
INTRODUCTION
Breast cancer is the most common cancer in women, affecting 12% of females worldwide [1], and is the leading cause of cancer related deaths in women [2]. Breast cancer most frequently originates from the lobe or the milk duct, both of which highly express estrogen receptor alpha (ERα). Thus, the majority of breast cancers are ERα-positive, which makes them suitable for selective ERα modulators, such as tamoxifen. Importantly, in 30% to 40% of patients receiving adjuvant tamoxifen therapy the tumor eventually relapses, and this is a significant clinical problem [3]. Clearly, additional and/or complementary approaches are necessary to more accurately define patients who will benefit from the above therapy and to design novel treatment strategies. Interestingly, recent work implicates that activation of Hedgehog (HH) signaling may have a role in the development of tamoxifen resistance in breast cancer [4].
The HH signaling pathway has critical roles in embryonic development and tumorigenesis [5–7]. Aberrant activation of HH signaling is involved in several types of malignant tumors [8], including basal cell carcinoma, medulloblastoma, rhabdomyosarcoma and cancers of the pancreas, colon, stomach, lung and prostate. The pathway is initiated by HH ligand [Sonic HH (SHH), Indian HH (IHH), Desert HH (DHH)] [7, 9–11] binding to Patched (PTCH1, PTCH2), a twelve trans-membrane domain protein. In the absence of HH ligands, PTCH inhibits the signaling of the seven trans-membrane domain protein and proto-oncogene Smoothened (SMO). Upon HH ligand binding, the PTCH inhibition of SMO is released and the signal is transduced to the terminal effectors, the GLI, Glioma associated oncogene, proteins (GLI1, GLI2, GLI3) [11]. GLI1 is a transcription factor that acts not only as a signaling effector but also represents a pathway target gene [12], amplifying the HH signal. Its expression levels thus correlate directly with pathway activity [13]. GLI1 is known to function as an oncogene [14], promoting cell proliferation and angiogenesis [15].
Possible links between HH signaling activation and the development of breast cancer have been widely studied. An elevated expression of HH ligands was associated with a basal-like breast cancer phenotype and a poor prognosis [16]. This may be the result of hypomethylation of the SHH promoter [17]. Genetic alternations in components of the HH pathway, including loss of PTCH1 or GLI1 amplification, were suggested to result in breast cancer [18, 19]. Consistently, transgenic mice that conditionally expressed GLI1 in the mammary epithelium developed mammary tumors [20]. Constitutive activation of HH signaling in MMTV-SmoM2 transgenic mice caused alterations in mammary gland morphology, increased proliferation, and changed stem/progenitor cell numbers [21]. In ERα-positive breast cancer cells, estrogen was found to act via GLI1, promoting the development of cancer stem cells and epithelial to mesenchymal transition [22]. Interestingly, neuropilin 2 (NRP2) signaling increased GLI1 expression in breast tumor initiating cells, with GLI1 also inducing BMI-1, a key stem cell factor, and NRP2 expression, establishing an autocrine loop [23]. These studies provide insights into the mechanisms of HH signaling activation in the mammary gland and its possible role in breast tumorigenesis.
An additional connection between HH signaling and ERα-positive breast cancer was suggested in 2012, when Ramaswamy et al reported that the HH pathway can mediate tamoxifen resistance in breast cancer cells. In this work evidence was provided that the PI3K/Akt pathway activates HH signaling, bypassing the blockade of ERα signaling that was elicited by tamoxifen treatment [4].
Our current results indicate that GLI1 is expressed to a higher extent in tamoxifen resistant compared to sensitive breast cancer cells. Interestingly, we also find that depletion of GLI1 decreases ERα protein levels, with concomitant reduction of ERα signaling activity in both tamoxifen resistant and sensitive cells. Furthermore, GLI1 depletion enhances tamoxifen cytotoxicity in both resistant and sensitive cells. These observations indicate that GLI1 may have a role not only for tamoxifen resistance but can also modulate ERα signaling in both sensitive and resistant cells.
RESULTS
Hedgehog signaling activity in the tamoxifen resistant LCC2 and their parental, tamoxifen sensitive MCF7 cells
Expression analysis of key markers of the activity of the HH signaling pathway i.e. GLI1 and PTCH1, revealed higher expression in the tamoxifen resistant LCC2 breast cancer cells compared to the parental, tamoxifen sensitive MCF7 cells (Figure 1A and 1B). Notably, MCF7 and LCC2 cells showed similar expression of the ERα mRNA and protein [24], however the ERα target genes ADORA1 and pS2 were upregulated in the resistant cells (Figure 1A and 1B). Cell viability assays indicated that LCC2 but not MCF7 cells are resistant to 10 μM tamoxifen, however 20 μM tamoxifen kills both cell types (Figure 1C).
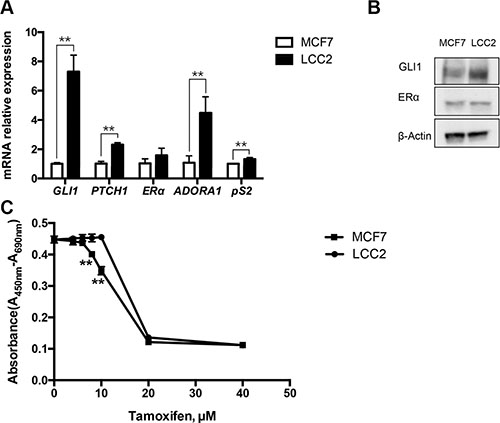
Figure 1: Characterization of tamoxifen sensitive MCF7 and tamoxifen resistant LCC2 breast cancer cells. (A) Endogenous expression of GLI1, PTCH1, ERα, ADORA1 and pS2 in MCF7 and LCC2 cells was determined by real-time PCR. Data are represented as relative expression (2−ΔΔCt values), calculated by subtracting the Ct value of the housekeeping gene TBP from the Ct value of the interrogated transcripts (ΔCt), and normalized to the ΔCt values obtained with MCF7. Representative data from one of three independent experiments are shown. Error bars indicate the standard deviation. **, Statistical significant, P < 0.01, compared to control, calculated by the Student’s t-test. (B) Protein levels of GLI1, ERα and β-Actin in MCF7 and LCC2 cells were analyzed by Western blot. β-Actin was used as the endogenous protein control. (C) The effects of tamoxifen on cell viability. MCF7 and LCC2 cells were treated with 0, 4, 6, 8, 10, 20 or 40 μM tamoxifen and after 48 hours cell viability was determined with the WST-1 assay. The absorbance at 450 nm was measured with the reference wavelength set at 690 nm. Shown are data from triplicate measurements. Representative data from one of three independent experiments are shown. Error bars indicate the standard deviation. The two-way ANOVA analysis using Sidak’s multiple comparisons was employed to calculate statistical significance (**P < 0.01).
This analysis demonstrates the higher HH signaling activity in the resistant cells and suggests that ERα activity may also be higher, despite the comparable ERα expression.
Depletion of ERα or GLI1 reduces cell proliferation
To investigate the role of ERα and GLI1 in breast cancer cell proliferation, we transfected MCF7 and LCC2 cells with siRNAs targeting ERα or GLI1. RNA expression analysis showed that the ERα and GLI1 siRNAs successfully knocked down the respective genes in both cell lines (Figure 2B and 2C). Western blot analysis also showed ERα to be dramatically decreased by ERα siRNA treatment, and GLI1 to be downregulated by GLI1 siRNA treatment (Figure 2D, Supplementary Figure S1). Depletion of ERα resulted in a major reduction of the cell proliferation in both cell lines (Figure 2A), highlighting their dependence on ERα. Depletion of GLI1 also reduced the cell proliferation of the two cell lines, but to a lesser extent (Figure 2A).
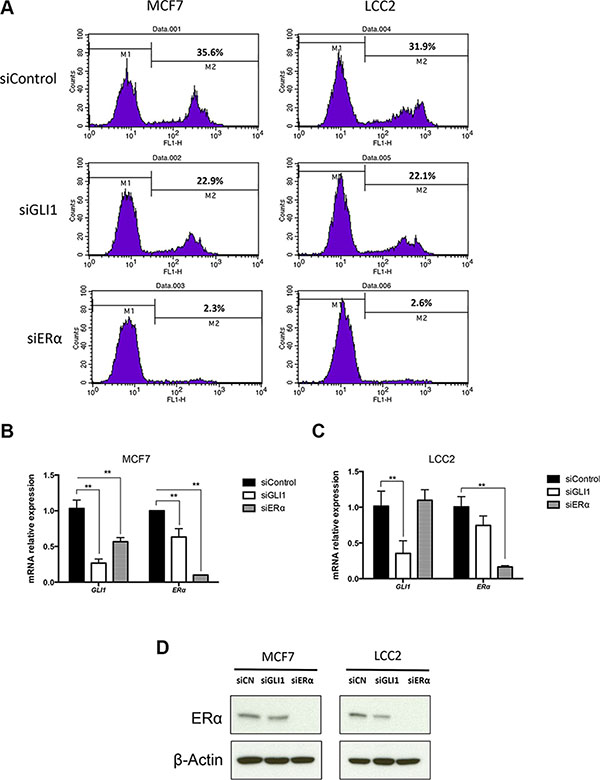
Figure 2: Depletion of GLI1 or ERα reduces the proliferation of MCF7 and LCC2 cells. (A) MCF7 and LCC2 cells, cultured for 48 hours following transfection with control siRNA (siControl), GLI1 siRNA (siGLI1) or ERα siRNA (siERα), were subjected to the EdU incorporation assay for 1 hour. The percentage of cells labeled with Alexa Fluor 488 azide was detected by flow cytometry. The expression of GLI1 and ERα in MCF7 (B) and LCC2 (C) cells, following siRNA knockdown of GLI1 or ERα, was determined by real-time PCR. Data are represented as relative expression (2-ΔΔCt values), calculated by subtracting the Ct value of the housekeeping gene TBP from the Ct value of the interrogated transcripts (ΔCt), and normalized to the ΔCt value obtained with control siRNA. Representative data from one of three independent experiments are shown. Error bars indicate the standard deviation. **, Statistical significant, P < 0.01, compared to control, calculated by the Student’s t-test. (D) Protein levels of ERα in MCF7 and LCC2 cells, transfected with control siRNA (siCN), GLI1 siRNA (siGLI1) or ERα siRNA (siERα) for 48 hours, was determined by Western blot. β-Actin was used as the endogenous protein control.
These observations are in-line with the significance of ERα in breast cancer cells [3, 25, 26]. Moreover, they indicate that GLI1 can modulate proliferation not only in tamoxifen resistant but also in tamoxifen sensitive cells.
GLI1 depletion reduces ERα activity assayed through an Estrogen Response Element (ERE) reporter
To determine whether endogenous GLI1 expression may have an impact on ERα transcriptional activity, we used an Estrogen Response Element (ERE) luciferase reporter. GLI1 depletion reduced ERα activity both in MCF7 and LCC2 cells, irrespective of the presence or absence of estrogen (Figure 3, Supplementary Figure S2). Importantly, the basal level of the ERα transcriptional activity was higher in LCC2 compared to MCF7 cells, an observation in-line with the expression pattern of the ERα target genes ADORA1 and pS2 (Figure 1A).
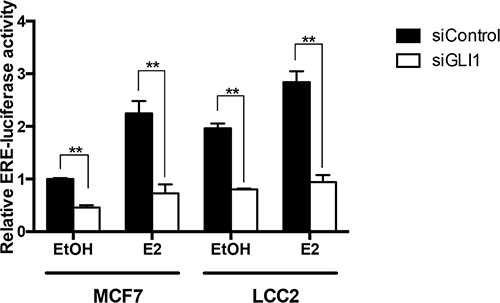
Figure 3: GLI1 depletion reduces the activity of an ERα reporter. MCF7 and LCC2 cells were transfected with control siRNA (siControl) or GLI1 siRNA (siGLI1) and after 24 hours were co-transfected with the reporter plasmid ERE-TK-Luc and the pRL-TK control plasmid. Subsequently, both cell lines were treated with 10 nM E2 or ethanol (EtOH) for 24 hours in serum-deprived medium before harvesting. Luciferase expression was measured 48 hours after plasmid transfection. Shown are data from two independent experiments. Error bars indicate the standard error of the mean (SEM). **, Statistical significant, P < 0.01, compared to control, calculated by the Student’s t-test.
These findings suggest an interplay of GLI1 with ERα signaling in both tamoxifen resistant and sensitive cells.
GLI1 depletion decreases the expression of ERα and its target genes
To address the functional consequences of the suggested GLI1 and ERα interplay, RNA expression analysis was used following GLI1 knockdown. GLI1 depletion was first confirmed and also shown to decrease the expression of the GLI1 target gene PTCH1. Moreover, the expression of ERα and its target genes IL20, ADORA1 and pS2 were also reduced in the context of estrogen treatment, while limited effects were observed without addition of estrogen (Figure 4A). The same assay was also performed using two additional ERα-positive breast cancer cell lines, ZR751 and T47D, resulting in a similar downregulation of ERα, IL20 and pS2 by GLI1 knockdown (Supplementary Figure S3A). Western blot analysis demonstrated that GLI1 depletion downregulated ERα in both MCF7 and LCC2 cells, irrespective of the absence or presence of estrogen for 6 or 12 hours (Figure 4B, Supplementary Figure S1). As noted before, estrogen treatment reduced ERα protein expression [24]. Consistently, ChIP analysis revealed decreased ERα binding at the promoter region of its target gene pS2 [27–29] following GLI1 depletion in the presence of estrogen (Figure 4C).
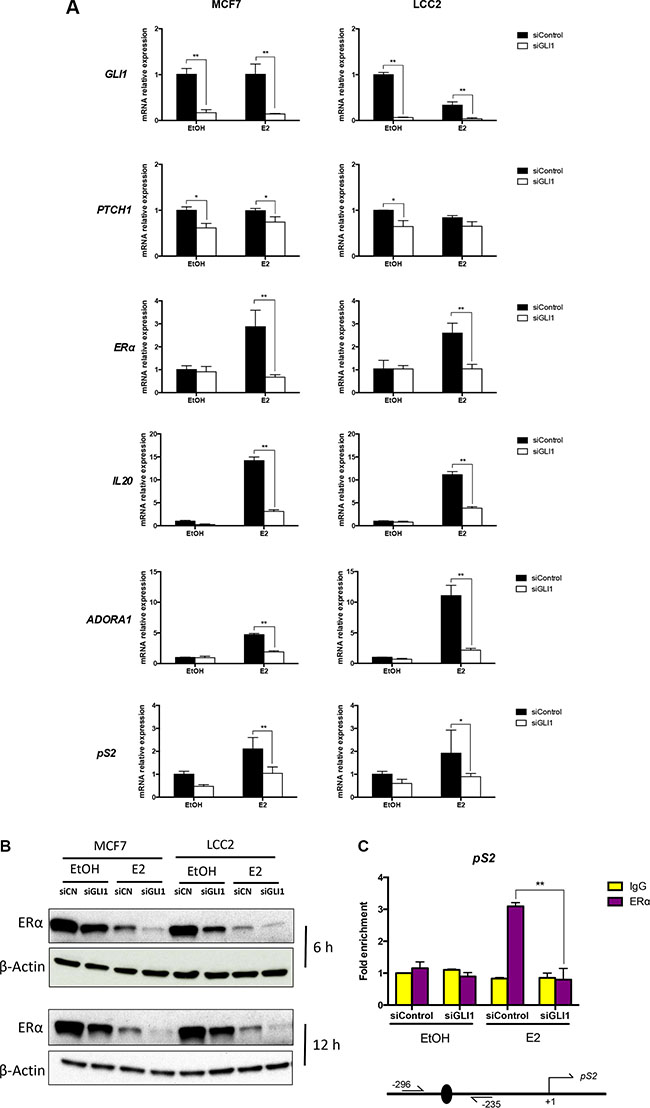
Figure 4: GLI1 depletion reduces the expression of ERα, its target genes and the binding to its targets. (A) The expression of GLI1, PTCH1, ERα and its target genes, IL20, ADORA1 and pS2 in MCF7 and LCC2 cells treated with 10 nM E2 or ethanol (EtOH) for 3 hours in serum-deprived medium, following siRNA knockdown of GLI1, was determined by real-time PCR. Data are represented as relative expression (2−ΔΔCt values), calculated by subtracting the Ct value of the housekeeping gene TBP from the Ct value of the interrogated transcripts (ΔCt), and normalized to the ΔCt value obtained with control siRNA in MCF7/LCC2 cells. Representative data from one of three independent experiments are shown. Error bars indicate the standard deviation. * or **, Statistical significant, P < 0.05 or P < 0.01 respectively, compared to control siRNA, calculated by the Student’s t-test. (B) Protein levels of ERα and β-Actin in MCF7 and LCC2 cells treated with 10 nM E2 or ethanol (EtOH) for 6 and 12 hours in serum-deprived medium, 24 hours after transfection with control siRNA (siCN) or GLI1 siRNA (siGLI1), was determined by Western blot. β-Actin was used as the endogenous protein control. (C) Recruitment of ERα to the promoter of the pS2 gene is diminished following GLI1 depletion. MCF7 cells were transfected with control siRNA or GLI1 siRNA and after 48 hours treated with 10 nM E2 or ethanol (EtOH) for 30 min in serum-deprived medium before harvesting and subjected to ChIP-qPCR analysis with an ERα antibody and PCR primers spanning the ERα binding site at the pS2 gene promoter. The data presented are normalized to input DNA and expressed as fold enrichment over IgG. Shown are data from two independent experiments. Error bars indicate the standard error of the mean (SEM). **, Statistical significant, P < 0.01, compared to IgG control, calculated by the Student’s t-test. A schematic diagram of the promoter region of the pS2 gene, with the transcriptional start sites (+1) indicated by an arrow, the ERα binding site by a black oval and the position of the primers (−296, −235) is also shown. Note the increased ERα binding at the promoter following E2 treatment, which is eliminated by GLI1 depletion.
The GLI inhibitor GANT61 increases the cytotoxicity of tamoxifen on MCF7 and LCC2 cells, with or without addition of estrogen
To examine possible therapeutic applications of the HH signaling interplay with ERα, we investigated whether treatment of MCF7 and LCC2 cells with the GLI inhibitor GANT61 [30] may enhance tamoxifen cytotoxicity. First, we tested the effects of only GANT61 administration on cell viability and cell proliferation. As expected, GANT61 treatment resulted in a dose-dependent reduction of the viability of MCF7 and LCC2 cells (Figure 5A and 5B). Additionally, the proliferation of both cell lines was inhibited (Figure 5C) and the mRNA expression of ERα and its corresponding target genes were downregulated by 48-hour GANT61 treatment (Figure 5D and 5E). Interestingly, a 24-hour GANT61 treatment also had an obvious impact on cell proliferation (Supplementary Figure S4A) and mRNA expression (Supplementary Figure S4B).
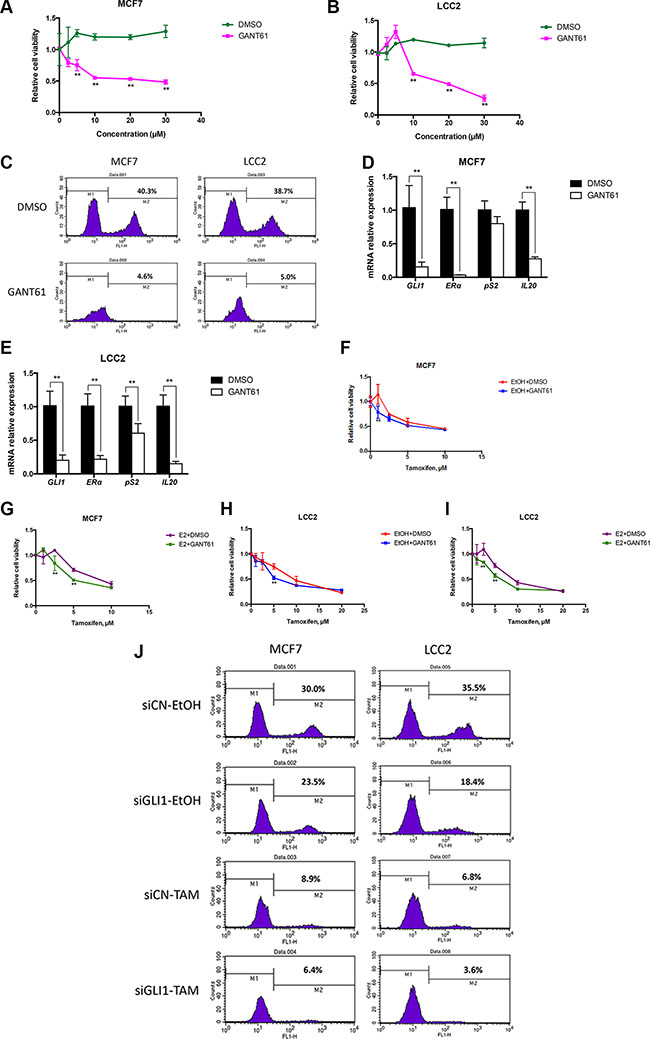
Figure 5: GANT61 increases tamoxifen cytotoxicity, irrespective of the presence or absence of estrogen. (A), (B) GANT61 suppresses the cell viability of MCF7 and LCC2 cells. Both cell lines were treated with 0, 2.5, 5, 10, 20 and 30 μM GANT61 or DMSO as a control. After 48 hours cell viability was determined with the WST-1 assay. Error bars indicate the standard deviation. The two-way ANOVA analysis using Sidak’s multiple comparisons was employed to calculate statistical significance (**P < 0.01). (C) GANT61 treatment reduces the cell proliferation of MCF7 and LCC2 cells. Both cell lines were treated with 10 μM GANT61 or DMSO as a control. After 48 hours cell proliferation was determined by the EdU incorporation assay using flow cytometry. (D), (E) The expression of GLI1, ERα, pS2 and IL20 in MCF7 and LCC2 cells treated with GANT61 or DMSO for 48 hours were measured by real-time PCR. Error bars indicate the standard deviation. **, Statistical significant, P < 0.01 respectively, compared to the DMSO control, calculated by the Student’s t-test. (F–I) MCF7 and LCC2 cells were treated with 10 nM E2 or EtOH and 10 μM GANT61 or DMSO in the presence of different concentrations (0, 1, 2.5, 5, 10 or 20 μM) of tamoxifen, for 48 hours and the number of viable cells was measured with the WST-1 assay using a TECAN plate spectrophotometer. Shown are data from triplicate measurements expressed as percentage of control. Representative data from one of three independent experiments are shown. Error bars indicate the standard deviation. The two-way ANOVA analysis using Sidak’s multiple comparisons was employed to calculate statistical significance (**P < 0.01). (J) MCF7 and LCC2 cells, cultured for 24 hours following transfection with control siRNA (siCN) or GLI1 siRNA (siGLI1) and treated with 10 μM tamoxifen (TAM) or ethanol (EtOH) for 48 hours, were subjected to the EdU incorporation assay for 1 hour. The percentage of cells labeled with Alexa Fluor 488 azide was detected by flow cytometry.
Moreover, GANT61 co-administration with tamoxifen further decreased the cell growth of MCF7 and LCC2 cells, and this was irrespective of the presence or absence of estrogen (Figure 5F–5I). SiRNA depletion of GLI1 also enhanced the impact of tamoxifen in reducing the proliferation of the two cell lines (Figure 5J). Similar enhancement of the tamoxifen impact by GLI1 depletion was also observed in ZR751 and T47D cells (Supplementary Figure S3B). However, in ZR751 cells GLI1 depletion reduced cell proliferation to a comparable extent as tamoxifen treatment, suggesting an increased significance of GLI1 in this cellular context.
Thus, the role of GLI1 for the proliferation of ERα-positive breast cancer cells may be exploited for therapeutic purposes, and drug targeting of GLI1 could enhance the tamoxifen efficacy in the treatment of breast cancer.
Correlation between GLI1 and ERα/ERα target gene expression in breast cancer - Impact of GLI1 expression in distant metastasis-free survival
To explore the clinical relevance of the effect of GLI1 on ERα signaling and breast cancer, we examined the expression of GLI1, ESR1 (the gene encoding ERα) and known ERα target genes in a dataset of breast cancer samples from 286 individuals [31]. Our analysis revealed that the expression of GLI1 positively correlates with ESR1 (Figure 6A) and the ERα targets genes pS2 (Figure 6B) and GREB1 (Figure 6C). Then we examined the prognostic role of GLI1 expression for breast cancer patients using the Kaplan-Meier Plotter dataset [32]. We observed that high GLI1 expression is associated with poor distant metastasis-free survival (DMFS) in 126 patients with Grade 1, ERα-positive breast cancer (Figure 6D).
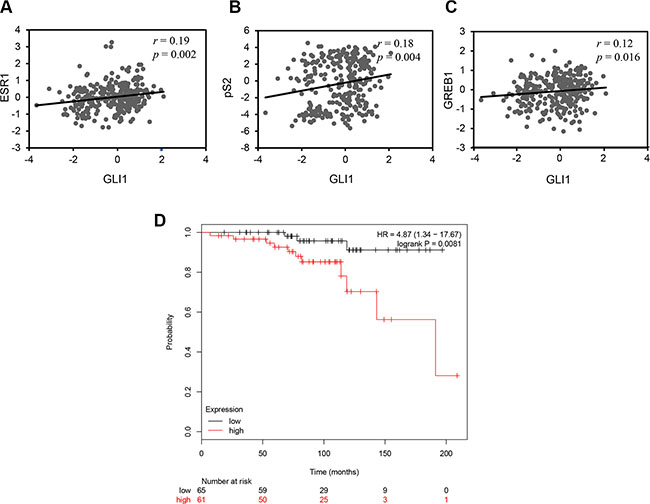
Figure 6: GLI1 expression positively correlates with the expression of ESR1 and its target genes and is a negative prognostic marker in breast cancer. (A–C) Scatter plots showing significant correlation between GLI1 and ESR1 (A), GLI1 and pS2 (B), GLI1 and GREB1 (C) expression in a publically available breast cancer dataset, test statistics were from Pearson product-moment correlation. (D) Kaplan-Meier plot showing that high GLI1 expression correlates with worse distant metastasis-free survival (DMFS) in patients with ERα-positive, Grade 1 breast cancer. The Kaplan-Meier plot is stratified for high (red) and low (black) GLI1 expression (n = 126; P = 0.0081).
These findings suggest that GLI1 may represent not only a therapeutic target but could also be a valuable prognostic marker for breast cancer patients.
DISCUSSION
Our data indicate that GLI1 depletion reduces the proliferation of both tamoxifen resistant and sensitive breast cancer cells (Figure 2). Moreover, the GLI inhibitor GANT61 increases the cytotoxicity of tamoxifen on both resistant and sensitive cells and this is irrespective of the activation of ERα signaling by estrogen (Figure 5F–5I). Additionally, GLI1 knockdown enhanced the effect of tamoxifen in reducing the proliferation of four breast cancer cell lines (Figure 5I, Supplementary Figure S3B). These data contrast earlier observations on tamoxifen and cyclopamine, a HH signaling inhibitor acting upstream of the GLI factors, co-treatments, which indicated that the two drugs counter each other’s effect [33]. Interestingly, the GLI1 depletion confers a reduction of ERα transcriptional activity in the LCC2 and MCF7 cells, with or without estrogen treatment (Figure 3). Note that LCC2 cells are less sensitive than MCF7 to estrogen-mediated increase in proliferation [24]. Further analysis demonstrated that GLI1 depletion reduces ERα protein expression (Figure 4B). Additionally, in the context of ERα activation, GLI1 depletion elicited a consistent reduction in the mRNA expression of ERα and the ERα target genes analyzed (Figure 4A). These findings may suggest that GLI1, acting as transcription factor, regulates ERα expression when ERα signaling is on. However, exogenous expression of GLI1 did not increase the mRNA and protein levels of ERα (Supplementary Figure S5A and S5B), implying that the impact of GLI1 on ERα is more complicated than a typical direct GLI1 target, e.g. PTCH1 (Supplementary Figure S5A). To further test the hypothesis of transcriptional regulation of ERα by GLI1 we applied ConSite [34], a web-based tool for binding sites prediction, and took advantage of the Position Specific Frequency Matrices (PSFM) of the GLI1 binding sites [35]. Combined with the predicted GLI1 binding consensus sequence [36] and following BLAST analysis with the estrogen receptor 1 gene (ERS1), three common hits were identified. However, we were unable to detect convincing GLI1 binding on the ERS1 gene promoter, possibly reflecting limitations of ChIP validated GLI1 antibodies. Unsuccessful were also our efforts to detect possible GLI1/ERα protein interactions using immunoprecipitations assays.
The mechanisms of tamoxifen resistance are not entirely clear, although this phenomenon is commonly observed. Tamoxifen resistance may be mediated by altered ERα function/ERα signaling. Loss of ERα function [37–39] and ERα mutations [40–42] are well-studied mechanisms that induce resistance to tamoxifen. Moreover, the complexity of ERα signaling is indicative of additional pathways that may be involved in tamoxifen resistance [43]. These include PI3K (phosphoinositide 3-kinase) [4], HER2 (human epidermal growth factor receptor 2) [44], MAPK (mitogen-activated protein kinase) [45], and NOTCH [5, 46]. We specifically examined the activity of PI3K/Akt signaling in MCF7 and LCC2 cells. The basal level of phosphorylated Akt (p-Akt) was found to be comparable. Moreover, PI3K/Akt signaling in both cell lines could be activated by insulin treatment (Supplementary Figure S6.) Although Ramaswamy et al [4] showed that treatment with the PI3K inhibitor LY294002 elicited a 50% decrease in GLI-dependent luciferase activity in tamoxifen resistant but not in tamoxifen sensitive MCF7 cells, they did not directly demonstate that the PI3K/Akt signaling pathway is more active in the tamoxifen resistant cells.
There is strong evidence that the ability of tamoxifen to function as an ERα agonist or antagonist is dependent on whether it recruits co-activators or co-repressors to the ERα transcription complex [47–50]. This may partly explain the observation of a tamoxifen-induced increase in the proliferation of the ERα-positive ZR751 and BT474 breast cancer cells, which moreover, was accompanied by a sustained upregulation of GLI1 expression [51].
Resistance to tamoxifen is a major therapeutic concern for the treatment of breast cancer. The clinical and experimental evidence on both intrinsic and acquired resistance are well documented in many reviews [52–54]. Changes in the expression of ERα, ERα pathway components or in signaling cascades interacting with ERα are observed in experimental models of tamoxifen resistance. Some findings are consistent with the clinical data, while others are not. To create effective therapeutic approaches, a further focus on the tumor itself and the detailed classification of breast tumor subtypes, with personalized treatment options should be considered. Due to the complexity of tumor heterogeneity and tumor environment, emerging high throughput technologies will be indispensable to study and potentially overcome tamoxifen resistance.
Our data highlighting that the expression of GLI1 correlates with ESR1, pS2 and GREB1 using a publicly available dataset (Figure 6A, 6B and 6C), suggest that GLI1 may represent a gene with implications in breast cancer. Indeed, we observe that high GLI1 expression predicts worse DMFS in Grade 1, ERα-positive breast cancer patients (Figure 6D). It would be interesting but also challenging to verify GLI1 as prognostic marker in a larger number of patients and/or other subtypes of breast cancer.
Taken together, in this work we have demonstrated that tamoxifen cytotoxicity can be enhanced by blockade of the HH pathway, reflecting a cross-talk between ERα and GLI1 signaling, both in tamoxifen resistant and sensitive breast cancer cells. Therefore GLI1 may be a potential therapeutic target and moreover, could also act as a prognostic marker in breast cancer.
MATERIALS AND METHODS
Cell culture
Tamoxifen resistant cell line LCC2 and its parental, tamoxifen sensitive cell line MCF7 were kind gifts of Staffan Strömblad (Karolinska Institutet, Stockholm, Sweden). Both cell lines were cultured in DMEM high glucose medium with 10% fetal calf serum (FCS) supplemented with 100 IU/ml penicillin/streptomycin and maintained in a 5% CO2 humidified incubator. DMEM, penicillin/streptomycin, and trypsin were purchased from Sigma-Aldrich. 4-Hydroxytamoxifen (T176-10MG) and 17β-Estradiol (E2, E2758-250MG) were obtained from Sigma-Aldrich. For the experiments evaluating E2 or tamoxifen treatment, FCS with dextran-coated charcoal and DMEM without phenol red were used. Ethanol was the vehicle control.
SiRNA transfection
Cells were transfected with 50 nM siRNA. GLI1 siRNAs and control siRNAs were purchased from Sigma-Aldrich, ERα siRNA (sc-44204) was purchased from Santa Cruz Biotechnology. Lipofectamine® RNAiMAX Transfection Reagent (Invitrogen) was used according to the manufacturer’s protocol.
Cell proliferation
Cell proliferation assays were performed essentially as previously described [55]. Briefly, 5 × 105 cells per well were seeded in 6-well plates, treated with siRNAs for 48 hours, followed by an 1 hour 10 μM EdU (5-ethynyl- 2′-deoxyuridine) incubation. EdU was detected by a fluorescent-azide coupling reaction (Click-iT, Invitrogen). For each treatment, 10 000 cells were analyzed on a FACS calibur machine (BD Biosciences, Stockholm, Sweden). Cell cycle distribution was calculated using the CellQuest software (BD Bioscience).
Cell viability
Cells were seeded into 96-well plates 24 hours before starvation. GANT61 was dissolved in DMSO (dimethyl sulfoxide), whereas E2 in ethanol. Cells were treated with 10 nM E2 or EtOH and 10nM GANT61 or DMSO in the presence of different concentrations of tamoxifen, and then incubated with the indicated combination of drugs for 48 hours. Metabolic activity was measured with the WST-1 (Water Soluble Tetrazolium salt 1) cell proliferation reagent (Roche), and the number of viable cells was quantified at 450 nm using a TECAN plate spectrophotometer, with the reference wavelength set at 690 nm. Each measurement represents the mean of triplicates. The ordinary two-way ANOVA test was performed using the GraphPad Prism version 6.0d.
RNA preparation, cDNA synthesis and real-time PCR
Total RNA was isolated with the RNeasy mini kit (Qiagen, Hamburg, Germany) according to the manufacturer’s protocol. cDNA synthesis was performed with random N6 primers (New England Biolabs) and Superscript III (Invitrogen). Real-time PCR was carried out with the FastStart Universal SYBR Green Master (Rox) (Roche) on a 7500 fast real-time PCR system (Applied Biosystems), with primers designed to detect GLI1, PTCH1, ERα, IL20, ADORA1, pS2 and TBP (Table 1). All amplifications were run at least in triplicate and the fold change was normalized to the expression of TBP. The relative expression was determined by the ΔCt method. All RNA expression experiments were done at least in triplicate and representative experiments are shown.
Table 1: Primer sequences for qRT-PCR analysis
TBP-E3 forward |
5′ GCCAGCTTCGGAGAGTTCTGGGATT |
TBP-E4 reverse |
5′ CGGGCACGAAGTGCAATGGTCTTTA |
GLI1-E11 forward |
5′ CAGCTACATCAACTCCGGCCAATAGGG |
GLI1-E12 reverse |
5′ TGCTGCGGCGTTCAAGAGAGACTG |
PTCH1-E17 forward |
5′ AATGGGTCCACGACAAAGCCGACTA |
PTCH1-E18 reverse |
5′ TCCCGCAAGCCGTTGAGGTAGAAAG |
ERα forward |
5′ GCTACGAAGTGGGAATGATGAAAG |
ERα reverse |
5′ TCTGGCGCTTGTGTTTCAAC |
IL20 forward |
5′ CTGCCTCCTGCGCCATTTGC |
IL20 reverse |
5′ TCATGTGGGCATGACAGAGC |
ADORA1 forward |
5′ TTCCACACCTGCCTCATGGT |
ADORA1 reverse |
5′ GCGGTCCACAGCAATTGC |
pS2 forward |
5′ CATCGACGTCCCTCCAGAAGAG |
pS2 reverse |
5′ CTCTGGGACTAATCACCGTGCTG |
Western blot
For Western blot analysis, cells were lysed with RIPA buffer (150 mM NaCl, 50 mM Tris base pH 8.0, 1 mM EDTA, 0.5% sodium deoxycholate, 1% NP-40, 0.1% sodium dodecyl sulfate, 1 mM DTT, 1 mM PMSF, and 1 mM Na3VO4) supplemented with Complete Protease Inhibitor Tablets (Roche) and Phosphatase Inhibitor Cocktail 3 (Sigma). Proteins were separated on a 7.5% sodium dodecyl sulfate polyacrylamide gel electrophoresis (PAGE) followed by transfer (220 mA for 1 hour) to an Immobilon-P membrane (Millipore). The membrane was incubated at 4°C overnight in StartingBlock™ T20 (TBS) Blocking Buffer (#37543, Thermo Scientific) with monoclonal anti-mouse β-Actin antibody (A5441, Sigma-Aldrich), anti-rabbit GLI1 Ab (#2553, Cell Signaling Technology) or ERα antibody (sc-543, Santa Cruz Biotechnology), followed by incubation with goat anti-rabbit or anti-mouse secondary antibodies for 1 hour in StartingBlock™ T20 (TBS) Blocking Buffer and visualized using Pierce ECL chemiluminescent substrate (Thermo Scientific).
Luciferase reporter assay
Cells were transfected with 50 nM GLI1 siRNAs or control siRNA. After 24 hours cells were co-transfected with the reporter plasmid ERE-TK-Luc and the pRL-TK control plasmid, which contains the Renilla luciferase gene, for normalizing transfection efficiency. Plasmid transfection was done using Lipofectamine 3000 (Invitrogen). After 24-hour plasmid transfection, the cells were changed to serum-deprived medium, incubated overnight, and then treated with 10 nM E2 or vehicle for 24 hours prior to harvesting. Luciferase activity was measured using the Dual-Luciferase Reporter Assay (Promega). The reporter plasmid ERE-TK-Luc has been described previously [56]. Multiple t-test analysis (corrected for multiple comparisons using the Holm-Sidak method) was performed with GraphPad Prism 6.0d.
Chromatin immunoprecipitation (ChIP)
Cells were seeded in 150 mm dishes and were transfected with GLI1 siRNA or control siRNA for 48 hours. Cells were then treated with vehicle or 10 nM E2 for 30 min before harvesting and chromatin preparation. ChIP assays were performed essentially as described [57]. Briefly, 5 μg of ERα antibody (sc-543, Santa Cruz Biotechnology) or normal rabbit IgG (sc-2027, Santa Cruz Biotechnology) were conjugated to Dynabeads® Protein A beads (Life Technologies), then antibody-bound beads were incubated with sonicated cell lysates. Immunoprecipitated DNA was purified using the QIAquick PCR Purification Kit and quantified by PCR. Input DNA was used to produce standard curves and the ChIP data were converted to percentages of total input. The PCR primer sequences are given in Table 2, with the negative control primer set (ERα ChIP NC) originating from the ESR1 gene, 11 kb downstream of the transcription start site.
Table 2: Primer sequences for ChIP analysis
pS2 ChIP forward |
5′ CCGGCCATCTCTCACTATGAA |
pS2 ChIP reverse |
5′ CCTCCCGCCAGGGTAAATAC |
ERα ChIP NC forward |
5′ CCTGATCTGGTTCTTCCTCTGCAT |
ERα ChIP NC reverse |
5′ CCAAATACAAGGGCTTGATTGCCA |
Statistics
Multiple t-test and ordinary two-way ANOVA test were performed using GraphPad Prism 6.0d. P values lower than 0.05 were considered as significant.
ACKNOWLEDGMENTS
We thank Saioa Goñi and Ning Liang, Department of Biosciences and Nutrition, Karolinska Institutet, for their help in establishing the ChIP assay protocol. We thank Philip Jonsson, Human Oncology and Pathogenesis Program, Memorial Sloan Kettering Cancer Center, NY, USA, for bioinformatics analysis.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was funded by the Swedish Childhood Cancer Foundation and the AFA Insurance. Y.D. was supported by the China Scholarship Council and A.A by the Karolinska Institutet Doctoral (KID) funding program.
REFERENCES
1. McGuire A, Brown JA, Malone C, McLaughlin R, Kerin MJ. Effects of age on the detection and management of breast cancer. Cancers. 2015; 7:908–929.
2. McGuire S. World Cancer Report 2014. Geneva, Switzerland: World Health Organization, International Agency for Research on Cancer, WHO Press, 2015. Adv Nutr. 2016; 7:418–419.
3. Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009; 9:631–643.
4. Ramaswamy B, Lu YZ, Teng KY, Nuovo G, Li XB, Shapiro CL, Majumder S. Hedgehog signaling is a novel therapeutic target in tamoxifen-tesistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012; 72:5048–5059.
5. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang S, Ivy SP. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015; 12:445–464.
6. Levanat S. Hedgehog signaling and cross-talk therapeutic potential. Period Biol. 2014; 116:371–379.
7. Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat Med. 2013; 19:1410–1422.
8. Ng JM, Curran T. The Hedgehog’s tale: developing strategies for targeting cancer. Nat Rev Cancer. 2011; 11:493–501.
9. Ingham PW. Hedgehog signalling. Curr Biol. 2008; 18:R238–241.
10. Teglund S, Toftgård R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010; 1805:181–208.
11. Briscoe J, Thérond PP. The mechanisms of Hedgehog signalling and its roles in development and disease. Nature reviews Molecular cell biology. 2013; 14:416–429.
12. Aberger F, Altaba ARI. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin Cell Dev Biol. 2014; 33:93–104.
13. Hui CC, Angers S. Gli proteins in development and disease. Annual review of cell and developmental biology. 2011; 27:513–537.
14. Nilsson M, Undén AB, Krause D, Malmqwist U, Raza K, Zaphiropoulos PG, Toftgård R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97:3438–3443.
15. Carpenter RL, Lo HW. Hedgehog pathway and GLI1 isoforms in human cancer. Discov Med. 2012; 13:105–113.
16. O’Toole SA, Machalek DA, Shearer RF, Millar EKA, Nair R, Schofield P, McLeod D, Cooper CL, McNeil CM, McFarland A, Nguyen A, Ormandy CJ, Qiu MR, et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011; 71:4002–4014.
17. Wolf I, Bose S, Desmond JC, Lin BT, Williamson EA, Karlan BY, Koeffler HP. Unmasking of epigenetically silenced genes reveals DNA promoter methylation and reduced expression of PTCH in breast cancer. Breast Cancer Res Treat. 2007; 105:139–155.
18. Nessling M, Richter K, Schwaenen C, Roerig P, Wrobel G, Wessendorf S, Fritz B, Bentz M, Sinn HP, Radlwimmer B, Lichter P. Candidate genes in breast cancer revealed by microarray-based comparative genomic hybridization of archived tissue. Cancer Res. 2005; 65:439–447.
19. Naylor TL, Greshock J, Wang Y, Colligon T, Yu QC, Clemmer V, Zaks TZ, Weber BL. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005; 7:R1186-R1198.
20. Fiaschi M, Rozell B, Bergström Å, Toftgård R. Development of mammary tumors by conditional expression of GLI1. Cancer Res. 2009; 69:4810–4817.
21. Visbal AP, LaMarca HL, Villanueva H, Toneff MJ, Li Y, Rosen JM, Lewis MT. Altered differentiation and paracrine stimulation of mammary epithelial cell proliferation by conditionally activated Smoothened. Developmental biology. 2011; 352:116–127.
22. Sun Y, Wang YS, Fan C, Gao P, Wang XW, Wei GW, Wei JM. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol Cancer. 2014; 13:137.
23. Goel HL, Pursell B, Chang C, Shaw LM, Mao J, Simin K, Kumar P, Vander Kooi CW, Shultz LD, Greiner DL, Norum JH, Toftgård R, Kuperwasser C, et al. GLI1 regulates a novel neuropilin-2/alpha6beta1 integrin based autocrine pathway that contributes to breast cancer initiation. EMBO molecular medicine. 2013; 5:488–508.
24. Johansson HJ, Sanchez BC, Mundt F, Forshed J, Kovacs A, Panizza E, Hultin-Rosenberg L, Lundgren B, Martens U, Máthé G, Yakhini Z, Helou K, Krawiec K, et al. Retinoic acid receptor alpha is associated with tamoxifen resistance in breast cancer. Nat Commun. 2013; 4:2175.
25. Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006; 116:561–570.
26. Thomas C, Gustafsson JA. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011; 11:597–608.
27. Matthews J, Wihlén B, Tujague M, Wan J, Ström A, Gustafsson JA. Estrogen receptor (ER) beta modulates ERalpha-mediated transcriptional activation by altering the recruitment of c-Fos and c-Jun to estrogen-responsive promoters. Molecular endocrinology. 2006; 20:534–543.
28. Zhao C, Matthews J, Tujague M, Wan J, Ström A, Toresson G, Lam EW, Cheng G, Gustafsson JA, Dahlman-Wright K. Estrogen receptor beta2 negatively regulates the transactivation of estrogen receptor alpha in human breast cancer cells. Cancer Res. 2007; 67:3955–3962.
29. Cai W, Kramarova TV, Berg P, Korbonits M, Pongratz I. The immunophilin-like Protein XAP2 is a negative regulator of estrogen signaling through interaction with estrogen receptor alpha. Plos One. 2011; 6:e25201.
30. Lauth M, Bergström Å, Shimokawa T, Toftgård R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:8455–8460.
31. Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005; 365:671–679.
32. Györffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010; 123:725–731.
33. Sabol M, Trnski D, Uzarevic Z, Ozretic P, Musani V, Rafaj M, Cindric M, Levanat S. Combination of cyclopamine and tamoxifen promotes survival and migration of mcf-7 breast cancer cells—interaction of hedgehog-gli and estrogen receptor signaling pathways. Plos One. 2014; 9:e114510.
34. Sandelin A, Wasserman WW, Lenhard B. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004; 32:249–252.
35. Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006; 124:47–59.
36. Kinzler KW, Vogelstein B. The GLI gene encodes a nuclear protein which binds specific sequences in the human genome. Mol Cell Biol. 1990; 10:634–642.
37. de Leeuw R, Neefjes J, Michalides R. A role for estrogen receptor phosphorylation in the resistance to tamoxifen. International journal of breast cancer. 2011; 2011:232435.
38. Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001; 61:7025–7029.
39. Parl FF. Multiple mechanisms of estrogen receptor gene repression contribute to ER-negative breast cancer. The pharmacogenomics journal. 2003; 3:251–253.
40. Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, Dvir A, Soussan-Gutman L, Jeselsohn R, Yelensky R, Brown M, Miller VA, Sarid D, Rizel S, Klein B, Rubinek T, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res. 2013; 73:6856–6864.
41. Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, Kalyana-Sundaram S, Wang R, Ning Y, Hodges L, Gursky A, Siddiqui J, Tomlins SA, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nature genetics. 2013; 45:1446–1451.
42. Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, Li Z, Gala K, Fanning S, King TA, Hudis C, Chen D, Taran T, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nature genetics. 2013; 45:1439–1445.
43. Rondón-Lagos M, Villegas VE, Rangel N, Sánchez MC, Zaphiropoulos PG. Tamoxifen resistance: Emerging molecular targets. International journal of molecular sciences. 2016; 17:1357.
44. Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. Journal of the National Cancer Institute. 2004; 96:926–935.
45. Levin ER. Integration of the extranuclear and nuclear actions of estrogen. Molecular endocrinology. 2005; 19:1951–1959.
46. Roma J, Almazán-Moga A, Sánchez de Toledo J, Gallego S. Notch, wnt, and hedgehog pathways in rhabdomyosarcoma: from single pathways to an integrated network. Sarcoma. 2012; 2012:695603.
47. Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engström O, Öhman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997; 389:753–758.
48. Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor′s ligand binding domains. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:5998–6003.
49. Pike ACW, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structural insights into the mode of action of a pure antiestrogen. Structure. 2001; 9:145–153.
50. Burris TP, Solt LA, Wang YJ, Crumbley C, Banerjee S, Griffett K, Lundasen T, Hughes T, Kojetin DJ. Nuclear receptors and their selective pharmacologic modulators. Pharmacol Rev. 2013; 65:710–778.
51. Villegas VE, Rondón-Lagos M, Annaratone L, Castellano I, Grismaldo A, Sapino A, Zaphiropoulos PG. Tamoxifen treatment of breast cancer cells: impact on Hedgehog/GLI1 signaling. International journal of molecular sciences. 2016; 17:308.
52. Garcia-Becerra R, Santos N, Diaz L, Camacho J. Mechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistance. International journal of molecular sciences. 2013; 14:108–145.
53. Nass N, Kalinski T. Tamoxifen resistance: From cell culture experiments towards novel biomarkers. Pathol Res Pract. 2015; 211:189–197.
54. Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene. 2015; 34:3617–3626.
55. Diao Y, Rahman MF, Villegas VE, Wickström M, Johnsen JI, Zaphiropoulos PG. The impact of S6K1 kinase on neuroblastoma cell proliferation is independent of GLI1 signaling. BMC cancer. 2014; 14:600.
56. Zhu J, Zhao C, Kharman-Biz A, Zhuang T, Jonsson P, Liang N, Williams C, Lin CY, Qiao Y, Zendehdel K, Strömblad S, Treuter E, Dahlman-Wright K. The atypical ubiquitin ligase RNF31 stabilizes estrogen receptor alpha and modulates estrogen-stimulated breast cancer cell proliferation. Oncogene. 2014; 33:4340–4351.
57. Mohammed H, Taylor C, Brown GD, Papachristou EK, Carroll JS, D′Santos CS. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc. 2016; 11:316–326.