
INTRODUCTION
Cervical cancer (CC) is one of the most common gynecologic cancers worldwide [1], with an estimated 527,600 new cases and 265,700 deaths each year [2]. The development of CC is considered as a continuous process from normal epithelium to squamous intra-epithelial lesion (SIL) and ultimately to invasive carcinoma [3]. SIL, the precursor lesions of CC, can be further divided into low-grade SIL (LSIL) and high-grade SIL (HSIL) depending on the risk of cancer progression [4]. Although infection with human papillomavirus (HPV) is a widely accepted risk factor for SIL and CC [5], the evidence that only a small subset of HPV-induced lesions progress to CC [6], suggests that HPV infection is essential but insufficient for cervical carcinogenesis [4].
DNA hypermethylation, the major epigenetic event in humans, can occur at CPG islands within promoter regions of tumor suppressor genes (TSGs), and consequently silence the TSGs’ transcription [7]. P16INK4a gene, a well known TSG, has been widely investigated in cervical cancer due to its downregulation in cell cycle [8]. Impaired P16INK4a gene function caused by promoter hypermethylation could result in uncontrolled cell proliferation and eventually oncogenesis [9-11]. In 1999, Wong et al. first reported that P16INK4a promoter hypermethylation was correlated with the advanced stage of CC [11]. Thereafter, numerous studies were carried out to assess the associations of P16INK4a hypermethylation with the development of SIL and CC. However, most of these studies only included relatively small sample size, leading to inconsistent results and a broad range of P16INK4a hypermethylation rates (from 2% to 93%) in cancer tissues [12, 13]. Moreover, the effect of P16INK4a promoter hypermethylation on different phases of cervical carcinogenesis (from LSIL to CC) is less summarized. Thus, a meta-analysis was conducted to systematically appraise the associations of P16INK4a methylation status with LSIL, HSIL, CC and their clinicopathological features.
RESULTS
Study characteristics
According to the definitions of the 2001 Bethesda System [14], LSIL encompassed cytopathic effects of HPV, mild dysplasia and cervical intraepithelial neoplasia (CIN) 1; HSIL contained moderate or severe dysplasia, carcinoma in situ (CIS) and CIN 2 or 3; CC encompassed squamous cell carcinoma (SCC) and adenocarcinoma (AdC). Based on these definitions, 43 articles were initially selected. Then, 19 articles were excluded due to in vitro experiments (n = 3), family-based designs (n = 2), abstracts (n = 2) or reviews (n = 8), non-English papers (n = 2) and insufficient data (n = 2). Manual search of references cited in the published articles identified four additional articles [15-18]. One article [19] contained data from two independent studies. Hence, 28 articles with 29 studies were finally included [11-13, 15-39]. Among these studies, all studies were eligible to estimate the P16INK4a hypermethylation rates; 20 studies (1 cross-sectional [13] and 19 case-control designs [16, 17, 19, 21-28, 30-35, 37, 38]) investigated the associations of P16INK4a methylation status with the risk of LSIL, HSIL and CC; 1254 SIL/CC patients from 18 studies (11 case-control studies [19, 21, 23, 25, 26, 31, 32, 35-38] and 7 case-only studies [11, 12, 15, 18, 20, 29, 39]) were eligible to assess the associations between P16INK4a methylation status and clinicopathological features. For most of these studies (26 studies), the methylation detection was based on methylation-specific PCR (MSP) (including MSP, nested MSP and MSP with another method (sequencing, prosequencing and BSP) for quality control). Only one study used plasma samples to detect methylation status [19]; other studies involved cervical tissues. Fifteen studies were conducted on Asians, 9 studies on Caucasians, 5 studies on other ethnicities (Brazilians, Moroccans and Senegalese). The flowchart for the study selection procedure was shown in Figure 1. The characteristics of included studies were summarized in Table 1.
Table 1: Characteristics of included studies in this meta-analysis
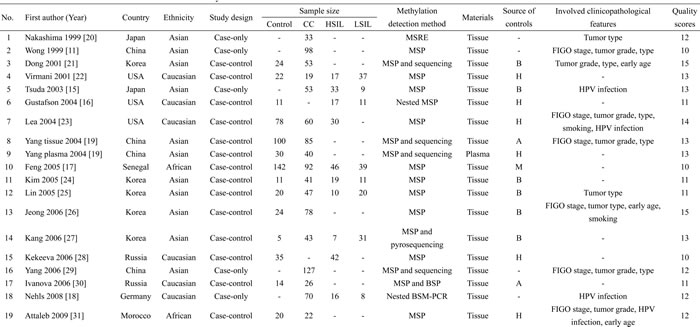
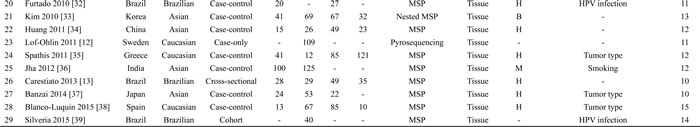
Abbreviations: CC, cervical cancer; LSIL, low-grade squamous intra-epithelial lesion; HSIL, high-grade squamous intra-epithelial lesion; MSRE, methylation-sensitive restriction endonucleases; MSP, methylation-specific PCR; BSP, bisculfite sequencing PCR; H, healthy controls; B, controls with benign gynecological diseases; A, autologous controls; M, mixed controls..
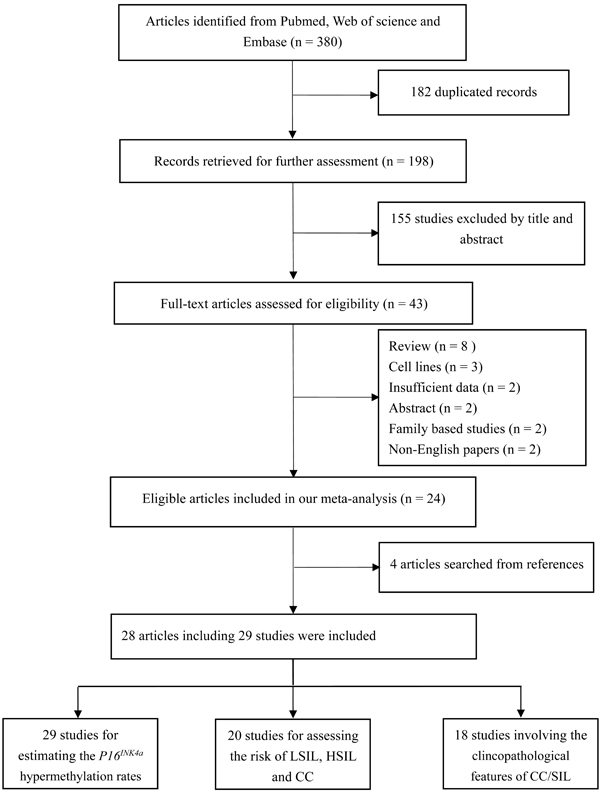
Figure 1: Flowchart for the study selection procedures in this meta-analysis.
Pooled rates of P16INK4a hypermethylation in patients with LSIL, HSIL and CC
A total of 388 LSIL [13, 15-18, 22, 24, 25, 27, 33-35, 38, 39], 636 HSIL [13, 15-18, 22-25, 27, 28, 32-35, 37, 38] and 1439 CC [11-13, 15, 17-26, 29-31, 33-38] specimens were included in this meta-analysis. As summarized in Table 2, the pooled rates of P16INK4a hypermethylation showed an increasing trend (p < 0.001 for the differences in pooled rates ) from LSIL tissues (21.4%, 95% confidence interval (CI): 15.0-29.7%) to HSIL tissues (30.9%, 95% CI: 21.9-41.7%) and ultimately to CC specimens (35.0%, 95%CI: 27.6-43.3%). The respective P16INK4a hypermethylation rates for Asians and Caucasians were similar: 24.6% and 21.5% in LSIL tissues; 31.9% and 27.2% in HSIL tissues; 33.7% and 38.2% in CC specimens. In CC specimens, the pooled rates did not significantly change after excluding one study using plasma samples (35.6%, 95% CI: 28.0-44.1%).
Table 2: Pooled hypermethylation rates of P16INK4a in LSIL, HSIL and CC specimens
Comparison |
Studies (N) |
Specimens (N) |
Heterogeneity |
Model a |
Methylation rates (%) |
|
I2(%) |
PQ-test |
|||||
LSIL |
||||||
Total |
14 |
388 |
47 |
0.025 |
R |
21.4 (15.0-29.7) |
Asian |
6 |
86 |
21 |
0.278 |
F |
24.6 (16.1-35.5) |
Caucasian |
5 |
193 |
67 |
0.016 |
R |
21.5 (9.8-41.0) |
Others |
3 |
109 |
59 |
0.088 |
R |
13.8 (5.1-31.9) |
HSIL |
||||||
Total |
17 |
636 |
82 |
< 0.001 |
R |
30.9 (21.9-41.7) |
Asian |
7 |
231 |
81 |
< 0.001 |
R |
31.9 (18.2-49.7) |
Caucasian |
7 |
286 |
76 |
< 0.001 |
R |
27.2 (16.6-41.2) |
Others |
3 |
119 |
88 |
< 0.001 |
R |
34.5 (9.9-71.6) |
CC |
||||||
Total |
24 |
1439 |
88 |
< 0.001 |
R |
35.0 (27.6-43.3) |
Asian |
14 |
941 |
87 |
< 0.001 |
R |
33.7 (25.5-43.3) |
Caucasian |
6 |
363 |
85 |
0.006 |
R |
38.2 (27.1-50.6) |
Others |
3 |
135 |
96 |
< 0.001 |
R |
39.7 (26.7-54.3) |
a When significant heterogeneity was found (I2 ≥ 50% or PQ-test ≤ 0.1), the random-effects model (DerSimonian-Laird method) was used to pool the results; otherwise, the fixed-effects model (Mantel-Haenszel method) was applied.
Abbreviations: N, number; LSIL, low-grade squamous intra-epithelial lesion; HSIL, high-grade squamous intra-epithelial lesion; CC, cervical cancer; R, random-effects model.
Association of P16INK4a methylation status with LSIL risk
Eleven studies [13, 16, 17, 22, 24, 25, 27, 33-35, 38], involving 336 LSIL patients and 334 controls, were included to assess the association between P16INK4a methylation status and LSIL risk. Overall, P16INK4a promoter hypermethylation was associated with a 3.26-fold (95% CI: 1.86-5.71, p < 0.001) increased risk of LSIL (Figure 2 and Table 3). This association remained significant in almost all subgroups, except for the “other ethnicities” subgroup (Table 3). No significant heterogeneity was found in all comparisons (I2: 0-42%).
Table 3: Pooled results for the association between P16INK4a promoter hypermethylation and LSIL risk.
Comparisons |
Studies (N) |
Sample size (LSIL/controls) |
Heterogeneity |
Model a |
Effect size |
||
I2(%) |
PQ-test |
OR (95% CI) |
P |
||||
Total |
11 |
336/334 |
0 |
0.499 |
F |
3.26 (1.86-5.71) |
< 0.001 |
Ethnicity |
|||||||
Asian |
5 |
77/88 |
0 |
0.817 |
F |
7.76 (2.39-25.15) |
0.001 |
Caucasian |
4 |
185/87 |
4 |
0.374 |
F |
2.98 (1.29-6.91) |
0.011 |
Other ethnicities |
2 |
74/159 |
42 |
0.190 |
F |
1.39 (0.45-4.27) |
0.565 |
Source of controls |
|||||||
Healthy |
6 |
237/126 |
0 |
0.677 |
F |
2.79 (1.39-5.57) |
0.004 |
Non-healthyb |
5 |
99/208 |
23 |
0.266 |
F |
4.52 (1.78-11.47) |
0.001 |
Quality of studies |
|||||||
High (≥ 12) |
6 |
224/133 |
0 |
0.489 |
F |
3.37 (1.58-7.21) |
0.002 |
Low (< 12) |
5 |
112/201 |
20 |
0.290 |
F |
3.09 (1.35-7.09) |
0.008 |
a When significant heterogeneity was found (I2 ≥ 50% or PQ-test ≤ 0.1), the random-effects model (DerSimonian-Laird method) was used to pool the results; otherwise, the fixed-effects model (Mantel-Haenszel method) was applied.
b Non-healthy controls included autologous controls (normal tissues adjacent to LSIL specimens), controls with benign gynecological diseases and mixed controls.
Abbreviations: N, number; LSIL, low-grade squamous intra-epithelial lesion; F, fixed-effects model.
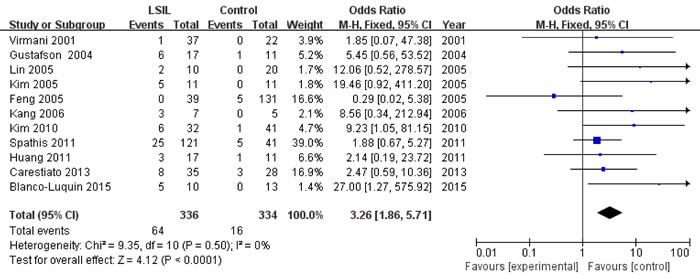
Figure 2: Forest plot for the association between P16INK4a promoter hypermethylation and LSIL risk.
Association of P16INK4a methylation status with HSIL risk
Fifteen studies [13, 16, 17, 22-25, 27, 28, 32-35, 37, 38] with 587 HSIL patients and 491 controls were eligible to evaluate the association of P16INK4a methylation status with HSIL risk. A significant association was found between P16INK4a promoter hypermethylation and increased HSIL risk, with an odds ratio (OR) of 5.80 (95% CI: 3.80-8.84) and a p value of < 0.001 (Figure 3 and Table 4). This association remained significant in all subgroups (Table 4). We did not find significant heterogeneity in all comparisons (I2: 0-43%).
Table 4: Pooled results for the association between P16INK4a promoter hypermethylation and HSIL risk.
Comparisons |
Studies (N) |
Sample size (HSIL/controls) |
Heterogeneity |
Model a |
Effect size |
||
I2(%) |
PQ-test |
OR (95% CI) |
P |
||||
Total |
15 |
587/491 |
18 |
0.253 |
F |
5.80 (3.80-8.84) |
< 0.001 |
Ethnicity |
|||||||
Asian |
6 |
198/112 |
0 |
0.869 |
F |
9.70 (3.85-24.42) |
< 0.001 |
Caucasian |
6 |
270/200 |
38 |
0.374 |
F |
4.61 (2.50-8.52) |
< 0.001 |
Other ethnicities |
3 |
119/179 |
43 |
0.167 |
F |
5.25 (2.46-11.18) |
< 0.001 |
Source of controls |
|||||||
Healthy |
9 |
393/272 |
22 |
0.247 |
F |
5.74 (3.51-9.36) |
< 0.001 |
Non-healthy b |
6 |
194/219 |
27 |
0.236 |
F |
5.99 (2.61-13.74) |
< 0.001 |
Quality of studies |
|||||||
High (≥ 12) |
7 |
354/211 |
0 |
0.453 |
F |
4.08 (2.16-7.73) |
< 0.001 |
Low (< 12) |
8 |
233/280 |
17 |
0.298 |
F |
7.80 (4.47-13.62) |
< 0.001 |
a When significant heterogeneity was found (I2 ≥ 50% or PQ-test ≤ 0.1), the random-effects model (DerSimonian-Laird method) was used to pool the results; otherwise, the fixed-effects model (Mantel-Haenszel method) was applied.
b Non-healthy controls included autologous controls (normal tissues adjacent to HSIL specimens), controls with benign gynecological diseases and mixed controls.
Abbreviations: N, number; HSIL, high-grade squamous intra-epithelial lesion; F, fixed-effects model.
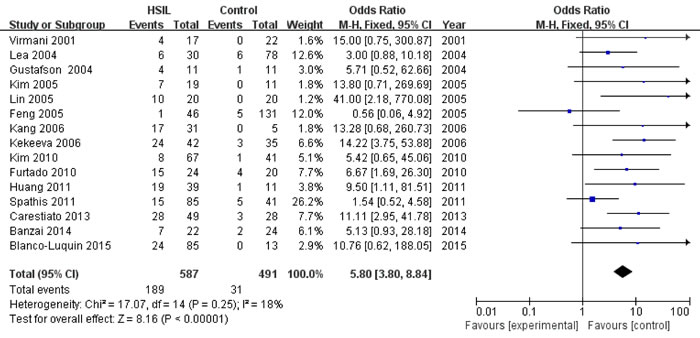
Figure 3: Forest plot for the association between P16INK4a promoter hypermethylation and HSIL risk.
Association of P16INK4a methylation status with CC risk
Eighteen studies [13, 17, 19, 21-26, 30, 31, 33-38] with 950 CC patients and 732 controls were included to appraise the effect of P16INK4a promoter hypermethylation on CC risk. There was a significant association between P16INK4a promoter hypermethylation and increased CC risk, with an OR of 12.17 (95% CI: 5.86-25.27) and a p value of < 0.001 (Figure 4 and Table 5). Consistent with the increasing rates of P16INK4a hypermethylation in LSIL, HSIL and CC specimens, we also found an increasing trend (p < 0.001) in effects of P16INK4a promoter hypermethylation on the risk of LSIL (OR = 3.26), HSIL (OR = 5.80) and CC (OR = 12.17).
Since moderate heterogeneity was observed in the overall comparison (I2 = 58%), subgroup, meta-regression and Galbraith plot analyses were performed to seek the potential sources of heterogeneity. In subgroup analyses, P16INK4a promoter hypermethylation was consistently associated with increased CC risk in all subgroups (Table 5). However, moderate heterogeneity remained in most of the subgroups, except for the subgroups involving high-quality studies (I2 = 0%), Asians (I2 = 19%) and healthy controls (I2 = 44%). The results of meta-regression analyses indicated that ethnicity (p = 0.668), source of controls (p = 0.678) and quality of studies (p = 0.289) were not major sources of heterogeneity (Supplementary Table 1). The subsequent Galbraith plot depicted three outliers [13, 17, 30] as the potential origins of heterogeneity (Supplementary Figure 1). When we excluded these three studies, the association between P16INK4a methylation status and CC risk remained significant (OR = 17.36, 95% CI: 10.61-28.42, p < 0.001), followed by an effective reduction in I2 value from 58% to 12%.
Table 5: Pooled results for the association between P16INK4a promoter hypermethylation and CC risk.
Comparisons |
Studies (N) |
Sample size (CC/controls) |
Heterogeneity |
Model a |
Effect size |
||
I2(%) |
PQ-test |
OR (95% CI) |
P |
||||
Total |
18 |
950/732 |
58 |
0.001 |
R |
12.17 (5.86-25.27) |
< 0.001 |
Ethnicity |
|||||||
Asian |
10 |
631/385 |
19 |
0.272 |
F |
18.94 (9.75-36.81) |
< 0.001 |
Caucasian |
5 |
270/200 |
60 |
0.039 |
R |
6.83 (1.98-23.55) |
0.002 |
Other ethnicities |
3 |
135/179 |
88 |
< 0.001 |
R |
9.87 (4.45-21.90) |
< 0.001 |
Source of controls |
|||||||
Healthy |
9 |
322/267 |
44 |
0.073 |
R |
13.67 (5.64-33.10) |
< 0.001 |
Non-healthy |
9 |
628/465 |
69 |
0.001 |
R |
11.32 (3.28-39.05) |
< 0.001 |
Quality of studies |
|||||||
High (≥ 12) |
11 |
583/491 |
0 |
0.495 |
F |
18.81 (10.84-32.63) |
< 0.001 |
Low (< 12) |
7 |
427/311 |
77 |
< 0.001 |
R |
8.83 (1.85-42.11) |
0.006 |
a When significant heterogeneity was found (I2 ≥ 50% or PQ-test ≤ 0.1), the random-effects model (DerSimonian-Laird method) was used to pool the results; otherwise, the fixed-effects model (Mantel-Haenszel method) was applied.
b Non-healthy controls included autologous controls (normal tissues adjacent to HSIL specimens), controls with benign gynecological diseases and mixed controls.
Abbreviations: N, number; CC, cervical cancer; R, random-effects model; F, fixed-effects model.
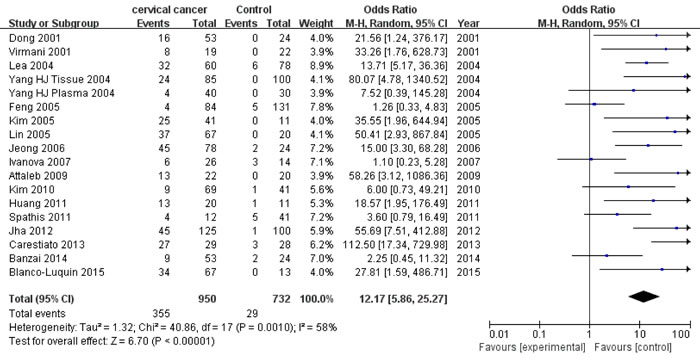
Figure 4: Forest plot for the association between P16INK4a promoter hypermethylation and CC risk.
Association of P16INK4a methylation status with clinicopathological features of SIL/CC
We first evaluated the associations of P16INK4a methylation status with several risk factors for SIL/CC, including HPV infection (Positive vs Negative), smoking habit (Smoker vs Nonsmoker) and early age at diagnosis ( < 50 vs ≥ 50) (Table 6), and observed that P16INK4a promoter hypermethylation was significantly associated with smoking habit, (OR = 3.88, 95% CI: 2.13-7.08, P < 0.001) (Figure 5), but was not correlated with HPV infection and early age at diagnosis (Supplementary Figure 2 and 3). In meta-analyses for the effects of P16INK4a methylation status on histological types (SCC vs AdC), clinical stages (FIGO stage: III + IV vs I + II) and tumor grades (Grade 2 + 3 vs Grade 1) in CC patients, no significant association was found (Table 6 and Supplementary Figure 4-6).
Table 6: Pooled results for the associations between P16INK4a hypermethylation and clinicopathological features of CC/SIL.
Clinicopathological features |
Studies (N) |
Patients (N) |
Heterogeneity |
Model a |
Effect size |
||
I2 (%) |
PQ-test |
OR (95% CI) |
P |
||||
Risk factors for SIL/CC |
|||||||
HPV infection (Positive vs Negative) |
6 |
288 |
0 |
0.974 |
F |
1.06 (0.49-2.28) |
0.883 |
Smoking habit (Smoker vs Nonsmoker) |
3 |
323 |
0 |
0.751 |
F |
3.88 (2.13-7.08) |
< 0.001 |
Early age at diagnosis (<50 vs ≥ 50) |
3 |
153 |
0 |
0.380 |
F |
0.91 (0.47-1.76) |
0.774 |
Clinical and histological data of CC |
|||||||
Tumor type (SCC vs AdC) |
11 |
731 |
22 |
0.235 |
F |
1.00 (0.68-1.48) |
0.986 |
FIGO stage (III + IV vs I + II) |
6 |
470 |
62 |
0.020 |
R |
1.49 (0.62-3.56) |
0.368 |
Tumor grade (G2 + G3 vs G1) |
6 |
440 |
0 |
0.441 |
F |
0.76 (0.46-1.24) |
0.263 |
a When significant heterogeneity was found (I2 ≥ 50% or PQ-test ≤ 0.1), the random-effects model (DerSimonian-Laird method) was used to pool the results; otherwise, the fixed-effects model (Mantel-Haenszel method) was applied.
Abbreviations: N, number; CC, cervical cancer; SCC, squamous cell carcinoma; AdC, adenocarcinoma; SIL, squamous intra-epithelial lesion; F, fixed-effects model ; R,random-effects model.
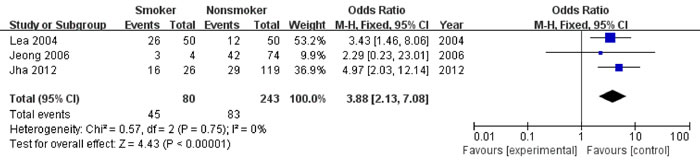
Figure 5: Forest plot for the association between P16INK4a promoter hypermethylation and smoking habit.
Evidence grading
Because all eligible studies were observational, the Grading of Recommendations Assessment, Development and Evaluation (GRADE) process for all comparisons began as “low quality” [40]. For the comparisons of CC risk, HPV infection, early age at diagnosis, tumor type and clinical stage, the quality of evidence was further downgraded to “very low quality”, due to study limitations, inconsistency or imprecision (Supplementary Table 2).
Sensitivity analyses for assessing the stability of pooled results
In all comparisons, sensitivity analyses by sequentially removing each study did not significantly change the pooled results, suggesting the stability of our meta-analyses (Supplementary Figure 7)
Analyses for publication bias
In all comparisons, funnel plots did not reveal obvious asymmetry (Supplementary Figure 8). These observations, combined with the results of Egger’s test (pEgger > 0.05 for all comparisons), suggested that no significant publication bias was found.
DISCUSSION
Previous studies have long aimed to seek methylation biomarkers associated with diagnosis, progression or prognosis of cervical neoplasia. Particularly, a bi-marker panel consisting of CADM1-M18 and MAL-M1 has been considered as a stable triage tool, which could be equally discriminatory for CIN3+ as cytology or cytology with HPV16/18 genotyping in HPV-positive women [41]. In contrast, although P16INK4a promoter hypermethylation has been linked to CC and SIL, the relatively small sample size of independent studies led to inconsistent results and a broad range of hypermethylation rates in cancer tissues. In this meta-analysis, on the basis of data from over 3000 subjects, we found that the hypermethylation rates in LSIL, HSIL and CC specimens were gradually increased, resulting in a growing trend in effects of P16INK4a hypermethylation on susceptibility to LSIL, HSIL and CC. These results, combined with the previous epidemiological evidence that P16INK4a hypermethylation was correlated with the progression of LSIL to HSIL [39, 42], suggest that P16INK4a promoter hypermethylation may be an epigenetic marker for the progression of cervical carcinogenesis. Hence, detecting P16INK4a hypermethylation may help clinicians to determine whether patients with cervical neoplasia are in disease regression, persistence or progression. Especially in patients with an initial diagnosis of LSIL, once P16INK4a hypermethylation is found, more effective clinical management for these patients are encouraged to conduct.
However, the existing evidence provides limited information on the prognostic value of P16INK4a hypermethylation in cervical neoplasia. In a case-series study from China, Yang et al. found no significant association between P16INK4a hypermethylation and overall survival [29]. In contrast, Blanco-Luquin et al. suggested that P16INK4a hypermethylation was correlated with improved disease-free survival [38]. Considering that these two studies involved relatively small sample sizes and inconsistent follow-up times, better designed studies are required to address this issue.
The interaction of P16INK4a hypermethylation with HPV infection is controversial in various HPV-related cancers. For HPV-related oral and oropharyngeal cancer (OSCC) [43], Schlecht et al. found four P16INK4a-specific CPG loci associated with HPV infection in OSCC tissues [44], while another study from Chile failed to replicate this association [45]. For cervical carcinoma, previous functional studies have suggested that P16INK4a promoter hypermethylation mainly occurred at early cervical tumor cell populations without HPV’s E7 transcription [46]. In this meta-analysis, HPV infection was not associated with P16INK4a hypermethylation in patients with SIL/CC. P16INK4a hypermethylation was associated with a 3.26-fold increased risk of LSIL, suggesting the effect of P16INK4a hypermethylation on early stage of cervical oncogenesis. All these findings may suggest that P16INK4a hypermethylation is an early event in cervical carcinogenesis, independent of HPV infection,.
In this meta-analysis, smoking habit was associated with increased P16INK4a hypermethylation rates in patients with SIL/CC. The correlation between smoking habit and P16INK4a hypermethylation has been revealed in several cancers, including non-small cell lung cancer (NSCLC) and esophageal squamous cell carcinoma (ESCC) [47, 48]. In a longitudinal study, Ma et al. [49] reported that smoking initiation was associated with a 3.76-fold increased risk of the appearance of P16INK4a hypermethylation in normal cervical smears, providing direct evidence for the relationship between smoke exposure and subsequent acquisition of P16INK4a hypermethylation in cervix. As a well known risk factor for CC [50], exposure to tobacco smoke, or to its key ingredients (such as nicotine or its derivative), is followed by overexpression of DNA methyltransferases 1, 3A or 3B [51, 52], which has been reported to cause hypermethylation of P16INK4a promoter in mice and cancer patients [53]. Considering that our pooled results were based on the data from relatively few studies, more studies with large sample size are required to repeat this finding.
Moderate heterogeneity was found in our meta-analysis for the association between P16INK4a hypermethylation and CC risk. Therefore, the results were first pooled by using the random-effects model, which cautiously estimates the study weights after accounting for the inter-study differences [54]. Then, by depicting the Galbraith plot, we found that three studies might be the major contributors to the existence of heterogeneity [13, 17, 30]. Notably, the hypermethylation rates of CC tissues enormously varied across these three studies (from 5% [17] to 23% [30] and to 95% [13]), suggesting the existence of inter-study differences. By appraising these three studies using our quality scoring system, we found some common flaws for these studies, including lack of biospecimen information [13, 17, 30], lack of information on conventional risk factors [17, 30], and lack of quality controls for methylation detection [13, 17]. Otherwise, two of three studies collected non-healthy samples (autologous tissues and samples with atypical squamous cells) as their controls [17, 30]. All these issues may lead to the heterogeneous results. Thus, to increase the stability of results, subsequent association analyses for P16INK4a hypermethylation and CC risk should collect healthy controls, and provide adequate information on related confounding factors.
The following limitations merit consideration. First, most of included studies used the MSP method to detect P16INK4a methylation status. As a qualitative method, MSP mainly relies on primer designs to guarantee its accuracy [55]. However, the included studies applied different primers to detect methylation status, causing the potential bias that the promoter regions detected by MSP might not always be uniform. Second, lack of clinical data for each participant limited our ability to adjust for other covariates, such as age at primiparity and menopausal status. Finally, most of included studies adopted case-control or case-only design. This might lead to some selection bias due to inherent drawback of retrospective studies. Therefore, large prospective studies should be carried out with consistent primer designs, quantitative methylation analyses and multiple clinical data.
In this meta-analysis, P16INK4a hypermethylation rates showed an increasing trend from LSIL to HSIL and ultimately to CC, causing the increasing effects of P16INK4a hypermethylation on susceptibility to LSIL, HSIL and CC. Moreover, P16INK4a hypermethylation was also correlated with smoking habit in patients with CC/SIL. Future studies are warranted to repeat these findings and elucidate the underlying mechanism.
MATERIALS AND METHODS
Literature search
This meta-analysis was reported based on the PRISMA statement [56]. Electronic databases, including Pubmed, EMBASE and Web of Science (up to April 19, 2016), were searched by using the combinations of following terms: (P16INK4a or P16 or CDKN2A) and (methylation or promoter methylation or DNA methylation) and (cervical cancer/cervical tumor/cervical neoplasia or SIL/LSIL/HSIL/ or cervical dysplasia/CIN/CIS). Reference lists in reviews and retrieved articles were also checked for other relevant studies.
Eligibility criteria
Eligible studies were required to meet the following criteria: (1) an observational design (cohort, case-control, case-only or cross-sectional studies); (2) studies assessing the associations of P16INK4a methylation status with LSIL, HSIL, CC or their clinicopathological features; (3) studies with sufficient data to calculate the hypermethylation rates, ORs and their 95% CI; (4) written in English.
Exclusion criteria were as follows: (1) reviews, letters, abstracts and case reports; (2) reports with insufficient data; (3) studies regarding in vitro or ex vivo experiments; (4) family-based studies; (5) studies focusing on benign gynecological diseases. For duplicated data, only the most recent or detailed data set was selected.
Data extraction
According to a predefined data collection form, data extraction was carried out by two independent authors (XBW and YDH), with any discrepancies resolved by consensus. The following information for eligible studies was collected: the first author’s name, publication year, study design, ethnicity (country), involved diseases (LSIL, HSIL or CC) or their clinicopathological features (tumor type, clinical stage and tumor grade; age at diagnosis, smoking habit and HPV status), sample size, methods for methylation detection, sample materials, source of controls, and quality of studies.
Quality assessment of eligible studies
According to a predefined system derived from the REMARK [57, 58] and BRISQ [59] guidelines, the quality of eligible studies was appraised by two independent authors (NHC and SZ). This quality scoring system involved 18 items, allowing for assessment of study design, study population, biospecimen information, methylation detection, clinicopathological features and results analysis (Supplementary Table 3). Studies that reported at least 12 items were considered as high-quality studies.
Evidence grading
Once data synthesis was complete, we used the GRADE process to rate the quality of evidence for each comparison as high, moderate, low or very low [40]. Each rating was mainly based on 8 factors, involving study limitations, inconsistency, indirectness, imprecision, reporting bias, magnitude of effect, dose-response gradient and handling of potential confounders [40] (appraised by XBW and NHC).
Statistical Methods
The P16INK4a hypermethylation rates in LSIL, HSIL and CC specimens were estimated using the inverse variance method [60]. Pooled ORs and their 95% CIs were calculated to assess the associations of P16INK4a methylation status with LSIL, HSIL, CC and their clinicopathological features. The heterogeneity across the included studies was evaluated by the χ2-based Q-test and I2 statistic. I2 values of 25%, 50% and 75% were set as the cutoff values for mild, moderate and extensive heterogeneity, respectively [61]. When significant heterogeneity was found (I2 ≥ 50% or PQ-test ≤ 0.1), the random-effects model (DerSimonian-Laird method) was used to pool the results; otherwise, the fixed-effects model (Mantel-Haenszel method) was applied. To further seek the potential sources of heterogeneity, meta-regression and subgroup analyses were performed based on ethnicity, source of controls and quality of studies. Then, a Galbraith plot was depicted to visualize the contribution of individual studies to the overall heterogeneity. To further appraise the stability of the pooled results, sensitivity analyses were performed by sequentially omitting each study or removing the outliers depicted by the Galbraith plot [62]. Publication bias was assessed qualitatively by funnel plots and quantitatively by the Egger’s test [63]. An asymmetric funnel plot and PEgger ≤ 0.05 suggested the existence of publication bias. All the above analyses were conducted by RevMan 5.2 (The Nordic Cochrane Centre, The Cochrane Collaboration) and STATA 12.0 (Stata, College, TX, USA).
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
GRANT SUPPORT
This study was supported by grants from the National Basic Research Program of China (973 Program, 2012CB720600) and the Science and Technology Research Plan of Wuhan City (2015060101010057).
REFERENCES
1. Ferlay J, Shin HR, Bray F, Forman D, Mathers C and Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010; 127(12):2893-2917.
2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J and Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015; 65(2):87-108.
3. Schiffman M and Wentzensen N. Human papillomavirus infection and the multistage carcinogenesis of cervical cancer. Cancer Epidemiol Biomarkers Prev. 2013; 22(4):553-560.
4. Apostolidou S, Hadwin R, Burnell M, Jones A, Baff D, Pyndiah N, Mould T, Jacobs IJ, Beddows S, Kocjan G and Widschwendter M. DNA methylation analysis in liquid-based cytology for cervical cancer screening. Int J Cancer. 2009; 125(12):2995-3002.
5. Guan P, Howell-Jones R, Li N, Bruni L, de Sanjose S, Franceschi S and Clifford GM. Human papillomavirus types in 115,789 HPV-positive women: a meta-analysis from cervical infection to cancer. Int J Cancer. 2012; 131(10):2349-2359.
6. zur Hausen H. Papillomaviruses causing cancer: evasion from host-cell control in early events in carcinogenesis. J Natl Cancer Inst. 2000; 92(9):690-698.
7. Kazanets A, Shorstova T, Hilmi K, Marques M and Witcher M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles and potential. Biochim Biophys Acta. 2016.
8. Sherr CJ, Beach D and Shapiro GI. Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov. 2016; 6(4):353-367.
9. Tang B, Li Y, Qi G, Yuan S, Wang Z, Yu S, Li B and He S. Clinicopathological Significance of CDKN2A Promoter Hypermethylation Frequency with Pancreatic Cancer. Sci Rep. 2015; 5:13563.
10. Xing X, Cai W, Shi H, Wang Y, Li M, Jiao J and Chen M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: a meta-analysis. Br J Cancer. 2013; 108(12):2542-2548.
11. Wong YF, Chung TK, Cheung TH, Nobori T, Yu AL, Yu J, Batova A, Lai KW and Chang AM. Methylation of p16INK4A in primary gynecologic malignancy. Cancer Lett. 1999; 136(2):231-235.
12. Lof-Ohlin ZM, Sorbe B, Wingren S and Nilsson TK. Hypermethylation of promoter regions of the APC1A and p16INK4a genes in relation to prognosis and tumor characteristics in cervical cancer patients. Int J Oncol. 2011; 39(3):683-688.
13. Carestiato FN, Afonso LA, Moyses N, Almeida Filho GL, Velarde LG and Cavalcanti SM. An upward trend in DNA p16ink4a methylation pattern and high risk HPV infection according to the severity of the cervical lesion. Rev Inst Med Trop Sao Paulo. 2013; 55(5):329-334.
14. Solomon D, Davey D, Kurman R, Moriarty A, O’Connor D, Prey M, Raab S, Sherman M, Wilbur D, Wright T, Jr. and Young N. The 2001 Bethesda System: terminology for reporting results of cervical cytology. Jama. 2002; 287(16):2114-2119.
15. Tsuda H, Hashiguchi Y, Nishimura S, Kawamura N, Inoue T and Yamamoto K. Relationship between HPV typing and abnormality of G1 cell cycle regulators in cervical neoplasm. Gynecol Oncol. 2003; 91(3):476-485.
16. Gustafson KS, Furth EE, Heitjan DF, Fansler ZB and Clark DP. DNA methylation profiling of cervical squamous intraepithelial lesions using liquid-based cytology specimens: an approach that utilizes receiver-operating characteristic analysis. Cancer. 2004; 102(4):259-268.
17. Feng Q, Balasubramanian A, Hawes SE, Toure P, Sow PS, Dem A, Dembele B, Critchlow CW, Xi L, Lu H, McIntosh MW, Young AM and Kiviat NB. Detection of hypermethylated genes in women with and without cervical neoplasia. J Natl Cancer Inst. 2005; 97(4):273-282.
18. Nehls K, Vinokurova S, Schmidt D, Kommoss F, Reuschenbach M, Kisseljov F, Einenkel J, von Knebel Doeberitz M and Wentzensen N. p16 methylation does not affect protein expression in cervical carcinogenesis. Eur J Cancer. 2008; 44(16):2496-2505.
19. Yang HJ, Liu VW, Wang Y, Chan KY, Tsang PC, Khoo US, Cheung AN and Ngan HY. Detection of hypermethylated genes in tumor and plasma of cervical cancer patients. Gynecol Oncol. 2004; 93(2):435-440.
20. Nakashima R, Fujita M, Enomoto T, Haba T, Yoshino K, Wada H, Kurachi H, Sasaki M, Wakasa K, Inoue M, Buzard G and Murata Y. Alteration of p16 and p15 genes in human uterine tumours. Br J Cancer. 1999; 80(3-4):458-467.
21. Dong SM, Kim HS, Rha SH and Sidransky D. Promoter hypermethylation of multiple genes in carcinoma of the uterine cervix. Clin Cancer Res. 2001; 7(7):1982-1986.
22. Virmani AK, Muller C, Rathi A, Zoechbauer-Mueller S, Mathis M and Gazdar AF. Aberrant methylation during cervical carcinogenesis. Clin Cancer Res. 2001; 7(3):584-589.
23. Lea JS, Coleman R, Kurien A, Schorge JO, Miller DS, Minna JD and Muller CY. Aberrant p16 methylation is a biomarker for tobacco exposure in cervical squamous cell carcinogenesis. Am J Obstet Gynecol. 2004; 190(3):674-679.
24. Kim NR, Lin Z, Kim KR, Cho HY and Kim I. Epstein-Barr virus and p16INK4A methylation in squamous cell carcinoma and precancerous lesions of the cervix uteri. J Korean Med Sci. 2005; 20(4):636-642.
25. Lin Z, Gao M, Zhang X, Kim YS, Lee ES, Kim HK and Kim I. The hypermethylation and protein expression of p16 INK4A and DNA repair gene O6-methylguanine-DNA methyltransferase in various uterine cervical lesions. J Cancer Res Clin Oncol. 2005; 131(6):364-370.
26. Jeong DH, Youm MY, Kim YN, Lee KB, Sung MS, Yoon HK and Kim KT. Promoter methylation of p16, DAPK, CDH1, and TIMP-3 genes in cervical cancer: correlation with clinicopathologic characteristics. Int J Gynecol Cancer. 2006; 16(3):1234-1240.
27. Kang S, Kim J, Kim HB, Shim JW, Nam E, Kim SH, Ahn HJ, Choi YP, Ding B, Song K and Cho NH. Methylation of p16INK4a is a non-rare event in cervical intraepithelial neoplasia. Diagn Mol Pathol. 2006; 15(2):74-82.
28. Kekeeva TV, Zhevlova AI, Podisov Iu I, Solov’eva Iu V, Zaletataev DV and Nemtsova MV. Aberrant methylation of tumor suppressor genes and allelic imbalance in cervical intraepitelial neoplasia. Mol Biol (Mosk). 2006; 40(2):224-230.
29. Yang HJ, Liu VW, Wang Y, Tsang PC and Ngan HY. Differential DNA methylation profiles in gynecological cancers and correlation with clinico-pathological data. BMC Cancer. 2006; 6:212.
30. Ivanova TA, Golovina DA, Zavalishina LE, Volgareva GM, Katargin AN, Andreeva YY, Frank GA, Kisseljov FL and Kisseljova NP. Up-regulation of expression and lack of 5’ CpG island hypermethylation of p16 INK4a in HPV-positive cervical carcinomas. BMC Cancer. 2007; 7:47.
31. Attaleb M, El hamadani W, Khyatti M, Benbacer L, Benchekroun N, Benider A, Amrani M and El Mzibri M. Status of p16(INK4a) and E-cadherin gene promoter methylation in Moroccan patients with cervical carcinoma. Oncol Res. 2009; 18(4):185-192.
32. Furtado YL, Almeida G, Lattario F, Silva KS, Maldonado P, Silveira FA, do Val IC, Fonseca R and Carvalho Mda G. The presence of methylation of the p16INK4A gene and human papillomavirus in high-grade cervical squamous intraepithelial lesions. Diagn Mol Pathol. 2010; 19(1):15-19.
33. Kim JH, Choi YD, Lee JS, Lee JH, Nam JH and Choi C. Assessment of DNA methylation for the detection of cervical neoplasia in liquid-based cytology specimens. Gynecol Oncol. 2010; 116(1):99-104.
34. Huang LW, Pan HS, Lin YH, Seow KM, Chen HJ and Hwang JL. P16 methylation is an early event in cervical carcinogenesis. Int J Gynecol Cancer. 2011; 21(3):452-456.
35. Spathis A, Aga E, Alepaki M, Chranioti A, Meristoudis C, Panayiotides I, Kassanos D and Karakitsos P. Promoter methylation of p16(INK4A), hMLH1, and MGMT in liquid-based cervical cytology samples compared with clinicopathological findings and HPV presence. Infect Dis Obstet Gynecol. 2011; 2011:927861.
36. Jha AK, Nikbakht M, Jain V, Capalash N and Kaur J. p16(INK4a) and p15(INK4b) gene promoter methylation in cervical cancer patients. Oncol Lett. 2012; 3(6):1331-1335.
37. Banzai C, Nishino K, Quan J, Yoshihara K, Sekine M, Yahata T and Tanaka K. Promoter methylation of DAPK1, FHIT, MGMT, and CDKN2A genes in cervical carcinoma. Int J Clin Oncol. 2014; 19(1):127-132.
38. Blanco-Luquin I, Guarch R, Ojer A, Perez-Janices N, Martin-Sanchez E, Maria-Ruiz S, Monreal-Santesteban I, Blanco-Fernandez L, Pernaut-Leza E, Escors D and Guerrero-Setas D. Differential role of gene hypermethylation in adenocarcinomas, squamous cell carcinomas and cervical intraepithelial lesions of the uterine cervix. Pathol Int. 2015; 65(9):476-485.
39. Silveira FA, Almeida G, Furtado Y, Silva KS, Maldonado P, Cavalcanti S and Carvalho Mda G. HPV DNA genotyping and methylation of gene p16 INK4A in cervical LSIL. Exp Mol Pathol. 2015; 98(2):308-311.
40. Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P and Schunemann HJ. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. Bmj. 2008; 336(7650):924-926.
41. Hesselink AT, Heideman DA, Steenbergen RD, Coupe VM, Overmeer RM, Rijkaart D, Berkhof J, Meijer CJ and Snijders PJ. Combined promoter methylation analysis of CADM1 and MAL: an objective triage tool for high-risk human papillomavirus DNA-positive women. Clin Cancer Res. 2011; 17(8):2459-2465.
42. Lee H and Lee EJ. HPV infection and p16 promoter methylation as predictors of ASC-US/LSIL progression. Cancer Cytopathol. 2016; 124(1):58-65.
43. Al-Kaabi A, van Bockel LW, Pothen AJ and Willems SM. p16INK4A and p14ARF gene promoter hypermethylation as prognostic biomarker in oral and oropharyngeal squamous cell carcinoma: a review. Dis Markers. 2014; 2014:260549.
44. Schlecht NF, Ben-Dayan M, Anayannis N, Lleras RA, Thomas C, Wang Y, Smith RV, Burk RD, Harris TM, Childs G, Ow TJ, Prystowsky MB and Belbin TJ. Epigenetic changes in the CDKN2A locus are associated with differential expression of P16INK4A and P14ARF in HPV-positive oropharyngeal squamous cell carcinoma. Cancer Med. 2015; 4(3):342-353.
45. Reyes M, Rojas-Alcayaga G, Pennacchiotti G, Carrillo D, Munoz JP, Pena N, Montes R, Lobos N and Aguayo F. Human papillomavirus infection in oral squamous cell carcinomas from Chilean patients. Exp Mol Pathol. 2015; 99(1):95-99.
46. Nuovo GJ, Plaia TW, Belinsky SA, Baylin SB and Herman JG. In situ detection of the hypermethylation-induced inactivation of the p16 gene as an early event in oncogenesis. Proc Natl Acad Sci U S A. 1999; 96(22):12754-12759.
47. Huang T, Chen X, Hong Q, Deng Z, Ma H, Xin Y, Fang Y, Ye H, Wang R, Zhang C, Ye M and Duan S. Meta-analyses of gene methylation and smoking behavior in non-small cell lung cancer patients. Sci Rep. 2015; 5:8897.
48. Talukdar FR, Ghosh SK, Laskar RS and Mondal R. Epigenetic, genetic and environmental interactions in esophageal squamous cell carcinoma from northeast India. PLoS One. 2013; 8(4):e60996.
49. Ma YT, Collins SI, Young LS, Murray PG and Woodman CB. Smoking initiation is followed by the early acquisition of epigenetic change in cervical epithelium: a longitudinal study. Br J Cancer. 2011; 104(9):1500-1504.
50. Appleby P, Beral V, Berrington de Gonzalez A, Colin D, Franceschi S, Goodill A, Green J, Peto J, Plummer M and Sweetland S. Carcinoma of the cervix and tobacco smoking: collaborative reanalysis of individual data on 13,541 women with carcinoma of the cervix and 23,017 women without carcinoma of the cervix from 23 epidemiological studies. Int J Cancer. 2006; 118(6):1481-1495.
51. Soma T, Kaganoi J, Kawabe A, Kondo K, Imamura M and Shimada Y. Nicotine induces the fragile histidine triad methylation in human esophageal squamous epithelial cells. Int J Cancer. 2006; 119(5):1023-1027.
52. Liu H, Zhou Y, Boggs SE, Belinsky SA and Liu J. Cigarette smoke induces demethylation of prometastatic oncogene synuclein-gamma in lung cancer cells by downregulation of DNMT3B. Oncogene. 2007; 26(40):5900-5910.
53. Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, Lee CF and Wang YC. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest. 2010; 120(2):521-532.
54. DerSimonian R and Kacker R. Random-effects model for meta-analysis of clinical trials: an update. Contemp Clin Trials. 2007; 28(2):105-114.
55. Shen L and Waterland RA. Methods of DNA methylation analysis. Curr Opin Clin Nutr Metab Care. 2007; 10(5):576-581.
56. Moher D, Liberati A, Tetzlaff J and Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Bmj. 2009; 339:b2535.
57. Altman DG, McShane LM, Sauerbrei W and Taube SE. Reporting Recommendations for Tumor Marker Prognostic Studies (REMARK): explanation and elaboration. PLoS Med. 2012; 9(5):e1001216.
58. Chen M, Cai E, Huang J, Yu P and Li K. Prognostic value of vascular endothelial growth factor expression in patients with esophageal cancer: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2012; 21(7):1126-1134.
59. Moore HM, Kelly AB, Jewell SD, McShane LM, Clark DP, Greenspan R, Hayes DF, Hainaut P, Kim P, Mansfield EA, Potapova O, Riegman P, Rubinstein Y, et al. Biospecimen reporting for improved study quality (BRISQ). Cancer Cytopathol. 2011; 119(2):92-101.
60. Thakkinstian A, McEvoy M, Minelli C, Gibson P, Hancox B, Duffy D, Thompson J, Hall I, Kaufman J, Leung TF, Helms PJ, Hakonarson H, Halpi E, et al. Systematic review and meta-analysis of the association between {beta}2-adrenoceptor polymorphisms and asthma: a HuGE review. Am J Epidemiol. 2005; 162(3):201-211.
61. Higgins JP, Thompson SG, Deeks JJ and Altman DG. Measuring inconsistency in meta-analyses. Bmj. 2003; 327(7414):557-560.
62. Wang B, Huang G, Wang D, Li A, Xu Z, Dong R, Zhang D and Zhou W. Null genotypes of GSTM1 and GSTT1 contribute to hepatocellular carcinoma risk: evidence from an updated meta-analysis. J Hepatol. 2010; 53(3):508-518.
63. Egger M, Davey Smith G, Schneider M and Minder C. Bias in meta-analysis detected by a simple, graphical test. Bmj. 1997; 315(7109):629-634.