
Introduction
High hyperdiploidy (HD), characterized by a non-random pattern of chromosomal gains (51 to 67 chromosomes), comprises the largest cytogenetic subgroup of B-cell acute lymphoblastic leukemia (B-ALL) and is one of the most common malignancies in children [1]. Although HD usually confers a favorable prognosis, survivors of childhood ALL have long-term treatment-related morbidity and mortality, including secondary cancers, cardiovascular disease and pulmonary disease [2, 3]. A fuller understanding of the genomic landscape driving leukemogenesis in HD-ALL may inform the biology of this disease and potentially reveal novel therapeutic strategies.
Hyperdiploidy has been shown to arise prenatally [4, 5] and is likely initiated by a single abnormal mitotic event [6, 7], implicating it as the primary genetic lesion. This is followed by additional somatic alterations, arising pre- or postnatally, and these are believed to be required for overt leukemia. Distinct characteristics of HD-ALL have been highlighted both by studies of heritable and acquired genetic alterations. Genome-wide association studies (GWAS) have identified SNPs that distinguish HD-ALL from other subtypes, for example in ARID5B, CEBPE, and PIP4K2A [8-11]. Similarly, a distinct pattern of somatic mutations characterizes HD-ALL. For instance, frequency of common childhood ALL gene deletions, including CDKN2A, IKZF1, and PAX5, is relatively low [12], whereas activating mutations in receptor tyrosine kinase (RTK)/Ras/MAPK signaling genes KRAS, NRAS, FLT3, and PTPN11, are relatively common [13-15]. Indeed, recent next-generation sequencing analyses of HD-ALL confirmed that mutations in these genes, and in the epigenetic regulatory gene CREBBP, are the most frequent somatic alterations in this leukemia subtype [16, 17]. Here, we describe targeted sequencing of 538 cancer-relevant genes using a clinical sequencing platform in a set of 57 HD-ALL patients lacking the common KRAS and NRAS codon 12 and 13 hotspot mutations and common gene deletions, to enrich for discovery of novel driver genes and to assess recurrently-mutated pathways in HD-ALL tumorigenesis. This deep-sequencing enabled identification of putative novel drivers and permitted an in-depth assessment of intratumoral heterogeneity.
Results
Deep-sequencing of 538 cancer-relevant genes (Table S1) was carried out in 57 HD-ALL patients lacking the common KRAS and NRAS codon 12 or 13 hotspot mutations and common B-cell ALL deletions. High quality sequencing data were obtained for each patient, with a mean coverage of 596X and an average of 94.3% reads with > 100X coverage. After removal of low quality variants, known polymorphisms, and likely germline mutations (based on mutant allele fraction, MAF), there were 108 variants predicted to be damaging (CADD Phred score ≥ 20) at 97 unique loci, including 90 SNVs and 7 INDELs (Figure 1). On average, patients harbored a median of only 1 mutation (range: 0-8) (Table S2), with no correlation between age-at-diagnosis and total number of mutations (R2 < 0.01). The most recurrently mutated genes included the RTK/Ras/MAPK signaling pathway genes FLT3 (n = 12), PTPN11 (n = 12), KRAS (n = 9), NRAS (n = 5), and the epigenetic regulator CREBBP (n = 5) (Figure 2). No novel hotspot mutations (i.e. recurrently mutated nucleotides) were identified. A descriptive summary of the predicted damaging mutation loci can be found in Table S2.
Based on DNA availability, we attempted to validate 62 mutations in recurrently mutated genes in the RTK/Ras/MAPK signaling pathway and involved in epigenetic regulation, including likely germline mutations. We successfully validated 60 out of 62 mutations (96.8%) in tumor DNA by PCR and Sanger sequencing. The 2 mutations that were not validated had MAF < 15% in the deep-sequencing, potentially explaining our inability to detect them via Sanger sequencing. Nineteen of the 62 mutations were also confirmed in matched germline DNA samples and, except for one mutation with MAF = 0.43, these had been predicted “likely germline” based on MAF.
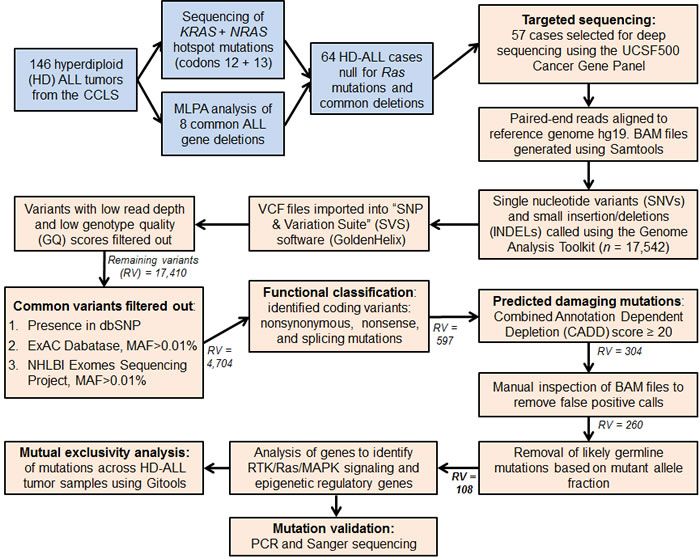
Figure 1 : Flow diagram summarizing the analysis pipeline. Blue colored boxes represent subject selection criteria. Peach colored boxes include sequencing data and variant filtering analyses. MAF refers to the SNP minor allele frequency. Remaining variants (RV) refers to the number of combined SNVs and INDELs remaining in the analysis pipeline following each filtering step. The final number of variants highlighted in bold (RV = 108) refers to the predicted damaging and likely somatic variants detected in the 57 HD-ALL patients.
RTK/Ras/MAPK signaling pathway genes
In total, two-thirds (38/57) of HD-ALL patients carried a predicted damaging and somatic mutation in at least one RTK/Ras/MAPK signaling pathway gene (Figure S1). Twelve patients harbored a total of 15 mutations in FLT3, including 12 point mutations, one in-frame deletion of codon I836, and 2 internal tandem duplications (ITD) at the juxta-membrane region in exon 14. Six of the point mutations detected within FLT3 were novel mutations in ALL (Figure S2), of which 2 (I867N and M664I) were in codons in which different amino acid-altering mutations were recently identified in HD-ALL [17], with the remaining 4 located in codons known to be mutated in AML. These novel mutations all lie within known mutational hotspot regions within or just upstream of the tyrosine kinase domain. In addition, we identified a FLT3 mutation Y842C with MAF = 0.49 that was confirmed as a germline mutation via Sanger sequencing of remission DNA (Figure S2).
In PTPN11, one of the 9 mutation loci (F71L) was previously found in AML but not ALL according to COSMIC, though this mutation lies within one of the mutational hotspot regions in the N-terminal SH2 domain (Figure S3). The mutation P491L had MAF = 0.49 and was possibly germline, however constitutive DNA was not available for this patient to confirm this. In KRAS, we identified mutations at codon 146 (n = 4) and codon 117 (n = 1) (Figure 2, Figure S4). Subclonal mutations (MAF < 0.30) were also detected at codons 12 and 13 in 4 patients. In NRAS, mutations were found at known ALL hotspots at codons 61 (n = 4) and 146 (n = 1) (Figure S5). Additional likely-somatic damaging mutations were identified in the Ras homolog gene MRAS (n = 1), the leukemia oncogene CBL (n = 1), the MYC oncogene (n = 1), and in the receptor tyrosine kinase genes ROS1 (n = 2), ERBB2 (n = 1), EPHB1 (n = 1), FGFR1 (n = 1), FGFR4 (n = 1), and IGF1R (n = 1). Mutations in KRAS, NRAS, FLT3, and PTPN11 were largely mutually exclusive, with 38 mutations detected across 34 patients compared with a predicted distribution of 26±3.7 based on 10,000 permutations (p = 1.4x10-5; Z-score = 4.18). Of 38 patients with RTK/Ras/MAPK signaling pathway mutations, there were a total of 52 unique mutations. These were significantly mutually exclusive, with a distribution of 38 compared with a predicted distribution of 29.9±4.6 (p = 8.2x10-5; Z-score = 3.77) (Figure S1).
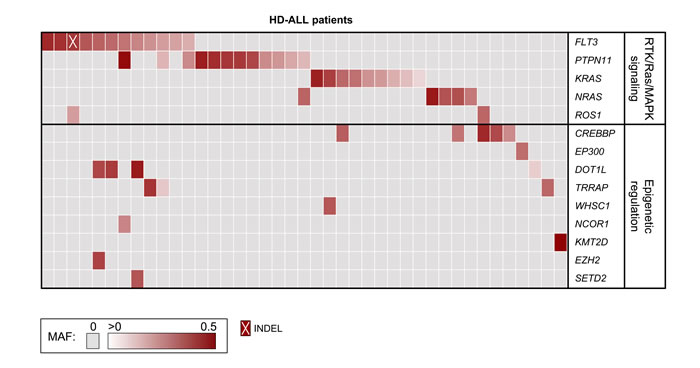
Figure 2 :Tiling plot of mutations in high hyperdiploid ALL. Predicted damaging mutations are included for recurrently affected genes and for genes previously identified as mutated in HD-ALL (rows) across 41 of 57 HD-ALL patients (columns) in the study. Patients without mutations in such genes were excluded from the figure. Mutations are color-coded according to their mutant allele fraction (MAF), and INDELs are highlighted as shown at the bottom of the figure. MAF was adjusted for chromosome copy number. Where > 1 mutation was present in the same gene in a patient, the color of the cell represents the most clonal mutation.
Epigenetic regulatory genes
Predicted damaging and somatic mutations in epigenetic regulators were detected in one-third (19/57) of HD-ALL patients (Figure S6). CREBBP mutations were the most frequent (n = 5), including previously unreported nonsense and missense mutations in the histone acetyltransferase (HAT) domain. Another patient had a splicing mutation within the HAT domain in the closely related gene EP300 (Figure 3). DOT1L was also recurrently mutated (n = 4), including 3 mutations in the highly-conserved S-adenosylmethionine (AdoMet)-dependent methyltransferase domain with CADD Phred scores > 30, with an additional mutation adjacent to the AdoMet domain and also predicted to be highly deleterious (CADD Phred = 29.5) (Figure 3). Predicted damaging somatic mutations were also identified in TRAPP (n = 3) (Figure S7), and one each in WHSC1, NCOR1, KMT2D, DNMT3A, EZH2, HIST1H1C, SETD2, and TAF1. There was significant mutual exclusivity for epigenetic regulatory gene mutations, with 21 predicted damaging mutations detected across 19 patients compared with a predicted distribution of only 16.2±2.6 based on 10,000 permutations (p = 0.04; Z-score = 1.75) (Figure S6).
Epigenetic gene mutations were more frequent in patients with somatic FLT3 mutations than in patients with PTPN11 mutations, with 50% (6/12) of FLT3 mutant patients harboring ≥ 1 epigenetic gene mutation, compared with only 9.1% (1/11) of patients with PTPN11 mutations (p = 0.067, Fisher’s exact test). FLT3 mutations significantly co-occurred with DOT1L mutations, with 16 mutations across 13 patients (Figure 2) compared with a predicted distribution of 14.6±0.8 (p = 0.04; Z-score = -1.76). Further, PTPN11 mutations were significantly mutually exclusive of epigenetic gene mutations (as a group), with 31 mutations across 29 patients compared with a predicted distribution of only 25.0±2.2 (p = 3.4x10-3; Z-score = 2.70). A similar trend was seen for KRAS (p = 0.12) and NRAS (p = 0.07) but not for FLT3 (p = 0.49).
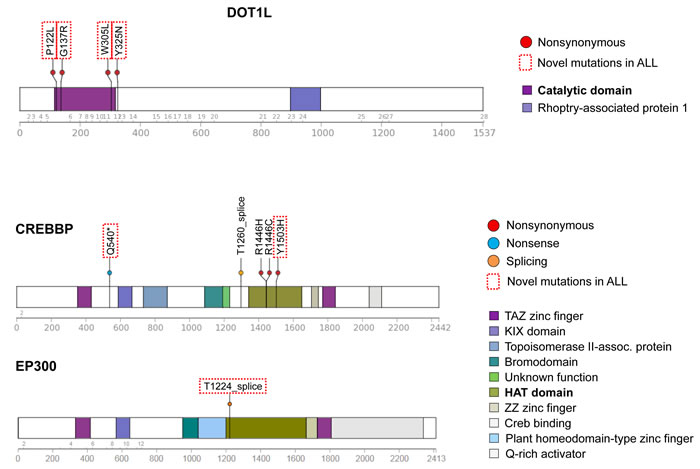
Figure 3 : Novel mutations in epigenetic regulatory genes. Schematic diagrams of DOT1L, CREBBP, and EP300 proteins showing positions of detected mutations in HD-ALL patients. Types of mutation (i.e. nonsynonymous, nonsense or splicing) are shown, along with color-coded functional domains as determined using the Protein Painter tool (http://explore.pediatriccancergenomeproject.org/proteinPainter). Bolded functional domains are ones affected by mutations. In DOT1L, nonsynonymous mutations cluster at the AdoMet catalytic domain. In CREBBP, nonsynonymous mutations cluster at the histone acetyltransferase (HAT) domain. Red dotted line boxes highlight novel mutations not previously reported in ALL.
Tumor microheterogeneity
We identified 65 minor subclonal, or “microclonal”, hotspot mutations at known mutation hotspot loci in KRAS (patient n = 21), NRAS (n = 17), FLT3 (n = 4), PTPN11 (n = 7), and CREBBP (n = 3) (Figure 4, Figure S8, Table S3). Median MAF of these mutations was 0.012 (range: 0.003-0.096). Of the 16 mutation loci assessed, only FLT3 codon 663, PTPN11 codon 69, and the negative control NRAS codon 117 did not harbor any microclonal mutations. Analysis of flanking sequences at the 65 microclonal hotspot mutations revealed only 5 additional mutations that qualified as microclonal in 3250bp (65 x 50bp) of sequence, thus 99.8% of flanking sequences did not harbor such mutations.
In total, there were 32 patients with microclonal mutations, of which 31 (54.4%) had at least one mutation in KRAS, NRAS, FLT3, and/or PTPN11. We assessed intra-sample tumor microheterogeneity and identified 16 patients with multiple microclonal mutations, including 10 patients carrying mutations in both KRAS and NRAS and 4 patients with mutations in at least 3 of the genes analyzed (Figure 4, Figure S8, Table S3). There was a highly significant co-occurrence of microclonal mutations in KRAS and NRAS codons 12 and 13, with 28 mutations found in only 19 patients compared with a predicted distribution of 23.8±2.1 (p = 4.8x10-4; Z-score = -3.3) (Figure S8). There was no significant difference between age-at-diagnosis in patients with microclonal hotspot mutations (mean = 4.67 yrs) and those without (mean = 4.89 yrs) (p = 0.77).
In addition, there were 8 patients with multiple microclonal mutations at the same hotspot locus, either in adjacent nucleotides within the same codon or in adjacent codons (Figure S9, Table S4). Five patients had adjacent KRAS hotspot mutations and 3 patients had adjacent NRAS mutations, including one patient with two codon 12 mutations (MAF = 2.9% and 5.1%) and one codon 13 mutation (MAF = 5.5%). In all 8 patients, these concurrent nucleotide changes occurred on different sequencing reads (p = 7.8x10-3, binomial significance test, 2-tailed), indicating that they were part of distinct tumor subclones (Table S4).
Including all identified clonal, subclonal, and microclonal mutations, there were 43 HD-ALL patients (75.4%) with a damaging mutation in KRAS, NRAS, FLT3, and/or PTPN11, and 8 patients (14.0%) with a damaging CREBBP mutation (Figure 4).
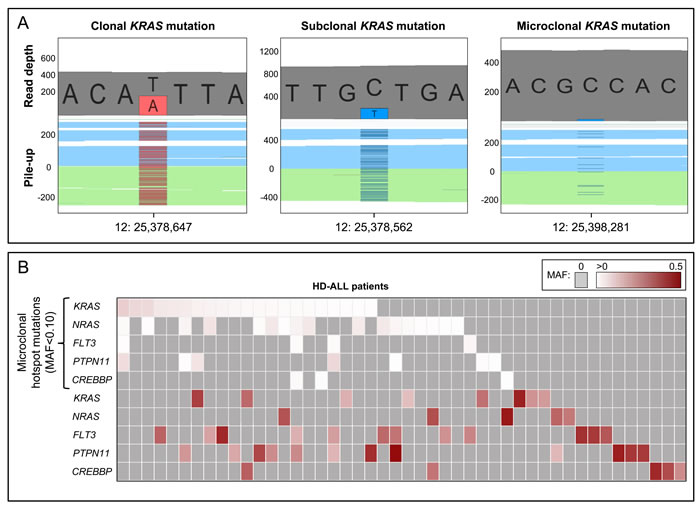
Figure 4 : Clonal and microclonal tumor heterogeneity at HD-ALL hotspot loci. A. GenomeBrowse screenshots showing clonal, subclonal, and microclonal KRAS mutations. In each screenshot, the top plot shows the total read depth and relative read depths of the reference (grey) and alternate alleles (red or blue). Bottom plots show the sequence read pile-up, split into forward (blue) and reverse (green) strands, with presence of mutant alleles shown in red or blue. The left screenshot shows a clonal KRAS T > A mutation at chr12:25,378,647 (K117N) with MAF = 0.44. The middle screenshot shows a subclonal KRAS C > T mutation at chr12:25,378,562 (A146T) with MAF = 0.20. The right screenshot shows a microclonal KRAS C > T mutation at chr12:25,398,281 (G13D) with MAF = 0.025. B. Heatmap showing the clonal and microclonal heterogeneity of HD-ALL hotspot mutations at KRAS, NRAS, FLT3, PTPN11, and CREBBP. The top 5 rows show the presence of microclonal (MAF < 0.10) hotspot mutations in codons 12, 13, 61, 117, and 146 of KRAS and NRAS; codon 835 of FLT3; codons 61 and 72 of PTPN11; and codon 1446 of CREBBP, across HD-ALL patients (columns). There were 21 patients with microclonal KRAS mutations (median MAF: 1.08%; range: 0.44-9.57%), 17 patients with microclonal NRAS mutations (median MAF: 1.28%; range: 0.39-5.47%), 4 patients with microclonal FLT3 mutations (median MAF: 1.03%; range: 0.66-1.97%), 7 patients with microclonal PTPN11 mutations (median MAF: 0.80%; range: 0.34-7.97%), and 3 patients with microclonal CREBBP mutations (median MAF: 0.68%; range: 0.48-1.13%). Clonal and subclonal mutations (MAF > 0.10) in the 5 genes are shown below, and include damaging mutations at additional codons. Mutations are color-coded according to their MAF as shown at the top right of the figure. Grey boxes represent patients with zero mutations. Where > 1 mutation was present in the same gene in a patient, the color of the cell represents the most clonal mutation. Patients without mutations in KRAS, NRAS, FLT3, PTPN11, or CREBBP were excluded from the figure.
Discussion
Targeted deep-sequencing of over 500 cancer-relevant genes in 57 HD-ALL patients revealed extensive tumor heterogeneity in the RTK/Ras/MAPK signaling pathway, with the majority of patients harboring clonal, subclonal, and/or microclonal mutations at hotspot loci in KRAS, NRAS, FLT3, or PTPN11. Further, we found a substantial proportion of patients with epigenetic regulatory gene mutations, including in the putative novel driver gene DOT1L, and these typically co-occurred with mutations in FLT3.
We confirmed the high frequency and mutual exclusivity of mutations in FLT3, PTPN11, KRAS, and NRAS [13]. In KRAS, we verified that codon 146 and 117 mutations, recently identified by Paulsson et al. (2015) [16], are hotspot loci in HD-ALL. Indeed, 7 patients harbored microclonal mutations at one of these two loci (Figure S8). FLT3 and PTPN11 mutations were more frequent than previously reported, likely due to our patient selection criteria. We identified 6 novel mutations in FLT3 and one novel mutation in PTPN11, in known activating hotspot regions but not previously detected in ALL. We also made the unexpected discovery of a likely causal germline FLT3 mutation at Y842C. This same amino acid change was previously identified in tumor DNA from a non-hyperdiploid ALL patient [18], and was reported as an activating mutation in AML [19]. Recent analysis of germline mutations in childhood cancers identified a FLT3 frameshift mutation in a child with E2A-fusion ALL [20]. However, to our knowledge this is the first report of a germline FLT3 mutation in HD-ALL. Germline PTPN11 mutations cause Noonan syndrome [21], therefore this patient may present with RASopathy-related phenotypes given that somatic mutations in FLT3 and PTPN11 activate the Ras/MAPK pathway and are largely mutually exclusive in ALL. However, clinical data were not available to confirm this.
Though we enriched for discovery of novel driver genes in HD-ALL, few novel recurrently mutated RTK/Ras/MAPK signaling genes were identified. Somatic alterations of ROS1 have previously been detected in lung cancer and glioblastoma [22, 23], and we found ROS1 mutations in a small fraction of HD-ALL patients. ROS1 protein mediates phosphorylation and increases activation of the PTPN11-encoded protein SHP2, and ROS1 activation can induce tumorigenesis [24]. Thus, the mutations we identified are potentially gain-of-function, suggesting that a subset of ALL patients could be treated with ROS1 inhibitors such as crizotinib [25] or foretinib [26].
The identification of subclonal KRAS codon 12 and 13 mutations in our initial analyses led us to investigate microclonal mutations (MAF < 10%) at ALL hotspot loci in KRAS, NRAS, FLT3, PTPN11, and CREBBP. Utilizing the high-coverage sequencing data we revealed an extraordinary level of intra- and inter-tumoral heterogeneity, with microclonal Ras pathway hotspot mutations detected in over 50% of patients. Adjacent microclonal mutations never appeared on the same sequencing reads and, therefore, appear to have arisen in distinct clones within tumor samples, supporting that the formation and selection of such hotspot mutations is ongoing. Further, the significant co-occurrence of KRAS and NRAS codon 12 and 13 microclonal mutations suggests the existence of a highly-heterogeneous subset of Ras-driven leukemias. Alternatively, these may not be driven by Ras pathway activation but by clonal mutations in other pathways that reduced the selective advantage of Ras mutant clones. These two possibilities are evidenced by a lack of clonal RTK/Ras/MAPK signaling gene mutations in the 9 patients with concurrent microclonal Ras codon 12/13 mutations, which included 2 patients harboring microclonal mutations at 4 different loci (Figure S8).
Microclonal Ras mutations have previously been reported in ALL and other tumor types [27, 28]. In a study of relapsed ALL, microclonal KRAS and NRAS mutations were detected in diagnostic DNA in over 50% of patients found to be wildtype by Sanger sequencing [27]. These patients had Ras mutations at relapse, suggesting that the microclonal mutations evaded initial treatment and drove the relapsed ALL [27]. Similarly, CREBBP HAT domain mutations are enriched in relapsed HD-ALL [17, 29-31]. Whether microclonal Ras or CREBBP mutations confer a similar risk of relapse to the more clonal mutations remains to be determined.
Mutations in epigenetic regulatory genes comprise a substantial portion of the mutational landscape of several hematologic malignancies, including T-ALL [32], AML [33], and B-cell lymphomas (reviewed in Lunning and Green [34]). We identified a high frequency and mutual exclusivity of epigenetic regulator mutations in HD-ALL, supporting a biological role for epigenetic dysregulation in this leukemia subtype [35] and that multiple possible genetic lesions may generate this disruption. Clustered damaging mutations discovered in the histone 3 lysine 79 (H3K79) methyltransferase gene DOT1L strongly support this as a driver of leukemogenesis. The only previously reported DOT1L mutation in childhood ALL was also in a HD patient, with a V114F mutation in close proximity to the AdoMet domain [36], suggesting DOT1L mutations may be specific to the HD subtype. In infant ALL, MLL-fusion proteins recruit DOT1L protein leading to H3K79 hypermethylation and increased expression of MLL target genes [37]. In contrast, DOT1L deficiency is associated with hyperploidy in vitro, likely due to aberrant mitotic spindle formation [38-40]. In mice, DOT1L knockout resulted in downregulation of GATA2 and differentiation towards the myeloid lineage during hematopoiesis [41]. We recently proposed that heritable risk alleles in CEBPE, a modulator of myelopoiesis, may increase ALL risk via lineage confusion [11]. Polymorphisms in CEBPE were more strongly associated with HD-ALL, supporting that de-differentiation of pre-B cells towards the myeloid lineage may be a hallmark of hyperdiploidy. DOT1L is also thought to play a role in double-strand break DNA repair and cell cycle control [42], thus more research is required to elucidate the role of DOT1L in HD-ALL.
Recent studies have shown that mutations in other epigenetic regulator genes, in particular the histone acetyltransferase CREBBP, are common in childhood ALL [16, 17]. We identified novel nonsense and HAT domain mutations in CREBBP, one of which (Y1503H) was previously reported in lymphoma [43]. Mutations in the H3K36 methyltransferase gene WHSC1 were also recently reported in HD-ALL [16, 17]. In our patients, we did not detect the SET domain hotspot mutation at E1099 [16] but did find another SET domain mutation T1150A that was recently reported in B-ALL [30] and shown to have increased methyltransferase activity in mantle cell lymphoma [44]. We also identified novel mutations in TRRAP, a component of HAT complexes.
Only a fraction of known epigenetic regulatory genes were sequenced, thus it is likely that a higher proportion of HD-ALL tumors may suffer epigenetic perturbation. A recent study of over 1000 pediatric cancer genomes, including leukemias but not HD-ALL, revealed significant variation in frequency of epigenetic regulator mutations across tumor types [45]. Our data suggest that epigenetic dysregulation may be prominent in HD-ALL. One explanation is that hyperdiploidy leads to overexpression of tumor suppressor genes, which requires downregulation via epigenetic silencing. Indeed, some genes positioned on gained chromosomes in HD-ALL show decreased expression that is associated with hypermethylation [46, 47]. Alternatively, epigenetic dysregulation may be required for oncogene overexpression, supported by lower mean methylation levels in HD-ALL relative to other subtypes [46, 48], in particular on gained chromosomes [48, 49]. Drugs targeting epigenetic modifications, such as histone deacetylase (HDAC) inhibitors [50], are being explored as treatment options in ALL and might prove particularly effective against the HD-ALL subtype.
It was interesting to note that DOT1L mutations significantly co-occurred with mutations in FLT3. Moreover, epigenetic regulatory gene mutations were more frequent in patients harboring FLT3 mutations than in patients with PTPN11, KRAS, or NRAS mutations, which tended to be mutually exclusive of epigenetic mutations. These could be a chance finding given the relatively small numbers, although FLT3 mutations in AML frequently co-occur and functionally cooperate with mutations in epigenetic regulatory genes [33, 51-53]. Future studies should investigate whether FLT3 activation cooperates with epigenetic dysregulation in ALL, which might suggest a potential use for combination treatment using FLT3 inhibitors with epigenetic therapies, as recently proposed for AML [54].
There are some caveats to consider when interpreting results from this study. First, the sequencing was limited to 538 genes included in the UCSF500 Cancer Gene panel and, though these are thought to be the most frequently mutated cancer genes, we may have missed potential driver mutations in genes not included on the panel. However, recent whole-genome and exome sequencing studies of HD-ALL did not identify any driver mutations in genes missing from our analyses [16, 17]. Clinical information, such as rate of relapse and outcome data, was unfortunately not available for the patients in this study. In addition, the exclusion of patients harboring common gene deletions precluded analysis of whether copy number alterations co-operate with mutations in Ras pathway or epigenetic regulatory genes in ALL. Therefore, next-generation sequencing of large numbers of unselected HD-ALL patients with available clinical data will be required to elucidate all possible leukemogenic pathways and the implications of particular clonal and microclonal mutations on patient outcomes.
Conclusions
Deep-sequencing of HD-ALL genomes has revealed extensive tumor heterogeneity at both the clonal and microclonal level, highlighting the importance of RTK/Ras/MAPK signaling pathway activation and epigenetic dysregulation in HD leukemogenesis. The identification of putative novel driver genes DOT1L and ROS1 demands further investigation into potential novel and targeted treatment strategies, such as ROS1 inhibition [26]. Targeting epigenetic modifications has been suggested as a new therapeutic option in ALL [30, 50]. Moreover, future work is required to investigate whether tumor microheterogeneity should impact therapeutic regimens in childhood ALL.
materials and Methods
Ethics statement
This study was reviewed and approved by institutional review committees at the University of California Berkeley, the California Department of Public Health (CDPH), and all collaborating institutions. Written informed consent was obtained from all parents of participants.
Study subjects
Subjects included in this study were enrolled in the California Childhood Leukemia Study (CCLS) as previously described [55]. The initial set of patients comprised 457 newly-diagnosed B-ALL patients with cytogenetic subtype information, who were born in California and had diagnostic bone marrow samples available. These included 146 HD-ALL patients, of which 52.6% of patients were Hispanic and 33.6% were non-Hispanic white (the remainder included a mixture of African-American, Asian, and other ethnicities). Median age-at-diagnosis was 4.12 years (range 1.1-13.5). DNA was extracted from leukemia bone marrow collected at diagnosis using the QIAamp DNA Blood Mini Kit (QIAGEN, Germany).
Determination of immuno-phenotype and cytogenetic profiles
Immuno-phenotype was determined for ALL patients using flow cytometry profiles, with those expressing CD10 or CD19 ( ≥ 20%) classified as B-lineage ALL, as described previously [56]. Ploidy was determined using FISH or G-banding, with HD patients classed as having > 50 chromosomes.
Targeted sequencing of cancer genes
A summary of subject selection is included in the Supplementary Methods. In brief, 57 HD B-ALL patients with neither Ras codon 12/13 hotspot mutations nor common ALL deletions, as assessed by Sanger sequencing and multiplex ligation-dependent probe amplification respectively, were selected for deep-sequencing of cancer-relevant genes to enrich for discovery of novel driver mutations (Figure 1). NimbleGen SeqCap EZ libraries were used for capture of 538 genes included in the “UCSF500 Cancer Gene Panel” (Table S1). Library preparation was carried out according to manufacturer’s protocol. DNA samples were barcoded and pooled for multiplexed sequencing on the Illumina HiSeq 2500 platform, with sequencing carried out to ~600X depth. Sequencing data analysis and variant filtering for predicted damaging SNVs and INDELs are described in detail in the Supplementary Methods and summarized in Figure 1. Briefly, to exclude likely germline polymorphisms we filtered out variants present in dbSNP or with a minor allele frequency > 0.01% (i.e. > 0.0001) in the Exome Aggregation Consortium (ExAC) Database. Predicted damaging mutations were identified using the Combined Annotation Dependent Depletion (CADD) tool version 1.3 (http://cadd.gs.washington.edu/score) [57]. A CADD Phred score threshold of ≥ 20 (i.e. top 1% deleterious variants in the genome) was used, as recommended for discovery of causal variants.
Given that targeted deep-sequencing data was not created from matched germline DNA samples, our variant filtering methods would not exclude rare germline variants. Thus, we used a mutant allele fraction (MAF) threshold of ≥ 0.45 to define “likely germline” variants. Due to the high frequency of chromosomal gains in HD-ALL, chromosome copy number must be considered when assessing clonality of mutations. For each patient, chromosomal copy number was inferred from sequencing data using the CNVkit software [58]. Whole chromosome gains were determined using mean log2 copy ratio and visual confirmation of copy number scatter plots. We then adjusted the MAF of each mutation based on copy number of the chromosome carrying that mutation, and based on the assumption that chromosomal gains occurred prior to development of somatic mutations. Hence, a mutation with MAF = 0.33 that was located on a chromosome with copy number of 3 would have an adjusted MAF of ~0.49 and, thus, was likely a germline variant that was present on a non-gained chromosome.
Sanger sequencing was carried out to validate a subset of mutations of interest (see Supplementary Methods and Table S2). Where available, remission DNA or neonatal bloodspot DNA was used to determine whether mutations were germline or somatic. Mutations were assessed in the Catalogue of Somatic Mutations in Cancer (COSMIC, http://cancer.sanger.ac.uk) [59] and in data from two recent sequencing studies of HD-ALL [16, 17], to determine whether they were previously identified in ALL or other cancer types. Clonal mutations were defined as those with MAF ≥ 0.30, and subclonal mutations with MAF ≥ 0.10 and < 0.30, following adjustment for chromosome copy number. Minor subclonal (i.e. microclonal) mutations were defined as having MAF < 0.10.
Mutual exclusivity analysis
Mutual exclusivity analysis was carried out using Gitools (version 2.2.3) [60]. We included all genes in which ≥ 1 patient carried a predicted damaging somatic mutation. Mutation data were imported for all 57 patients, with a linear score corresponding to the mutant allele fraction as calculated from the reference:alternative allele ratio. Six patients carried 2 mutations in the same gene, and these mutations were grouped for each patient. Mutual exclusion analysis was carried out for genes in the RTK/Ras/MAPK signaling pathway and for those involved in epigenetic regulation, as well as for microclonal mutations. Observed distribution of mutations across patients (i.e. number of columns with a mutation) was compared to the expected distribution, and a Z-score obtained through 10,000 permutations, maintaining the number of “events” (i.e. mutations) per row and with weighted permutations for columns, as developed by Perez-Llamas, et al. [60]. P-values reflect the significance of mutual exclusivity derived from Z-scores.
Tumor microheterogeneity analysis
To investigate tumor microheterogeneity, we assessed the frequency of microclonal mutations (i.e. MAF < 0.10). In this analysis, we included 15 known ALL hotspot loci and/or loci found to be recurrently mutated ( ≥ 1 patient) in our initial analysis of VCF data, including: KRAS codons 12, 13, 61, 117, and 146; NRAS codons 12, 13, 61, and 146; FLT3 codons 663 and 835; PTPN11 codons 61, 69, and 72; and CREBBP codon 1446. We also included NRAS codon 117 as a negative control, as this is not known to be mutated in ALL. Each patient BAM file was imported into the GenomeBrowse software (Golden Helix), and visual examination of the hotspot mutation loci was carried out. Inclusion criteria for microclonal mutations were nucleotide changes that were: 1) Nonsynonymous; 2) Present in ≥ 3 reads (i.e. > approximately 0.5% reads); 3) Present in both forward and reverse sequencing directions; and 4) Reads with mean base Phred quality score ≥ 30. To assess the validity of our methods, we counted the number of “non-hotspot” microclonal mutations in the 50bp immediately flanking each microclonal hotspot mutation (25bp up-/downstream) in patients harboring these mutations, relative to the total number of basepairs examined.
We observed several instances where ≥ 2 microclonal mutations in the same patient were present at adjacent nucleotides and/or in adjacent codons at the KRAS or NRAS codon 12/13 locus. To determine whether these mutations were likely in the same tumor clone or not, we exported sequencing read data and examined whether the adjacent nucleotide changes were present in the same reads or in different reads.
Acknowledgments
The authors gratefully acknowledge the families for their participation. We thank the clinical investigators at the following collaborating hospitals for help in recruiting patients: University of California Davis Medical Center (Dr. Jonathan Ducore), University of California San Francisco (Drs. Mignon Loh and Katherine Matthay), Children’s Hospital of Central California (Dr. Vonda Crouse), Lucile Packard Children’s Hospital (Dr. Gary Dahl), Children’s Hospital Oakland (Dr. James Feusner), Kaiser Permanente Roseville (formerly Sacramento) (Drs. Kent Jolly and Vincent Kiley), Kaiser Permanente Santa Clara (Drs. Carolyn Russo, Alan Wong, and Denah Taggart), Kaiser Permanente San Francisco (Dr. Kenneth Leung), and Kaiser Permanente Oakland (Drs. Daniel Kronish and Stacy Month). We thank Dr. Luoping Zhang at University of California Berkeley for performing FISH experiments to determine tumor cell ploidy. The authors would also like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Conflicts of Interest
The authors declare no conflicts of interest.
Grant support
This work was supported by the 500 Cancer Gene Panel Pilot Award from the UCSF Helen Diller Family Comprehensive Cancer Center (A.J.D.), and by Alex’s Lemonade Stand Foundation ‘A’ Awards (A.J.D., K.M.W.). This work was also supported by National Institute of Health grants R01ES009137 (C.M., J.L.W.), P01ES018172 (C.M., J.L.W.), and R01CA155461 (J.L.W.), and National Cancer Institute grant 1T32CA151022-01 (S.S.F). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or its subsidiary Institutes and Centers.
Authorship
Contribution: A.J.D., E.S., A.A.E. and H.M.H. performed experiments; A.J.D., J.O., S.S.F. and I.S. carried out bioinformatic analyses; J.L.W., C.M. and A.T. provided essential materials and patient samples; A.J.D., J.L.W., C.M. and K.M.W. designed the research and interpreted data; A.J.D., J.L.W. and K.M.W. wrote the paper; all authors reviewed and approved the manuscript.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Paulsson K, Johansson B. High hyperdiploid childhood acute lymphoblastic leukemia. Genes, chromosomes & cancer. 2009; 48: 637-660.
2. Hudson MM, Mertens AC, Yasui Y, Hobbie W, Chen H, Gurney JG, Yeazel M, Recklitis CJ, Marina N, Robison LR, Oeffinger KC, Childhood Cancer Survivor Study Investigators. Health status of adult long-term survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. Jama. 2003; 290: 1583-1592.
3. Mody R, Li S, Dover DC, Sallan S, Leisenring W, Oeffinger KC, Yasui Y, Robison LL, Neglia JP. Twenty-five-year follow-up among survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Blood. 2008; 111: 5515-5523.
4. Gruhn B, Taub JW, Ge Y, Beck JF, Zell R, Hafer R, Hermann FH, Debatin KM, Steinbach D. Prenatal origin of childhood acute lymphoblastic leukemia, association with birth weight and hyperdiploidy. Leukemia. 2008; 22: 1692-1697.
5. Panzer-Grumayer ER, Fasching K, Panzer S, Hettinger K, Schmitt K, Stockler-Ipsiroglu S, Haas OA. Nondisjunction of chromosomes leading to hyperdiploid childhood B-cell precursor acute lymphoblastic leukemia is an early event during leukemogenesis. Blood. 2002; 100: 347-349.
6. Paulsson K, Panagopoulos I, Knuutila S, Jee KJ, Garwicz S, Fioretos T, Mitelman F, Johansson B. Formation of trisomies and their parental origin in hyperdiploid childhood acute lymphoblastic leukemia. Blood. 2003; 102: 3010-3015.
7. Onodera N, McCabe NR, Rubin CM. Formation of a hyperdiploid karyotype in childhood acute lymphoblastic leukemia. Blood. 1992; 80: 203-208.
8. Migliorini G, Fiege B, Hosking FJ, Ma Y, Kumar R, Sherborne AL, da Silva Filho MI, Vijayakrishnan J, Koehler R, Thomsen H, Irving JA, Allan JM, Lightfoot T, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013; 122: 3298-3307.
9. Trevino LR, Yang W, French D, Hunger SP, Carroll WL, Devidas M, Willman C, Neale G, Downing J, Raimondi SC, Pui CH, Evans WE, Relling MV. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nature genetics. 2009; 41: 1001-1005.
10. Walsh KM, de Smith AJ, Chokkalingam AP, Metayer C, Dahl GV, Hsu LI, Barcellos LF, Wiemels JL, Buffler PA. Novel childhood ALL susceptibility locus BMI1-PIP4K2A is specifically associated with the hyperdiploid subtype. Blood. 2013; 121: 4808-4809.
11. Wiemels JL, de Smith AJ, Xiao J, Lee ST, Muench MO, Fomin ME, Zhou M, Hansen HM, Termuhlen A, Metayer C, Walsh KM. A functional polymorphism in the CEBPE gene promoter influences acute lymphoblastic leukemia risk through interaction with the hematopoietic transcription factor Ikaros. Leukemia. 2015.
12. Paulsson K, Forestier E, Lilljebjorn H, Heldrup J, Behrendtz M, Young BD, Johansson B. Genetic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107: 21719-21724.
13. Paulsson K, Horvat A, Strombeck B, Nilsson F, Heldrup J, Behrendtz M, Forestier E, Andersson A, Fioretos T, Johansson B. Mutations of FLT3, NRAS, KRAS, and PTPN11 are frequent and possibly mutually exclusive in high hyperdiploid childhood acute lymphoblastic leukemia. Genes, chromosomes & cancer. 2008; 47: 26-33.
14. Wiemels JL, Zhang Y, Chang J, Zheng S, Metayer C, Zhang L, Smith MT, Ma X, Selvin S, Buffler PA, Wiencke JK. RAS mutation is associated with hyperdiploidy and parental characteristics in pediatric acute lymphoblastic leukemia. Leukemia. 2005; 19: 415-419.
15. Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, Palmi C, Carta C, Pession A, Arico M, Masera G, Basso G, Sorcini M, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004; 104: 307-313.
16. Paulsson K, Lilljebjorn H, Biloglav A, Olsson L, Rissler M, Castor A, Barbany G, Fogelstrand L, Nordgren A, Sjogren H, Fioretos T, Johansson B. The genomic landscape of high hyperdiploid childhood acute lymphoblastic leukemia. Nature genetics. 2015; 47: 672-676.
17. Malinowska-Ozdowy K, Frech C, Schonegger A, Eckert C, Cazzaniga G, Stanulla M, zur Stadt U, Mecklenbrauker A, Schuster M, Kneidinger D, von Stackelberg A, Locatelli F, Schrappe M, et al. KRAS and CREBBP mutations: a relapse-linked malicious liaison in childhood high hyperdiploid acute lymphoblastic leukemia. Leukemia. 2015; 29: 1656-1667.
18. Chang P, Kang M, Xiao A, Chang J, Feusner J, Buffler P, Wiemels J. FLT3 mutation incidence and timing of origin in a population case series of pediatric leukemia. BMC cancer. 2010; 10: 513-2407-10-513.
19. Kindler T, Breitenbuecher F, Kasper S, Estey E, Giles F, Feldman E, Ehninger G, Schiller G, Klimek V, Nimer SD, Gratwohl A, Choudhary CR, Mueller-Tidow C, et al. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood. 2005; 105: 335-340.
20. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA, Wilkinson MR, Vadodaria B, Chen X, et al. Germline Mutations in Predisposition Genes in Pediatric Cancer. The New England journal of medicine. 2015; 373: 2336-2346.
21. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A, Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nature genetics. 2001; 29: 465-468.
22. Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, Nardone J, Lee K, Reeves C, Li Y, Hu Y, Tan Z, Stokes M, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007; 131: 1190-1203.
23. Charest A, Kheifets V, Park J, Lane K, McMahon K, Nutt CL, Housman D. Oncogenic targeting of an activated tyrosine kinase to the Golgi apparatus in a glioblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100: 916-921.
24. Charest A, Wilker EW, McLaughlin ME, Lane K, Gowda R, Coven S, McMahon K, Kovach S, Feng Y, Yaffe MB, Jacks T, Housman D. ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer research. 2006; 66: 7473-7481.
25. Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clinical cancer research. 2013; 19: 4040-4045.
26. Davare MA, Saborowski A, Eide CA, Tognon C, Smith RL, Elferich J, Agarwal A, Tyner JW, Shinde UP, Lowe SW, Druker BJ. Foretinib is a potent inhibitor of oncogenic ROS1 fusion proteins. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110: 19519-19524.
27. Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, Swidenbank I, Ponthan F, Kirschner-Schwabe R, Groeneveld-Krentz S, Hof J, Allan J, Harrison C, et al. Ras pathway mutations are prevalent in relapsed childhood acute lymphoblastic leukemia and confer sensitivity to MEK inhibition. Blood. 2014; 124: 3420-3430.
28. Laurent-Puig P, Pekin D, Normand C, Kotsopoulos SK, Nizard P, Perez-Toralla K, Rowell R, Olson J, Srinivasan P, Le Corre D, Hor T, El Harrak Z, Li X, et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clinical cancer research. 2015; 21: 1087-1097.
29. Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J, Buetow KH, Pui CH, Baker SD, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011; 471: 235-239.
30. Ma X, Edmonson M, Yergeau D, Muzny DM, Hampton OA, Rusch M, Song G, Easton J, Harvey RC, Wheeler DA, Ma J, Doddapaneni H, Vadodaria B, et al. Rise and fall of subclones from diagnosis to relapse in pediatric B-acute lymphoblastic leukaemia. Nature communications. 2015; 6: 6604.
31. Inthal A, Zeitlhofer P, Zeginigg M, Morak M, Grausenburger R, Fronkova E, Fahrner B, Mann G, Haas OA, Panzer-Grumayer R. CREBBP HAT domain mutations prevail in relapse cases of high hyperdiploid childhood acute lymphoblastic leukemia. Leukemia. 2012; 26: 1797-1803.
32. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012; 481: 157-163.
33. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, et al. DNMT3A mutations in acute myeloid leukemia. The New England journal of medicine. 2010; 363: 2424-2433.
34. Lunning MA, Green MR. Mutation of chromatin modifiers; an emerging hallmark of germinal center B-cell lymphomas. Blood cancer journal. 2015; 5: e361.
35. Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, Lee JC, Nicoletti R, Hatton C, et al. High-throughput oncogene mutation profiling in human cancer. Nature genetics. 2007; 39: 347-351.
36. Spinella JF, Cassart P, Garnier N, Rousseau P, Drullion C, Richer C, Ouimet M, Saillour V, Healy J, Autexier C, Sinnett D. A novel somatic mutation in ACD induces telomere lengthening and apoptosis resistance in leukemia cells. BMC cancer. 2015; 15: 621-015-1639-5.
37. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer cell. 2011; 20: 66-78.
38. Kim W, Kim R, Park G, Park JW, Kim JE. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. The Journal of biological chemistry. 2012; 287: 5588-5599.
39. Barry ER, Krueger W, Jakuba CM, Veilleux E, Ambrosi DJ, Nelson CE, Rasmussen TP. ES cell cycle progression and differentiation require the action of the histone methyltransferase Dot1L. Stem cells (Dayton, Ohio). 2009; 27: 1538-1547.
40. Fu H, Maunakea AK, Martin MM, Huang L, Zhang Y, Ryan M, Kim R, Lin CM, Zhao K, Aladjem MI. Methylation of histone H3 on lysine 79 associates with a group of replication origins and helps limit DNA replication once per cell cycle. PLoS genetics. 2013; 9: e1003542.
41. Feng Y, Yang Y, Ortega MM, Copeland JN, Zhang M, Jacob JB, Fields TA, Vivian JL, Fields PE. Early mammalian erythropoiesis requires the Dot1L methyltransferase. Blood. 2010; 116: 4483-4491.
42. Kim W, Choi M, Kim JE. The histone methyltransferase Dot1/DOT1L as a critical regulator of the cell cycle. Cell cycle (Georgetown, Tex.). 2014; 13: 726-738.
43. Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, Ouillette P, Trifonov V, Rossi D, Tabbo F, Ponzoni M, Chadburn A, Murty VV, et al. Genetics of follicular lymphoma transformation. Cell reports. 2014; 6: 130-140.
44. Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, Gine E, Pinyol M, Royo C, Nadeu F, Conde L, Juan M, Clot G, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110: 18250-18255.
45. Huether R, Dong L, Chen X, Wu G, Parker M, Wei L, Ma J, Edmonson MN, Hedlund EK, Rusch MC, Shurtleff SA, Mulder HL, Boggs K, et al. The landscape of somatic mutations in epigenetic regulators across 1,000 paediatric cancer genomes. Nature communications. 2014; 5: 3630.
46. Figueroa ME, Chen SC, Andersson AK, Phillips LA, Li Y, Sotzen J, Kundu M, Downing JR, Melnick A, Mullighan CG. Integrated genetic and epigenetic analysis of childhood acute lymphoblastic leukemia. The Journal of clinical investigation. 2013; 123: 3099-3111.
47. Andersson A, Olofsson T, Lindgren D, Nilsson B, Ritz C, Eden P, Lassen C, Rade J, Fontes M, Morse H, Heldrup J, Behrendtz M, Mitelman F, et al. Molecular signatures in childhood acute leukemia and their correlations to expression patterns in normal hematopoietic subpopulations. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 19069-19074.
48. Lee ST, Muench MO, Fomin ME, Xiao J, Zhou M, de Smith A, Martin-Subero JI, Heath S, Houseman EA, Roy R, Wrensch M, Wiencke J, Metayer C, et al. Epigenetic remodeling in B-cell acute lymphoblastic leukemia occurs in two tracks and employs embryonic stem cell-like signatures. Nucleic acids research. 2015; 43: 2590-2602.
49. Davidsson J, Lilljebjorn H, Andersson A, Veerla S, Heldrup J, Behrendtz M, Fioretos T, Johansson B. The DNA methylome of pediatric acute lymphoblastic leukemia. Human molecular genetics. 2009; 18: 4054-4065.
50. Stubbs MC, Kim W, Bariteau M, Davis T, Vempati S, Minehart J, Witkin M, Qi J, Krivtsov AV, Bradner JE, Kung AL, Armstrong SA. Selective Inhibition of HDAC1 and HDAC2 as a Potential Therapeutic Option for B-ALL. Clinical cancer research. 2015; 21: 2348-2358.
51. Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, Van Vlierberghe P, Dolgalev I, Thomas S, Aminova O, Huberman K, Cheng J, Viale A, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. The New England journal of medicine. 2012; 366: 1079-1089.
52. Yang L, Rodriguez B, Mayle A, Park HJ, Lin X, Luo M, Jeong M, Curry CV, Kim SB, Ruau D, Zhang X, Zhou T, Zhou M, et al. DNMT3A Loss Drives Enhancer Hypomethylation in FLT3-ITD-Associated Leukemias. Cancer cell. 2016; 30: 363-365.
53. Shih AH, Jiang Y, Meydan C, Shank K, Pandey S, Barreyro L, Antony-Debre I, Viale A, Socci N, Sun Y, Robertson A, Cavatore M, de Stanchina E, et al. Mutational cooperativity linked to combinatorial epigenetic gain of function in acute myeloid leukemia. Cancer cell. 2015; 27: 502-515.
54. Chang E, Ganguly S, Rajkhowa T, Gocke CD, Levis M, Konig H. The combination of FLT3 and DNA methyltransferase inhibition is synergistically cytotoxic to FLT3/ITD acute myeloid leukemia cells. Leukemia. 2016; 30: 1025-1032.
55. Bartley K, Metayer C, Selvin S, Ducore J, Buffler P. Diagnostic X-rays and risk of childhood leukaemia. International journal of epidemiology. 2010; 39: 1628-1637.
56. Aldrich MC, Zhang L, Wiemels JL, Ma X, Loh ML, Metayer C, Selvin S, Feusner J, Smith MT, Buffler PA. Cytogenetics of Hispanic and White children with acute lymphoblastic leukemia in California. Cancer epidemiology, biomarkers & prevention. 2006; 15: 578-581.
57. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014; 46: 310-315.
58. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS computational biology. 2016; 12: e1004873.
59. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic acids research. 2015; 43: D805-11.
60. Perez-Llamas C, Lopez-Bigas N. Gitools: analysis and visualisation of genomic data using interactive heat-maps. PloS one. 2011; 6: e19541.