Regulated by MiR-204 and Promotes the Self-Renewal Potential of Breast Cancer Cells by Activating the Wnt/Beta-Catenin Signaling Pathway. Medicine (Baltimore). 2015; 94:e2228.
17. Richard S, Torabi N, Franco GV, Tremblay GA, Chen T, Vogel G, Morel M, Cleroux P, Forget-Richard A, Komarova S, Tremblay ML, Li W, Li A, et al. Ablation of the Sam68 RNA binding protein protects mice from age-related bone loss. PLoS Genet. 2005; 1:e74.
18. Huot ME, Vogel G, Zabarauskas A, Ngo CT, Coulombe-Huntington J, Majewski J, Richard S. The Sam68 STAR RNA-binding protein regulates mTOR alternative splicing during adipogenesis. Molecular cell. 2012; 46:187–199.
19. Lukong KE, Richard S. Motor coordination defects in mice deficient for the Sam68 RNA-binding protein. Behav Brain Res. 2008; 189:357–363.
20. Iijima T, Wu K, Witte H, Hanno-Iijima Y, Glatter T, Richard S, Scheiffele P. SAM68 regulates neuronal activity-dependent alternative splicing of neurexin-1. Cell. 2011; 147:1601–1614.
21. Klein ME, Castillo PE, Jordan BA. Coordination between Translation and Degradation Regulates Inducibility of mGluR-LTD. Cell reports. 2015; pii:S2211–124700166–7.
22. Klein ME, Younts TJ, Castillo PE, Jordan BA. RNA-binding protein Sam68 controls synapse number and local beta-actin mRNA metabolism in dendrites. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110:3125–3130.
23. Pagliarini V, Pelosi L, Bustamante MB, Nobili A, Berardinelli MG, D’Amelio M, Musaro A, Sette C. SAM68 is a physiological regulator of SMN2 splicing in spinal muscular atrophy. The Journal of cell biology. 2015; 211:71–90.
24. Lukong KE, Richard S. Targeting the RNA-binding protein Sam68 as a treatment for cancer? Future oncology (London, England). 2007; 3:539–544.
25. Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nature reviews Cancer. 2009; 9:749–758.
26. Richard S, Vogel G, Huot ME, Guo T, Muller WJ, Lukong KE. Sam68 haploinsufficiency delays onset of mammary tumorigenesis and metastasis. Oncogene. 2008; 27:548–556.
27. Huot ME, Brown CM, Lamarche-Vane N, Richard S. An adaptor role for cytoplasmic Sam68 in modulating Src activity during cell polarization. Molecular and cellular biology. 2009; 29:1933–1943.
28. Matter N, Herrlich P, Konig H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature. 2002; 420:691–695.
29. Paronetto MP, Cappellari M, Busa R, Pedrotti S, Vitali R, Comstock C, Hyslop T, Knudsen KE, Sette C. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer research. 2010; 70:229–239.
30. Liu K, Li L, Nisson PE, Gruber C, Jessee J, Cohen SN. Neoplastic transformation and tumorigenesis associated with sam68 protein deficiency in cultured murine fibroblasts. The Journal of biological chemistry. 2000; 275:40195–40201.
31. Taylor SJ, Resnick RJ, Shalloway D. Sam68 exerts separable effects on cell cycle progression and apoptosis. BMC Cell Biol. 2004; 5:5.
32. Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. The Journal of cell biology. 2007; 176:929–939.
33. DeOcesano-Pereira C, Amaral MS, Parreira KS, Ayupe AC, Jacysyn JF, Amarante-Mendes GP, Reis EM, Verjovski-Almeida S. Long non-coding RNA INXS is a critical mediator of BCL-XS induced apoptosis. Nucleic acids research. 2014; 42:8343–8355.
34. Frese KK, Tuveson DA. Maximizing mouse cancer models. Nature reviews Cancer. 2007; 7:645–658.
35. Cheon DJ, Orsulic S. Mouse models of cancer. Annu Rev Pathol. 2011; 6:95–119.
36. Macleod KF, Jacks T. Insights into cancer from transgenic mouse models. The Journal of pathology. 1999; 187:43–60.
37. Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007; 2:1998–2004.
38. Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992; 356:215–221.
39. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science (New York, NY). 1991; 253:49–53.
40. Donehower LA. The p53-deficient mouse: a model for basic and applied cancer studies. Semin Cancer Biol. 1996; 7:269–278.
41. Harvey M, McArthur MJM, C. A., Jr., Butel JS, Bradley A, Donehower LA. Spontaneous and carcinogen-induced tumorigenesis in p53-deficient mice. Nat Genet. 1993; 5:225–229.
42. Li N, Richard S. Sam68 functions as a transcriptional coactivator of the p53 tumor suppressor. Nucleic acids research. 2016; pii:gkw582
43. Erdman SH, Wu HD, Hixson LJ, Ahnen DJ, Gerner EW. Assessment of mutations in Ki-ras and p53 in colon cancers from azoxymethane- and dimethylhydrazine-treated rats. Mol Carcinog. 1997; 19:137–144.
44. Paronetto MP, Messina V, Bianchi E, Barchi M, Vogel G, Moretti C, Palombi F, Stefanini M, Geremia R, Richard S, Sette C. Sam68 regulates translation of target mRNAs in male germ cells, necessary for mouse spermatogenesis. The Journal of cell biology. 2009; 185:235–249.
45. Kechavarzi B, Janga SC. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome biology. 2014; 15:R14.
46. Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Genet. 2008; 24:416–425.
47. Kemp CJ, Donehower LA, Bradley A, Balmain A. Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell. 1993; 74:813–822.
48. Valente LJ, Gray DH, Michalak EM, Pinon-Hofbauer J, Egle A, Scott CL, Janic A, Strasser A. p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell reports. 2013; 3:1339–1345.
49. Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell. 1995; 82:675.
50. Shiohara M, el-Deiry WS, Wada M, Nakamaki T, Takeuchi S, Yang R, Chen DL, Vogelstein B, Koeffler HP. Absence of WAF1 mutations in a variety of human malignancies. Blood. 1994; 84:3781–3784.
51. Di Fruscio M, Chen T, Richard S. Characterization of Sam68-like mammalian proteins SLM-1 and SLM-2: SLM-1 is a Src substrate during mitosis. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96:2710–2715.
52. Ehrmann I, Dalgliesh C, Liu Y, Danilenko M, Crosier M, Overman L, Arthur HM, Lindsay S, Clowry GJ, Venables JP, Fort P, Elliott DJ. The tissue-specific RNA binding protein T-STAR controls regional splicing patterns of neurexin pre-mRNAs in the brain. PLoS Genet. 2013; 9:e1003474.
53. Fu K, Sun X, Wier EM, Hodgson A, Liu Y, Sears CL, Wan F. Sam68/KHDRBS1 is critical for colon tumorigenesis by regulating genotoxic stress-induced NF-kappaB activation. Elife. 2016; 5:e15018.
54. Chen ZY, Cai L, Zhu J, Chen M, Chen J, Li ZH, Liu XD, Wang SG, Bie P, Jiang P, Dong JH, Li XW. Fyn requires HnRNPA2B1 and Sam68 to synergistically regulate apoptosis in pancreatic cancer. Carcinogenesis. 2011; 32:1419–1426.
55. Nielsen LL, Gurnani M, Shi B, Terracina G, Johnson RC, Carroll J, Mathis JM, Hajian G. Derivation and initial characterization of a mouse mammary tumor cell line carrying the polyomavirus middle T antigen: utility in the development of novel cancer therapeutics. Cancer research. 2000; 60:7066–7074.
56. Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004; 10:33–39.
57. Fu K, Sun X, Zheng W, Wier EM, Hodgson A, Tran DQ, Richard S, Wan F. Sam68 modulates the promoter specificity of NF-κB and mediates expression of CD25 in activated T cells. Nat Commun. 2013; 4:1909.
58. Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nature cell biology. 2007; 9:1208–1215.
59. Aubele M, Walch AK, Ludyga N, Braselmann H, Atkinson MJ, Luber B, Auer G, Tapio S, Cooke T, Bartlett JM. Prognostic value of protein tyrosine kinase 6 (PTK6) for long-term survival of breast cancer patients. Br J Cancer. 2008; 99:1089–1095.
60. Chen T, Damaj BB, Herrera C, Lasko P, Richard S. Self-association of the single-KH-domain family members Sam68, GRP33, GLD-1, and Qk1: role of the KH domain. Molecular and cellular biology. 1997; 17:5707–5718.
61. Lukong KE, Larocque D, Tyner AL, Richard S. Tyrosine phosphorylation of sam68 by breast tumor kinase regulates intranuclear localization and cell cycle progression. The Journal of biological chemistry. 2005; 280:38639–38647.
62. Paronetto MP, Farini D, Sammarco I, Maturo G, Vespasiani G, Geremia R, Rossi P, Sette C. Expression of a truncated form of the c-Kit tyrosine kinase receptor and activation of Src kinase in human prostatic cancer. Am J Pathol. 2004; 164:1243–1251.
63. Meunier C, Cai J, Fortin A, Kwan T, Marquis JF, Turbide C, Van Der Kraak L, Jothy S, Beauchemin N, Gros P. Characterization of a major colon cancer susceptibility locus (Ccs3) on mouse chromosome 3. Oncogene. 2010; 29:647–661.
64. Van Der Kraak L, Meunier C, Turbide C, Jothy S, Gaboury L, Marcus V, Chang SY, Beauchemin N, Gros P. A two-locus system controls susceptibility to colitis-associated colon cancer in mice. Oncotarget. 2010; 1:436–446. doi: 10.18632/oncotarget.101009.
65. Mallette FA, Richard S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell reports. 2012; 2:1233–1243.