
INTRODUCTION
Male breast cancer (MBC) is a rare disease, representing less than 1% of all breast cancers (BCs) and less than 1% of all cancers in men [1]. Germ-line mutations in the high-penetrance BC genes, BRCA1 and, mainly, BRCA2 play a major role in MBC susceptibility and about 10% of MBCs are associated with mutations in these genes [2].
Due to its rarity, MBC research and patient management are mostly based upon data derived from its largely known female counterpart. To date treatment strategies for MBC patients generally follow those for female patients. As most breast cancers in men are hormone receptor-positive, current therapeutic options mainly include hormone therapy that is usually recommended for MBC patients following the same recommendations as hormone-dependent-BC in women [3].
MBC shares some similarities with post-menopausal estrogen receptor (ER)-positive female breast cancer (FBC), however increasing evidence indicates that, on clinical and molecular level, MBC may be a heterogeneous disease, different from FBC [4–6]. Compared to FBC, MBC occurs later in life, with higher stage, lower grade and more estrogen/progesterone receptor (ER/PR) positivity [7–8].
To date, the majority of MBC studies focused on germ-line mutational analysis and gene expression profiling, while there are only few studies investigating somatic alterations in MBCs [4–5, 9]. By contrast, accumulating data have provided a landscape of somatic alterations with possible clinical relevance and several somatic mutations with potential prognostic and therapeutic significance are known in FBC [10–13]. In addition to gene mutations, gene copy number variations (CNVs) have been reported as an important mechanism in the development and progression of cancer and could serve as potential prognostic biomarkers and molecular therapeutic targets [14–18].
A comprehensive portrait of the genetic landscape of FBCs has demonstrated that Phosphatidylinositol 3-kinase gene (PIK3CA) is the most commonly mutated gene and two mutation hotspot regions in exons 9 and 20 have been identified [19–20]. Mutations of PIK3CA have been suggested to have prognostic value and to confer resistance to hormone therapy and to downstream mTOR inhibitors [11, 21]. The proportion of FBCs exhibiting PIK3CA mutations ranges between 20-25%, and, notably, mutations occur approximately in 40% of ER-positive FBCs [11, 19–23]. To date four studies investigated PIK3CA mutations in MBC [6, 24–26]. Overall PIK3CA mutation frequency observed in MBC was lower than FBC. In addition to somatic mutations, gene amplification of PIK3CA is reported in about 10% of FBC [27–29]. Among targetable genes, the epidermal growth factor receptor (EGFR) is one of the first important molecular targets identified in BC [10, 30–31]. EGFR gene CNVs in FBC range from 8 to 10%, [32–33] while the proportion of FBCs exhibiting EGFR somatic mutations ranges from 2 to 15% [34–36]. Notably a higher rate of activating EGFR mutations is detected in BRCA1/2 mutation positive tumors, supporting the hypothesis that carcinogenic processes may be dependent on the germ-line phenotype [34]. CCND1 is an estrogen-responsive gene that enhances ER-mediated gene transcription. The product of CCND1, cyclin D1, provide growth advantage to cancer cells and contribute toward resistance to endocrine therapy in ER-positive cancers. In FBC, CCND1 is amplified in 5–20% of primary BCs, typically ER-positive BC, and is a prognostic biomarker with a potential therapeutic role [12, 13, 15, 37, 38]. As MBC is an estrogen-driven disease and is most frequently ER-positive, the investigation of CCND1 alterations might be relevant in male breast tumors [8]. Recently, CCND1 amplification has been shown to be an independent prognostic factor in MBC [39]. Somatic mutations of CCND1 are not a frequent alteration in cancer, and, in particular have not been reported in BC [19, 40].
Interestingly, crosstalk between ER and PI3K pathway may increase estrogen-induced and ligand-independent ER transcriptional activity [41]. Recurrent activating mutations of ESR1, the gene encoding the estrogen receptor alpha (ERα), have been identified in hormone therapy-refractory ER-positive metastatic FBC [42–44]. CNVs of ESR1 have been largely investigated in FBC, although with controversial results [45]. Data emerging from studies evaluating ESR1 somatic alterations in MBC suggest that ESR1 alterations may be gender specific [39].
Although knowledge on somatic landscape of MBC is increasing, comprehension on the role of somatic alterations of specific genes with potential prognostic and therapeutic significance need to be further investigated in MBCs characterized for BRCA1/2 mutations. In this study, we focused on somatic mutations and CNVs of PIK3CA, EGFR, ESR1 and CCND1 genes by examining a large series of MBCs screened for BRCA1/2 germ-line mutations.
RESULTS
Mutational screening
Mutational screening of PIK3CA was performed in 102 primary breast cancers and one lymph-node metastasis. Two mutations, both located in exon 9, were detected: the c.1633G>A (p.E545K) was identified in two MBC cases and the c.1634A>C (p.E545A) in two additional cases (Table 1). In these latter cases, a second mutation, c.1658_1659delGTinsC (p.S553Tfs*7), also located in exon 9, was detected (Figure 1).
Table 1: Summary of results from PIK3CA, EGFR and ESR1 mutational analysis
Gene |
Exon |
Nucleotide change |
Amino acid change |
Sample |
|---|---|---|---|---|
PIK3CA |
9 |
c.1633G>A |
p.E545Ka |
primary tumor |
PIK3CA |
9 |
c.1634A>C; c.1658_1659delGTinsC |
p.E545Aa; p.S553Tfs*7a |
primary tumor |
PIK3CA |
20 |
c.3140A>G |
p.H1047Ra |
metastatic lymph node |
EGFR |
20 |
c.2361G>A |
p.Q787Qb |
primary tumor |
EGFR |
23 |
c.2748C>T |
p.N916Nb |
primary tumor |
ESR1 |
4 |
c.975G>C |
p.P325Pb |
primary tumor |
a pathogenic mutation;
b synonymous variant.
All mutations and variants identified are recorded on COSMIC database (http://cancer.sanger.ac.uk/cosmic).
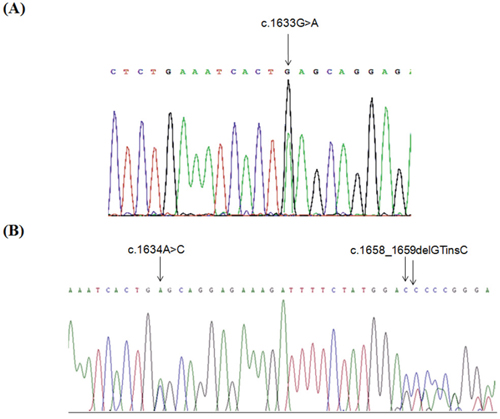
Figure 1: PIK3CA mutations identified in MBCs. Partial electropherograms from direct sequencing of PIK3CA exon 9 in a case harboring the c.1633G>A (p.E545K) mutation A. and in a case harboring c.1634A>C (p.E545A) and 1658delGTinsC (p.S553Tfs*7) mutations B.
To exclude that PIK3CA pseudogene, retaining high homology with the sequence of exon 9 (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch), might affect PIK3CA mutation screening, we performed amplification and sequencing analysis using primers specific for the PIK3CA gene and pseudogene (Supplementary Table S1). This analysis showed that both c.1634G>A and c.1658_1659delGTinsC were due to the pseudogene amplification (Figure 2).
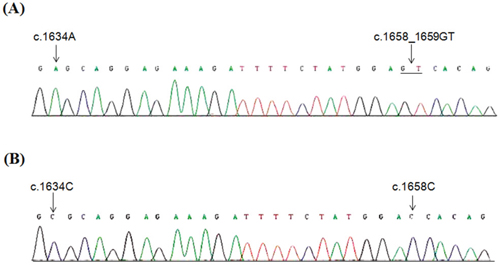
Figure 2: Specific sequencing of PIK3CA exon 9 and pseudogene. Partial electropherograms from direct sequencing of DNA region using primers specific for PIK3CA exon 9 A. and for pseudogene B.
Overall, PIK3CA mutations were identified in 2 out of 102 (2%) primary MBCs examined. Both these MBCs were from BRCA1/2 mutation negative patients with no family history of BC, were invasive ductal carcinomas, and presented with high grade (G3), ER-positive, HER2-negative and lymph node positive status.
An additional PIK3CA mutation located in exon 20, the c.3140A>G (p.H1047R), was detected in the metastatic lymph node sample examined from a MBC case whose primary tumor was PIK3CA mutation negative (Table 1).
Mutational analysis of EGFR identified two synonymous variants (Table 1): the c.2361G>A (p.Q787Q) occurring in 15 of 102 cases (14.7%), and c.2748C>T (p.N916N) occurring in one case. Mutational analysis of ESR1 identified one synonymous variant c.975G>C (p.P325P) in 15/70 cases (21.4%). No pathogenic mutations were identified in ESR1 and EGFR genes (Table 1).
Gene copy number variation analysis
Overall, gene CNV analysis showed amplification of PIK3CA, EGFR and CCND1, and deletion of ESR1. Specifically, amplification of PIK3CA was detected in 6/74 (8.1%), EGFR in 5/74 (6.8%) and CCND1 in 10/62 (16%) of the MBC cases analyzed. Overall, gene amplifications were detected in MBCs from BRCA1/2 mutation negative patients. Deletion of ESR1 was shown in 11/73 (15%) of the cases, including one case with BRCA2 germ-line mutation. Co-amplification of PIK3CA, EGFR and CCND1 was observed in two MBC cases and co-amplification of EGFR and CCND1 in one case. Furthermore, one case showed both PIK3CA mutation and CCND1 amplification.
The overall CNV profile was investigated by unsupervised hierarchical clustering analysis performed on 41 MBC cases, for which data on CNVs of all genes examined were available. Hierarchical cluster analysis revealed two groups of cases (Figure 3). One group characterized by no CNV or CNV in only one gene, mainly CCND1 amplification, and a second group characterized by CNVs in multiple genes. Notably, this second group included cases characterized by aggressive pathologic features, including high tumor grade (G3), large tumor size (T4) and lymph node involvement.
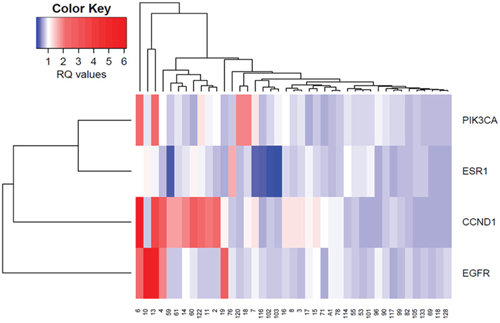
Figure 3: Unsupervised hierarchical clustering analysis of CNVs in EGFR, PIK3CA, ESR1 and CCND1 genes in male breast tumors.
Associations between somatic alterations and clinical-pathologic features
As shown in Table 2, statistically significant differences emerged in the distribution of EGFR, CCND1 and ESR1 CNVs according to ER, HER2, MIB1 and tumor size (T) status. In particular, CNVs of EGFR were associated to ER, HER2, MIB1 and T status, of CCND1 to HER2 and MIB1 status and of ESR1 to ER status.
Table 2: Frequencies of clinical-pathologic features considered in 103 primary male breast tumors crossed with gene copy number variations (CNVs)
Parameter |
Total cases (%) |
Gene CNVs |
|||||||
|---|---|---|---|---|---|---|---|---|---|
PIK3CA |
EGFR |
CCND1 |
ESR1 |
||||||
amp/Na |
pb |
amp/Na |
pb |
amp/Na |
pb |
del/Na |
pb |
||
FHc of breast/ovarian cancer |
|
|
|
|
|
|
|
|
|
Negative |
75 (72.8) |
6/53 |
0.108 |
5/56 |
0.189 |
6/49 |
0.106 |
7/52 |
0.546 |
Positive |
28 (27.2) |
0/21 |
0/18 |
4/13 |
4/21 |
||||
Personal history of cancer |
|
|
|
|
|
|
|
|
|
Negative |
77 (74.8) |
5/56 |
0.648 |
5/56 |
0.189 |
8/46 |
0.647 |
9/50 |
0.302 |
Positive |
26 (25.2) |
1/18 |
0/18 |
2/16 |
2/23 |
||||
BRCA1/2 status |
|
|
|
|
|
|
|
|
|
BRCA1 mutated |
2 (2) |
0/1 |
0.673 |
0/1 |
0.789 |
0/1 |
0.528 |
0/1 |
0.872 |
BRCA2 mutated |
9 (8.7) |
0/7 |
0/5 |
0/5 |
1/5 |
||||
BRCA1/2 wild-type |
92 (89.3) |
6/66 |
5/68 |
10/56 |
10/67 |
||||
Hystological type |
|
|
|
|
|
|
|
|
|
Infiltrating ductal carcinoma |
82 (83.7) |
5/59 |
0.277 |
5/60 |
0.352 |
10/52 |
0.15 |
9/64 |
0.702 |
Other |
16 (6.3) |
0/13 |
0/10 |
0/9 |
1/10 |
||||
ER |
|
|
|
|
|
|
|
|
|
Negative |
15 (14.8) |
2/11 |
0.192 |
3/10 |
0.002 |
2/10 |
0.736 |
4/9 |
0.009 |
Positive |
86 (85.2) |
4/62 |
2/62 |
8/55 |
7/64 |
||||
PR |
|
|
|
|
|
|
|
|
|
Negative |
24 (23.8) |
1/20 |
0.538 |
3/18 |
0.061 |
5/42 |
0.159 |
3/16 |
0.641 |
Positive |
77 (76.2) |
5/53 |
2/54 |
5/19 |
8/57 |
||||
HER2 |
|
|
|
|
|
|
|
|
|
Negative |
69 (74.2) |
2/46 |
0.051 |
1/46 |
0.012 |
4/41 |
0.019 |
10/53 |
0.201 |
Positive |
24 (25.8) |
4/21 |
4/20 |
6/17 |
1/17 |
||||
MIB1 (Ki67) |
|
|
|
|
|
|
|
|
|
Negative |
60 (61.2) |
1/38 |
0.064 |
0/38 |
0.013 |
2/30 |
0.032 |
8/44 |
0.293 |
Positive |
38(38.8) |
5/34 |
5/33 |
8/29 |
3/30 |
||||
Hystological grade |
|
|
|
|
|
|
|
|
|
G1+G2 |
61 (64.2) |
4/43 |
0.818 |
2/45 |
0.199 |
4/37 |
0.135 |
8/47 |
0.383 |
G3 |
34 (35.8) |
2/26 |
3/23 |
5/19 |
2/22 |
||||
Lymph nodes |
|
|
|
|
|
|
|
|
|
Not involved |
43 (55.8) |
1/29 |
0.249 |
2/34 |
0.487 |
3/24 |
0.517 |
3/31 |
0.513 |
Involved |
34 (44.2) |
3/26 |
3/28 |
5/26 |
4/26 |
||||
Tumor size |
|
|
|
|
|
|
|
|
|
T1-T2 |
67 (83.6) |
3/49 |
0.151 |
2/56 |
0.006 |
6/44 |
0.21 |
9/51 |
0.132 |
T4 |
13 (16.4) |
2/10 |
3/11 |
3/10 |
0/11 |
||||
a amp: amplified; del: deleted; N: number of tumors that were successfully analyzed;
b p value from Chi-square test;
c FH: Family history of breast and/or ovarian cancer.
In bold p-value <0.05, considered statistically significant.
By linear logistic regression analysis a statistically significant association was confirmed between EGFR amplification and ER-negative status (p=0.01), HER2-positive status (p=0.03) and T4 (p=0.01). By using a stepwise logistic regression, in which ER, HER2, MIB1 and T status were included, the dependency of these four characteristics in relation to EGFR amplification was analyzed, and only ER was selected as a significant effect (p=0.044). Furthermore, by linear logistic regression analysis significant association was confirmed between CCND1 amplification and HER2-positive (p=0.0005) and MIB1-positive (p=0.04) status, and between ESR1 deletion and ER-negative status (p=0.01).
DISCUSSION
In this study we investigated somatic mutations and CNVs of PIK3CA, EGFR, CCND1 and ESR1 in a large series of MBCs characterized for germ-line BRCA1 and BRCA2 mutations. We found that gene CNVs are frequent in BRCA1/2 mutation negative MBCs and allow for the identification of MBC subsets characterized by pathologic characteristics suggestive of aggressive phenotype. By contrast, somatic mutations were rarely detected and were identified only in PIK3CA gene. We did not detect somatic mutations in ESR1 and EGFR, indicating that EGFR and ESR1 mutations are infrequent in MBC and confirming recent data showing no deleterious somatic mutations of these genes in MBC [6].
In our series PIK3CA mutations were identified in 2% of MBCs. Our data indicate a lower PIK3CA mutation frequency compared with previous data on MBC. PIK3CA mutations have been reported in 18-20% of MBCs, not characterized for BRCA1/2 mutation status, and in about 10% of familial BRCA1/2 mutation negative MBCs [6, 24–26]. Though differences in PIK3CA mutation frequency may be due to different screening methods and sample selection criteria, it is worth noting that our results refer to a MBC series that represents the twice of those previously reported and included hereditary, familial and sporadic cases, all characterized for BRCA1/2 mutation status. Overall, in our series PIK3CA mutations were identified in BRCA1/2 mutation negative MBCs, thus confirming the hypothesis that PIK3CA mutation status is dependent on germ-line genotypes of MBC [25–26].
We detected the co-existence of two PIK3CA mutations, c.1634G>A (p.E545A) and c.1658_1659delGTinsC (p.S553Tfs*7) both located in exon 9, in two independent MBC cases. The concurrence of these two somatic mutations has been previously reported in large B cell lymphoma and non-small cell lung cancer [46–47]. Moreover, the concurrence of these mutations as germ-line mutations has been suggested to predispose to Cowden and Cowden like syndromes [48]. Double mutations in PIK3CA are rare phenomena [49]. To exclude that a pseudogene of PIK3CA located on chromosome 22q11.2 and that retains about 95% homology with the sequence of PIK3CA exons 9-13 (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch), might have influenced the accuracy of mutation detection, we performed true-specific PIK3CA and pseudogene sequencing analysis. These analyses showed that both c.1634A>C (p.E545A) and c.1658_1659delGTinsC (p.S553Tfs*7) were due to the pseudogene amplification. Similar observations have been reported in previous studies [50–52]. Therefore, when interpreting data corresponding to PIK3CA mutations, attention should be given to the possible co-amplification of PIK3CA gene and its pseudogene.
Interestingly, we detected PIK3CA c.3140A>G (p.H1047R) hotspot activating mutation in a metastatic lymph node of a MBC case that did not harbor PIK3CA mutation in the corresponding primary breast tumor. A different PIK3CA mutation status between primary tumors and corresponding metastatic lesions has been previously reported in FBC [53–54], indicating that PIK3CA mutations may be acquired during tumor progression. This finding may be relevant for clinical management, suggesting the need of assessing PIK3CA mutation status in metastatic lesions for the selection of PIK3CA inhibitor therapy.
We reported the occurrence of PIK3CA amplification at higher frequency than that previously reported in MBC [26]. The frequency of PIK3CA amplification found in our study is consistent with that reported in FBC [27–29]. As for PIK3CA mutations, PIK3CA amplification was found in BRCA1/2 mutation negative MBCs.
In our MBC series EGFR amplification was found at a higher frequency compared to previous data on MBC [26, 39] and FBC [32–33]. Furthermore, we showed that EGFR amplification was associated with ER-negative status, HER2-positive status and large tumor size (T4). These findings suggest that EGFR amplification could represent a prognostic marker in MBC and could identify a subset of MBC patients at high risk for adverse prognosis. Our data also suggest that this subset of MBCs (i.e. ER-negative and HER2-positive) might benefit from target therapy. Interestingly, there is evidence showing that therapies combining HER2 inhibitors with EGFR inhibitors, such as trastuzumab with lapatinib, may produce promising results in MBC treatment [55]. Overall, the association between EGFR amplification and ER-negative status suggest that EGFR signaling pathway may play a role in male breast carcinogenesis, mainly in estrogen-insensitive breast tumors. Interestingly, association between EGFR amplification and ER-negative status was also reported in FBC [32].
CCND1 amplification was the most frequent CNV observed in our MBC series, thus supporting the hypothesis that this gene may play a relevant role in male breast carcinogenesis, particularly in BRCA1/2 mutation negative cases. Our results are in agreement with previous data showing that CCND1 was one of the genes that most frequently showed copy number gain in MBC [39]. In our series, CCND1 amplification identified a subset of MBCs characterized by HER2-positive and MIB1-positive status, pathologic features suggestive of aggressive phenotype. These findings are in line with the function of CCND1 protein product and are consistent with previous data showing that MBCs with CCND1 copy number gain tended to have a higher mean mitotic count compared to tumors without CCND1 amplification [39]. Notably, CCND1 amplification seems to be an independent prognostic factor in MBC correlating with poor survival [39]. Amplification of CCND1 has also been associated with poor prognosis in ER-positive FBC [12, 37]. As CCND1 is an estrogen-responsive gene, one possible mechanism by which its amplification could lead to a poor prognosis is by conferring resistance to hormone therapy [13, 56]. These findings may acquire great importance in MBC, for which endocrine therapy is the most common treatment [3].
ESR1 deletion was reported at high frequency in our MBC series. Our results are consistent with data reporting gene deletion in MBC [39] and confirm that ESR1 amplification is not a characteristic alteration of MBC. By contrast, ESR1 amplification has been reported, although with controversial results, in FBC in which ESR1 deletion does not appear to be a significant event [45]. In our series, a significant association emerged between ESR1 gene deletion and ER-negative status. Interestingly several data suggest that low-level of ESR1 expression may be one of the mechanisms responsible for tamoxifen resistance in ER-positive FBC [57]. These observations confirm the utility of our results in the identification of MBC subsets that may need specific therapeutic approach.
In agreement with the hypothesis that tumors rising on gene amplification are more likely to develop multiple amplifications as they have greater genetic instability [15], in this study we identified co-amplification of genes analyzed. By cluster analysis, we identified a group of cases characterized by co-amplification of two or more genes, suggesting the need to develop new therapeutic strategies involving multiple targets.
Overall, our data indicate that genes known to be important prognostic biomarkers and molecular therapeutic targets in FBC are also important in MBC. For instance, adjuvant trastuzumab, a monoclonal antibody that selectively targeted HER2 and is used as therapeutic agent in HER2-positive FBCs, can be considered for high-risk HER2-positive MBCs [58]. However, differences emerged between male and female breast cancers, emphasizing the importance of identifying biomarkers based on research in MBC.
In conclusion our results showed that CNVs of PIK3CA, EGFR, CCND1 and ESR1 genes may play a relevant role in male breast carcinogenesis and could lead to the identification of specific molecular profiles. In particular, amplification of targetable oncogenes is frequent in BRCA1/2 mutation negative MBCs and may identify biomarkers relevant for clinical management and targeted therapeutic approach.
MATERIALS AND METHODS
Patients and genomic DNA
A series of 103 primary male breast tumors was included in the study. A metastatic lymph node sample was available for one case and was also included in the analyses. All MBC cases were characterized for germ-line BRCA1 and BRCA2 mutations and for the main clinical-pathologic features, including: age at diagnosis, family and personal history of cancer, tumor histological type, grade (G), nodal status, tumor size (T), estrogen receptor (ER), progesterone receptor (PR), MIB1 (Ki67) and HER2 expression (Table 2). The expression of ER, PR, and Ki67/MIB1 was scored based on the percentages of positive nuclei (ER/PR positive if >10%; Ki67/MIB1 high if >20%) over the total number of counted cancer cell nuclei HER2 positivity was defined as a score of 3+ using immunohistochemistry (IHC) test, or amplification shown by fluorescence in situ hybridization (FISH), in equivocal cases [59–60].
Genomic DNA was isolated from microdissected formalin fixed paraffin-embedded (FFPE) and fresh frozen tumor sections. DNA from FFPE sections was extracted using DNA/RNA FFPE kit (Qiagen Inc., Venlo, Netherlands) according to the manufacturer’s instructions. DNA from fresh frozen samples was extracted by following standard phenol-chloroform extraction protocol. For some cases the amount of DNA was inadequate to carry out all molecular analyses.
The participants signed an informed consent form with a detailed description of the study protocol. The study was approved by The Local Ethical Committee (Sapienza University of Rome, Protocol 264/12).
Mutational screening
Mutational analysis of PIK3CA, EGFR and ESR1 was performed mainly by direct sequencing and by SSCP (Single Strand Conformation Polymorphism). Genomic DNA from each tumor sample was amplified using primers covering exons in which is reported to cluster the great majority of mutations (Supplementary Table S1), in particular exons 9 and 20, coding the helical and kinase domains of PIK3CA, respectively [61]; exons from 18 to 24, coding kinase domain of EGFR; exons from 4 to 8, coding the ligand binding domain, of ESR1.
To overcome the interference of a pseudogene of PIK3CA, we also designed specific primers for both PIK3CA gene and pseudogene (Supplementary Table S1). DNA samples from cell lines (HCC15 and HCT16) with known mutations in exons 9 (c.1633G>A, p.E545K) and 20 (HCT-116 c.3140 A>G, p.H1047R) of PIK3CA, were used as positive control.
Gene copy number variation analysis
PIK3CA, EGFR, ESR1 and CCND1 gene copy number variations (CNVs) were analyzed by Real-Time PCR, using TaqMan probe-based technology (Applied Biosystems, Foster City California, USA). The RNaseP as housekeeping gene and a normal male breast tissue sample as calibrator were used. TaqMan Copy number Assays (Hs02708380_cn for PIK3CA, Hs00756893_cn for EGFR, Hs02577150_cn for ESR1 and Hs01818912_cn for CCND1) and TaqMan Copy number Reference Assay human RNaseP, containing primers and probe mix for target and housekeeping genes respectively, were used (Applied Biosystems, Foster City California, USA). The analysis of relative gene expression data was performed, using the 2-ΔΔCt method. The fold change in studied gene copy number, normalized to endogenous control, was calculated using RQ=2-ΔΔCt [62–63]. Values of RQ ≥2 were considered amplified and values of RQ <0.5 were considered deleted.
Statistical analysis
Unsupervised hierarchical clustering was performed to analyze relevant clusters characterized by co-amplification/deletion in EGFR, PIK3CA, ESR1 and CCND1.
Chi square test and logistic regression models were performed in order to evaluate the potential associations between each gene alteration (mutations and CNVs) and clinical-pathologic features. A P value ≤0.05 was considered statistically significant. All statistical analyses were performed with the R software (www.r-project.org).
ACKNOWLEDGMENTS
The authors thank all the individuals who participated in this study and all the institutions and their staff who supported the recruitment of patients and the collection of samples and data.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
Study supported by Associazione Italiana per la Ricerca sul Cancro (AIRC IG 16933) to L.O., Istituto Toscano Tumori (ITT-2010) to D.P. and Fondazione Umberto Veronesi (Annual Post-doctoral Fellowships) to P.R.
REFERENCES
1. Ly D, Forman D, Ferlay J, Brinton LA, Cook MB. An international comparison of male and female breast cancer incidence rates. Int J Cancer. 2013; 132:1918-26.
2. Rizzolo P, Silvestri V, Tommasi S, Pinto R, Danza K, Falchetti M, Gulino M, Frati P, Ottini L. Male breast cancer: genetics, epigenetics, and ethical aspects. Ann Oncol. 2013; 24 Suppl 8:viii75-viii82. Review.
3. Khan MH, Allerton R, Pettit L. Hormone Therapy for Breast Cancer in Men. Clin Breast Cancer. 2015; 15:245-50.
4. Johansson I, Killander F, Linderholm B, Hedenfalk I. Molecular profiling of male breast cancer - lost in translation? Int J Biochem Cell Biol. 2014; 53:526-35.
5. Callari M, Cappelletti V, De Cecco L, Musella V, Miodini P, Veneroni S, Gariboldi M, Pierotti MA, Daidone MG Gene expression analysis reveals a different transcriptomic landscape in female and male breast cancer. Breast Cancer Res Treat. 2011; 127:601-10.
6. Piscuoglio S, Ng CK, Murray MP, Guerini-Rocco E, Martelotto LG, Geyer FC, Bidard FC, Berman S, et al. The Genomic Landscape of Male Breast Cancers. Clin Cancer Res. 2016; 22:4045-56.
7. Korde LA, Zujewski JA, Kamin L, Giordano S, Domchek S, Anderson WF, Bartlett JM, Gelmon K, Nahleh Z, Bergh J, Cutuli B, Pruneri G, McCaskill-Stevens W et al. Multidisciplinary meeting on male breast cancer: summary and research recommendations. J Clin Oncol. 2010; 28:2114-22.
8. Brinton LA, Cook MB, McCormack V, Johnson KC, Olsson H, Casagrande JT, Cooke R, Falk RT, Gapstur SM, Gaudet MM, Gaziano JM, Gkiokas G, Guénel P, et al. Anthropometric and hormonal risk factors for male breast cancer: male breast cancer pooling project results. J Natl Cancer Inst. 2014; 106:djt465. Erratum in: J Natl Cancer Inst. 2014; 106:dju117.
9. Deb S, Lakhani SR, Ottini L, Fox SB The cancer genetics and pathology of male breast cancer. Histopathology. 2016; 68:110-8.
10. Normanno N, Morabito A, De Luca A, Piccirillo MC, Gallo M, Maiello MR, Perrone F. Target-based therapies in breast cancer: current status and future perspectives. Endocr Relat Cancer. 2009; 16:675-702. Review.
11. Baselga J. Targeting the phosphoinositide-3 (PI3) kinase pathway in breast cancer. Oncologist. 2011; 16 Suppl 1:12-9. Review.
12. Roy PG, Pratt N, Purdie CA, Baker L, Ashfield A, Quinlan P, Thompson AM. High CCND1 amplification identifies a group of poor prognosis women with estrogen receptor positive breast cancer. Int J Cancer 2010; 127: 355-360.
13. Tabarestani S, Ghaderian SM, Rezvani H, Mirfakhraie R, Ebrahimi A, Attarian H, Rafat J, Ghadyani M, Alavi HA, Kamalian N, Rakhsha A, Azargashb E. Prognostic and predictive value of copy number alterations in invasive breast cancer as determined by multiplex ligation-dependent probe amplification. Cell Oncol (Dordr) 2014; 37: 107-118.
14. Ménard S, Fortis S, Castiglioni F, Agresti R, Balsari A. HER2 as a prognostic factor in breast cancer. Oncology. 2001; 61 Suppl 2:67-72.
15. Al-Kuraya K, Schraml P, Torhorst J, Tapia C, Zaharieva B, Novotny H, Spichtin H, Maurer R, Mirlacher M, Köchli O, Zuber M, Dieterich H, Mross F, et al. Prognostic relevance of gene amplifications and coamplifications in breast cancer. Cancer Res. 2004; 64:8534-40.
16. Choi YJ, Choi YL, Cho EY, Shin YK, Sung KW, Hwang YK, Lee SJ, Kong G, Lee JE, Kim JS, Kim JH, Yang JH, Nam SJ. Expression of Bmi-1 protein in tumor tissues is associated with favorable prognosis in breast cancer patients. Breast Cancer Res Treat. 2009; 113:83-93.
17. Nielsen KV, Ejlertsen B, Møller S, Jørgensen JT, Knoop A, Knudsen H, Mouridsen HT. The value of TOP2A gene copy number variation as a biomarker in breast cancer: Update of DBCG trial 89D. Acta Oncol. 2008; 47:725-34.
18. Rodriguez-Pinilla SM, Jones RL, Lambros MB, Arriola E, Savage K, James M, Pinder SE, Reis-Filho JS. MYC amplification in breast cancer: a chromogenic in situ hybridisation study. J Clin Pathol. 2007; 60:1017-23.
19. Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61-70.
20. Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, Yates LR, Papaemmanuil E, Beare D et al. The landscape of cancer genes and mutational processes in breast cancer. Nature 2012; 486:400-4.
21. Chandarlapaty S, Sakr RA, Giri D, Patil S, Heguy A, Morrow M, Modi S, Norton L, Rosen N, Hudis C, King TA. Frequent mutational activation of the PI3K-AKT pathway in trastuzumab-resistant breast cancer. Clin Cancer Res. 2012; 18:6784-91.
22. Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, Symmans WF, Pusztai L, Nolden LK, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008; 68:6084-91.
23. Mayer IA, Arteaga CL. PIK3CA activating mutations: a discordant role in early versus advanced hormone-dependent estrogen receptor–positive breast cancer? J Clin Oncol. 2014; 32:2932-4.
24. Benvenuti S, Frattini M, Arena S, Zanon C, Cappelletti V, Coradini D, Daidone MG, Pilotti S, Pierotti MA, Bardelli A. PIK3CA cancer mutations display gender and tissue specificity patterns. Hum Mutat. 2008; 29:284-8.
25. Deb S Do H, Byrne D, Jene N; kConFab Investigators, Dobrovic A, Fox SB. PIK3CA mutations are frequently observed in BRCAX but not BRCA2-associated male breast cancer. Breast Cancer Res. 2013; 15:R69.
26. Deb S, Wong SQ, Li J, Do H, Weiss J, Byrne D, Chakrabarti A, Bosma T; kConFab Investigators, Fellowes A, Dobrovic A, Fox SB. Mutational profiling of familial male breast cancers reveals similarities with luminal A female breast cancer with rare TP53 mutations. Br J Cancer. 2014; 111:2351-60.
27. Wu G, Xing M, Mambo E, Huang X, Liu J, Guo Z, Chatterjee A, Goldenberg D, Gollin SM, Sukumar S, Trink B, Sidransky D. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005; 7:R609-16.
28. Kadota M, Sato M, Duncan B, Ooshima A, Yang HH, Diaz-Meyer N, Gere S, Kageyama S, Fukuoka J, Nagata T, Tsukada K, Dunn BK, Wakefield LM, et al. Identification of novel gene amplifications in breast cancer and coexistence of gene amplification with an activating mutation of PIK3CA. Cancer Res. 2009; 69:7357-65.
29. Gonzalez-Angulo AM, Chen H, Karuturi MS, Chavez-MacGregor M, Tsavachidis S, Meric-Bernstam F, Do KA, Hortobagyi GN, Thompson PA, Mills GB, Bondy ML, Blumenschein GR Jr. Frequency of mesenchymal-epithelial transition factor gene (MET) and the catalytic subunit of phosphoinositide-3-kinase (PIK3CA) copy number elevation and correlation with outcome in patients with early stage breast cancer. Cancer. 2013; 119:7-15
30. Goffin JR, Zbuk K. Epidermal growth factor receptor: pathway, therapies, and pipeline. Clin Ther. 2013; 35:1282-303.
31. Roskoski R Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014; 79:34-74.
32. Moelans CB, de Weger RA, Monsuur HN, Vijzelaar R, van Diest PJ Molecular profiling of invasive breast cancer by multiplex ligation-dependent probe amplification-based copy number analysis of tumor suppressor and oncogenes. Mod Pathol. 2010; 23:1029-39.
33. Lee HJ, Seo AN, Kim EJ, Jang MH, Kim YJ, Kim JH, Kim SW, Ryu HS, Park IA, Im SA, Gong G, Jung KH, Kim HJ et al. Prognostic and predictive values of EGFR overexpression and EGFR copy number alteration in HER2-positive breast cancer. Br J Cancer. 2015; 112:103-11.
34. Weber F, Fukino K, Sawada T, Williams N, Sweet K, Brena RM, Plass C, Caldes T, Mutter GL, Villalona-Calero MA, Eng C. Variability in organ-specific EGFR mutational spectra in tumour epithelium and stroma may be the biological basis for differential responses to tyrosine kinase inhibitors. Br J Cancer. 2005; 92:1922-6.
35. Generali D, Leek R, Fox SB, Moore JW, Taylor C, Chambers P, Harris AL. EGFR mutations in exons 18-21 in sporadic breast cancer. Ann Oncol. 2007; 18:203-5.
36. Lv N, Xie X, Ge Q, Lin S, Wang X, Kong Y, Shi H, Xie X, Wei W. Epidermal growth factor receptor in breast carcinoma: association between gene copy number and mutations. Diagn Pathol. 2011; 6:118.
37. Brown LA, Johnson K, Leung S, Bismar TA, Benítez J, Foulkes WD, Huntsman DG. Co-amplification of CCND1 and EMSY is associated with an adverse outcome in ER-positive tamoxifen-treated breast cancers. Breast Cancer Res Treat. 2010; 121:347-54.
38. Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011; 11:558-72. Review.
39. Kornegoor R, Moelans CB, Verschuur-Maes AH, Hogenes MC, de Bruin PC, Oudejans JJ, Marchionni L, van Diest PJ. Oncogene amplification in male breast cancer: analysis by multiplex ligation-dependent probe amplification. Breast Cancer Res Treat. 2012; 135:49-58.
40. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, Moorhead M, Chaudhuri S, Tomsho LP et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869-73. 2007;12:798-807.
41. Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol 2011; 29:4452–61.
42. Segal CV, Dowsett M. Estrogen receptor mutations in breast cancer--new focus on an old target. Clin Cancer Res. 2014; 20:1724-6.
43. Alluri PG, Speers C, Chinnaiyan AM. Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res. 2014; 16:494.
44. Thomas C, Gustafsson JÅ. Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol Metab. 2015; 26:467-76. Review.
45. Holst F. Estrogen receptor alpha gene amplification in breast cancer: 25 years of debate. World J Clin Oncol. 2016; 7:160-73. Review.
46. Baohua Y, Xiaoyan Z, Tiecheng Z, Tao Q, Daren S. Mutations of the PIK3CA gene in diffuse large B cell lymphoma. Diagn Mol Pathol. 2008; 17:159-65.
47. Ekinci S, Ilgin-Ruhi H, Dogan M, Gursoy S, Dizbay-Sak S, Demirkazik A, Tukun A. Molecular spectrum of PIK3CA gene mutations in patients with nonsmall-cell lung cancer in Turkey. Genet Test Mol Biomarkers. 2015; 19:353-8.
48. Orloff MS, He X, Peterson C, Chen F, Chen JL, Mester JL, Eng C. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am J Hum Genet. 2013; 92:76-80.
49. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene 2008; 27:5497–5510. Review
50. Or YY, Hui AB, To KF, Lam CN, Lo KW. PIK3CA mutations in nasopharyngeal carcinoma. Int J Cancer. 2006; 118:1065-7.
51. Tanaka Y, Kanai F, Tada M, Asaoka Y, Guleng B, Jazag A, Ohta M, Ikenoue T, Tateishi K, Obi S, Kawabe T, Yokosuka O, Omata M. Absence of PIK3CA hotspot mutations in hepatocellular carcinoma in Japanese patients. Oncogene. 2006; 25:2950-2.
52. Müller CI, Miller CW, Hofmann WK, Gross ME, Walsh CS, Kawamata N, Luong QT, Koeffler HP. Rare mutations of the PIK3CA gene in malignancies of the hematopoietic system as well as endometrium, ovary, prostate and osteosarcomas, and discovery of a PIK3CA pseudogene. Leuk Res. 2007; 31:27-32.
53. Dupont Jensen J, Laenkholm AV, Knoop A, Ewertz M, Bandaru R, Liu W, Hackl W, Barrett JC, Gardner H. PIK3CA mutations may be discordant between primary and corresponding metastatic disease in breast cancer. Clin Cancer Res. 2011; 17:667-77.
54. Goswami RS, Patel KP, Singh RR, Meric-Bernstam F, Kopetz ES, Subbiah V, Alvarez RH, Davies MA, Jabbar KJ, Roy-Chowdhuri S, Lazar AJ, Medeiros LJ, Broaddus RR, et al. Hotspot mutation panel testing reveals clonal evolution in a study of 265 paired primary and metastatic tumors. Clin Cancer Res. 2015; 21:2644-51.
55. Saxena R, Dwivedi A. ErbB family receptor inhibitors as therapeutic agents in breast cancer: current status and future clinical perspective. Med Res Rev. 2012; 32:166-215.
56. Lundgren K, Brown M, Pineda S, Cuzick J, Salter J, Zabaglo L, Howell A, Dowsett M, Landberg G; TransATAC investigators. Effects of cyclin D1 gene amplification and protein expression on time to recurrence in postmenopausal breast cancer patients treated with anastrozole or tamoxifen: a TransATACstudy. Breast Cancer Res BCR. 2012; 14:R57.
57. Kim C, Tang G, Pogue-Geile KL, Costantino JP, Baehner FL, Baker J, Cronin MT, Watson D, Shak S, Bohn OL, Fumagalli D, Taniyama Y, Lee A, Reilly ML, et al. Estrogen receptor (ESR1) mRNA expression and benefit from tamoxifen in the treatment and prevention of estrogen receptor-positive breast cancer. J Clin Oncol. 2011; 29:4160-7.
58. Ottini L, Capalbo C, Rizzolo P, Silvestri V, Bronte G, Rizzo S, Russo A. HER2-positive male breast cancer: an update. Breast Cancer:Targets and Therapy. 2010; 2:45-58. Review.
59. Ottini L, Masala G, D'Amico C, Mancini B, Saieva C, Aceto G, Gestri D, Vezzosi V, Falchetti M, De Marco M, Paglierani M, Cama A, Bianchi S, et al. BRCA1 and BRCA2 mutation status and tumor characteristics in male breast cancer: a population-based study in Italy. Cancer Res. 2003; 63:342-7.
60. Bianchi S, Palli D, Falchetti M, Saieva C, Masala G, Mancini B, Lupi R, Noviello C, Omerovic J, Paglierani M, Vezzosi V, Alimandi M, Mariani-Costantini R, et al. ErbB-receptors expression and survival in breast carcinoma: a 15-year follow-up study. J Cell Physiol. 2006; 206:702-8.
61. Kang S, Bader AG, Vogt PK Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005; 102:802-7.
62. Holst F, Stahl PR, Ruiz C, Hellwinkel O, Jehan Z, Wendland M, Lebeau A, Terracciano L, Al-Kuraya K, Jänicke F, Sauter G, Simon R. Estrogen receptor alpha (ESR1) gene amplification is frequent in breast cancer. Nat Genet. 2007; 39:655-60.
63. Pu T, Guo P, Qiu Y, Chen S, Yang L, Sun L, Ye F, Bu H. Quantitative real-time polymerase chain reaction is an alternative method for the detection of HER-2 amplification in formalin-fixed paraffin-embedded breast cancer samples. Int J Clin Exp Pathol. 2015; 8:10565-74.