
Introduction
Breast cancer is the most commonly encountered form of cancer and the second leading cause of cancer-related mortality among women in the world [1]. Every year, an estimated 1 to 1.3 million breast cancer cases are diagnosed worldwide. Of these, approximately 15-20% belong to the triple-negative subtype [2]. TNBC is defined by the lack of expression of estrogen receptor (ER) and progesterone receptor (PR) and the lack of expression or amplification of human epidermal growth factor receptor 2 (HER2). TNBC is an important subject of intense investigation for both basic researchers and clinicians for several reasons [3]. First, there is a clustering of TNBC cases in premenopausal women and in women of African descent. Second, in spite of initial good response to chemotherapy, the prognosis of TNBC remains poor as compared to non-TNBC. Third, there is a significant overlap of BRCA-associated breast cancers with the TNBC phenotype. Lastly and most importantly, no effective specific targeted therapy is readily available for TNBC. In recent years, significant advances have been made in characterizing the molecular features of TNBC and in preclinical and clinical studies of novel therapeutic options for TNBC. In this review, we will focus on our current understanding of the characteristics of TNBC and the recent developments in the area of TNBC treatment.
TNBC vs. Basal-like Breast Cancer
DNA microarray analysis has led to the classification of breast cancer into the luminal A, luminal B, HER2-positive, basal-like, and normal-like subtypes [4]. Further refinement of the intrinsic subgroups has identified the claudin-low group, which is characterized by low-level expression of claudins 3, 4, and 7, occludin, and E-cadherin [5]. The normal-like breast carcinomas were later found to represent contamination of breast cancer samples by normal breast cells [6]. Basal-like breast cancers (BLBCs) were referred to as basal because of their expression of genes typically expressed in basal epithelial cells, such as cytokeratin 5, 6, or 17. BLBCs also express genes normally associated with normal basal-like myoepithelial cells of the breast ductal and lobular system, such as the epidermal growth factor receptor (EGFR, also known as HER1) [7].
In general, there is a significant overlap between TNBC and BLBC and many investigators have used the absence of hormone receptors as a characteristic feature to define BLBC. Roughly, approximately 70-84% of TNBCs are basal-like; conversely, about 70% of basal-like tumors are TNBCs [8-11]. In spite of the similarities between TNBC and BLBC; however, equating TNBC with BLBC is not fully supported by other studies [12-14]. TNBCs do not represent a homogeneous group when analyzed by gene expression profiling, whereas the basal-like subtype cancers do form a homogeneous group with a similar gene expression profile [15]. This indicates that the poor prognosis of TNBC may have resulted from the high percentage of triple-negative tumors which are actually basal-like. Therefore, the overall poor prognosis of TNBC may be a result of this basal-like subgroup, and triple negativity may be seen more as a symptom rather than as a separate entity of breast cancer. It should be noted that although TNBC and BLBC are not the same entity, practically, TNBC takes the place of BLBC in the application of clinical diagnosis and treatment because immunohistochemical characterization is more feasible compared to examination of the gene expression signature.
Although many molecules are involved in the development of BLBCs, changes of the breast cancer susceptibility gene BRCA-related pathway are the key event leading to the formation of the BLBC phenotype [4, 16]. If loss of hormone receptor expression in breast cancer develops following the disruption of BRCA without HER2 amplification, it might result in triple-negative BLBC (TN-BLBC). However, if HER2 gene amplification occurs by random mutation even in the presence of BRCA disruption, the cancer will no longer be triple-negative; instead, it will become non-triple-negative BLBC (NTN-BLBC). Non-basal-like TNBC (NB-TNBC) arises as a result of loss of expression of hormone receptors and HER2 without the involvement of BRCA (Figure 1).
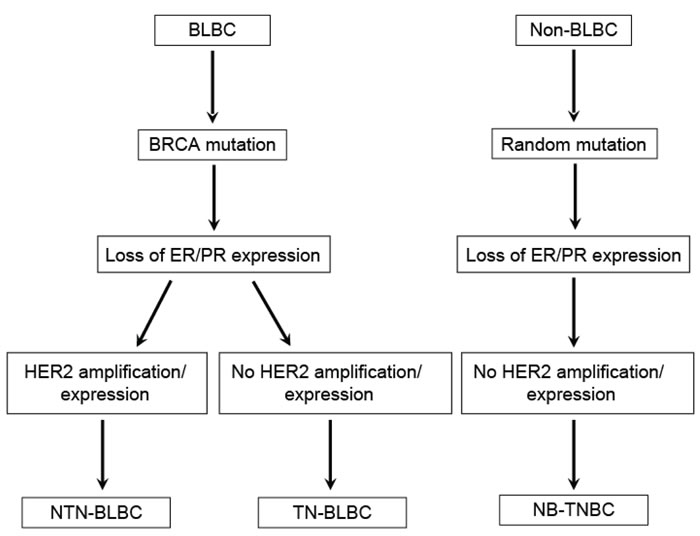
Figure 1: Origin of triple-negative and basal-like breast cancers. Non-triple-negative basal-like breast cancer (NTN-BLBC) and triple-negative basal-like breast cancer (TN-BLBC) originate from basal-like breast cancer (BLBC) depending on whether HER2 amplification/mutation occurs in ER/PR-negative cancers following BRCA mutation. Non-basal-like triple-negative breast cancer (NB-TNBC) may originate from non-basal-like breast cancer (Non-BLBC) without BRCA mutation. Adapted from de Ruijter TC et al: J Cancer Res Clin Oncol 2011;137:183-192.
Characteristics of TNBC
Clinical characteristics of TNBC
TNBC is more frequent in younger patients, in BRCA1 mutation carriers, and in specific ethnic groups (African American and Hispanic women). TNBC accounts for 39% of breast cancers in African-American women under the age of 50, but only 16% in Caucasian women of the same age group [17, 18]. Histologically, approximately 80-90% of TNBC tumors are invasive ductal carcinomas, with the rest classified as apocrine, lobular, adenoid cystic, medullary, and metaplastic [19, 20]. Besides, TNBCs have increased lymphocytic infiltration, and are high grade with large tumor size. TNBCs are associated with a 4-fold increase in the risk of distant metastases [21]. In contrast to the non-TNBCs which most frequently metastasize to the bone, TNBCs more frequently metastasize to the lungs and the central nervous system [22, 23]. It is estimated that approximately 15-30% of TNBC patients will develop brain metastases [2]. This aggressive metastatic behavior contributes to the overall shortened survival of patients with TNBC compared with non-TNBC. The prognosis of patients with TNBC is very poor, because these tumors are clinically more aggressive than other breast carcinoma subtypes and targeted therapy is ruled out in these patients [3, 24, 25].
Molecular characteristics of TNBC
Gene expression profiling of TNBC
The heterogeneity of TNBCs in terms of gene expression profiling and responses to therapeutic regimens is widely acknowledged. A study by Lehmann et al. has revealed 6 distinct subtypes of TNBCs defined by their gene expression profiles [26]. These subtypes identified are basal-like 1 and 2 (BL1 and BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem-like (MSL), and luminal androgen receptor (LAR). They differ in important biological pathways and prognosis. For example, BL1 and BL2 were highly proliferative and had a higher expression of the genes related with cell-cycle and DNA damage response. The M and MSL groups were enriched for genes of the epithelial-to-mesenchymal transition (EMT) pathway, whereas the IM subtype was characterized by immune cell signaling features. The LAR subtype was ER-negative but AR-positive and the LAR cell lines were sensitive to the AR antagonist bicalutamide [26]. This sub-classification of TNBC is useful not only in the understanding of the disease properties but also in the identification of the molecular targets for treatment.
Gene mutations in TNBC
Basal-like TNBCs are associated with mutation of the BRCA gene because the majority of BRCA germ-line mutation carriers develop BLBC [19]. The tumor suppressors TP53 and PTEN are more frequently lost or mutated in triple-negative BLBC than in non-TNBC [11, 27]. Other genes that tend to be frequently mutated in triple-negative BLBC compared to other breast tumors include the tumor suppressor gene Rb and the K-Ras oncogene [28]. It is believed that the combined loss of activity of TP53, Rb, and BRCA pathways is responsible for the high level of genomic instability observed in basal-like tumors [29].
The most frequently somatically mutated genes in TNBC include TP53, Rb, and PTEN among others [30]. Somatic mutations of TP53 are found in the majority of TNBCs (53.8-85.7%), and when combined with inferred pathway analysis there is evidence for loss of TP53 function in nearly all basal-like tumors. Interestingly, TP53 mutations in basal-like tumors were more of the nonsense and frame-shift type, in contrast to mutations in luminal tumors that more frequently were missense. Integrative pathway analysis comparing basal-like and luminal breast cancer identified hyperactivated FOXM1 as a transcriptional driver of proliferation and increased activity of MYC and HIF1-α/ARNT as a key regulator of this process [27].
Therapeutic Options
Generally speaking, patients with TNBC are treated similarly as women who present with non-TNBC, especially in terms of adjuvant and neoadjuvant settings. Surgery and radiotherapy are employed routinely in a similar way as with other types of breast cancer [3]. Patients with TNBC do not benefit from therapies that are designed to target the hormone receptors (such as tamoxifen) or HER2 (such as Herceptin). Currently, chemotherapy, individually or in combination with surgery and/or radiotherapy, is the standard treatment mode for TNBC. TNBCs can be chemo-sensitive particularly to cytotoxic agents, such as anthracyclines and taxanes, which are part of the standard therapy used for high-risk patients, for example patients with lymph node-positive disease [31].
Cytotoxic chemotherapy
Currently, taxane- and anthracycline-based combination chemotherapy remains the standard treatment approach for early-stage TNBC patients, and this approach has changed little during the last decade. To date, there are no specific guidelines for chemotherapeutic management of TNBC. The European Society for Medical Oncology (ESMO) states that cytotoxic chemotherapy is the standard of care for the treatment of TNBC and that the choice of the regimen should be made after consideration of disease-related factors (previous therapies and response, tumor burden, and need for rapid disease/symptom control) and patient-related factors (patient preferences, biological age, menopausal status, comorbidities and performance status, and socioeconomic and psychological factors).
Microtubule stabilizers
Microtubule stabilizers (such as taxanes) are a group of potent tubulin polymerizers that are available for the treatment of breast cancer. Many studies have demonstrated that taxanes (paclitaxel [Taxol], docetaxel [Taxotere], cabazitaxel) are more effective for TNBCs than receptor-positive cancers [32, 33]. A study by Martin et al. showed maximum benefit in TNBC patients when 4 cycles of 5-fluorouracil, epirubicin, cyclophosphamide (FEC) were followed by weekly paclitaxel for 8 weeks compared to just 6 cycles of FEC [32]. Shortening the administration interval from once every 3 weeks to once every 1-2 weeks substantially improved efficacy [33].
Ixabepilone (BMS-247550), another microtubule stabilizer, is actively used in patients with taxane-refractory and locally advanced breast cancer as well as TNBC patients. The clinical activity and toxicity of ixabepilone are similar to those of the taxanes, with neuropathy and myelosuppression as dose-limiting toxicities [34, 35]. Ixabepilone has been shown to bypass the resistance mechanisms associated with taxanes and anthracyclines and provides a treatment option to avoid platinum tolerance (discussed later). In patients with taxane- and/or anthracycline-resistant metastatic TNBC, a combination of ixabepilone and capecitabine (a prodrug of 5-fluorouracil) has an improved progression-free survival (PFS) compared to capecitabine alone (4.1 vs. 2.1 mo) [35]. The ixabepilone and capecitabine combination can be used in patients who do not tolerate cisplatin combinations or when renal functions are compromised.
Anthracyclines
Anthracyclines (doxorubicin and epirubicin) are among the most active drugs for breast cancer treatment. Many studies have shown that TNBC is sensitive to anthracycline-containing regimens [36, 37]. It is noteworthy that the benefit of anthracycline-based regimens in patients with TNBC remains controversial [38]. In a retrospective analysis, Liedtke et al. reported a pathological complete remission (pCR) rate of 22% in TNBCs compared to 11% in non-TNBCs with an epirubicin-containing regimen [36]. However, the 3-year disease-free survival (DFS) was similar in both groups. On the contrary, Carey et al. [37] showed that TNBC patients had a much higher clinical response to doxorubicin and cyclophosphamide than non-TNBC patients. Although the role of anthracyclines alone in TNBC is questionable, a definite benefit was observed when anthracyclines were used in combination with taxanes in node-positive TNBC patients [39].
Platinum agents
The intense interest in the role of platinum compounds including carboplatin and cisplatin in TNBC is based on phenotypic and molecular similarities between BRCA-associated breast cancer and the basal-like subtype. The platinums act by generating intra- and inter-strand double-stranded DNA crosslinks, preventing the formation of the replication fork and producing double-strand breaks and replication lesions. Due to BRCA mutation, which leads to the dysfunction of the DNA repair cascade, platinums produce cell death in BRCA-mutant breast cancer cells [40]. In a retrospective study, Staudacher et al. [41] reported that median overall survival (OS) and median PFS were improved in patients responding to platinum-based chemotherapy: 27 vs. 8 mo and 10 vs. 4 mo, respectively. Another retrospective investigation of a large cohort of metastatic TNBC by Zhang et al. revealed that platinum use as first-line chemotherapy resulted in longer PFS compared with patients without platinum therapy (7.8 vs. 4.9 mo), although no statistical difference of OS was observed. In the different platinum drugs administered, cisplatin-based regimens gave the best performance [42]. It should be noted, however, that platinums should be used in combination with other chemotherapeutic agents to increase response and survival rates. For example, when platinums are used in combination with epirubicin and 5-fluorouracil, a very high complete clinical response was achieved [43].
Targeted therapies
PARP inhibitors
Poly (ADP-ribose) polymerase (PARP) plays a vital role in the repair of single-stranded DNA breaks through the base excision repair pathway [44]. As mentioned above, TNBC has a high frequency of mutation of the breast cancer susceptibility gene BRCA. Therefore, PARP inhibitors can lead to cell death in BRCA-mutated TNBCs because of the inability of the cell to repair DNA damage due to BRCA mutation. It has been demonstrated that PARP inhibition potentiates the effects of ionizing radiation agents, DNA-methylating compounds, topoisomerase inhibitors, and platinums [40]. Several PARP inhibitors such as olaparib (AZD 2811) and BSI-201 are at different stages of clinical development [45]. Encouraging [46, 47] as well as discouraging [48] results have been reported for PARP inhibitors. Several mechanisms of PARP inhibition resistance in BRCA-associated tumors have been proposed. These include reversal of truncating mutations and stabilization of mutated BRCA proteins [49]. Several strategies to overcome these resistance mechanisms are currently under investigation.
Angiogenesis inhibitors
Expression of vascular endothelial growth factor (VEGF) is much higher in TNBC compared with non-TNBC [50]. Bevacizumab (Avastin), an anti-VEGF monoclonal antibody, has consistently exhibited improved PFS and response rate when used in combination with first-line chemotherapy in HER2-negative breast cancer. A meta-analysis of patients with HER2-negative metastatic breast cancer (n = 2447) demonstrated that bevacizumab improved efficacy, including 1-year OS rate, both overall and in subgroups of poor-prognosis patients [51].
EGFR inhibitors
Overexpression of EGFR has been observed in more than half of TNBCs and is correlated with a poor prognosis and decreased response to chemotherapy [52-54]. This observation has prompted a series of clinical trials incorporating anti-EGFR agents, such as cetuximab and lapatinib. Cetuximab binds specifically to the extracellular domain of EGFR, thus inhibiting its activation [55]. Clinical data point to a modest effect of EGFR-targeted therapies in at least a subset of TNBCs [56]. Several phase II studies of anti-EGFR therapy in combination with cytotoxic agents or with other targeted therapies are currently ongoing in metastatic TNBC [57, 58].
TK inhibitors
Tyrosine kinases (TKs), including the Src and Abl family and c-Kit, are overexpressed in breast cancer and associated with the progression of metastatic breast cancer. Many small-molecule agents, such as imatinib, erlotinib, gefitinib, lapatinib, dasatinib, and pazopanib, are used for treating a variety of solid tumors through targeting the phosphorylation of the receptor by acting at TKs. Dasatinib (previously known as BMS-354825) is an oral inhibitor of multiple TKs. Dasatinib has been shown to inhibit the growth of TNBC cell lines in vitro when used alone or in combination with chemotherapeutic agents such as cisplatin [59]. Currently, several studies are being carried out to evaluate dasatinib as monotherapy or in combination with chemotherapy in treating TNBC [60, 61]. Pazopanib, an anti-angiogenic TK inhibitor, which was approved in 2009 for the treatment of advanced renal cell carcinoma, has been evaluated alone or in combination with the microtubule stabilizer capecitabine in breast cancer patients [62, 63]. In a model of a mouse orthotopic implanted breast tumor model, Di Desidero et al. showed that the combination of pazopanib and topotecan significantly enhanced the anti-tumor activity of either drug alone and prolonged survival, with a marked decrease in tumor vascularity, proliferative index, and apoptosis induction [64]. However, whether this combination has selectivity on TNBC over non-TNBC is not known.
mTOR inhibitors
The mammalian target of rapamycin (mTOR) is a key component of the PI3K-Akt-mTOR pathway, which has recently been considered to play a critical role in tumor escape from hormone dependence in breast cancer [65]. The expression of Acyl-CoA synthetase 4 (ACSL4), an enzyme participating in arachidonic acid metabolism, drives the hyperactivation of the PI3K-Akt-mTOR pathway in in vitro transfection experiments in breast cancer cells [66]. ACSL4 has been shown to be associated with the aggressive phenotype of breast cancer [67, 68]. Orlando et al. smartly showed that inhibition of ACSL4 through siRNA or rosiglitazone, a small-molecule anti-diabetic drug, could reverse the ER-negative phenotype in the TNBC MDA-MB-231 cells. Therefore, through combination of rosiglitazone and tamoxifen, an ER inhibitor, could synergistically inhibit the growth of MDA-MB-231 cells both in vitro and in the nude mouse xenograft model [66].
Statins
Statins, inhibitors of 3-hydroxy-3-methyl-glutaryl coenzyme A (HMG-CoA) reductase, reduce the intracellular biosynthesis of cholesterol by reversibly inhibiting the conversion of HMG-CoA to mevalonate (Figure 2). These lipid-lowering drugs are commonly used to treat hypercholesterolemia, thereby reducing the mortality from cardiovascular disease. Recently, statins have also pleiotropic anti-cancer effects in a variety of cancers including breast cancer [69]. Preclinical studies have shown anti-proliferative, pro-apoptotic, anti-invasive, and radio- and chemo-sensitizing properties of statins. Given that statins are FDA-approved, well tolerated, and affordable, they provide the opportunities for accelerated repurposing as cancer therapeutics.
Consensus regarding the clinical effects of statins on breast cancer has not been reached, which has resulted in inconsistency in the relationship between statin use and the incidence of breast cancer. Many studies have demonstrated a decrease in the risk of a variety of cancers, including breast cancer, among statin users [70-74]. Conversely, several studies revealed that long-term use of statins did not significantly affect the risk of breast cancer [75-77]. However, through a systematic review and meta-analysis, Wu et al. found that although statin use may not influence the risk of breast cancer, it is associated with a decrease in mortality of breast cancer patients [78].
Preclinically, statin sensitivity has been found to be associated with NF-κB activation [79], lack of expression of ERα [79, 80], mutation of TP53 [81], and the status of PTEN-PI3K pathway [82]. Campbell et al. studied the effect of statins on the growth of breast cancer cells in vitro. Of the four statins tested, only lipophilic statins (fluvastatin, lovastatin, and simvastatin) could significantly inhibit the proliferation of TNBC MDA-MB-231 cells with an IC50 in the range of 1-2 μM. However, the IC50 was much higher in the HER2-positive SKBr3 cells (26-49 μM) and the ER-positive MCF-7 cells (85-138 μM) [79]. Goard et al. characterized fluvastatin sensitivity in 19 breast cancer cell lines and found that fluvastatin sensitivity was strongly associated with an ERα-negative status and the basal-like phenotype [83]. Xenografts of ERα-negative tumor cells have also been shown to respond to treatment with lipophilic statins, including simvastatin and fluvastatin [79, 81].
Table 1: Growth-Inhibitory Effect of Lovastatin on Breast Cancer Cell Lines.
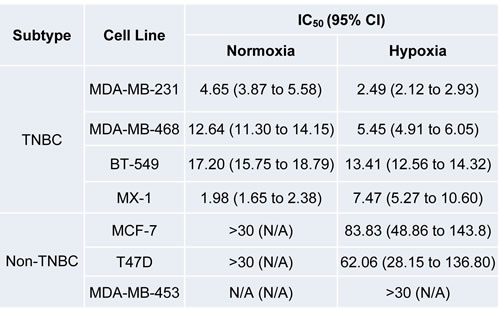
CI: Confidence Interval; N/A: Not Applicable
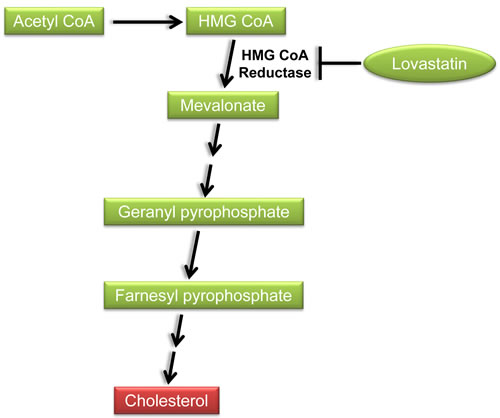
Figure 2: Inhibition of the cholesterol biosynthetic pathway by lovastatin. HMG-CoA: 3-hydroxy-3-methyl-glutaryl coenzyme A.
We extended this observation to TNBC vs. non-TNBC cell lines and confirmed that lovastatin, a natural and lipophilic statin derived from Monascus ruber-fermented rice or from Oyster mushroom [84], preferentially inhibited cell proliferation and migration of TNBC cells compared to non-TNBC cells (Table 1) . A nude mouse xenograft model also showed that lovastatin, at its clinically relevant concentration (2 or 10 mg/kg body weight), inhibited the in vivo tumor growth of triple-negative MDA-MB-231 cells (data not shown). The molecular mechanisms underlying lovastatin’s effect on MDA-MB-231 cells included modulation of the proteins involved in apoptosis, differentiation, cell proliferation, signal transduction, epithelial-to-mesenchymal transition (EMT) and tumor metastasis (ref. [85] and Figure 3).
Therefore, statins have the potential to prevent or treat a subset of breast cancers, such as TNBC. The lipophilicity of statins also affects their role in breast cancer. Only lipophilic statins are able to permeate the cell membrane and affect cellular functions. This has been demonstrated in the study by Mueck et al. in which showed that all lipophilic statins, i.e., lovastatin, atorvastatin, fluvastatin, and simvastatin, but not a hydrophilic statin, i.e., pravastatin, significantly inhibited the cell proliferation of breast cancer cell lines [80].
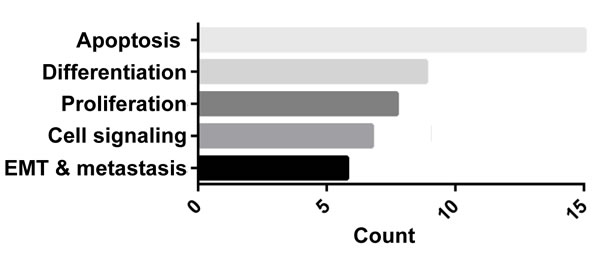
Figure 3: GO enrichment analysis of proteins regulated by lovastatin in MDA-MB-231 cells. MDA-MB-231 cells were treated with lovastatin or vehicle under hypoxia for 48 h and subjected to antibody microarray analysis followed by GO enrichment analysis as described in ref. [85]. A complete list of proteins regulated by lovastatin in MDA-MB-231 cells is available in that reference.
Perspectives
In spite of the general susceptibility to standard chemotherapy, TNBCs exhibit an overall poorer survival compared to non-TNBCs. The benefits of targeted therapies have eluded patients with TNBC due to the absence of well-defined molecular targets. Novel therapeutic targets that are being actively explored and new agents with therapeutic potential that are under development are summarized in Figure 4. Two important areas need in-depth investigations that may bring about significant changes in the management of TNBC.
First, the identification of molecular targets will be crucial to identifying actionable targets in patients with TNBC. Within the TNBC subtypes, there are some potentially targetable pathways such as the BRCA-mediated pathway, the Wnt/β-catenin, Notch, Hedgehog, and JAK2 pathways, which could be exploited for future therapeutic strategies. Unfortunately, many years of study have failed to identify a single pathway that is targetable in TNBC. A major obstacle to this area is the inter- and intra-tumoral heterogeneity. Better understanding of the molecular basis of the heterogeneity of TNBC and development of more robust therapeutic strategies are desired.
Secondly, agents that selectively or preferentially target TNBC are urgently needed. In this regard, the lipid-lowering statins have shown great promise. The confusing results obtained from clinical use of statins in breast cancer prevention may have resulted from lack of distinction between TNBC and non-TNBC. Preclinical data from several independent groups have shown that lipophilic statins exhibit preference for ER-negative or basal-like breast cancer. All lines of evidence obtained up to now clearly point to an obvious preference of statins for TNBCs. Future studies about the use of statins in TNBC should focus on: 1) exploring the role of statins in breast cancer stem cells; 2) optimizing the formulation of statins, for example using novel nanoparticles to encapsulate the statins; and 3) identifying the molecular mechanisms underlying statins’ preference for TNBC and the possible drug targets of statins in TNBC. With the understanding of the molecular basis for the preference of statins for TNBC and more investigations in clinical trials, statins may find their avenue to becoming clinically useful drugs against TNBC.
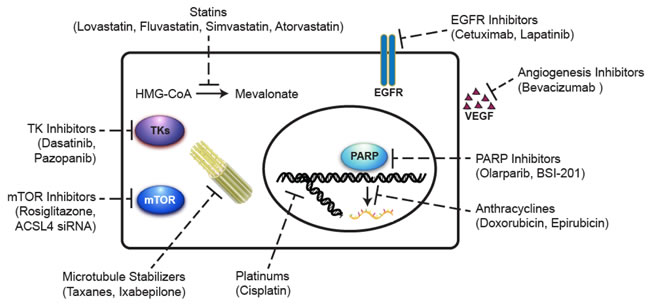
Figure 4: Summary of the potential agents under development for the treatment of triple-negative breast cancer. Microtubule stabilizers polymerize tubulin in the microtubule, thereby inhibiting cell division. Anthracyclines inhibit RNA synthesis by intercalating between base pairs of the DNA/RNA strand, thus preventing the replication of rapidly growing cancer cells. Platinums generate intra- and inter-strand double-stranded DNA crosslinks, preventing the formation of the replication fork and inhibiting cell division. PARP inhibitors prevent the repair of single-strand breaks that occur during cell cycle especially in BRCA-mutated cells. Angiogenesis inhibitors block the growth of new blood vessels by inhibiting VEGF. EGFR inhibitors bind to EGFR and turn off the uncontrolled growth of cancer cells with EGFR mutations. TK inhibitors block tumor growth through inhibiting intracellular tyrosine kinase activity. mTOR inhibitors suppress cancer cell growth and proliferation through targeting the PI3K/Akt/mTOR signaling pathway. Statins inhibit the conversion of HMG-CoA to mevalonate in the cholesterol biosynthesis pathway. The anti-cancer effects of statins may involve the inhibition of multiple signaling pathways important for the malignant phenotype of cancer cells. EGFR, epidermal growth factor receptor; HMG-CoA, 3-hydroxy-3-methylglutaryl-coenzyme A; mTOR, mammalian target of rapamycin; PARP, poly(ADP-ribose) polymerase; TK, tyrosine kinase.
Abbreviations
AR-Androgen receptor , BLBC-Basal-like breast cancer, BRCA-Breast cancer susceptibility gene, cPR-Complete pathological remission, DFS-Disease-free survival , EGFR-Epidermal growth factor receptor, ER -Estrogen receptor, FEC-5-Fluorouracil, epirubicin, cyclo-phosphamide, HER2-Human epidermal growth factor receptor 2, LV-Lovastatin, OS-Overall survival, PARP-Poly(ADP-ribose) polymerase, pCR-Pathological complete remission, PFS-Progression-free survival, PR-Progesterone receptor, PTEN-Phosphatase and tensin homolog deleted on chromosome 10, Rb--Retinoblastoma gene, TNBC-Triple-negative breast cancer, TK-Tyrosine kinase, VEGF-Vascular endothelial growth factor.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Project 81472496), the Key Project of Department of Education of Hunan (14A089), and the Research Project of the S&T Bureau of Changsha (k1403024-31). We thank Mr. Linquan Zhang of the class of 2013 medical students and Ms. Lu Lu of the class of 2016 master’s students for assistance in preparing the manuscript.
CONFLICTs OF INTEREST
The authors declare that no conflict of interest exists.
References
1. Wahba HA, and El-Hadaad HA. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med. 2015;12:106-16.
2. Anders C, and Carey LA. Understanding and treating triple-negative breast cancer. Oncology (Williston Park). 2008;22:1233-9; discussion 9-40, 43.
3. Nofech-Mozes S, Trudeau M, Kahn HK, Dent R, Rawlinson E, Sun P, Narod SA, and Hanna WM. Patterns of recurrence in the basal and non-basal subtypes of triple-negative breast cancers. Breast Cancer Res Treat. 2009;118:131-7.
4. de Ruijter TC, Veeck J, de Hoon JP, van Engeland M, and Tjan-Heijnen VC. Characteristics of triple-negative breast cancer. J Cancer Res Clin Oncol. 2011;137:183-92.
5. Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, Backlund MG, Yin Y, Khramtsov AI, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76.
6. Morris SR, and Carey LA. Gene expression profiling in breast cancer. Curr Opin Oncol. 2007;19:547-51.
7. Rakha EA, Reis-Filho JS, and Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26:2568-81.
8. Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346-52.
9. Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, and Perou CM. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist. 2013;18:123-33.
10. Prat A, and Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5:5-23.
11. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, Hernandez-Boussard T, Livasy C, Cowan D, Dressler L, Akslen LA, Ragaz J, Gown AM, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10:5367-74.
12. Rakha EA, Elsheikh SE, Aleskandarany MA, Habashi HO, Green AR, Powe DG, El-Sayed ME, Benhasouna A, Brunet JS, Akslen LA, Evans AJ, Blamey R, Reis-Filho JS, et al. Triple-negative breast cancer: distinguishing between basal and nonbasal subtypes. Clin Cancer Res. 2009;15:2302-10.
13. Cheang MC, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, Perou CM, and Nielsen TO. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14:1368-76.
14. Bertucci F, Finetti P, Cervera N, Esterni B, Hermitte F, Viens P, and Birnbaum D. How basal are triple-negative breast cancers? Int J Cancer. 2008;123:236-40.
15. Weigelt B, Horlings HM, Kreike B, Hayes MM, Hauptmann M, Wessels LF, de Jong D, Van de Vijver MJ, Van’t Veer LJ, and Peterse JL. Refinement of breast cancer classification by molecular characterization of histological special types. J Pathol. 2008;216:141-50.
16. Diaz LK, Cryns VL, Symmans WF, and Sneige N. Triple negative breast carcinoma and the basal phenotype: from expression profiling to clinical practice. Adv Anat Pathol. 2007;14:419-30.
17. Morris GJ, Naidu S, Topham AK, Guiles F, Xu Y, McCue P, Schwartz GF, Park PK, Rosenberg AL, Brill K, and Mitchell EP. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer. 2007;110:876-84.
18. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, Karaca G, Troester MA, Tse CK, Edmiston S, Deming SL, Geradts J, Cheang MC, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295:2492-502.
19. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, Demeter J, Perou CM, Lønning PE, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418-23.
20. Yuan N, Meng M, Liu C, Feng L, Hou L, Ning Q, Xin G, Pei L, Gu S, Li X, and Zhao X. Clinical characteristics and prognostic analysis of triple-negative breast cancer patients. Mol Clin Oncol. 2014;2:245-51.
21. Koeneman KS, Yeung F, and Chung LW. Osteomimetic properties of prostate cancer cells: a hypothesis supporting the predilection of prostate cancer metastasis and growth in the bone environment. Prostate. 1999;39:246-61.
22. Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, and Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113:2638-45.
23. Sihto H, Lundin J, Lundin M, Lehtimaki T, Ristimaki A, Holli K, Sailas L, Kataja V, Turpeenniemi-Hujanen T, Isola J, Heikkilä P, and Joensuu H. Breast cancer biological subtypes and protein expression predict for the preferential distant metastasis sites: a nationwide cohort study. Breast Cancer Res. 2011;13:R87.
24. Anders CK, and Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009;9 Suppl 2:S73-81.
25. Chacon RD, and Costanzo MV. Triple-negative breast cancer. Breast Cancer Res. 2010;12 Suppl 2:S3.
26. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, and Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750-67.
27. Koboldt DC, Fulton RS, McLellan MD, and Palchik JD. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61-70.
28. Hu X, Stern HM, Ge L, O’Brien C, Haydu L, Honchell CD, Haverty PM, Peters BA, Wu TD, Amler LC, Chant J, Stokoe D, Lackner MR, et al. Genetic alterations and oncogenic pathways associated with breast cancer subtypes. Mol Cancer Res. 2009;7:511-22.
29. Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2010;15 Suppl 5:39-48.
30. Engebraaten O, Vollan HK, and Borresen-Dale AL. Triple-negative breast cancer and the need for new therapeutic targets. Am J Pathol. 2013;183:1064-74.
31. Ismail-Khan R, and Bui MM. A review of triple-negative breast cancer. Cancer Control. 2010;17:173-6.
32. Martin M, Rodriguez-Lescure A, Ruiz A, Alba E, Calvo L, Ruiz-Borrego M, Santaballa A, Rodríguez CA, Crespo C, Abad M, Domínguez S, Florián J, Llorca C, et al. Molecular predictors of efficacy of adjuvant weekly paclitaxel in early breast cancer. Breast Cancer Res Treat. 2010;123:149-57.
33. Sparano JA, Wang M, Martino S, Jones V, Perez EA, Saphner T, Wolff AC, Sledge GW Jr, Wood WC, and Davidson NE. Weekly paclitaxel in the adjuvant treatment of breast cancer. N Engl J Med. 2008;358:1663-71.
34. Baselga J, Zambetti M, Llombart-Cussac A, Manikhas G, Kubista E, Steger GG, Makhson A, Tjulandin S, Ludwig H, Verrill M, Ciruelos E, Egyhazi S, Xu LA, et al. Phase II genomics study of ixabepilone as neoadjuvant treatment for breast cancer. J Clin Oncol. 2009;27:526-34.
35. Pivot XB, Li RK, Thomas ES, Chung HC, Fein LE, Chan VF, Jassem J, de Mendoza FH, Mukhopadyay P, and Roché HH. Activity of ixabepilone in oestrogen receptor-negative and oestrogen receptor-progesterone receptor-human epidermal growth factor receptor 2-negative metastatic breast cancer. Eur J Cancer. 2009;45:2940-6.
36. Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, and Pusztai L. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26:1275-81.
37. Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, and Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13:2329-34.
38. Gennari A, Sormani MP, Pronzato P, Puntoni M, Colozza M, Pfeffer U, and Bruzzi P. HER2 status and efficacy of adjuvant anthracyclines in early breast cancer: a pooled analysis of randomized trials. J Natl Cancer Inst. 2008;100:14-20.
39. Esserman LJ, Berry DA, DeMichele A, Carey L, Davis SE, Buxton M, Hudis C, Gray JW, Perou C, Yau C, Livasy C, Krontiras H, Montgomery L, et al. Pathologic complete response predicts recurrence-free survival more effectively by cancer subset: results from the I-SPY 1 TRIAL—CALGB 150007/150012, ACRIN 6657. J Clin Oncol. 2012;30:3242-9.
40. Hastak K, Alli E, and Ford JM. Synergistic chemosensitivity of triple-negative breast cancer cell lines to poly(ADP-Ribose) polymerase inhibition, gemcitabine, and cisplatin. Cancer Res. 2010;70:7970-80.
41. Staudacher L, Cottu PH, Dieras V, Vincent-Salomon A, Guilhaume MN, Escalup L, Dorval T, Beuzeboc P, Mignot L, and Pierga JY. Platinum-based chemotherapy in metastatic triple-negative breast cancer: the Institut Curie experience. Ann Oncol. 2011;22:848-56.
42. Zhang J, Fan M, Xie J, Wang Z, Wang B, Zhang S, Wang L, Cao J, Tao Z, Li T, and Hu X. Chemotherapy of metastatic triple negative breast cancer: Experience of using platinum-based chemotherapy. Oncotarget. 2015;6:43135-43. doi: 10.18632/oncotarget.5654.
43. Sirohi B, Arnedos M, Popat S, Ashley S, Nerurkar A, Walsh G, Johnston S, and Smith IE. Platinum-based chemotherapy in triple-negative breast cancer. Ann Oncol. 2008;19:1847-52.
44. Dantzer F, de La Rubia G, Menissier-De Murcia J, Hostomsky Z, de Murcia G, and Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39:7559-69.
45. Audeh MW. Novel treatment strategies in triple-negative breast cancer: specific role of poly(adenosine diphosphate-ribose) polymerase inhibition. Pharmgenomics Pers Med. 2014;7:307-16.
46. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123-34.
47. Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK, Wardley A, Mitchell G, Earl H, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235-44.
48. Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, Hirte H, Huntsman D, Clemons M, Gilks B, Yerushalmi R, Macpherson E, Carmichael J, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12:852-61.
49. Bouwman P, and Jonkers J. Molecular pathways: how can BRCA-mutated tumors become resistant to PARP inhibitors? Clin Cancer Res. 2014;20:540-7.
50. Linderholm BK, Hellborg H, Johansson U, Elmberger G, Skoog L, Lehtio J, and Lewensohn R. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann Oncol. 2009;20:1639-46.
51. Miles DW, Dieras V, Cortes J, Duenne AA, Yi J, and O’Shaughnessy J. First-line bevacizumab in combination with chemotherapy for HER2-negative metastatic breast cancer: pooled and subgroup analyses of data from 2447 patients. Ann Oncol. 2013;24:2773-80.
52. Viale G, Rotmensz N, Maisonneuve P, Bottiglieri L, Montagna E, Luini A, Veronesi P, Intra M, Torrisi R, Cardillo A, Campagnoli E, Goldhirsch A, and Colleoni M. Invasive ductal carcinoma of the breast with the “triple-negative” phenotype: prognostic implications of EGFR immunoreactivity. Breast Cancer Res Treat. 2009;116:317-28.
53. Nogi H, Kobayashi T, Suzuki M, Tabei I, Kawase K, Toriumi Y, Fukushima H, and Uchida K. EGFR as paradoxical predictor of chemosensitivity and outcome among triple-negative breast cancer. Oncol Rep. 2009;21:413-7.
54. Banerjee S, Reis-Filho JS, Ashley S, Steele D, Ashworth A, Lakhani SR, and Smith IE. Basal-like breast carcinomas: clinical outcome and response to chemotherapy. J Clin Pathol. 2006;59:729-35.
55. Herbst RS, and Shin DM. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy. Cancer. 2002;94:1593-611.
56. Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, Liu MC, Storniolo AM, Rimawi MF, Forero-Torres A, Wolff AC, Hobday TJ, Ivanova A, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30:2615-23.
57. Crozier JA, Advani PP, LaPlant B, Hobday T, Jaslowski AJ, Moreno-Aspitia A, and Perez EA. N0436 (Alliance): A Phase II Trial of Irinotecan With Cetuximab in Patients With Metastatic Breast Cancer Previously Exposed to Anthracycline and/or Taxane-Containing Therapy. Clin Breast Cancer. 2016;16:23-30.
58. Nabholtz JM, Chalabi N, Radosevic-Robin N, Dauplat MM, Mouret-Reynier MA, Van Praagh I, Servent V, Jacquin JP, Benmammar KE, Kullab S, Bahadoor MR, Kwiatkowski F, Cayre A, et al. Multicentric neoadjuvant pilot Phase II study of cetuximab combined with docetaxel in operable triple negative breast cancer. Int J Cancer. 2016;138:2274-80.
59. Finn RS, Dering J, Ginther C, Wilson CA, Glaspy P, Tchekmedyian N, and Slamon DJ. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/”triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat. 2007;105:319-26.
60. Yadav BS, Sharma SC, Chanana P, and Jhamb S. Systemic treatment strategies for triple-negative breast cancer. World J Clin Oncol. 2014;5:125-33.
61. Brower V. Search for new treatments intensifies for triple-negative breast cancer. J Natl Cancer Inst. 2009;101:1536-7.
62. Pazopanib in Combination With Capecitabine in Patients With Metastatic Breast Cancer (PazoX). NCT01498458. https://clinicaltrials.gov/ct2/show/NCT01498458?term=NCT01498458&rank=1.
63. Treatment With Pazopanib for Neoadjuvant Breast Cancer. NCT00849472. https://clinicaltrials.gov/ct2/show/NCT00849472?term=NCT00849472&rank=1.
64. Di Desidero T, Xu P, Man S, Bocci G, and Kerbel RS. Potent efficacy of metronomic topotecan and pazopanib combination therapy in preclinical models of primary or late stage metastatic triple-negative breast cancer. Oncotarget. 2015;6:42396-410. doi: 10.18632/oncotarget.6377.
65. Nicolini A, Ferrari P, Kotlarova L, Rossi G, and Biava PM. The PI3K-AKt-mTOR Pathway and New Tools to Prevent Acquired Hormone Resistance in Breast Cancer. Curr Pharm Biotechnol. 2015;16:804-15.
66. Orlando UD, Castillo AF, Dattilo MA, Solano AR, Maloberti PM, and Podesta EJ. Acyl-CoA synthetase-4, a new regulator of mTOR and a potential therapeutic target for enhanced estrogen receptor function in receptor-positive and -negative breast cancer. Oncotarget. 2015;6:42632-50. doi: 10.18632/oncotarget.5822.
67. Maloberti PM, Duarte AB, Orlando UD, Pasqualini ME, Solano AR, López-Otín C, and Podestá EJ. Functional interaction between acyl-CoA synthetase 4, lipooxygenases and cyclooxygenase-2 in the aggressive phenotype of breast cancer cells. PLoS One. 2010;5:e15540.
68. Monaco ME, Creighton CJ, Lee P, Zou X, Topham MK, and Stafforini DM. Expression of Long-chain Fatty Acyl-CoA Synthetase 4 in Breast and Prostate Cancers Is Associated with Sex Steroid Hormone Receptor Negativity. Transl Oncol. 2010;3:91-8.
69. Gazzerro P, Proto MC, Gangemi G, Malfitano AM, Ciaglia E, Pisanti S, Santoro A, Laezza C, and Bifulco M. Pharmacological actions of statins: a critical appraisal in the management of cancer. Pharmacol Rev. 2012;64:102-46.
70. Coogan PF, Rosenberg L, Palmer JR, Strom BL, Zauber AG, and Shapiro S. Statin use and the risk of breast and prostate cancer. Epidemiology. 2002;13:262-7.
71. Jacobs EJ, Newton CC, Thun MJ, and Gapstur SM. Long-term use of cholesterol-lowering drugs and cancer incidence in a large United States cohort. Cancer Res. 2011;71:1763-71.
72. Friis S, Poulsen AH, Johnsen SP, McLaughlin JK, Fryzek JP, Dalton SO, Sørensen HT, and Olsen JH. Cancer risk among statin users: a population-based cohort study. Int J Cancer. 2005;114:643-7.
73. Cauley JA, McTiernan A, Rodabough RJ, LaCroix A, Bauer DC, Margolis KL, Paskett ED, Vitolins MZ, Furberg CD, Chlebowski RT, and Women’s Health Initiative Research Group. Statin use and breast cancer: prospective results from the Women’s Health Initiative. J Natl Cancer Inst. 2006;98:700-7.
74. Ahern TP, Pedersen L, Tarp M, Cronin-Fenton DP, Garne JP, Silliman RA, Sørensen HT, and Lash TL. Statin prescriptions and breast cancer recurrence risk: a Danish nationwide prospective cohort study. J Natl Cancer Inst. 2011;103:1461-8.
75. Undela K, Srikanth V, and Bansal D. Statin use and risk of breast cancer: a meta-analysis of observational studies. Breast Cancer Res Treat. 2012;135:261-9.
76. Bonovas S, Filioussi K, Tsavaris N, and Sitaras NM. Use of statins and breast cancer: a meta-analysis of seven randomized clinical trials and nine observational studies. J Clin Oncol. 2005;23:8606-12.
77. Nickels S, Vrieling A, Seibold P, Heinz J, Obi N, Flesch-Janys D, and Chang-Claude J. Mortality and recurrence risk in relation to the use of lipid-lowering drugs in a prospective breast cancer patient cohort. PLoS One. 2013;8:e75088.
78. Wu QJ, Tu C, Li YY, Zhu J, Qian KQ, Li WJ, and Wu L. Statin use and breast cancer survival and risk: a systematic review and meta-analysis. Oncotarget. 2015;6:42988-3004. doi: 10.18632/oncotarget.5557.
79. Campbell MJ, Esserman LJ, Zhou Y, Shoemaker M, Lobo M, Borman E, Baehner F, Kumar AS, Adduci K, Marx C, Petricoin EF, Liotta LA, Winters M, et al. Breast cancer growth prevention by statins. Cancer Res. 2006;66:8707-14.
80. Mueck AO, Seeger H, and Wallwiener D. Effect of statins combined with estradiol on the proliferation of human receptor-positive and receptor-negative breast cancer cells. Menopause. 2003;10:332-6.
81. Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A, Bissell MJ, Osborne TF, Tian B, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244-58.
82. Park YH, Jung HH, Ahn JS, and Im YH. Statin induces inhibition of triple negative breast cancer (TNBC) cells via PI3K pathway. Biochem Biophys Res Commun. 2013;439:275-9.
83. Goard CA, Chan-Seng-Yue M, Mullen PJ, Quiroga AD, Wasylishen AR, Clendening JW, Sendorek DH, Haider S, Lehner R, Boutros PC, and Penn LZ. Identifying molecular features that distinguish fluvastatin-sensitive breast tumor cells. Breast Cancer Res Treat. 2014;143:301-12.
84. Yang T, Liu J, Luo F, Lin Q, Rosol TJ, and Deng X. Anticancer properties of Monascus metabolites. Anticancer Drugs. 2014;25:735-44.
85. Yang T, Yao H, He G, Song L, Liu N, Wang Y, Yang Y, Keller ET, and Deng X. Effects of Lovastatin on MDA-MB-231 Breast Cancer Cells: An Antibody Microarray Analysis. J Cancer. 2016;7:192-9.