
INTRODUCTION
In order to grow and disseminate, tumors depend on an adequate blood supply [1, 2]. Based on the assumption that tumors arrange their own blood supply via induction of angiogenesis, in the last decade angiogenesis inhibitors (e.g. the anti-VEGF-A antibody bevacizumab or VEGFR2-specific tyrosine kinase inhibitors) have been widely implemented in clinical practice for a number of tumor types [3, 4]. Effects of anti-angiogenesis (mostly in combination with chemotherapy) are however mostly transient [5, 6] and cancer patients who undergo anti-angiogenic treatment mostly experience recurrences, one possible cause being that tumors adopt an invasive phenotype to accommodate their metabolic needs [7–10]. Resistance to anti-angiogenic treatment may also be related to heterogeneity of the tumor vasculature. Vessel formation in cancer is a multistep process and consequently cancers contain neovessels in different stages of development [11, 12]. Because endothelial cells in matured vessels are less dependent on VEGF-VEGFR2 signalling, these are also more refractory to VEGF inhibition [13].
Vessel heterogeneity is especially prominent in glioblastoma, a highly malignant brain tumor of glial origin [14]. One hallmark of glioblastoma is the presence of large areas of diffuse infiltrative growth in which tumor cells thrive on pre-existent vessels (co-option) without the need for angiogenesis [15, 16]. Another characteristic of glioblastoma is focal angiogenesis in the vicinity of areas of necrosis, and this has been rationale for preclinical and clinical testing of anti-VEGF therapy for recurrent and primary diagnosed glioblastoma. Such studies have shown effective inhibition of angiogenesis, however without preventing tumor progression in areas of diffuse infiltrative growth [17–19]. This growth pattern thus provides tumors with a route of escape from anti-angiogenic therapies [5, 20, 21] and recent clinical trials confirmed a lack of prolonged survival of glioblastoma patients upon treatment with bevacizumab [22, 23].
An alternative approach to deprive cancers from blood is direct anti-vascular therapy that aims at tumor-specific thrombosis and infarction. Such an approach requires that tumor vascular targeting agents (TVTAs) are developed with high enough specificity for tumor vasculature. Available TVTAs (e.g. RGD peptides or cilengitide, targeting endothelial αvβ3 integrin [24, 25], the L19 single chain antibody that targets the ED-B fragment of fibronectin [26], anti-V-CAM antibodies [27], anti-plexinD1 antibodies [28]) are directed against newly formed vessels and do not target tumor vessels in more matured stages of development.
Biopanning of peptide or single chain-antibody phage display libraries is a powerful technique that allows identification and isolation of tumor endothelium binding partners [29–32]. Nanobodies® (Variable Heavy chain domains of Heavy chain antibodies or VHHs) are recombinant antibodies, cloned from cameloid IgG2 and IgG3 heavy chain-only antibodies (V-H) and consist of a single polypeptide chain, making this class of antibodies suitable for display on phages without significant loss of affinity [33, 34]. Their small size (15–18 kDa) and stability make nanobodies to an attractive class of diagnostic and therapeutic compounds [35]. Nanobodies against epidermal growth factor receptor (EGFR) or carcinoembryonic antigen (CEA) have already successfully been used for in vivo diagnosis and therapy [36–38].
In a search for novel relevant nanobody-based TVTAs we performed in vivo biopannings with a nanobody phage display library [29]. As a tumor model we utilized mice carrying orthotopic E98 human glioma xenografts that characteristically display both angiogenesis-dependent growth and diffuse infiltrative growth [18, 39]. We identified a novel nanobody, C-C7, that targets a subpopulation of tumor blood vessels. Using C-C7 as a bait protein in yeast-2-hybrid screens we identified dynactin-1-p150Glued as its binding partner.
RESULTS
In vivo selection of tumor vessel binding phages in cerebral E98 xenografts
A nanobody-displaying phage library [28] was intravenously injected in mice carrying intracerebral E98 xenografts and irrelevant phages were removed from the circulation by cardiac perfusion. We chose to use mice carrying orthotopic E98 xenografts because these tumors display both areas of angiogenesis and diffuse infiltrative growth [39]. Similarly to our previous work using different tumor xenograft models and other phage libraries [29], anti-M13 immunostainings demonstrated already a tumor-specific vessel localization of phages after the first round of biopanning (Figure 1, compare the anti-M13 immunostaining in panel A to the endothelial cell CD34 staining in panel B). After collection of tumor areas from brain sections by laser capture dissection microscopy and subsequent trypsin treatment, a total of 453 colony-forming phages was rescued of which 192 clones were randomly picked and analysed for full length nanobody expression and diversity. Dot blot analysis revealed that 95% of clones expressed nanobodies and restriction enzyme finger print analysis resulted in five different restriction patterns (not shown). As there is some chance of nanobody clones with similar restriction patterns being different on the nucleotide level, we arbitrarily chose to analyze from each group the 30% of clones with highest nanobody expression levels.
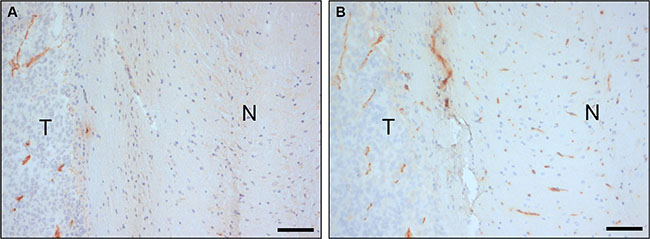
Figure 1: In vivo biopanning of a Llama phage library in an animal model of orthotopic glioma. Anti-M13-p8 (A) and anti-CD34 (B) immunostainings of sections of E98 xenografts in mouse brain after intravenous injection of 1012 phages of the nanobody-phage display library, and cardiac perfusion. Note that phages are associated with tumor vasculature, but to a lesser extent with blood vessels in normal brain. N = normal, T = tumor. Bars correspond to 100 μm.
Immunohistochemistry
Immunohistochemical stainings were performed on sections of intracerebral E98 xenografts to select for nanobodies that specifically recognize tumor vessels. Because interpretation of staining of delicate capillaries requires optimal morphology, we chose to perform immunostainings on sections of FFPE-tissue blocks instead of cryosections, and accepted that potentially interesting nanobodies (recognizing conformational epitopes that are disrupted during formalin fixation) could be lost during analyses. Positive staining of blood vessels was observed with 27 of the 39 analyzed nanobodies of which 10 were not tumor-specific. The remaining 17 nanobodies stained subsets of tumor vessels. Of these, nanobody C-C7 showed most prominent staining of tumor vasculature in E98 xenografts and was analyzed in more detail. C-C7 stained both small and medium-sized tumor vessels in the diffuse infiltrative component of E98 xenografts [39, 40] (Figure 2A). Interestingly, not all tumor associated vessels were recognized (compare the C-C7 staining pattern with the anti-CD34 staining of the serial sections in the inset in Figure 2A). Nanobody C-C7 did not stain normal vasculature in mouse brain (Figure 2B). The observation that C-C7 stained only a subpopulation of tumor vessels prompted us to further characterize C-C7 by immunohistochemistry in other mouse tumor models. Nanobody C-C7 showed delicate staining of the vasculature in colorectal cancer xenografts (Supplementary Figure S1). Interestingly, blood vessels in angiogenic brain metastases of the human melanoma cell line Mel57-VEGF165 [41] were homogeneously positive (see representative example in Figure 2C) while staining was absent in Mel57-VEGF165 tumors after treatment with the VEGFR2 inhibitor vandetanib [42] (Figure 2D). This suggests that C-C7 recognizes VEGF-activated endothelial cells.
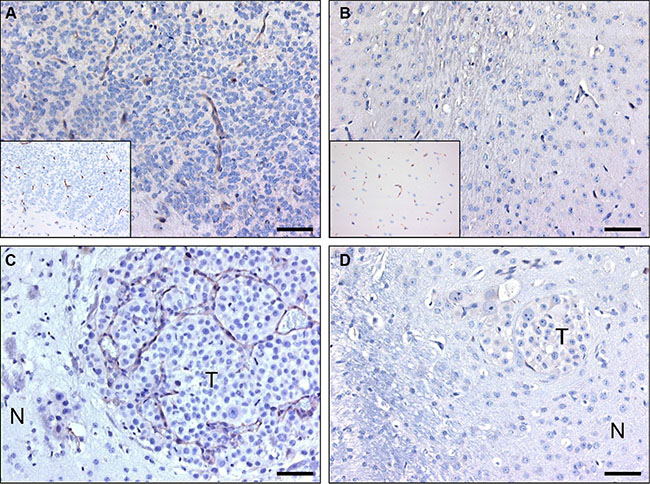
Figure 2: C-C7 recognizes tumor vessels in glioma xenografts and brain metastasis. Immunohistochemical analysis of nanobody C-C7 in a diffuse-invasive part of cerebral E98 xenografts (A), normal mouse brain parenchyma (B), a representative brain metastasis of Mel57-VEGF165 melanoma (C) and a Mel57-VEGF-A165 xenograft after treatment with the VEGFR2 inhibitor vandetanib [42] (D). C-C7 recognizes subsets of tumor vessels in cerebral E98 lesions, while normal mouse brain vessels are negative. Insets in A and B show CD34 immunostainings of serial sections. Note that inhibition of VEGFR2 activity in panel D results in loss of C-C7 reactivity. N = normal, T = tumor. Bars correspond to 50 μm.
To investigate whether nanobody C-C7 might have clinical relevance, we performed immunostainings on 9 clinical glioblastoma samples as well as normal human brain tissue. Subpopulations of tumor vessels in most glioma tissues stained positive for C-C7 (see representative examples in Figure 3A–3C and 3E) while C-C7 did not stain vasculature in normal human brain (inset in Figure 3A). In this set only one grade II glioma did not show any immunoreactivity towards C-C7 (Figure 3D). Of interest, paired glioma samples collected before and after avastin treatment [21], showed that vascular expression of the C-C7 ligand was absent after anti-angiogenic treatment (compare Figure 3E and 3F).
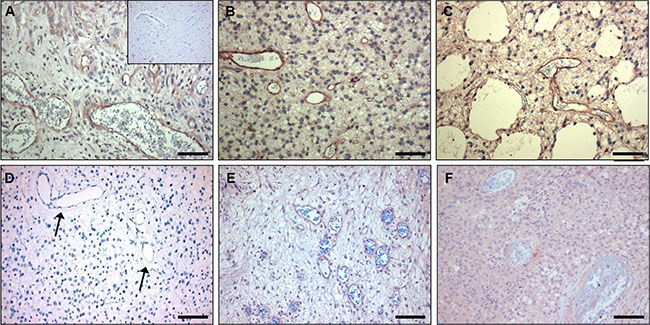
Figure 3: C-C7 recognizes tumor vessels in human glioblastoma multiforme. Subsets of vessels in human glioma are strongly positive for C-C7 (A–C, E). Panels E and F represent a paired primary and recurrent tumor from a patient before (E) and after (F) treatment with bevacizumab and temozolomide. Note the absence of C-C7 reactivity on vasculature a low grade glioma (D) (arrow). The inset in panel A illustrates absence of C-C7 reactivity in the vasculature of normal brain. Bars in A-C correspond to 50 μm, in panels D–F to 100 μm.
Vessel activation is not exclusive for cancers but also occurs in a number of other pathologies, e.g. atherosclerosis. Also in atherosclerotic lesions obtained from human carotid artery we observed CC7 immunostaining of endothelial cells, but also prominent staining of macrophages (compare C-C7 stainings in Figure 4A, 4D to CD68 stainings in Figure 4B, 4E and the CD31 staining in Figure 4C). Staining with anti-dynactin-1-p150Glued (Figure 4F, see later for identification of dynactin-1-p150Glued as the C-C7 target) showed clear colocalization with C-C7 (compare 4E and 4F).
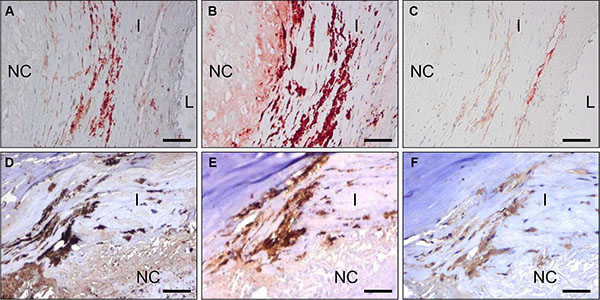
Figure 4: C-C7 recognizes blood vessels and macrophages in human atherosclerosis. Immunostainings of surgical samples of atherosclerotic lesions from carotid arteries with CC7 (A, D) the macrophage marker CD68 (B, E) and the endothelial marker CD31 (C) and anti-dynactin-1 p150Glued (F, the anti-dynactin antibody was implemented in thee staining based on identification of dynactin as a binding partner of C-C7). (A–C) and (D–F) represent serial sections from two different lesions. Note the similarity in staining profiles for C-C7 and dynactin P150Glued (D and F, respectively). Tissues in A–C were stained with AEC, tissues in (D–F) with DAB. NC = nectrotic core, I = intima, L = lumen. Bars correspond to 100 μM in panels (A–C) and to 50 μm in panels (D–F).
To further investigate whether macrophage staining could be reproduced, we stained in vitro differentiated mouse HoxB8 macrophages with C-C7 and anti-dynactin-1-p150Glued. Both antibodies showed similar staining profiles. Control VHH A9 did not stain these cells (Figure 5).
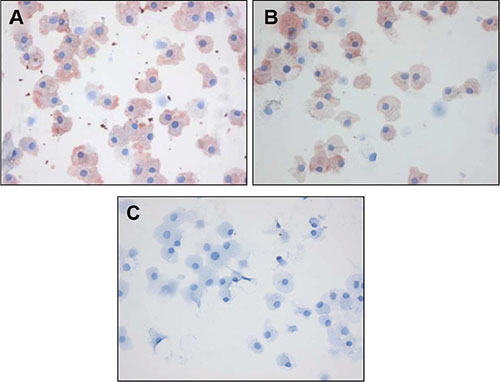
Figure 5: C-C7 recognizes dynactin-1 epitopes on macrophages. Immunostainings with C-C7, anti-dynactin-p150Glued antibody and an irrelevant control nanobody A9 on differentiated HoxB8 mouse macrophages. Note the similarity in staining profiles of C-C7 and dynactin-1 -p150Glued stainings.
C-C7 can be used for targeting of cancer and atherosclerosis
C-C7 was isolated from an in vivo biopanning experiment after intravenous injection, suggesting that it recognizes epitopes that are accessible to circulating nanobodies. To confirm the ability of C-C7 to target tumor vasculature, we injected monoclonal C-C7-phages in mice carrying orthotopic E98 xenografts, using the in vivo biopanning protocol which was used for its selection. C-C7-phages accumulated in subsets of vessels in E98 xenografts, but not in normal brain blood vessels (compare the phage localization in Figure 6A and 6C with the anti-CD34 staining of serial sections in Figure 6B and 6D). Control phages did not show specific tumor vessel localization, although some aspecific extravasation from leaky vessels was detected (data not shown). Similar experiments in LDLR-/- ApoB100/100 mice carrying atherosclerotic lesions showed prominent homing of C-C7-phages to luminal endothelium, intraplaque neovasculature and macrophages (Figure 6E). C-C7-phages colocalized with dynactin-1-p150Glued as shown by immunostainings (compare panels 6E and 6F).
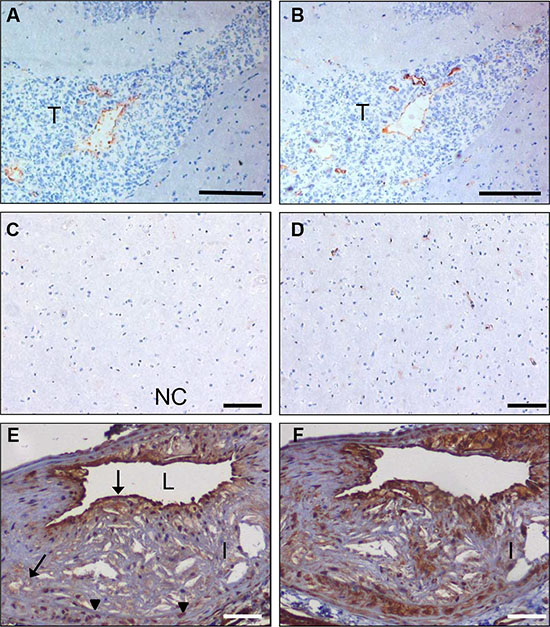
Figure 6: In vivo targeting of glioma xenografts and atherosclerotic lesions by C-C7-displaying M13 phages. Monoclonal C-C7 phages were injected intravenously in mice carrying orthotopic E98 xenografts and phage distribution was analyzed by M13-p8 immunostaining after cardiac perfusion (A). A serial section was stained with CD34 to highlight vasculature (B). Phages displaying C-C7 home to a subpopulation of tumor vessels in E98 xenografts. Phages do not accumulate in normal vasculature in non-affected brain parts, as illustrated by M13-p8 (C) and CD34 (D) immunostainings. (E) M13-p8 immunostaining of atherosclerotic lesions of LDLR−/− ApoB100/100 mice after an in vivo biodistribution experiment. Note that C-C7-phages home to luminal endothelium, intraplaque neovasculature (arrows) and macrophages (arrowheads). The M13-p8 immunostaining colocalizes with anti-Dynactin-1 immunostaining (F). T = tumor, I = intima, L = lumen. Bars in panels A,B correspond to 100 μm, in panels C-F: 50 μm
C-C7 recognizes the carboxyterminal domain of dynactin-1-p150Glued
Since endothelial proteins are greatly underrepresented in tumor extracts, we argued that a proteomic approach to identify the C-C7 ligand would be unlikely to succeed. Therefore we decided to use nanobody C-C7 as bait protein in a Y2H screen against a large yeast expression library, using histidine (HIS3), adenine and β-galactosidase (LacZ) as reporter genes [43]. Three independent clones were identified from these screens which represented the carboxyterminal part of the p150Glued subunit of dynactin-1. Clones C-C7-INT1 and C-C7-INT3 were composed of the carboxyterminal 432 amino acids of p150Glued (aa 808–1240) and included 150 nucleotides of the 3’-untranslated region, while clone C-C7-INT2 spanned aa 890–1238 of the p150Glued sequence (Figure 7A). A comparison of bovine dynactin-1-p150Glued with the human homologue revealed 96% sequence identity in this region (not shown).
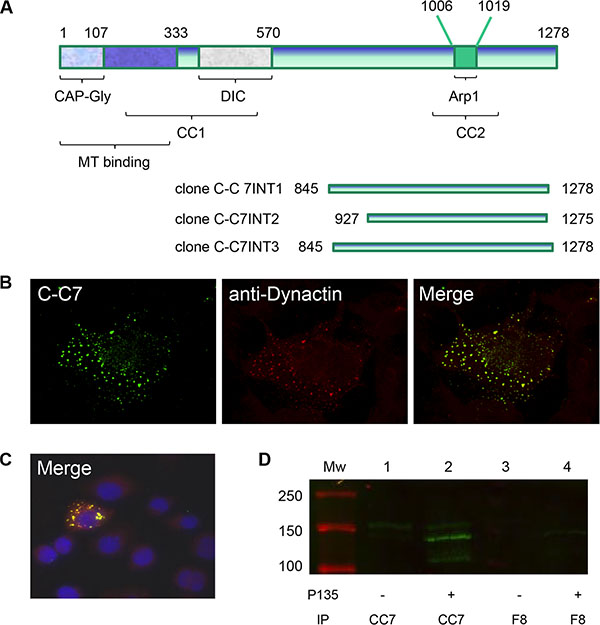
Figure 7: Dynactin-1-p150Glued is identified as a C-C7 binding partner by a yeast-2-hybrid screen. Structural domains of dynactin-1-p150Glued and p150 domains identified by yeast-2-hybrid screens as C-C7 interactants (A). The aminoterminal domain contains a CAP-Gly domain and a coiled-coil domain which are responsible for microtubule (MT) binding and dynein binding (DIC = dynein intermediate chain). A second coiled-coil domain (CC2), encompassing a binding site for Arp1, is present in the carboxyterminal part of the protein and mediates binding to membrane components. Confocal microscopy showed colocalization of C-C7 (green) and commercial anti-dynactin-1-p150Glued (red) in COS-1 cells, transfected with the recombinant carboxyterminal domain of dynactin-1-p150Glued (B). Non-transfected cells did not stain with both antibodies (C), see negative DAPI-stained cells surrounding one transfected cell in this panel). C-C7 immunoprecipitates from extracts from CHO-s cells transfected with recombinant p135Glued were analyzed on western blot using commercial anti-dynactin-1-p150Glued as indicated (D). Note that p135 is readily precipitated by C-C7 (lane 2) but not by an irrelevant nanobody F8 (lane 4).
To confirm the interaction between C-C7 and human dynactin-1-p150Glued, the region encompassing aa 816–1278 (human numbering) was RT-PCR-cloned from human glioblastoma tissue and transiently expressed in COS-1 cells. Confocal microscopy of transfected cells, co-stained with nanobody C-C7 (visualized using FITC) and anti-dynactin-1 p150Glued (visualized using TRITC) showed that both antibodies co-localized to cytoplasmic vesicular structures (Figure 7B). Non-transfected cells did not stain with both antibodies (see Figure 7C).
C-C7 specifically precipitates dynactin-1-p150Glued and recombinant dynactin-1-p135Glued
Because colocalization of C-C7 with commercial antibodies as shown in Figure 7B and 7C formally does not exclude indirect interactions with other proteins than p150Glued in the same vesicular structures, we wanted to further validate binding via immunoprecipitation. Under the conditions of cell lysis used in our experiments, the majority of carboxyterminal p150Glued 816–1278 protein ended up in the pellet fraction (data not shown). As this problem did not occur with the p135-isoform of p150Glued [44] we performed immunoprecipitation on extracts of p135-overexpressing CHO-s cells. P135 differs from p150 only in the N-terminal region, and hence contains the binding sites for both C-C7 and the commercial antibody. Furthermore, this variant can be readily distinguished from endogenous p150Glued based on molecular weight. Figure 7D shows effective precipitation of p135 by C-C7 but not by irrelevant nanobody F8. Also, small amounts of endogenous p150Glued were precipitated. In summary, these results confirm binding of C-C7 to p150Glued and indicate that it is not an artefact of the Y2H screen.
To further validate that p150Glued is expressed in tumor vasculature we performed immunostainings on clinical glioma samples with a commercially available rabbit anti-dynactin-1-p150Glued antibody. In all tumors examined this antibody gave a similar staining profile as nanobody C-C7 (see Figure 8A and 8B respectively, note that these figures contain serial sections of the tissues presented in the C-C7 stainings in Figure 3E and 3F). Normal brain vasculature did not stain with anti-dynactin although positivity could be found on neurons, consistent with literature data [44] (Figure 8C).
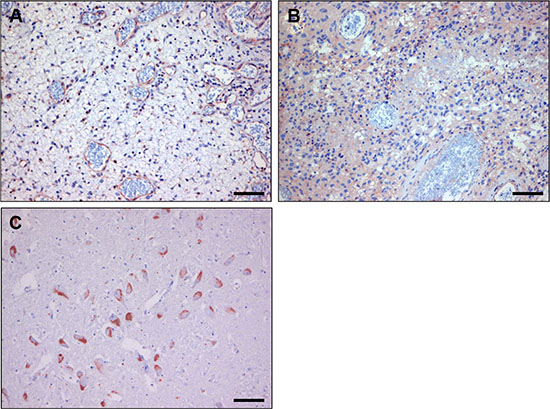
Figure 8: Immunostaining with anti-dynactin-1 -p150Glued of human glioblastoma before (A) and after bevacizumab treatment (B). Note the similarity with C-C7 immunostainings in Figure 3E and 3F. (Panel C) shows an anti-dynactin staining of normal brain, showing positive neurons, confirming literature data. Bars correspond to 100 μm.
DISCUSSION
In human tumors the vasculature is in general highly heterogeneous with glioblastomas as a prime example [45, 46]. The diffuse infiltrative growth of human gliomas and the relatively undisturbed blood-brain barrier in these areas contribute to their poor response to surgery and radio- and chemotherapy [14, 47]. Gliomas do not adequately respond to anti-angiogenic therapies either [16, 21–23, 48] which is likely a consequence of angiogenesis-independent progression of diffusely infiltrating tumor cells. Therefore, a strategy of tumor-vessel targeting aiming at the induction of tumor infarctions is worth investigating. To this end (combinations of) targeting agents that specifically and selectively recognize tumor vasculature are needed.
Here we employed in vivo biopanning of a nanobody phage display library in the E98 orthotopic mouse model of diffuse glioma [19, 39]. This approach resulted in the isolation of a set of nanobodies of which 44% specifically recognized tumor blood vessels, indicating tumor-specific enrichment already during one round of biopanning. Presumably during the in vivo selection procedure, phages that bind to the surplus of common epitopes on quiescent endothelial cells in the circulation are competitively depleted, enriching for tumor-vessel specific binders in the tumor tissue. In vivo biopanning of phage display libraries therefore appears to be a relatively quick and efficient method to identify tumor targeting nanobodies.
In this study we concentrated on nanobody C-C7 because it proved to be the most suitable for immunostainings on formalin-fixed tissues. In line with the complexity of neovascularization and the resulting molecular heterogeneity of tumor blood vessels, we found that C-C7 stained only a subpopulation of blood vessels in glioma xenografts and human glioma, but also neovasculature and macrophages in atherosclerotic plaques. Interestingly, C-C7 stained VEGF-A165- activated endothelial cells in cerebral Mel57-VEGF-A165 xenografts, but not endothelial cells in blood vessels in these xenografts after treatment with the VEGFR2 inhibitor vandetanib [49]. In line, cerebral metastases of Mel57 xenografts that do not express VEGF-A and grow entirely via vessel co-option [41] also did not show vascular staining with C-C7 (not shown). This suggests that VEGF activation of endothelial cells is involved in expression of the C-C7 ligand, and this was corroborated in staining of paired surgical samples of a human glioblastoma before and after bevacizumab treatment. Of note, these data predict that tumor-vascular targeting with C-C7-like TVTAs should not be combined with anti-angiogenesis, as this would reduce the ‘activation-status’ of the vasculature and reduce targeting potential. Whatever the molecular underpinnings of C-C7 ligand expression, the approach of in vivo biopanning with nanobody-phage display libraries may yield proper candidates for further development of anti-vascular therapies, e.g. by generating nanobodies fused to truncated tissue factor, inducing tumor-vessel specific thrombosis [27, 50, 51]. A similar approach has been employed using phage libraries displaying peptides [52].
Using yeast-2-hybrid screens we identified dynactin-1-p150Glued as the binding partner of C-C7 and we verified this interaction via co-immunostainings with nanobody C-C7 and commercial anti-dynactin-1- p150Glued antibodies on tumor tissues, on atherosclerotic plaque tissues and p150Glued- transfected cells. Further validation came from immunoprecipitations of recombinantly expressed domains of p150Glued.
Dynactin-1-p150Glued is involved in vesicular trafficking along microtubules, mitotic spindle assembly, cell migration and nuclear envelope breakdown during mitosis [53–55]. Co-immunostaining of COS-1 cells transiently overexpressing the carboxyterminal part of dynactin-1- p150Glued, with nanobody C-C7 and a commercial anti-dynactin-1- p150Glued antibody showed a vesicular pattern, in agreement with a previous study on a dynactin-p150Glued mutant that is defective in microtubule binding [56].
Since our biopanning protocol was designed to identify tumor-vessel associated proteins that are targetable via intravenous injection, the binding of nanobody C-C7 to the alleged intracellular protein p150Glued was unexpected. Which mechanisms are involved in extracellular presentation of dynactin-1-p150Glued making it accessible to intravenously administered phages, is unclear. However, a number of proteins have been shown to use non-conventional ways of trafficking in the cell [57]. Extracellular presentation of otherwise cytoplasmic or even nuclear proteins in a tumor context has been described before, examples being transglutaminase, fatty acid binding protein 3, and GRP78, although also for these targets the molecular basis for their extracellular localization is still enigmatic [52, 58–61]. A possible mechanism explaining extracellular presentation of p150Glued involves exocytosis of dynactin-decorated vesicles via multivesicular endosomes [62, 63]. After exocytosis of these vesicles, fusion with activated endothelial cells may lead to extracellular p150Glued presentation. This resembles the horizontal transmission of tumor targets via exosomes that has been proposed as an explanation for endothelial expression of tumor-derived EGFRvIII [64–66]. Of note, we also observed high expression of dynactin-1-p150Glued in macrophages in atherosclerotic lesions. Based on our immunohistochemical analyses we conclude that dynactin-1-p150Glued is not expressed at high levels in tumor cells. The actual source of dynactin-1-150Glued may therefore be activated macrophages/ microglial cells. These issues are currently under investigation.
In conclusion, in vivo biopanning of phage nanobody-display libraries in animal models of cancer in combination with Y2H technology represents a powerful platform to identify novel TVTAs and their binding ligands. We identified C-C7 as a targeting nanobody against dynactin-1-p150Glued in a glioma xenograft model and in a model of atherosclerosis. It remains to be seen whether this nanobody can be used for therapeutic anti-vascular targeting purposes.
MATERIALS AND METHODS
Tumor models
All animal experiments were approved by the Animal Experiment Committee of the RadboudUMC. Orthotopic E98 glioma xenografts were established in 6–8 week old Balb/c nude mice by intracerebral injections of E98 tumor cell suspensions as described previously [18]. In this model mice characteristically start to display weight loss and neurological symptoms due to tumor growth 3–4 weeks after tumor implantation. When tumor-related symptoms were apparent, mice were used for in vivo biopanning.
In vivo biopanning
Hundred μl of phosphate buffered saline (PBS) containing 1012 phages of a nanobody-displaying phage library (cloned from lymphocytes of non-immunized Llama glama in phagemid pHENIX-HIS-VSV [28]) was injected in the tail vein of nude mice, carrying cerebral E98 tumors (n = 2). Phages were allowed to circulate for 15 minutes. Mice were subsequently put under deep anaesthesia with a mixture of isoflurane/N2O and chests were opened. Cardiac perfusion was performed with 10 ml sterile 0.9% NaCl solution to remove unbound phages from the circulation. Brains were removed and snap frozen in liquid nitrogen. Sections of 4 μm were stained with anti-M13 p8 antibody (Abcam Limited, Cambridge, UK) to assess phage distribution. Using a laser dissection microscope (Leica AS LMD, Wetzlar, Germany) tumor areas were dissected from 10 μm brain sections. Phages were eluted from these sections by incubation with trypsin (10 mg/ml in PBS) for 30 min at room temperature. Following infection in 10 ml of log-phase E. coli TG1 culture, bacteria were seeded on 2xTY plates containing 100 mg/L ampicillin and 2% glucose. After overnight incubation, individual clones were picked and analyzed via colony-PCR for the presence of full-length nanobody insert using flanking primers M13rev (5′-TCA CAC AGG AAA CAG CTA TGA-3′) and Fedseq3 (5′-GTA ACG ATC TAA AGT TTT GTC G-3′). Subsequently, PCR products were digested with the 4-cutter restriction enzyme BstNI (New England Biolabs, Ipswich, MA, USA) to analyze the diversity of the phage population. Small scale production of soluble nanobodies by independent clones was induced in log-phase TG1 cells by culturing at 30°C in 2 × TY medium, containing 100 mg/L ampicillin and 1 mmol/L isopropylthiogalactoside (IPTG, Serva, Heidelberg, Germany). Expression of nanobodies was verified by dot blot analysis of medium using anti-VSV antibodies, as previously described [28]. Based on combined data of full length PCR, BstNI restriction digestion and dot blot analysis, expression of selected individual nanobodies was induced in 50 ml log-phase TG1 cells with 1 mM IPTG for 3 hours at 30°C. Nanobodies were isolated from the periplasm by osmotic lysis and purified by Ni-NTA sepharose (IBA Life Sciences, Gottingen, Germany) as described previously [28].
Immunohistochemistry
Nanobodies were tested in immunohistochemical stainings on 4 μm sections of formalin-fixed paraffin-embedded (FFPE) tissues and tumor xenografts of different origin (intracerebral E98 xenografts; brain metastases of Mel57-VEGF165 melanoma after treatment with vandetanib or controls [49]; subcutaneous Mel57-VEGF165 xenografts and subcutaneous xenografts of the colon carcinoma cell lines C26 and C38 [67]). Human gliomas, classified by an experienced neuropathologist as grade II (n = 2) and grade IV (n = 7) gliomas, as well as normal brain tissue (n = 2) were retrieved from the archives of the departments of Pathology of RadboudUMC and of the Academic Medical Center. GBM patients provided written informed consent, and the study was conducted in accordance with the Declaration of Helsinki and was approved by the Academic Medical Center Institutional Review Board and the Dutch Central Committee on Research investigating Human Subjects (ISRCTN23008679). Use of the other archival tissues was in accordance with institutional guide lines.
Following deparaffinization, endogenous peroxidase activity was blocked by incubation with 3% H2O2. Antigen retrieval was performed by boiling in 10 mmol/L citrate buffer (pH 6.0). Sections were pre-incubated with normal goat serum to block non-specific binding sites, followed by overnight incubation with 3 μM nanobody in PBS/2%BSA at 4°C. Detection was performed by sequential incubations with rabbit anti-VSV-G antibody (for mouse tissues, Sigma-Aldrich Chemie B.V., Zwijndrecht, The Netherlands,) or anti-VSV-G P5D4 (for human tissues), biotinylated anti-rabbit antibody (Vector, Burlingame, CA) and avidin-biotin peroxidase complex (Vector). Peroxidase was visualized with 3-amino-9-ethylcarbazole (AEC, ScyTek, Utah, USA) with haematoxylin as counterstain. Tumor vessels in mouse xenograft tissues were visualized in serial sections with rat-anti-mouse CD34 antibody (Hycult, Uden, The Netherlands). Rabbit antibodies against dynactin-1-p150Glued were from Abcam (Cambridge, UK).
C-C7 in atherosclerosis
Carotid artery atherosclerotic tissue from patients at risk for ischemic stroke was obtained from routine surgical procedures at the department of Vascular Surgery of RadboudUMC (kindly provided by Dr. J. Pol). Directly after surgery tissues were processed to FFPE blocks. Four μm sections were deparrafinized and pretreated as described, and incubated with 3 μM C-C7 in normal antibody diluent (Immunologic, Duiven, The Netherlands). Then slides were incubated with anti-VSV-G P5D4, PolyHRP-anti-Ms/Rt/Rb (Immunologic, Duiven, The Netherlands), and peroxidase was visualized with bright 3′3Diaminobenzidine (DAB) (Immunologic, Duiven, The Netherlands).
Furthermore, specificity of C-C7 was assessed by staining on mouse macrophages, obtained by differentiating ER-HoxB8 myeloid precursor cells by 25 ng/ml GM-CSF on 8-well Lab-Tek Chamber Slide™ System (Nunc, Roskilde, Denmark) [68]. Cells were fixed with 2% paraformaldehyde for 10 min, and pre-incubated with normal goat serum. Then cells were incubated with 3 μM C-C7 or rabbit-anti-Dynactin-1-P150Glued in PBS/2%BSA. Detection was performed as described above in immunohistochemistry.
In another set of experiments hypercholesterolemic (low-density lipoprotein receptor (LDLR)-/- apolipoprotein B (ApoB)100/100) mice [69] were subjected to high-cholesterol diet (TD 88137, Harlan Laboratories, Indianapolis, IN, USA) for 8 weeks after which a peri-adventitial collar was placed around a carotid artery. Hundred μl PBS containing 1012 M13-phages displaying nanobody C-C7 or helper phages as control, was injected via the tail vein and allowed to circulate for 15 minutes after which a cardiac perfusion with 10 ml PBS was performed. Lesions from the carotid artery were harvested and processed to FFPE blocks, sections of which were stained with anti-M13 p8 or with anti-dynactin-pGlued150 antibody.
Y2H screens
All cloning steps were performed using the Gateway System (Invitrogen, Carlsbad, CA). The cDNA encoding nanobody C-C7 (without pelB leader) was flanked by attB sites via PCR and cloned in vector pDONR201 before transfer into destination vector pBD-GAL4-CAM/DEST, generating a C-C7 fusion protein with the GAL4 DNA binding domain. After sequence-verification this vector was transfected into yeast strain PJ69 (A-mating type) using standard protocols [43]. Generation of a randomly primed bovine retina cDNA library in pAD-GAL4, transfected in PJ69 (α-mating type, 2 × 106 clones) has been previously described [43]. The yeast strain containing pBD-GAL4-C-C7 was mated with this library and diploid cells in which C-C7-prey interactions led to functional GAL4 transcription factor, were selected based on histidine and adenine prototrophy and transactivation of ß-galactosidase activity. Production of ß-galactosidase by activation of the LacZ reporter gene was detected by a filter-lift assay [43]. From positive yeast clones, pAD-GAL4 library expression plasmids were rescued and amplified in E. coli. Plasmids were sequenced using flanking forward and reverse primers. Sequences were blasted (http://www.ncbi.nlm.nih.gov/blast/Blast.cgi) and aligned with multalin software (http://bioinfo.genotoul.fr/multalin/multalin.html). All sequenced clones contained intact reading frames in fusion with the GAL4-activation domain.
Verification of the C-C7-dynactin-1 interaction
The carboxyterminal part of dynactin-1-p150Glued was RT-PCR-cloned from human glioblastoma cDNA using primers attB1-dyn 2532 (5′-AAA AAG CAG GCT TCA CCA TGG CAG CTG CTG CTG CC-3′, sense) and attB2-dyn 3837 (AGA AAG CTG GGT GTT AGG AGA TGA GGC GAC TGT G-3′, antisense). AttB1 and attB2 sites were extended in a second PCR and products were cloned via pDONR201 in p3xflg-CMV (Invitrogen, Carlsbad, CA, USA) to generate p3xflg-CMV-dyn 2532–3837. Plasmids were transfected into COS1 cells in 8-well glass slides (Lab-Tek Chamber Slide™ System, Nunc, Roskilde, Denmark). Forty-eight hours later, cells were fixed with ice-cold acetone and incubated with nanobody C-C7 for 1 hr, followed by sequential incubations with mouse anti-VSV and goat-anti-mouse FITC (Invitrogen, Carlsbad, CA, USA). After washing, slides were stained with rabbit-anti-dynactin-1 which was detected with donkey-anti-rabbit TRITC (Invitrogen, Carlsbad, CA, USA). Images were processed on a Leica Discovery Fluorescence Microscope and a Leica confocal microscope.
Immunoprecipitation studies
In another set of experiments, the p135 variant of dynactin [44] was obtained via RT-PCR from glioblastoma RNA using primers P135fw-ClaI (5′-CATCGATACCATGATGAGACAGGCACC-3′) and P135rev-NotI (5′ CAGCGGCCGCTTAGGAGATGA GGCGACTG-3′) and digested and cloned in ClaI-NotI digested pIRESneo (Clontech, Mountain View, CA). Suspension cultures of Chinese Hamster Ovary cells (CHO-s) in SFM4CHO medium with L-glutamin (ThermoFisher, Landsmeer, The Netherlands) were transfected with pIRES-Dynactin-p135 using the Amaxa Nucleofactor kit according to the manufacturer’s instructions (Lonza, Cologne, Germany). Cells were harvested 48 hours later and lysates prepared in lysis buffer [50 mM Tris-HCl, pH 7.4, 1% Triton-X100, 150 mM NaCl and protease inhibitors (CompleteTM protease inhibitor cocktail, Roche, Mannheim, Germany)] with untransfected CHO-s cells serving as control. Lysates were clarified at 13,000 g for 10 min at 4°C. Dynactin-p135 expression was verified on western blot using rabbit-anti-Dynactin (AbCam Cambridge, UK).
For immunoprecipitations, periplasmic extracts of His-tagged C-C7 or irrelevant control nanobody F8 were diluted five times in IPP500 (50 mM phosphate buffer pH 8.0, 500 mM NaCl) and incubated with Ni-NTA sepharose for 2 hours at 4°C. After washing three times with IPP500 and three times with IPP150 (50 mM phosphate buffer, pH 8.0, 150 mM NaCl) beads with immobilized C-C7 were incubated with cleared cell lysates for 2 hours at RT. Beads were washed two times with IPP150 incubated with 5 mM imidazole in IPP150 for 10 min at RT, boiled for 5 minutes in reducing sample buffer and loaded on a 7% SDS-PAGE gel for Western blot analysis with rabbit anti-dynactin and goat anti-rabbit IRDye800 (LI-COR, Lincoln, NE, USA) using the Odyssey infrared imaging system (LI-COR, Lincoln, NE, USA).
ACKNOWLEDGMENTS
The authors are grateful to Jillis Pol and Michiel Warle for providing us with surgically derived atherosclerotic carotid artery tissue, and Wouter de Munter and Peter van Lent for providing us with HoxB8 macrophages. We thank Bianca Lemmers-van de Weem and Maarten Brom for their excellent technical assistance with the animal work.
CONFLICTS OF INTEREST
None of the authors have conflicts of interest to disclose.
GRANT SUPPORT
This work was supported by grants from the Hersenstichting Nederland and a RadboudUMC grant.
REFERENCES
1. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995; 1:27–31.
2. Krishna Priya S, Nagare RP, Sneha VS, Sidhanth C, Bindhya S, Manasa P, Ganesan TS. Tumour angiogenesis - origin of blood vessels. Int J Cancer. 2016.
3. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004; 350:2335–2342.
4. Tirumani SH, Fairchild A, Krajewski KM, Nishino M, Howard SA, Baheti AD, Rosenthal MH, Jagannathan JP, Shinagare AB, Ramaiya NH. Anti-VEGF molecular targeted therapies in common solid malignancies: comprehensive update for radiologists. Radiographics. 2015; 35:455–474.
5. Kerbel RS. Tumor angiogenesis. N Engl J Med. 2008; 358:2039–2049.
6. Roodink I, Leenders WP. Targeted therapies of cancer: angiogenesis inhibition seems not enough. Cancer Lett. 2010; 299:1–10.
7. Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009; 15:232–239.
8. Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009; 15:220–231.
9. Kats-Ugurlu G, Oosterwijk E, Muselaers S, Oosterwijk-Wakka J, Hulsbergen-van de Kaa C, de Weijert M, van Krieken H, Desar I, van Herpen C, Maass C, de Waal R, Mulders P, Leenders W. Neoadjuvant sorafenib treatment of clear cell renal cell carcinoma and release of circulating tumor fragments. Neoplasia. 2014; 16:221–228.
10. Verhoeff JJ, van Tellingen O, Claes A, Stalpers LJ, van Linde ME, Richel DJ, Leenders WP, van Furth WR. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer. 2009; 9:444.
11. Jin Y, Kaluza D, Jakobsson L. VEGF, Notch and TGFbeta/BMPs in regulation of sprouting angiogenesis and vascular patterning. Biochem Soc Trans. 2014; 42:1576–1583.
12. Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009; 10:165–177.
13. Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003; 111:1287–1295.
14. Wesseling P, Ruiter DJ, Burger PC. Angiogenesis in brain tumors; pathobiological and clinical aspects. J Neurooncol. 1997; 32:253–265.
15. Claes A, Idema AJ, Wesseling P. Diffuse glioma growth: a guerilla war. Acta Neuropathol. 2007; 114:443–458.
16. Claes A, Wesseling P, Jeuken J, Maass C, Heerschap A, Leenders WP. Antiangiogenic compounds interfere with chemotherapy of brain tumors due to vessel normalization. Mol Cancer Ther. 2008; 7:71–78.
17. Norden AD, Drappatz J, Wen PY. Antiangiogenic therapy in malignant gliomas. Curr Opin Oncol. 2008; 20:652–661.
18. Navis AC, Bourgonje A, Wesseling P, Wright A, Hendriks W, Verrijp K, van der Laak JA, Heerschap A, Leenders WP. Effects of dual targeting of tumor cells and stroma in human glioblastoma xenografts with a tyrosine kinase inhibitor against c-MET and VEGFR2. PLoS One. 2013; 8:e58262.
19. Navis AC, Hamans BC, Claes A, Heerschap A, Jeuken JWM, Wesseling P, Leenders WPJ. Effects of targeting the VEGF and PDGF pathways in diffuse orthotopic glioma models. Journal of Pathology. 2011; 223:626–634.
20. Narayana A, Kelly P, Golfinos J, Parker E, Johnson G, Knopp E, Zagzag D, Fischer I, Raza S, Medabalmi P, Eagan P, Gruber ML. Antiangiogenic therapy using bevacizumab in recurrent high-grade glioma: impact on local control and patient survival. J Neurosurg. 2009; 110:173–180.
21. Verhoeff JJ, van Tellingen O, Claes A, Stalpers LJ, van Linde ME, Richel DJ, Leenders WP, van Furth WR. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer. 2009; 9:444.
22. Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014; 370:699–708.
23. Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014; 370:709–722.
24. Holig P, Bach M, Volkel T, Nahde T, Hoffmann S, Muller R, Kontermann RE. Novel RGD lipopeptides for the targeting of liposomes to integrin-expressing endothelial and melanoma cells. Protein Eng Des Sel. 2004; 17:433–441.
25. Tabatabai G, Tonn JC, Stupp R, Weller M. The role of integrins in glioma biology and anti-glioma therapies. Curr Pharm Des. 2011; 17:2402–2410.
26. Palumbo A, Hauler F, Dziunycz P, Schwager K, Soltermann A, Pretto F, Alonso C, Hofbauer GF, Boyle RW, Neri D. A chemically modified antibody mediates complete eradication of tumours by selective disruption of tumour blood vessels. Br J Cancer. 2011; 104:1106–1115.
27. Dienst A, Grunow A, Unruh M, Rabausch B, Nor JE, Fries JW, Gottstein C. Specific occlusion of murine and human tumor vasculature by VCAM-1-targeted recombinant fusion proteins. J Natl Cancer Inst. 2005; 97:733–747.
28. Roodink I, Raats J, van der Zwaag B, Verrijp K, Kusters B, van Bokhoven H, Linkels M, de Waal RM, Leenders WP. Plexin D1 expression is induced on tumor vasculature and tumor cells: a novel target for diagnosis and therapy? Cancer Res. 2005; 65:8317–8323.
29. Roodink I, Franssen M, Zuidscherwoude M, Verrijp K, van der Donk T, Raats J, Leenders WP. Isolation of targeting nanobodies against co-opted tumor vasculature. Lab Invest. 2009; 90:61–67.
30. Kolonin MG, Sun J, Do KA, Vidal CI, Ji Y, Baggerly KA, Pasqualini R, Arap W. Synchronous selection of homing peptides for multiple tissues by in vivo phage display. Faseb J. 2006; 20:979–981.
31. Rajotte D, Arap W, Hagedorn M, Koivunen E, Pasqualini R, Ruoslahti E. Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J Clin Invest. 1998; 102:430–437.
32. Valadon P, Garnett JD, Testa JE, Bauerle M, Oh P, Schnitzer JE. Screening phage display libraries for organ-specific vascular immunotargeting in vivo. Proc Natl Acad Sci USA. 2006; 103:407–412.
33. Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005; 23:1105–1116.
34. Rahbarizadeh F, Ahmadvand D, Sharifzadeh Z. Nanobody; an old concept and new vehicle for immunotargeting. Immunol Invest. 2011; 40:299–338.
35. Unciti-Broceta JD, Del Castillo T, Soriano M, Magez S, Garcia-Salcedo JA. Novel therapy based on camelid nanobodies. Ther Deliv. 2013; 4:1321–1336.
36. Huang L, Gainkam LO, Caveliers V, Vanhove C, Keyaerts M, De Baetselier P, Bossuyt A, Revets H, Lahoutte T. SPECT imaging with 99mTc-labeled EGFR-specific nanobody for in vivo monitoring of EGFR expression. Mol Imaging Biol. 2008; 10:167–175.
37. Cortez-Retamozo V, Backmann N, Senter PD, Wernery U, De Baetselier P, Muyldermans S, Revets H. Efficient cancer therapy with a nanobody-based conjugate. Cancer Res. 2004; 64:2853–2857.
38. van Driel PB, van der Vorst JR, Verbeek FP, Oliveira S, Snoeks TJ, Keereweer S, Chan B, Boonstra MC, Frangioni JV, van Bergen en Henegouwen PM, Vahrmeijer AL, Lowik CW. Intraoperative fluorescence delineation of head and neck cancer with a fluorescent anti-epidermal growth factor receptor nanobody. Int J Cancer. 2014; 134:2663–2673.
39. Claes A, Schuuring J, Boots-Sprenger S, Hendriks-Cornelissen S, Dekkers M, van der Kogel AJ, Leenders WP, Wesseling P, Jeuken JW. Phenotypic and genotypic characterization of orthotopic human glioma models and its relevance for the study of anti-glioma therapy. Brain Pathol. 2008; 18:423–433.
40. Navis AC, Hamans BC, Claes A, Heerschap A, Jeuken JW, Wesseling P, Leenders WP. Effects of targeting the VEGF and PDGF pathways in diffuse orthotopic glioma models. J Pathol. 2011; 223:626–634.
41. Kusters B, de Waal RM, Wesseling P, Verrijp K, Maass C, Heerschap A, Barentsz JO, Sweep F, Ruiter DJ, Leenders WP. Differential effects of vascular endothelial growth factor A isoforms in a mouse brain metastasis model of human melanoma. Cancer Res. 2003; 63:5408–5413.
42. Leenders W, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, Ruiter D, Ryan A, de Waal R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clinical Cancer Research. 2004; 10:6222–6230.
43. Roepman R, Schick D, Ferreira PA. Isolation of retinal proteins that interact with retinitis pigmentosa GTPase regulator by interaction trap screen in yeast. Methods Enzymol. 2000; 316:688–704.
44. Tokito MK, Howland DS, Lee VM, Holzbaur EL. Functionally distinct isoforms of dynactin are expressed in human neurons. Mol Biol Cell. 1996; 7:1167–1180.
45. Roodink I, van der Laak J, Kusters B, Wesseling P, Verrijp K, de Waal R, Leenders W. Development of the tumor vascular bed in response to hypoxia-induced VEGF-A differs from that in tumors with constitutive VEGF-A expression. Int J Cancer. 2006; 119:2054–2062.
46. Leenders W, Kusters B, De Waal R. Vessel co-option: How tumors obtain blood supply in the absence of sprouting angiogenesis. Endothelium. 2002; 9:83–87.
47. Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002; 61:215–225; discussion 226–219.
48. van Kempen LC, Leenders, W. P. Tumours can adapt to anti-angiogenic therapy depending on the stromal context: lessons from endothelial cell biology. Eur J Cell Biol. 2006; 85:61–68.
49. Leenders WP, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, Ruiter D, Ryan A, de Waal R. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res. 2004; 10:6222–6230.
50. Lv S, Ye M, Wang X, Li Z, Chen X, Dou X, Dai Y, Zeng F, Luo L, Wang C, Li K, Luo X, Yan J, et al. A recombined fusion protein SP5.2/tTF induce thrombosis in tumor blood vessel. Neoplasma. 2015; 62:531–540.
51. Hu P, Yan J, Sharifi J, Bai T, Khawli LA, Epstein AL. Comparison of three different targeted tissue factor fusion proteins for inducing tumor vessel thrombosis. Cancer Res. 2003; 63:5046–5053.
52. Hyvonen M, Enback J, Huhtala T, Lammi J, Sihto H, Weisell J, Joensuu H, Rosenthal-Aizman K, El-Andaloussi S, Langel U, Narvanen A, Bergers G, Laakkonen P. Novel target for peptide-based imaging and treatment of brain tumors. Mol Cancer Ther. 2014; 13:996–1007.
53. Schroer TA. Dynactin. Annu Rev Cell Dev Biol. 2004; 20:759–779.
54. Vaughan KT. Microtubule plus ends, motors, and traffic of Golgi membranes. Biochim Biophys Acta. 2005; 1744:316–324.
55. Li H, Liu XS, Yang X, Song B, Wang Y, Liu X. Polo-like kinase 1 phosphorylation of p150Glued facilitates nuclear envelope breakdown during prophase. Proc Natl Acad Sci USA. 2010; 107:14633–14638.
56. Dixit R, Levy JR, Tokito M, Ligon LA, Holzbaur EL. Regulation of dynactin through the differential expression of p150Glued isoforms. J Biol Chem. 2008; 283:33611–33619.
57. Butler GS, Overall CM. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat Rev Drug Discov. 2009; 8:935–948.
58. Roesli C, Borgia B, Schliemann C, Gunthert M, Wunderli-Allenspach H, Giavazzi R, Neri D. Comparative analysis of the membrane proteome of closely related metastatic and nonmetastatic tumor cells. Cancer Res. 2009; 69:5406–5414.
59. Zemskov EA, Janiak A, Hang J, Waghray A, Belkin AM. The role of tissue transglutaminase in cell-matrix interactions. Front Biosci. 2006; 11:1057–1076.
60. Jakobsen CG, Rasmussen N, Laenkholm AV, Ditzel HJ. Phage display derived human monoclonal antibodies isolated by binding to the surface of live primary breast cancer cells recognize GRP78. Cancer Res. 2007; 67:9507–9517.
61. Gonzalez-Gronow M, Selim MA, Papalas J, Pizzo SV. GRP78: a multifunctional receptor on the cell surface. Antioxid Redox Signal. 2009; 11:2299–2306.
62. Lo Cicero A, Stahl PD, Raposo G. Extracellular vesicles shuffling intercellular messages: for good or for bad. Curr Opin Cell Biol. 2015; 35:69–77.
63. Rak J, Guha A. Extracellular vesicles--vehicles that spread cancer genes. Bioessays. 2012; 34:489–497.
64. Deregibus MC, Cantaluppi V, Calogero R, Lo Iacono M, Tetta C, Biancone L, Bruno S, Bussolati B, Camussi G. Endothelial progenitor cell derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood. 2007; 110:2440–2448.
65. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT, Jr., Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008; 10:1470–1476.
66. D’Asti E, Garnier D, Lee TH, Montermini L, Meehan B, Rak J. Oncogenic extracellular vesicles in brain tumor progression. Frontiers in physiology. 2012; 3:294.
67. van Laarhoven HW, Bussink J, Lok J, Punt CJ, Heerschap A, van Der Kogel AJ. Effects of nicotinamide and carbogen in different murine colon carcinomas: immunohistochemical analysis of vascular architecture and microenvironmental parameters. Int J Radiat Oncol Biol Phys. 2004; 60:310–321.
68. Rosas M, Osorio F, Robinson MJ, Davies LC, Dierkes N, Jones SA, Reis e Sousa C, Taylor PR. Hoxb8 conditionally immortalised macrophage lines model inflammatory monocytic cells with important similarity to dendritic cells. Eur J Immunol. 2011; 41:356–365.
69. Theelen TL, Lappalainen JP, Sluimer JC, Gurzeler E, Cleutjens JP, Gijbels MJ, Biessen EA, Daemen MJ, Alitalo K, Yla-Herttuala S. Angiopoietin-2 blocking antibodies reduce early atherosclerotic plaque development in mice. Atherosclerosis. 2015; 241:297–304.