
INTRODUCTION
Human chromogranin A (CgA) is a 439 residue-long protein stored in the dense-core granules of many neuroendocrine cells and neurons, and exocytotically released into the blood to reach about 0.5 nM levels [1, 2]. CgA is also expressed in lower amounts by granulocytes, cardiomyocytes and, in certain conditions, by keratinocyte [1, 3-5]. An increasing number of studies have shown that CgA and certain CgA-derived peptides, called vasostatin-1, pancreastatin, catestatin and serpinin, can regulate the cardiovascular system and metabolism, suggesting that this protein can exert extra-cellular functions [1]. Increased levels of CgA have been immunologically detected in the blood of patients with neuroendocrine tumors or with tumors that undergo neuroendocrine differentiation, including prostate, breast and non-small cell lung cancer [2, 6-8]. For these reasons CgA is widely used as a serological marker for neuroendocrine tumor diagnosis or for monitoring tumor progression/regression after therapy. However, patients with non-neuroendocrine tumors may also have abnormal levels of circulating CgA. For example, increased levels of immunoreactive CgA have been observed in a subpopulation of patients with non-small cell lung cancer lacking neuroendocrine cells in tumor tissues and in cancer patients treated with proton pump inhibitors, a class of drugs commonly used to treat acid peptic disorders [8-11]. Furthermore, elevated serum levels of CgA have been observed in patients with renal failure, heart failure, hypertension, rheumatoid arthritis, atrophic gastritis, inflammatory bowel disease, sepsis and other inflammatory diseases [1, 2, 8, 12-23]. Using specific assays, we recently observed that CgA is present in the blood of normal subjects and cancer patients as a mixture of full-length protein and fragments, including the N-terminal fragment CgA1-76 (vasostatin-1) and other fragments lacking part or the entire C-terminal region [24, 25]. Interestingly, the plasma levels of the fragment CgA1-373, which is not or minimally present in normal subjects, is increased in the peripheral blood (and even more in the bone marrow plasma) of patients with multiple myeloma, while full-length CgA is decreased, pointing to increased CgA C-terminal fragmentation in these patients [25]. Thus, variable levels of full-length CgA and fragments are present in the blood of cancer patients for a variety of reasons.
A central question that we have yet to answer is whether the neuroendocrine release of CgA in circulation and its fragmentation can affect the biology and progression of non-neuroendocrine tumors.
Interestingly, recent studies have shown that CgA1-439 and the N-terminal fragment CgA1-76 inhibit angiogenesis, whereas the fragment CgA1-373 stimulates angiogenesis in various angiogenesis assays [24, 26]. Furthermore, full-length CgA1-439 and CgA1-78 can protect the endothelial barrier from the disassembly of vascular endothelial-cadherin adherence junctions, gap formation, and vascular leakage [27, 28], and reduce the traffic of tumor cells through the endothelium [29]. Although these findings suggest that CgA and some of its fragments might affect tumor biology, the pathophysiological relevance of circulating full-length CgA and the potential impact of CgA fragmentation on tumor progression remains to be established.
These notions prompted us to explore the potential role of circulating CgA as a regulator of tumor growth. As vascularized tumors are exposed to CgA released in circulation by the neuroendocrine system, we decided to perform the study using various murine models of non-neuroendocrine solid tumors, including subcutaneous fibrosarcomas, Lewis lung carcinoma and mammary adenocarcinomas, as well as orthotopic subcutaneous and metastatic (lung) melanomas.
We show that full-length CgA, but not fragments lacking the C-terminal region, can inhibit tumor growth in all these models, with U-shaped dose-response curves, and we provide evidence that an active site is located in the C-terminal region. Furthermore, we show that full-length CgA can impair angiogenesis, tumor perfusion and tumor growth through mechanisms depending on the induction of protease nexin-1, a serine protease inhibitor endowed of anti-angiogenic activity.
RESULTS
Exogenous CgA exerts anti-tumor effects with U-shaped dose-response curves in various murine models
The effect of exogenous recombinant and natural CgA (produced in E.coli cells and purified from pheochromocytomas, respectively) on tumor growth was investigated in different murine models, including WEHI-164 fibrosarcoma, TSA adenocarcinoma B16-F1 melanoma, and Lewis lung carcinoma (LLC), implanted subcutaneously in syngeneic mice. Furthermore, a metastasis model based on lung colonization by B16-F10 melanoma cells injected intravenously in mice was also used. Administration of 6 to 30 pmol of human recombinant full-length CgA to WEHI-164-bearing mice (i.p., every 3-4 days, three times) significantly delayed the growth of subcutaneous tumors in a dose-dependent manner (Figure 1A, upper panels). However, increasing the dose to 150 pmol resulted, paradoxically, in a lower effect (Figure 1A, lower panels). U-shaped dose response curves were also observed in the TS/A and B16-F1 subcutaneous models, after treatment (i.p.) with 0, 30, 90 or 0, 30, 740 pmol of recombinant CgA, respectively (Figure 1B and 1C) as well as in the B16-F10 lung-colonization model, after treatment (i.v.) with 0, 4, 20 and 100 pmol of CgA (Figure 1D). Of note, the 740 pmol dose, which was aimed at exploring an extremely high concentration of CgA, was completely inactive.
Recombinant and natural CgA (30 pmol, i.p.) exerted similar effects in the WEHI-164 and LLC models (Suppl. Figure S1). Furthermore, decreasing the dose to 6 pmol reduced the anti-tumor effects in both cases (not shown). Thus, natural CgA and recombinant CgA exerted similar anti-tumor effects in these models.
Optimal activity in all subcutaneous models was achieved with i.p. injection of 30 pmol of CgA (Figure 1A-1C), whereas i.v. injection of the same dose was less effective (data not shown). Pharmacokinetic analysis showed that the i.p. route generated circulating peak levels of about 3-4 nM CgA, whereas the i.v. route generated 17 nM levels (Suppl. Figure S2). This may explain the paradoxical lower activity of the i.v. versus the i.p. route. Accordingly, in the lung-colonization model, which was based on i.v. administration, maximal activity was obtained with a lower dose of recombinant CgA (4 pmol) (Figure 1D).
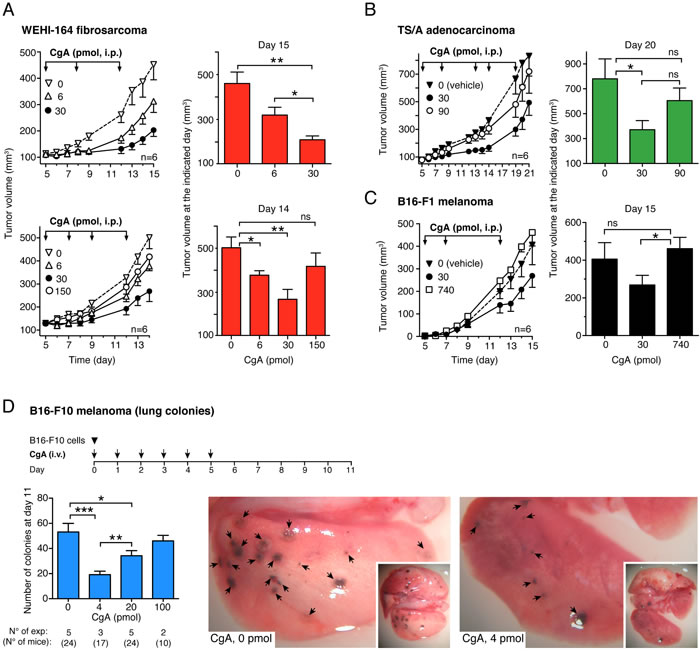
Figure 1: Effects of CgA on tumor growth in the WEHI-164 fibrosarcoma, TS/A adenocarcinoma and B16 melanoma models. A.-C. Effects of full-length CgA on the growth of subcutaneous tumors. Tumor-bearing mice were treated (i.p.) at the indicated time (arrows) after tumor implantation with the indicated doses of recombinant CgA. Tumor volumes are shown (mean±SE, n = 6 mice/group). *, P < 0.05; **, P < 0.01, by t test (2-tailed).D. Effects of CgA on lung colony formation by circulating melanoma cells.C57BL/6 mice were injected with 6x104 B16-F10 tumor cells (i.v.) and with recombinant CgA (i.v.) at the indicated time (arrows). At day 11, mice were sacrificed and the number of B16-F10 colonies in the lungs of mice was counted with the help of a stereomicroscope. Bars represent the number of colonies (mean±SE; number of mice/group reported below the graph). Representative photographs of lungs of mice treated with 0 and 4 pmol of CgA are also shown. *, P < 0.05; **, P < 0.01, and ***, P < 0.001 by t test (2-tailed).
The C-terminal region of CgA (residues 410-439) is crucial for its anti-tumor activity
We have previously shown that CgA contains an anti-angiogenic site in the C-terminal region 410-439 [24]. To assess whether the C-terminal region contributes to the anti-tumor activity of CgA we analyzed: a) the effect of antibodies against the region 410-439 on the anti-tumor activity of exogenous recombinant CgA, b) the anti-tumor activity of CgA1-409 and CgA1-373 (i.e. fragments lacking the C-terminal region), and c) the anti-tumor activity of CgA410-439 (a fragment corresponding to the C-terminal region). Anti-human CgA410-439 immunoglobulins (Igs), but not control Igs, could block the anti-tumor activity of exogenous CgA in the WEHI-164 model (Figure 2A). Furthermore, the C-terminal fragment CgA410-439, but not CgA1-373 and CgA1-409, could inhibit tumor growth (Figure 2B-2D). These findings suggest that the site responsible for the anti-tumor effects of full-length CgA is located in the C-terminal region.
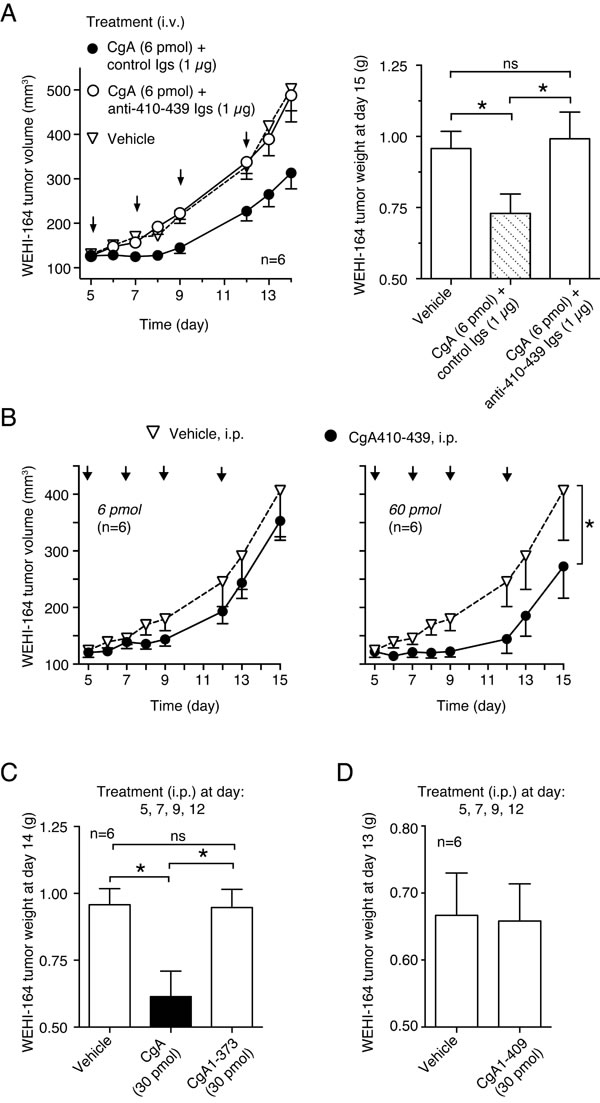
Figure 2: Role of CgA410-439 C-terminal region on the anti-tumor activity of CgA in the WEHI-164 model. A. Effect of anti-CgA410-439 antibodies on the anti-tumor activity of CgA. Tumor-bearing mice were treated (i.v.) at the indicated time (arrows) with 6 pmol of recombinant CgA, alone and in combination with purified anti-CgA410-439 immunoglobulins (Igs). Tumor volumes and weights are shown (mean±SE, n = 6 mice/group). *, P < 0.05, by t test (2-tailed). B. Anti-tumor activity of CgA410-439 peptide. Mice were treated (i.p., arrows) with the indicated doses of peptide. Tumor volume (mean ± SE). The area under the curve (from day 5 to day 13) for each mouse was calculated using the GraphPad Prism Software. Statistical analysis was performed by t test (2-tailed) on the calculated areas (6 animals per group; *, P < 0.05). C.-D. Anti-tumor activity of CgA, CgA1-409 and CgA1-373. Tumor bearing mice were treated at day 5, 7, 9, and 12 (i.p.) with 30 pmol of recombinant CgA or fragments lacking the C-terminal region CgA1-409 and CgA1-373. Tumor weights are shown (mean±SE, n = 6 mice/group). *, P < 0.05, by t test (2-tailed).
Neutralization of endogenous CgA with anti-CgA410-439 antibodies promotes tumor growth
To assess whether endogenous CgA, chronically produced and released in circulation by the neuroendocrine system, has a role in the regulation of tumor progression, we investigated the effect of CgA ablation on tumor growth in mice. Considering the crucial role of this protein in secretory granule biogenesis [30-34] and the consequent risk that genetic ablation of CgA might affect the release of several potentially relevant hormones, we decided to pursue an immunological approach based on systemic injection of antibodies against the C-terminal region of CgA (which are expected to neutralize extracellular CgA but not intra-granular CgA because of a different accessibility). Notably, the rabbit anti-human CgA410-439 Igs described above could efficiently recognize the CgA429-439 epitope of human and murine CgA (Figure 3A-3B), a sequence that is identical in these proteins (Figure 3C). Thus, this antibody was exploited to block the C-terminal region of endogenous circulating CgA in mice. Daily administration of this antibody (6 µg/mouse), but not normal rabbit Igs, significantly enhanced the growth rate of subcutaneous WEHI-164 tumors (Figure 3D). These results suggest that physiological levels of endogenous (natural) CgA can indeed reduce the growth rate of tumors.
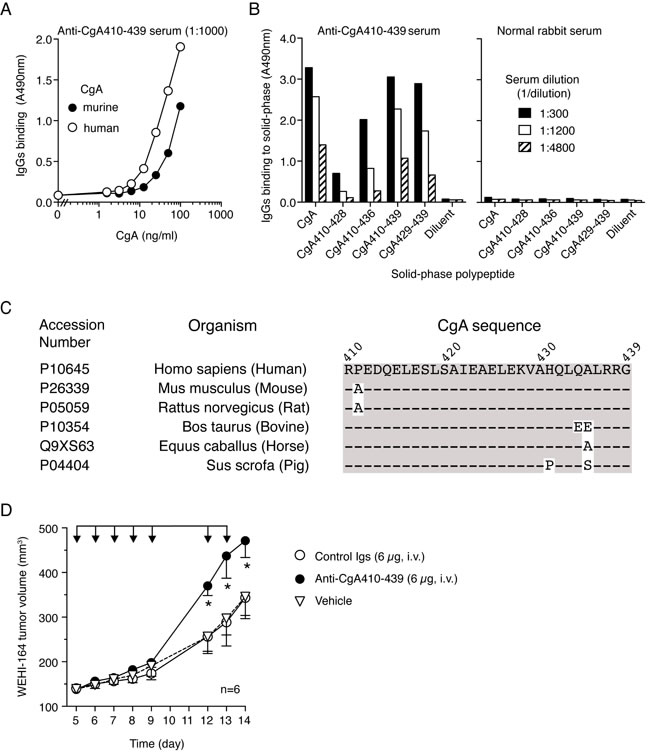
Figure 3: Neutralization of endogenous murine CgA with antibodies against the C-terminal region promotes the growth of subcutaneous WEHI-164 tumors. A. Cross-reactivity of anti-human CgA410-439 with human and murine CgA. Human and murine CgA binding assay was performed by sandwich ELISA with mAb 5A8 (cross-reactive) in the capture step and with anti-human CgA410-439 in the detection step. B. Epitope mapping of anti-CgA410-439 antibodies with synthetic peptides. Binding of anti-CgA410-439 antibodies to various peptides corresponding to the C-terminal region of CgA. Antibody binding was detected with peroxidase-labeled secondary goat-anti-rabbit antibodies. Normal rabbit serum was used as negative control. C. Alignment of CgA C-terminal sequence from the different species, showing high sequence conservation between human a murine CgA. D. Effect of anti-CgA410-439 antibodies on the growth of WEHI-164 fibrosarcomas in mice. Tumor-bearing mice were treated (i.v.) with 6 µg of purified anti-CgA410-439 or control immunoglobulins (Igs) as indicated (arrows). Tumor volumes (mean±SE, n = 6 mice/group) are shown. *, P < 0.05, by Mann Whitney test.
CgA inhibits angiogenesis in tumors with U-shaped dose-response curves
The mechanism of the anti-tumor activity of full-length CgA was then investigated. No effect on tumor cell proliferation and viability was observed with in vitro cultures of tumor cells (data not shown), suggesting the in vivo effect of CgA on tumor growth was indirect, possibly related to stromal components. To assess whether CgA could regulate tumor angiogenesis, we investigated the effect of 30 and 150 pmol doses of recombinant CgA on tumor microvessel density by immunofluorescence microscopy in the WEHI-164 model. Staining of tumor tissue sections with an anti-CD31 antibody (a marker of endothelial cells) at day 14 showed that 30 pmol, but not 150 pmol of CgA, could significantly reduce vascular density (Figure 4A-4C). Interestingly, 30 pmol CgA, but not 150 pmol (i.p.), could reduce bFGF in tumor tissue extracts, a proangiogenic cytokine (Figure 4D). These data support the hypothesis that low-dose CgA can trigger anti-angiogenic mechanisms in tumors.
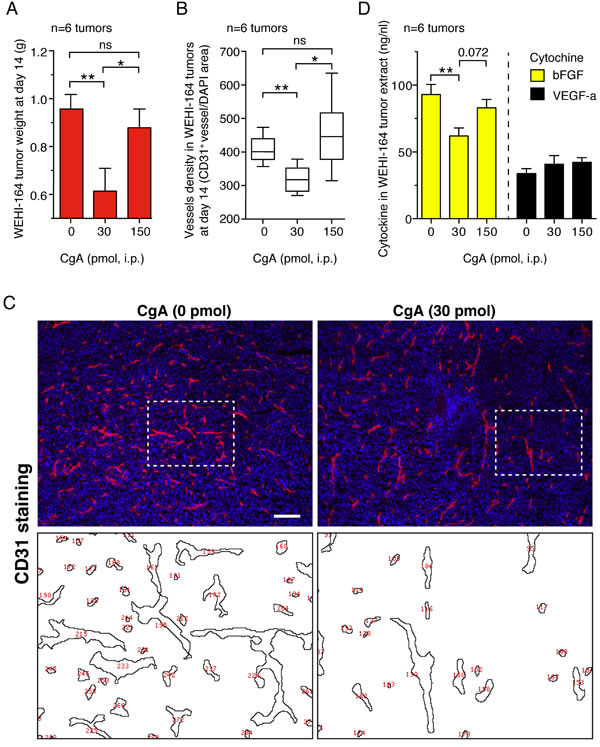
Figure 4: Effects of CgA on microvessel density and tumor growth in the murine WEHI-164 fibrosarcoma model. A.-C. Effects of CgA on tumor growth and vessel density. BALB/c mice (n = 6/group) were treated, i.p., at day 5, 7, 9, and 11 after tumor implantation, with the indicated doses of recombinant CgA. Tumors were excised and weighted. Tissue sections were stained with anti-CD31 antibody (mAb MEC 13.3) and AlexaFluor 546 goat anti-rat IgG (red, endothelial staining) and with 4,6-diamidino-2-phenylindole (DAPI) (blu, nuclear staining). Vessel density was quantified by counting the number of red spots (CD31+) in each field analyzed by fluorescence microscopy (10 fields/section, 3 sections/tumor, 6 tumors/group, see “Methods”). Tumor weight (mean±SE) (A) and vessel density (box-plots with median, interquartile and 5-95 percentile values) B. at day 14 are shown. *, P < 0.05; **, P < 0.01, by two-tailed t test. Representative images of CD31 staining (10x magnification; bar, 200 µm) C. Images obtained after elaboration for vessel counting (red numbers, corresponding to area delimited by dashed lines) are also shown. D. Effect of CgA on bFGF and VEGF-a expression in tumors. bFGF in tumor tissue extracts was analyzed by ELISA.
This view is further supported by the results of contrast enhanced ultrasound analysis of tumors (CEUS), performed on a different group of mice, showing that treatment with 30 pmol of CgA (i.p.), but not 150 pmol, could significantly reduce the tumor perfusion index (Figure 5A-5C).
The observed reduction of tumor weight, microvessel density, and perfusion by low-dose CgA suggests that regulation of angiogenesis is an important mechanism of action of its anti-tumor effects.
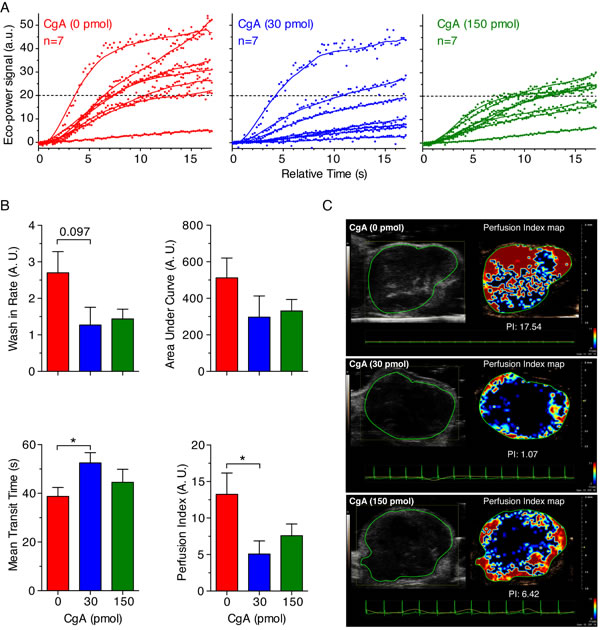
Figure 5: Effect of CgA on tumor perfusion as determined by Contrast Enhanced Ultrasound (CEUS) imaging. WEHI-tumor-bearing mice (n = 7/group) were treated, i.p., with the indicated dose of recombinant CgA at day 6, 8, and 10 after tumor implantation and analyzed by CEUS imaging at day 15-16. MicroMarker Contrast Agent was injected at time 0 and its uptake was recorded for at least 17 s. A. Wash-in curves of each mouse. B. Quantitative analysis of tumor perfusion. Quantitative analysis was performed using VevoCQTM software. Perfusion parameters (Wash-in Rates, Area Under Curve, Mean Transit Time) were calculated using a region of interest (ROI, green lines in panel C) corresponding to the entire tumor area. Perfusion index corresponds to the ratio Area Under Curve/Mean Transit Time. Bars: mean ± SE; *, P < 0.05, by Mann-Whitney test. C. Gray-scale tumor images (left) and color-coded Perfusion Index Maps of representative tumors treated with 0, 30 and 150 pmol of CgA (right). Red and blue areas correspond to high and low perfused tumor areas, respectively.
CgA and CgA410-439 inhibit capillary structure formations from cultured aorta rings with U-shaped dose-response curves
To provide further information on the mechanisms underlying the inhibition of angiogenesis by CgA in tumors, we evaluated the effect of various doses of CgA, CgA fragments and anti-CgA Igs on the spontaneous sprouting of capillary-like structures from rat aortic rings (RARs) cultured in collagen gels. Full-length CgA could exert significant inhibitory effects in this assay with a U-shaped dose-response curve, with the maximal activity being obtained with 0.2-1 nM recombinant CgA (Figure 6A). Notably, the anti-CgA410-439 Igs neutralized the effect of CgA (Figure 6B), as observed in the in vivo experiments. Furthermore, the peptide CgA410-439 recapitulated, albeit at higher doses, the U-shaped inhibitory effects of CgA (Figure 6C). Finally, no anti-angiogenic effects were observed with 1 nM CgA1-409 (Figure 6D). These data support the hypothesis that the C-terminal region of CgA contains the structural determinants of the anti-angiogenic activity and suggest that cleavage of this region causes a marked loss of anti-angiogenic potency, CgA410-439 being markedly less active than CgA1-439.
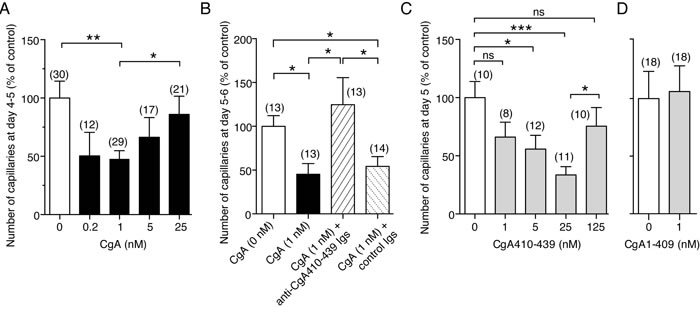
Figure 6: Effects of CgA and its C-terminal region on capillary sprouting from rat aortic rings (RAR). A.-D. Effect of CgA, CgA1-409, CgA410-439 and anti-CgA410-439 antibodies in the RAR assay. Bars represent the number of capillary-like structures emerging from the aorta rings treated as indicated, expressed as percentage of the untreated control (mean ± SE of 2-3 experiments). The number rings used (obtained from 2-4 rats) is indicated in each panel (n). *, P < 0.05; **, P < 0.01; ***, P < 0.001, by t test (2-tailed). RAR assays were performed as described previously [24, 43]. This method is based on the use of rat aorta rings cultured in three-dimensional collagen gels and on the measurement of the number of capillary-like structures sprouting from rat aorta rings after 4-5 days. Basal angiogenesis, obtained without addition of bFGF or VEGF, was examined in the absence or presence of various doses of recombinant CgA or CgA410-439. Anti-CgA410-349 and control Igs (20 µg/ml) were also added as indicated.
Endothelial cells are an important target of CgA and CgA410-439
To identify the cellular targets of the anti-tumor activity of CgA we then investigated its effects on cultured endothelial cells. Both natural and recombinant CgA could inhibit the sprouting of capillary-like structures from HUVEC spheroids cultured in three-dimensional collagen gels with U-shaped dose-response curves and similar potency (maximal activity: 0.04-0.2 nM CgA) (Figure 7A). Recombinant CgA and synthetic CgA410-439 exerted modest, albeit significant, inhibitory effects on the formation of capillary-like structures in a two-dimensional endothelial tube formation assay, again with U-shaped dose-response curves (Figure 7B). These data suggest that CgA can directly affect the physiology of endothelial cells. No effect on endothelial cell proliferation and viability was observed (not shown). Of note, the inhibitory effects observed in the tube formation assay were lower than those observed with RAR and HUVEC spheroid assays, suggesting that inhibition of three-dimensional matrix invasion is an important mechanism.
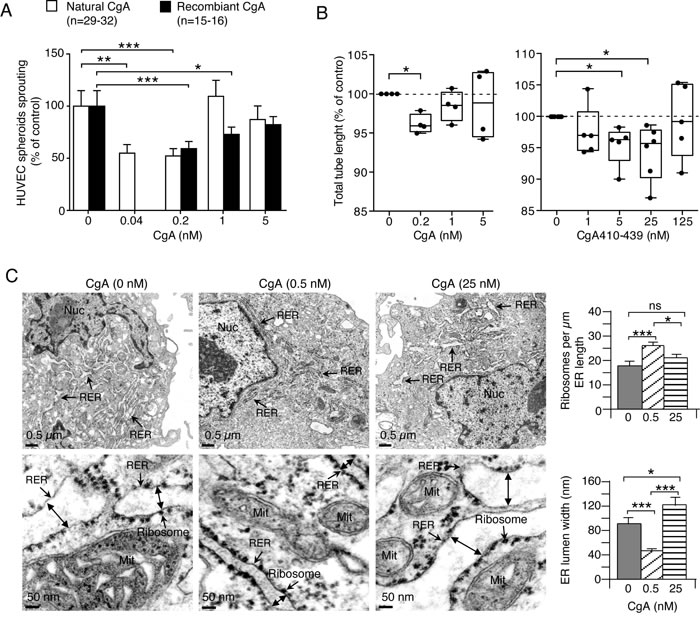
Figure 7: Effects of CgA and its C-terminal fragment on endothelial cells. A. Effect of CgA on endothelial spheroid sprouts formation. HUVEC spheroids were cultured in 3D-collagen gel in the presence of various concentrations of natural or recombinant CgA. After overnight incubation, sprouts were counted using an inverted-phase contrast microscope. Cumulative results of 2-3 experiments (8-16 spheroids/experiment). ***, P < 0.001; **, P < 0.01, *, P < 0.05, by t test. B. Effect of CgA and CgA410-439 on tube formation by cultured HUVECs. Cumulative results of 4-6 experiments (6 replicates/experiment). *, P < 0.05, by t-test (one-sample). Endothelial tube assays were performed with HUVECs and μ-Slide Angiogenesis ibiTreat plates (Ibidi) coated with 10 μl of reduced-growth factor Basement Membrane Extract (BME) (Cultrex). HUVECs (2 x 104 cells in EGM-2 medium, 50 µl/well) were seeded in the presence or absence of various doses of recombinant CgA or CgA410-439. After overnight incubation, the 2-dimensional tube organization was examined using an inverted-phase contrast microscope (LEICA, BM, IRB equipped with 10X objective lens and AxioVision acquisition software) and photographed. Photographs were then analyzed by ImageJ software using the Angiogenesis Analyzer tool (http://image.bio.methods.free.fr/ImageJ/?Angiogenesis-Analyzer-for-ImageJ.html&lang = en). C. Effect of CgA on the subcellular morphology of endothelial cells. TEM images of HUVECs after treatment with 0, 0.5, and 25 nM recombinant CgA for 12 h as indicated. Double-head arrows, ER lumen width; Mit, mitochondria; Nuc, nucleus; RER, rough endoplasmic reticulum. Histograms show quantitative descriptions of ER-bound ribosomes and ER-lumen size, as determined using NIH ImageJ software on 30 images from 15 cells (two images/cells) (right). *, P < 0.05: ***, P < 0.001, by t test (2-tailed).
CgA regulates endoplasmic-reticulum (ER) lumen width and number of ER-bound ribosomes in endothelial cells with bimodal-dose response curves
The effects of CgA at low and high concentrations on endothelial subcellular morphology were then analyzed by transmission electron microscopy. Low-dose recombinant CgA (0.5 nM) increased the number of ER-bound ribosomes/µm of ER length (by 44%), whereas high-dose CgA (25 nM) had no effect (Figure 7C). Furthermore, low-dose CgA reduced ER-lumen width (by 50%), as compared to saline-treated group, whereas high-dose CgA increased the lumen width (Figure 7C). Although the relationship between these changes in subcellular morphology and angiogenesis remains to be established, these results support the hypothesis that CgA can directly affect the physiology of endothelial cells with U-shaped dose-response curves.
CgA induces protease nexin-1 mRNA in endothelial cells with a bell-shaped dose response curve
To investigate the mechanism of the anti-angiogenic activity of CgA we then analyzed the effect of CgA on the endothelial expression of protease nexin-1 (PN-1), a serine protease inhibitor (serpin). The rationale for this study relies on the notion that a peptide corresponding to CgA411-436 can induce, at relatively high concentrations, PN-1 in AtT-20 pituitary cells [35], and that this protein is an important anti-angiogenic factor [36]. Interestingly, recombinant CgA induced the production of PN-1 mRNA in endothelial cells with a bell-shaped dose response curve, with the PN-1 mRNA expression being obtained after stimulation with 0.2 nM CgA, but not with 5 nM CgA (Figure 8A). This finding further support the concept that endothelial cells are important targets of CgA and suggest that PN-1 induction could be an important mechanism of its anti-tumor activity.
PN-1 gene silencing and anti-PN-1 antibodies inhibit the anti-angiogenic and anti-tumor activity of CgA
To assess whether the anti-angiogenic and anti-tumor activity of CgA was indeed mediated by PN-1, we investigated a) the effect of siRNA-mediated PN-1 gene silencing and of anti-PN-1 neutralizing antibodies on the anti-angiogenic activity of CgA in the endothelial-sprouting assay (spheroids), and b) the effect of anti-PN-1 neutralizing antibodies on the anti-tumor activity of CgA in the WEHI-164-fibrosarcoma and TS/A adenocarcinoma models, in vivo. PN-1-siRNA and anti-PN-1 neutralizing antibodies, but not control siRNA and antibodies, completely abolished the anti-angiogenic effect of 0.2 nM recombinant CgA (Figure 8B-8D). Furthermore, the anti-tumor effects of 30 pmol CgA were significantly inhibited by anti-PN1 antibodies, but not by control antibodies in both fibrosarcoma and adenocarcinoma models (Figure 8E). These results support the hypothesis that PN-1-induction by CgA is a crucial mechanism of its anti-tumor activity.
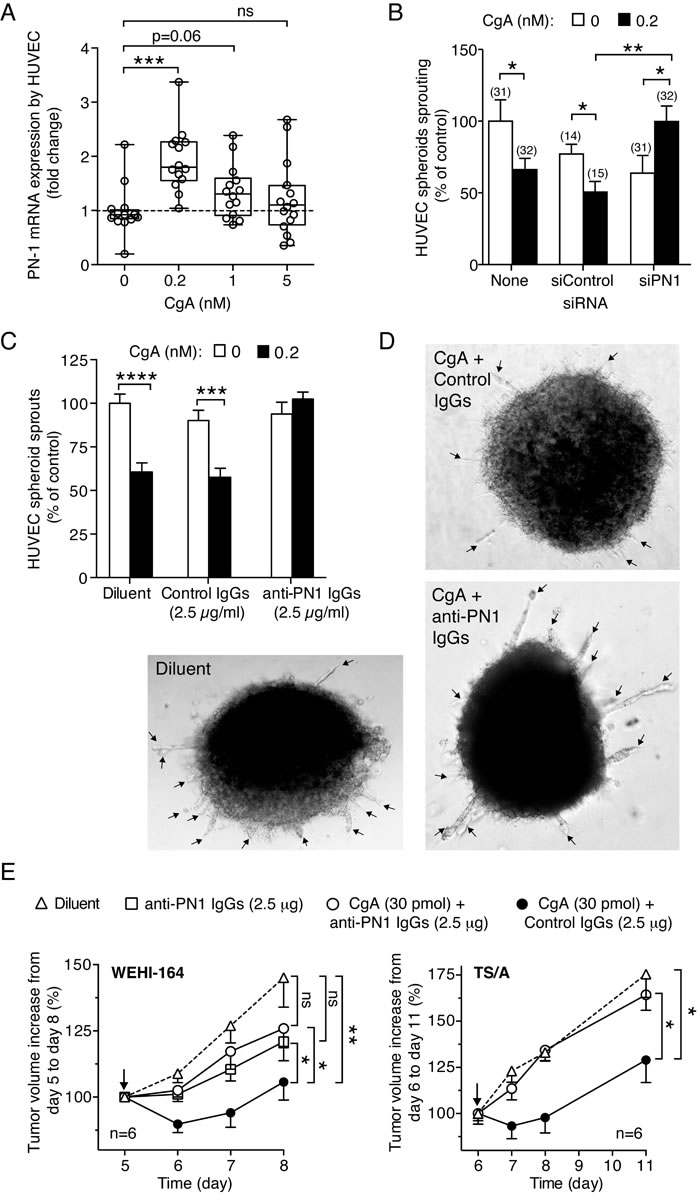
Figure 8: Role of PN-1 on CgA anti-tumor activity. A. Induction of protease nexin-1 mRNA in endothelial cells. HUVECs were treated with various doses of recombinant CgA for 20 h. The presence of PN-1 transcripts in cells was analyzed by real time PCR. PN-1 mRNA expression is reported as fold change compared to untreated cells. The cumulative results of 3 experiments are shown: in each experiment each sample was analyzed 5-6 times by real-time PCR (each in triplicate). Box-plots with median, interquartiles and 5-95 percentile values are shown (dotted line, mean value of controls). ***, P < 0.001, by t test (2-tailed). B. Effect of PN1 gene silencing on the activity of CgA in the endothelial spheroid sprouting assay. HUVEC cells spheroids were transfected with PN-1 siRNA mixture (siPN1) or with control siRNA (siCtrl) as described in Methods. After transfection, spheroids were treated with or without and 0.2 nM recombinant CgA1-439 for 16 hours. The number of sprouts was from each spheroid was then counted. Cumulative results of two independent experiments (8-16 spheroids/experiment). The number of spheroids used is indicated in each panel (n). *, P < 0.05; **, P < 0.01 by t test. C.-D. Effect of anti-PN-1 antibodies on the activity of CgA in the endothelial spheroid sprouting assay. Cumulative results of two independent experiments (8 spheroids/experiment, total n = 16). Spheroids were incubated with or without recombinant CgA (0.2 nM) in the absence or the presence of anti-PN-1 polyclonal immunoglobulins (goat IgGs anti-human Serpin E2/PN1), or control immunoglobulins (normal goat IgGs) as indicated. Bars: mean ± SE. ****, P < 0.0001, ***, P < 0.001, by t test (2-tailed). Representative microphotographs of spheroids are shown in (C) (arrows: endothelial cell sprouts). E. Effect of anti-PN-1 antibodies on the anti-tumor activity of CgA. WEHI-164- or TS/A-tumor-bearing mice were treated with recombinant CgA (i.p.) in combination with anti-PN-1, or with control immunoglobulins (normal goat IgGs) at the indicated doses. Antibodies were given 2.5 h before CgA. Arrows indicate the day of treatment. Tumor volume (mean ± SE). The area under the curve for each mouse was calculated using the GraphPad Prism Software. Statistical analysis was performed by Mann-Whitney test on the calculated areas (6 animals per group; **, P < 0.01; *, P < 0.05).
DISCUSSION
The results show that full-length CgA, a protein released in circulation by the neuroendocrine system, can regulate the growth of solid tumors. This view is supported by the results of in vivo studies performed in different murine models of non-neuroendocrine tumors, including fibrosarcoma, mammary adenocarcinoma, lung carcinoma, and primary and metastatic melanoma. In particular, the results obtained in the fibrosarcoma model show that neutralization of endogenous CgA with specific antibodies enhances tumor growth rate, whereas systemic administration of low amounts of exogenous full-length CgA reduces tumor growth. It appears, therefore, that circulating full-length CgA works as an inhibitor that delays tumor growth. Notably, dose-escalation studies showed that CgA affects tumor growth with a U-shaped dose-response curve in all tumor models studied, with the maximal inhibitory activity being obtained with biweekly injections of 30 pmol of CgA (i.p.). This dose, administered at 2-3 day intervals, generates peak plasma levels of about 3-4 nM that progressively declines to 0.5-1 nM in 7-8 h and to even lower concentrations at later time points. Considering that circulating levels of CgA in normal subject is 0.2-1 nM (most consisting of fragments lacking the C-terminal region and less than 10-20% consisting of full-length CgA) [25], the biological effects observed in our models are likely patho-physiologically relevant.
What is the mechanism of the anti-tumor activity of full-length CgA? The lack of effects on tumor cell proliferation and viability in vitro argues against the hypothesis that neoplastic cells are primary targets of CgA in our models. More likely, stromal components of the neoplastic tissues are affected. According to this view, the results of in vivo mechanistic studies (by immunohistochemistry and contrast enhanced ultrasound analysis) show that CgA can reduce microvessel density and tumor perfusion, suggesting that CgA targets the tumor vasculature. Notably, the U-shaped inhibitory effects on angiogenesis mirror those on tumor mass, supporting the hypothesis that reduction of angiogenesis is indeed an important mechanism of the anti-tumor activity of CgA. This view is also supported by the results of in vitro angiogenesis assays, showing that CgA inhibits capillary sprouting from rat aortic rings and HUVEC spheroids with U-shaped dose-response curve, maximal activity being obtained with 0.1-0.2 nM CgA (i.e. with physiologically relevant concentrations).
The results of studies aimed at elucidating the structure-function relationships of CgA show that the C-terminal region (residues 410-439) contains the active site. Notably, antibodies against this region, which is highly conserved in mice, humans and other species (see Figure 3C), could neutralize the anti-tumor activity of both endogenous and exogenous CgA. Furthermore, a synthetic peptide corresponding to this sequence could inhibit tumor growth and recapitulate the U-shaped inhibitory effects induced by recombinant full-length CgA in the in vitro angiogenesis assay. However, it is noteworthy that maximal activity of CgA410-439 in the RAR assay was achieved with 5-25 nM, i.e. with 25-fold higher concentration compared to full-length CgA. Possibly, the conformational structure of this peptide is less stable than that of the cognate sequence of full-length CgA. This finding may have important biological implications, as proteolytic cleavage of CgA C-terminal region might cause a marked loss of anti-angiogenic potency. These data also imply that the production of factors capable of promoting or inhibiting CgA cleavage in tumors, such as specific proteases or protease inhibitors, may be relevant for the anti-angiogenic activity of circulating CgA. Regarding C-terminal fragmentation of CgA it might be relevant also to note that the human and murine CgA sequence contains a proline or an alanine, respectively, at position 411, which may cause differential susceptibility to proteases and, consequently, generation of C-terminal peptides of different length and conformational stability.
What are the cellular targets of the CgA anti-angiogenic activity? The finding that CgA can inhibit endothelial cell sprouting and capillary tube formation with U-shaped dose-response curves suggests that the endothelium is an important target of CgA. Regulation of its physiology might be, therefore an important mechanism of its anti-tumor activity. Notably, in these cells CgA regulated, with U-shaped dose-response curves, the number of ER-bound ribosomes and the ER-lumen size. Although the relationships between these morphological changes with angiogenesis remains to be elucidated, these findings support the concept that CgA can directly affect the physiology of endothelial cells. This hypothesis is further supported by the observation that CgA can also induce in endothelial cells the protease nexin-1 (PN-1) mRNA transcript, a serine protease inhibitor (serpin), with a bell-shaped dose-response curve (maximal activity, 0.2 nM). Remarkably, recent studies have shown that PN-1 inhibits endothelial cell migration, capillary tube formation and angiogenesis in various in vitro and in vivo assays, independently of its serpin activity [36], and that overexpression of PN-1 inhibits angiogenesis and prostate adenocarcinomas growth in mice [37]. Thus, the induction of PN-1 by CgA may contribute to its anti-angiogenic and anti-tumor activities. This view is supported by our observation that anti-PN1 antibodies could neutralize the anti-angiogenic activity of CgA in vitro as well as the anti-tumor activity of CgA in different in vivo models. Moreover, considering that CgA is a good substrate of thrombin and plasmin [24, 25], often present in tumors, and that PN-1 is a potent inhibitor of these proteases [38], it is tempting to speculate that PN-1 may also serve to prevent the local cleavage of CgA in tumors (thereby preserving its anti-angiogenic activity) and/or other factors necessary for tumor progression and invasion. Finally, the bell-shaped dose-response curve and the consequent lack of PN1 induction by high concentrations of CgA may explain the lack of anti-tumor effects with high-dose CgA.
In conclusion, the results suggest that circulating full-length CgA can impair angiogenesis and tumor growth, and that cleavage of its C-terminal region markedly reduces its activity. Although CgA is a widely studied histopathological and serological marker for neuroendocrine tumors, this is the first evidence that circulating full-length CgA, released in the blood by the neuroendocrine system, has a functional role in non-neuroendocrine tumors. This may have important pathophysiological, prognostic and therapeutic implications. First, a decrease of CgA blood levels and/or an increase of its fragmentation might promote disease progression. Thus, pharmacological treatments aimed at restoring the balance of pro-/anti-angiogenic forms of CgA in patients might represent novel therapeutic strategies. Second, detection of full-length CgA levels and its fragmentation might have a diagnostic/prognostic value. At this regard, we have recently shown that fragmentation of the CgA C-terminal region in the bone marrow of multiple myeloma patients is associated with increased micro-vessel density, a known marker of disease activity [25], and is a prognostic indicator of poor outcome in patients with pancreatic adenocarcinoma (manuscript in preparation).
Finally, it is important to say that the observations reported here, obtained with non-neuroendocrine tumor models, cannot be extrapolated to neuroendocrine tumors, as neuroendocrine tumor-derived CgA might reach local concentrations out of the range studied here.
MATERIALS AND METHODS
Cell lines and reagents
Human umbilical vein endothelial cells (HUVECs) were from Lonza, Walkersville, MD) and cultured as recommended by the supplier. Murine WEHI-164 fibrosarcoma, B16-F1 and B16-F10 melanoma, and TS/A mammary adenocarcinoma cells were cultured in DMEM or RPMI with standard supplements. Recombinant human full-length CgA1-439, CgA1-409, CgA1-373, and synthetic CgA410-439 were prepared as described [24]. Natural human CgA was purified fron pheochromocytoma tissue extract as described [39]. Monoclonal antibodies (mAbs) B4E11 (epitope: CgA68-71) and 5A8 (epitope: CgA53-57) were prepared as described previously [40, 41]. Anti-human CgA and anti-CgA410-439, were raised in rabbits by immunization with human recombinant CgA1-439, or with a synthetic peptide encompassing the sequence CgA410-439 coupled to keyhole limpet hemocyanin (Primm, Italy). Rabbit anti-CgA410-439 immunoglobulins (Igs) were purified by affinity chromatography using the CgA410-439 peptide coupled to Activated CH-Sepharose (GE, Amersham). Goat IgGs anti-mouse serpin E2/PN1, goat IgGs anti-human serpin E2/PN1 and normal goat immunoglobulins were from R&D System.
ELISA
Endogenous CgA present in circulation in mice was detected by sandwich ELISA based on the use of mAb 5A8 (cross-reactive with murine CgA) and a rabbit polyclonal antibody against recombinant murine CgA in the capturing and detection steps, respectively. Exogenous human CgA in murine plasma samples was detected by a similar sandwich ELISA based on the use of mAb B4E11 (specific for human CgA) and rabbit polyclonal antibodies against human CgA, as described [12].
In vivo studies
Studies in animal models were approved by the Ethical Committee of the San Raffaele Scientific Institute and done according to the prescribed guidelines.
BALB/c and C57BL/6/N female mice (6-7 weeks old, from Charles River Laboratories, Calco, Italy) were challenged with s.c. injection in the left flank of 2x105 B16-F1 melanoma cells (C57BL/6/N), 2x105 TS/A cells (BALB/c), or 1.5x106 WEHI-164 cells (BALB/c). Mice were treated intra-peritoneally (i.p.) with full-length CgA1-439, CgA1-409, CgA1-373 or CgA410-439 in 0.9% sodium chloride containing 100 μg/ml endotoxin-free human serum albumin. Drug doses and treatment schedules for each experiment are reported in figures and figure legends. Tumor growth was monitored daily by measuring the tumor size with calipers [24].
Contrast enhanced ultrasound analysis of tumors
Contrast enhanced ultrasound (CEUS) was performed on BALB/c mice treated at day 6, 8, and 10 after tumor implantation, with 0, 30 and 150 pmol of CgA (i.p.), respectively. Analysis was performed using a high-performance ultrasonographic scanner designed for small animal imaging (Vevo 2100; Visual Sonics, Toronto, Canada). Mice were anesthetized with isoflurane (flurane; Isoba, Schering-Plough, USA, 4% in oxygen for induction, 2% for maintenance at a rate of 1 l/min). The tail vein was catheterized before imaging using a 27-gauge and one-half inch butterfly catheter. Mice were positioned in a supine position. ECG and respiration rate were monitored during the examination. CEUS studies were performed in contrast mode using the MS250 linear transducer (fc = 21 MHz, 13-24 MHz; Vevo 2100) during intravenous bolus injection of Vevo MicroMarker untargeted ultrasound contrast agent (CA) (Bracco, Geneva, Switzerland; VisualSonics), prepared in agreement with the specifications of the producer. A total volume of 50 µl of microbubble suspension (4.3x107 microbubbles/bolus) was injected in 4 seconds. Sonographic data acquisition started immediately after CA injection and 17 s cine loops of contrast wash-in were acquired. Imaging parameters were held constant throughout each experiment to reduce variability (transmit power 4%; dynamic range 40 dB; center frequency 18 MHz; frame rate 20 Hz; contrast gain 53 dB; gate 4; beam-width standard). Recorded cine loops were processed for time-intensity curve (TIC) analysis using VevoCQ™ Advanced Contrast Quantification Software (Visual Sonics). A region of interest (ROI) was drawn along the perimeter of each tumor. The time-intensity curve for each imaging protocol was plotted and a mathematical equation model based on log-normal distribution function was used to fit the contrast uptake time-intensity curve [42]. Perfusion parameters extracted from the fitted model were the Area Under the Curve (AUC), the wash-in rate (WiR) defined as the maximum slope of the fitted curve, the mean transit time (mTT) defined as the average time required for the contrast agent to pass through the ROI and perfusion index (PI) defined as AUC divided by mTT.
Tumor vasculature density analysis
Tumor vessel density was estimated by CD31-immunofluorescence staining of tumor tissue sections (7 µm, prepared by cutting tumors at three different levels) as previously described [24]. Each section was analyzed using an Axioplan2 fluorescence microscope (Zeiss, equipped with 10X objective lens and AxioVision acquisition software). Images of tissue sections (10 fields/section, 3 sections/tumor, 6 tumors/group) were randomly acquired. Vessel density was quantified by counting the number of red spots (CD31+) in each image using the ImageJ software (NIH, USA) as follows: each image was thresholded to highlight structures corresponding to CD31+ vessels; the resulting image was processed with the command “Binary” and “Close-” to link adjacent structures. The number of the resulting structures was then electronically counted and normalized on DAPI staining.
Real-time PCR
HUVECs were seeded in 12-well microtiter-plates. Six hours later, the cells were incubated for 20 h with various amounts of full-length CgA in cell culture medium consisting of 30% EGM-2 and 70% EBM-2 mixture. Total RNA was isolated from HUVECs using RNeasy Mini Kit and reverse transcribed with QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA). Real-time PCR was performed, in triplicate on each sample, using the 7900HT Fast Real-Time PCR System and SYBR® Green PCR Master Mix (Applied Biosystems®, Foster City, CA) according to the manufacturer’s instructions. Specific primer sequences were as follows: PN-1-forward 5’-CCGCTGTCTGCCATCATCC-3’, PN-1-reverse 5’-AGAACTTTCAGCGGCTCCTT-3’. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a reference gene employing the following primers: GAPDH-forward 5’-AACGGATTTGGTCGTATTG-3’, GAPDH-reverse 5’-GGAAGATGGTGATGGGATT-3’. Relative mRNA expression levels were calculated using the 2-ΔCt method.
Transmission electron microscopy (TEM)
HUVECs were fixed with Trump’s fixative (a mixture of 4% paraformaldehyde and 1% glutaraldehyde in 0.1 M cacodylate buffer) and post-fixed in 1% OsO4 in 0.1 M cacodylate buffer at 4°C. This was followed by staining with 2% uranyl acetate at 4oC, serial ethanol dehydrating, and lastly embedding as monolayers in Durcupan. Sections (50 to 60 nm) were cut using a Leica UCT ultra-microtome, and collected on 300 mesh grids. Grids were viewed using a JEOL 1200EX II (JEOL, Peabody, MA) TEM and photographed using a Gatan digital camera (Gatan, Pleasanton, CA). ER lumen width, length of the ER membrane on both sides of the ER lumen and the number of ER-bound ribosomes/µm of ER membrane were determined using NIH ImageJ software (30 images from 15 cells).
Endothelial spheroid sprouting assay
To generate multicellular spheroids, HUVEC were resuspended in EBM-2 medium containing 5% FBS and 5% (w/v) carboxymethylcellulose (Sigma Aldrich,), seeded in a round-bottom Corning Spheroid 96-well microplate (4x104 cells/well, 30 µl), and incubated overnight at 37°C, 5% CO2. Endothelial cell spheroids and their supernatant were transferred using a multichannel pipette into a 96-well flat-bottom microtiter plate (Corning™ 3631) containing CgA solutions (10 µl/well) in EBM-2 supplemented with 5% FBS. The spheroids were then mixed with a collagen solution (40 µl/well) consisting of EBM-2 supplemented with 0.5% carboxymethylcellulose, 10% Medium 199 (10x), 0.02 N sodium hydroxide, 5% of FBS, 0.2% of collagen-R (Serva). Spheroids were then incubated overnight at 37°C, 5% CO2, to allow sprout formation. Sprouts were counted using a ZEISS Axio Observer.Z1 microscope (10x magnification).
PN-1 gene silencing in HUVEC spheroids
Human PN1(rGrGrArAr CrArGrArCrUrCrGrArUrGrCrArArGrUrGrUrUTC andrUrGrUrArC rUrGrArGrGrArArUrGrArArUrArGrArArArGGC) (siPN1) and scrambled (rCrGrUrUr ArArUrCrGrCrGrUrArUrArArUrArCrGrCrGrUAT) (siCtrl) siRNAs were synthesized by Origene (Rockville, MD). Paired siRNAs were first heated to 94˚C for 2 min and then cooled to room temperature for annealing. HUVEC spheroids were mixed with 20 ml of Opti-MEM containing 20 nM siRNA, 0.35 ml lipofectamine RNAiMAX transfection reagent (Invitrogen) and incubated for 6 h (final volume of 50 ml). PN1 gene silencing was confirmed by PCR using the PN1 and housekeeping GAPDH (control) primers described above.
ACKNOWLEDGMENTS
TEM studies were carried out at the TEM Core Facility at the University of California, San Diego. We thank Sumana Mahata for editing the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This work was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC, Special Program Molecular Clinical Oncology 5x1000-9965 and IG-14338). S.K.M. is supported by the VA Research Career Scientist Award.
Author contributions
MB, AG, AD, AS, BC, MF and ED conducted the experiments and analyzed data. SS and SM performed transmission electron microscopy experiments and analyzed data. LP, MV, CT and AE performed CEUS experiments and analyzed data. RB discussed and analyzed data. FC and AC designed the research studies and wrote the manuscript with comments from all other authors.
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
REFERENCES
1. Helle KB, Corti A, Metz-Boutigue MH and Tota B. The endocrine role for chromogranin A: a prohormone for peptides with regulatory properties. Cell Mol Life Sci. 2007; 64:2863-2886.
2. Taupenot L, Harper KL and O’Connor DT. The chromogranin-secretogranin family. N Engl J Med. 2003; 348:1134-1149.
3. Pieroni M, Corti A, Tota B, Curnis F, Angelone T, Colombo B, Cerra MC, Bellocci F, Crea F and Maseri A. Myocardial production of chromogranin A in human heart: a new regulatory peptide of cardiac function. Eur Heart J. 2007; 28:1117-1127.
4. Radek KA, Lopez-Garcia B, Hupe M, Niesman IR, Elias PM, Taupenot L, Mahata SK, O’Connor DT and Gallo RL. The neuroendocrine peptide catestatin is a cutaneous antimicrobial and induced in the skin after injury. J Investig Dermatol. 2008; 128:1525-1534.
5. Curnis F, Gasparri AM, Longhi R, Colombo B, D’Alessio S, Pastorino F, Ponzoni M and Corti A. Chromogranin A binds to alphavbeta6-integrin and promotes wound healing in mice. Cell Mol Life Sci. 2012; 69:2791-2803.
6. Portela-Gomes GM, Grimelius L, Wilander E and Stridsberg M. Granins and granin-related peptides in neuroendocrine tumours. Regul Pept. 2010; 165:12-20.
7. Guerin M, Guillemot J, Thouennon E, Pierre A, El-Yamani FZ, Montero-Hadjadje M, Dubessy C, Magoul R, Lihrmann I, Anouar Y and Yon L. Granins and their derived peptides in normal and tumoral chromaffin tissue: Implications for the diagnosis and prognosis of pheochromocytoma. Regul Pept. 2010; 165:21-29.
8. Corti A. Chromogranin A and the tumor microenvironment. Cell Mol Neurobiol. 2010; 30:1163-1170.
9. Gregorc V, Spreafico A, Floriani I, Colombo B, Ludovini V, Pistola L, Bellezza G, Vigano MG, Villa E and Corti A. Prognostic value of circulating chromogranin A and soluble tumor necrosis factor receptors in advanced nonsmall cell lung cancer. Cancer. 2007; 110:845-853.
10. Giusti M, Sidoti M, Augeri C, Rabitti C and Minuto F. Effect of short-term treatment with low dosages of the proton-pump inhibitor omeprazole on serum chromogranin A levels in man. Eur J Endocrinol. 2004; 150:299-303.
11. Sanduleanu S, Stridsberg M, Jonkers D, Hameeteman W, Biemond I, Lundqvist G, Lamers C and Stockbrugger RW. Serum gastrin and chromogranin A during medium- and long-term acid suppressive therapy: a case-control study. Aliment Pharmacol Ther. 1999; 13:145-153.
12. Ceconi C, Ferrari R, Bachetti T, Opasich C, Volterrani M, Colombo B, Parrinello G and Corti A. Chromogranin A in heart failure; a novel neurohumoral factor and a predictor for mortality. Eur Heart J. 2002; 23:967-974.
13. Ligumsky M, Lysy J, Siguencia G and Friedlander Y. Effect of long-term, continuous versus alternate-day omeprazole therapy on serum gastrin in patients treated for reflux esophagitis. J Clin Gastroenterol. 2001; 33:32-35.
14. Waldum HL and Brenna E. Personal review: is profound acid inhibition safe? Aliment Pharmacol Ther. 2000; 14:15-22.
15. Peracchi M, Gebbia C, Basilisco G, Quatrini M, Tarantino C, Vescarelli C, Massironi S and Conte D. Plasma chromogranin A in patients with autoimmune chronic atrophic gastritis, enterochromaffin-like cell lesions and gastric carcinoids. Eur J Endocrinol. 2005; 152:443-448.
16. Di Comite G, Rossi CM, Marinosci A, Lolmede K, Baldissera E, Aiello P, Mueller RB, Herrmann M, Voll RE, Rovere-Querini P, Sabbadini MG, Corti A and Manfredi AA. Circulating chromogranin A reveals extra-articular involvement in patients with rheumatoid arthritis and curbs TNF-alpha-elicited endothelial activation. J Leukoc Biol. 2009; 85:81-87.
17. Chen Y, Rao F, Wen G, Gayen JR, Zhang K, Vaingankar SM, Biswas N, Mahata M, Friese RS, Fung MM, Salem RM, Nievergelt C, Bhatnagar V, et al. Naturally occurring genetic variants in human chromogranin A (CHGA) associated with hypertension as well as hypertensive renal disease. Cell Mol Neurobiol. 2010; 30:1395-1400.
18. Zissimopoulos A, Vradelis S, Konialis M, Chadolias D, Bampali A, Constantinidis T, Efremidou E and Kouklakis G. Chromogranin A as a biomarker of disease activity and biologic therapy in inflammatory bowel disease: a prospective observational study. Scand J Gastroenterol. 2014; 49:942-949.
19. Sciola V, Massironi S, Conte D, Caprioli F, Ferrero S, Ciafardini C, Peracchi M, Bardella MT and Piodi L. Plasma chromogranin a in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2009; 15:867-871.
20. Corti A and Ferrero E. Chromogranin A and the endothelial barrier function. Curr Med Chem. 2012; 19:4051-4058.
21. Corti A, Ferrari R and Ceconi C. Chromogranin A and tumor necrosis factor-alpha (TNF) in chronic heart failure. Adv Exp Med Biol. 2000; 482:351-359.
22. Castoldi G, Antolini L, Bombardi C, Perego L, Mariani P, Vigano MR, Torti G, Casati M, Corti A, Zerbini G, Valsecchi MG and Stella A. Oxidative stress biomarkers and chromogranin A in uremic patients: effects of dialytic treatment. Clin Biochem. 2010; 43:1387-1392.
23. Zhang D, Lavaux T, Sapin R, Lavigne T, Castelain V, Aunis D, Metz-Boutigue MH and Schneider F. Serum concentration of chromogranin A at admission: an early biomarker of severity in critically ill patients. Ann Med. 2009; 41:38-44.
24. Crippa L, Bianco M, Colombo B, Gasparri AM, Ferrero E, Loh YP, Curnis F and Corti A. A new chromogranin A-dependent angiogenic switch activated by thrombin. Blood. 2013; 121:392-402.
25. Bianco M, Gasparri AM, Colombo B, Curnis F, Girlanda S, Ponzoni M, Bertilaccio MTS, Calcinotto A, Sacchi A, Ferrero E, Ferrarini M, Chesi M, Bergsagel PL, et al. Chromogranin A Is Preferentially Cleaved into Proangiogenic Peptides in the Bone Marrow of Multiple Myeloma Patients. Cancer Res. 2016; 76:1781-1791.
26. Theurl M, Schgoer W, Albrecht K, Jeschke J, Egger M, Beer AG, Vasiljevic D, Rong S, Wolf AM, Bahlmann FH, Patsch JR, Wolf D, Schratzberger P, et al. The neuropeptide catestatin acts as a novel angiogenic cytokine via a basic fibroblast growth factor-dependent mechanism. Circ Res. 2010; 107:1326-1335.
27. Ferrero E, Scabini S, Magni E, Foglieni C, Belloni D, Colombo B, Curnis F, Villa A, Ferrero ME and Corti A. Chromogranin A protects vessels against tumor necrosis factor alpha-induced vascular leakage. FASEB J. 2004; 18:554-556.
28. Dondossola E, Gasparri AM, Colombo B, Sacchi A, Curnis F and Corti A. Chromogranin A restricts drug penetration and limits the ability of NGR-TNF to enhance chemotherapeutic efficacy. Cancer Res. 2011; 71:5881-5890.
29. Dondossola E, Crippa L, Colombo B, Ferrero E and Corti A. Chromogranin A regulates tumor self-seeding and dissemination. Cancer Res. 2012; 72:449-459.
30. Kim T, Tao-Cheng JH, Eiden LE and Loh YP. Chromogranin A, an “on/off”; switch controlling dense-core secretory granule biogenesis. Cell. 2001; 106:499-509.
31. Montesinos MS, Machado JD, Camacho M, Diaz J, Morales YG, Alvarez de la Rosa D, Carmona E, Castaneyra A, Viveros OH, O’Connor DT, Mahata SK and Borges R. The crucial role of chromogranins in storage and exocytosis revealed using chromaffin cells from chromogranin A null mouse. J Neurosci. 2008; 28:3350-3358.
32. Bartolomucci A, Possenti R, Mahata SK, Fischer-Colbrie R, Loh YP and Salton SR. The extended granin family: structure, function, and biomedical implications. Endocr Rev. 2011; 32:755-797.
33. Courel M, Rodemer C, Nguyen ST, Pance A, Jackson AP, O’Connor D T and Taupenot L. Secretory granule biogenesis in sympathoadrenal cells: identification of a granulogenic determinant in the secretory prohormone chromogranin A. J Biol Chem. 2006; 281:38038-38051.
34. Elias S, Delestre C, Ory S, Marais S, Courel M, Vazquez-Martinez R, Bernard S, Coquet L, Malagon MM, Driouich A, Chan P, Gasman S, Anouar Y, et al. Chromogranin A induces the biogenesis of granules with calcium- and actin-dependent dynamics and exocytosis in constitutively secreting cells. Endocrinology. 2012; 153:4444-4456.
35. Koshimizu H, Cawley NX, Kim T, Yergey AL and Loh YP. Serpinin: a novel chromogranin A-derived, secreted peptide up-regulates protease nexin-1 expression and granule biogenesis in endocrine cells. Mol Endocrinol. 2011; 25:732-744.
36. Selbonne S, Azibani F, Iatmanen S, Boulaftali Y, Richard B, Jandrot-Perrus M, Bouton MC and Arocas V. In vitro and in vivo antiangiogenic properties of the serpin protease nexin-1. Mol Cell Biol. 2012; 32:1496-1505.
37. McKee CM, Xu D, Cao Y, Kabraji S, Allen D, Kersemans V, Beech J, Smart S, Hamdy F, Ishkanian A, Sykes J, Pintile M, Milosevic M, et al. Protease nexin 1 inhibits hedgehog signaling in prostate adenocarcinoma. J Clin Investig. 2012; 122:4025-4036.
38. Bouton MC, Boulaftali Y, Richard B, Arocas V, Michel JB and Jandrot-Perrus M. Emerging role of serpinE2/protease nexin-1 in hemostasis and vascular biology. Blood. 2012; 119:2452-2457.
39. Corti A, Gasparri A, Chen FX, Pelagi M, Brandazza A, Sidoli A and Siccardi AG. Characterisation of circulating chromogranin A in human cancer patients. Br J Cancer. 1996; 73:924-932.
40. Corti A, Longhi R, Gasparri A, Chen F, Pelagi M and Siccardi AG. Antigenic regions of human chromogranin A and their topographic relationships with structural/functional domains. Eur J Biochem. 1996; 235:275-280.
41. Ratti S, Curnis F, Longhi R, Colombo B, Gasparri A, Magni F, Manera E, Metz-Boutigue MH and Corti A. Structure-activity relationships of chromogranin A in cell adhesion. Identification and characterization of an adhesion site for fibroblasts and smooth muscle cells. J Biol Chem. 2000; 275:29257-29263.
42. Needles A, Arditi M, Rognin NG, Mehi J, Coulthard T, Bilan-Tracey C, Gaud E, Frinking P, Hirson D and Foster FS. Nonlinear contrast imaging with an array-based micro-ultrasound system. Ultrasound Med Biol. 2010; 36:2097-2106.
43. Go RS and Owen WG. The rat aortic ring assay for in vitro study of angiogenesis. Methods Mol Med. 2003:59-64.