
INTRODUCTION
Ovarian clear cell carcinoma is one of four major histological types of epithelial ovarian cancer, the others being serous, mucinous, and endometrioid carcinomas [1–3]. Of these, high-grade serous carcinoma is the most common, and accounts for > 50-60% of cases [4, 5]. Thus, most genome-wide analyses, including whole-exome sequencing, target this form of ovarian cancer [6–8]. However, the ratio of clear cell carcinoma is high in Asia, especially in Japan, where it constitutes approximately 25% of cases [2]. Notably, clear cell carcinomas are characteristically resistant to platinum-based chemotherapy [9], and the 3-year survival rate in patients with ≥ 2 cm residual tumor is only 10.2%, which is significantly poorer than the 45.9% survival rate in patients with high-grade serous cancer [9]. Only a few molecular targeted therapies have been approved for ovarian cancers, none of which is specific for clear cell carcinoma. Therefore, novel therapeutic strategies are needed against this form of ovarian cancer.
TP53 mutations are characteristically infrequent, and are present in only 10% of ovarian clear cell carcinomas, with loss of heterozygosity in < 20% [10–12]. In contrast, TP53 mutations are present in 96% of high-grade serous tumors [6]. TP53 is a key tumor suppressor that induces cell cycle arrest, apoptosis, autophagy, and senescence while inhibiting angiogenesis and metastasis [13–15]. Notably, TP53 activity is determined not only by abundance, but also by phosphorylation. For instance, TP53 is activated by phosphorylation at Ser-46 to induce expression of apoptosis genes such as PUMA and TP53AIP1 in response to severe DNA damage or extreme TP53 overexpression [16]. TP53 activation also inhibits angiogenesis via suppression of hypoxia-inducible factor 1alpha (HIF-1a) [17]. Therefore, TP53 is expected to function as a tumor suppressor in cancers with wild type TP53, including clear cell carcinoma. Nevertheless, TP53 may be inactivated even in the absence of mutations, and this inactivation may promote cell survival in some of these cancers [18, 19].
Cellular TP53 levels are regulated by degradation pathways that require E3 ubiquitin ligases [20, 21]. Of these, murine double minute 2 (MDM2) and MDM4 (also known as MDMX, a structural homolog of MDM2) are the most essential [22]. Indeed, TP53 inactivation by overexpression of MDM2 and other E3 ligases has been reported in human cancers [23]. In addition, TP53 mutations are inversely correlated with abundant MDM2 expression [24]. In this light, MDM2 inhibitors such as Nutlin-3a and RG7112 were developed recently to block the interaction between TP53 and MDM2, and thereby stabilize TP53. Importantly, these compounds were reported to have in vitro and in vivo antitumor activity in human cancers with wild type TP53 [25–28], and are now in early-phase clinical trials [29–31]. Nevertheless, whether MDM2 and/or MDM4 are overexpressed in clear cell carcinoma remains to be established, along with whether MDM2 inhibitors are active against these forms of cancer. In this study, we investigated the expression of MDM2 and MDM4 in clear cell carcinomas, and evaluated the in vitro and in vivo activity of the MDM2 inhibitor RG7112 against clear cell tumors with wild type TP53.
RESULTS
High MDM2 expression is significantly associated with clear cell carcinoma histology and poor prognosis
MDM2 mRNA expression was analyzed by microarray in 75 clear cell carcinomas, 13 normal tissues, and 16 high-grade serous ovarian cancers. MDM2 expression was higher in 61 of 75 (81%) clear cell carcinomas than in normal ovarian tissue (Figure 1A and Supplementary Table 1). Indeed, MDM2 expression was significantly higher in clear cell carcinomas than in normal tissues (P = 0.035) and high-grade serous carcinomas (P = 0.0092, Figure 1B). However, expression of MDM4 was significantly lower in both cancer tissues than in normal tissues (Supplementary Figure 1A). Clear cell carcinomas were further stratified as MDM2-high (n = 25), MDM2-intermediate (n = 25), and MDM2-low (n = 25). TP53 mutations were detected by Sanger sequencing in 4 (5.6%) clear cell carcinomas (Supplementary Figure 1B), all of which were MDM2-low or intermediate (Supplementary Table 1). In clear cell carcinomas without TP53 mutations, high MDM2 expression was significantly associated with poor progression-free survival (PFS) (P = 0.0002 by log-rank test, Figure 1C), as was advanced stage (P = 0.0002 by log-rank test, Supplementary Figure 1C), but not age (Supplementary Figure 1D). MDM2-high was also associated with poor overall survival (OS) (P = 0.0008) (Supplementary Figure 2A). The prognosis (either PFS or OS) was comparable between MDM2-intermediate and MDMs-low (Supplementary Figure 2B and 2C). Similarly, univariate analysis demonstrated that advanced stage (HR = 5.05, 95% CI = 1.84-12.91, P = 0.0025) and high MDM2 expression (HR = 5.48, 95% CI = 2.10-15.97, P = 0.0005) were significantly associated with poor PFS (Table 1: upper rows) and with poor OS (Table 1: lower rows). In addition, multivariate analysis indicated that high MDM2 expression was a poor prognostic factor for PFS (HR = 5.61, 95% CI = 2.11-16.62, P = 0.0005) and OS (HR = 6.14, 95% CI = 1.85-24.32, P = 0.0028, independent of age and cancer stage (Table 1). We also performed real-time PCR in 4 normal ovarian tissues and 17 of the 75 clear cell carcinomas (Supplementary Figure 3A), and found that MDM2 expression was significantly higher in clear cell carcinomas than in normal ovaries (P = 0.039) (Supplementary Figure 3A), and that the expression level of MDM2 determined by microarray was highly associated with that determined by real-time PCR (Supplementary Figure 3B).
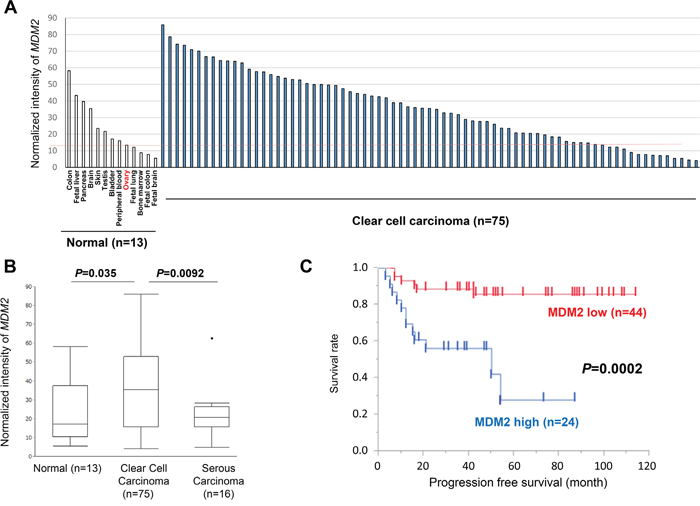
Figure 1: Expression of MDM2 in normal tissues and ovarian cancers. A. Expression of MDM2 in 13 normal tissues and 75 ovarian clear cell carcinomas, as determined by microarray analysis. B. Comparison (t-test) of MDM2 expression in normal tissues, clear cell, and high-grade serous carcinomas. C. Survival analysis (progression-free survival) using Kaplan-Meier method and log-rank test in clear cell carcinomas without TP53 mutations (n = 68). The upper 1/3 among clear cell carcinomas was defined as “MDM2 high” on the basis of the expression level determined by microarray.
Table 1: Univariate/multivariate analysis in CCC by MDM2 expression, age and stage
Progression Free Survival
Characteristics |
|
Univariate |
Multivariate |
||||
|---|---|---|---|---|---|---|---|
HR |
95%CI |
P-value |
HR |
95%CI |
P-value |
||
Age |
≥50 |
1.45 |
0.52-5.13 |
0.5 |
0.97 |
0.32-3.55 |
0.95 |
<50 (ref) |
|||||||
Stage |
III/IV |
5.05 |
1.84-12.91 |
0.0025 |
5.16 |
1.8-14.2 |
0.0032 |
I/II (ref) |
|||||||
MDM2 |
High |
5.48 |
2.1-15.97 |
0.0005 |
5.61 |
2.11-16.62 |
0.0005 |
Low (ref) |
|||||||
Overall Survival
Characteristics |
|
Univariate |
Multivariate |
||||
|---|---|---|---|---|---|---|---|
HR |
95%CI |
P-value |
HR |
95%CI |
P-value |
||
Age |
≥50 |
1.26 |
0.38-5.61 |
0.72 |
0.52 |
0.12-2.6 |
0.4 |
<50 (ref) |
|||||||
Stage |
III/IV |
8.6 |
2.83-26.99 |
0.0003 |
9.81 |
2.81-38.94 |
0.0004 |
I/II (ref) |
|||||||
MDM2 |
High |
6.078 |
1.95-22.76 |
0.0018 |
6.14 |
1.85-24.32 |
0.0028 |
Low (ref) |
|||||||
RG7112 inhibits cell viability in clear cell carcinomas with wild type TP53
MDM2 expression was assessed by immunoblotting in 7 clear cell carcinoma cell lines with wild-type (OVISE, OVTOKO, JHOC-7, and RMG-I) or mutated (SKOV3, JHOC-9, and ES-2) TP53 (Supplementary Figure 4). MDM2 expression was significantly higher in 3 of 4 TP53 mutant cell lines than in the control (immortalized cells derived from ovarian surface epithelium).
Next, the antiproliferative effect of RG7112, an MDM2 inhibitor, was evaluated in these clear cell carcinomas. RG7112 suppressed cell proliferation in dose-dependent fashion only in cells with wild type TP53, with half maximal inhibitory concentration (IC50) between 1.0 and 2.2 μM (Figure 2A). On the other hand, IC50 was > 10 μM in cell lines with mutant TP53 (Figure 2B). Indeed, differences in cell viability were statistically significant between tumors with wild type and mutated TP53 (P < 0.001 by paired t-test, Figure 2C). We also confirmed that the IC50 was > 10 μM in normal cell lines (COS-7 and 293T) (Supplementary Figure 5).
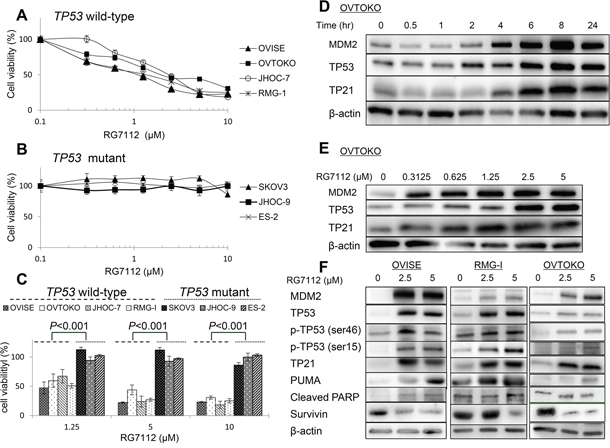
Figure 2: Cell viability and activation of TP53 target proteins in clear cell carcinomas treated with RG7112. A and B. Cell viability, as measured by MTT assay, in cells with wild type (A) or mutant (B) TP53. Experiments were repeated three times. C. Differences in cell viability in cell lines with wild type or mutant TP53 and exposed to indicated concentrations of RG7112. Group means were compared by paired t-test. D and E. Time- (D) and dose-dependent (E) accumulation of MDM2, TP53, and TP21, as measured by western blotting. RG7112 was added at 2.5 μM in (D). F. Induction of TP53 phosphorylation and TP53 target proteins (TP21 and PUMA), as determined by western blotting. Cleaved PARP and survivin were also assessed to detect proapoptotic signaling.
RG7112 increased the abundance of MDM2 and TP53 in OVTOKO cells in a time-dependent (at 2.5 μM) and dose-dependent fashion (Figure 2D and 2E), suggesting that interference with MDM2-TP53 binding resulted in accumulation of both proteins. Accordingly, expression of TP21, a well-known TP53 target gene, increased in time- and dose-dependent manner (Figure 2D and 2E). In particular, expression of TP53 and TP21 was induced within 2-4 h at RG7112 doses > 0.625 μM (Figure 2D and 2E). RG7112 also stimulated TP53-dependent apoptotic signaling in OVISE, RMG-I, and OVTOKO cells, as indicated by increased TP53 phosphorylation at Ser-46 and Ser-15, increased expression of the proapoptotic gene PUMA, and suppression of the antiapoptotic protein survivin (Figure 2F). Consequently, cleaved PARP accumulated following exposure to 2.5 or 5 μM RG7112 in OVISE cells (Figure 2F).
RG7112 and MDM2 siRNA induce apoptotic cell death in clear cell carcinoma with wild type TP53
To further examine whether activation of TP53 is associated with RG7112-induced cell death, we analyzed the cell cycle distribution of clear cell carcinomas with wild type TP53 and exposed to RG7112 for 72 h. The compound expanded the sub-G1 population in dose-dependent manner (Figure 3A). In particular, the proportion of cells in sub-G1 increased to 14-59% in cells treated with 5 μM RG7112, while the S phase population contracted. In addition, exposure to 2.5 μM or 5 μM RG7112 significantly increased the ratio of apoptotic cells by 12-22%, as measured by annexin V staining (Figure 3B). Suppression of MDM2 expression by siRNAs (Figure 3C) also significantly suppressed cell viability (Figure 3D) and increased apoptotic cell death, as measured by MTT assay and annexin V staining, respectively (Figure 3E).
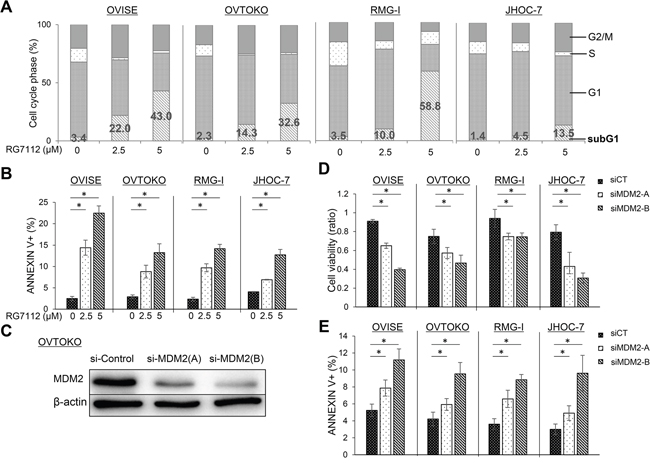
Figure 3: Induction of apoptosis after RG7112 treatment and MDM2 knockdown in clear cell carcinomas with wild type TP53. A. Cell cycle distribution after RG7112 treatment, as assessed by fluorescence-activated cell sorting. B. Apoptosis after RG7112 treatment, as evaluated by annexin V staining. C. siRNA knockdown of MDM2 in OVTOKO cells using MDM2-A and MDM2-B, as confirmed by western blotting. D. Cell viability after MDM2 knockdown was assessed by MTT assay. E. Apoptosis after MDM2 knockdown was assessed by annexin V assay. All experiments were repeated three times.
In vivo antitumor activity of RG7112
To examine the antitumor activity of RG7112 in vivo, we established subcutaneous tumor xenograft models in nude mice by using RMG-I and OVISE cells. Each mouse was treated daily for three weeks with an oral dose of 100 mg/kg RG7112 or vehicle. RG7112 significantly suppressed the growth of xenografted RMG-I cells (P = 0.033 by t-test, Figure 4A-4C) and tended to suppress the growth of the OVISE cells, although the growth suppression in OVISE cells did not reach statistical significance (P = 0.061, Supplementary Figure 6A). We note that mice lost < 10% body weight throughout the study (Figure 4A and Supplementary Figure 6A). Western blotting of three tumors from RG7112-treated mice showed upregulation of MDM2, TP53, and TP53 phosphorylation at Ser15 and Ser46 (Figure 4D and 4E, and Supplementary Figure 6B and 6C). In addition, RG7112 also induced TP53 target genes such as TP21, PUMA, and BAX. In agreement with in vitro results, cleaved PARP accumulated while survivin diminished with RG7112 treatment (Figure 4D and 4E, and Supplementary Figure 6B and 6C).
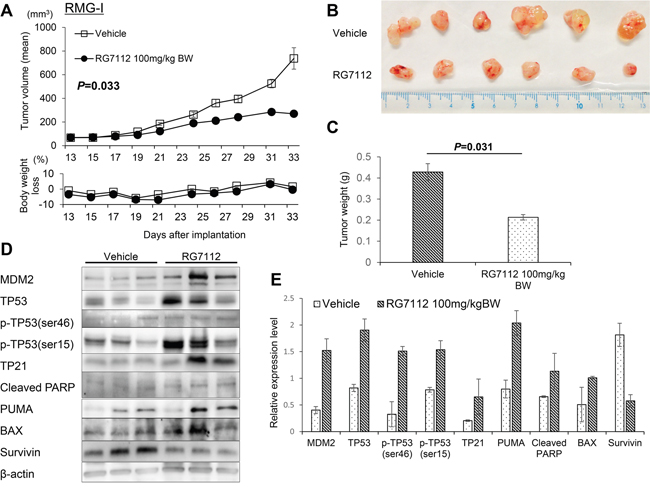
Figure 4: In vivo evaluation of RG7112 efficacy using xenografted RMG-I cells. A-C. Tumor size and body weight in xenograft mouse models orally treated with RG7112 were measured after the start of RG7112 treatment 13 days post implantation (A). Tumors were collected from mice sacrificed after 21 days of RG7112 treatment (B). Tumor weights were compared by t-test between control and RG7112-treated animals (C). D. TP53 phosphorylation and expression of TP53 target proteins were compared between tumors from control and RG7112-treated animals (n = 3 per group). E. Expression of each protein relative to beta-actin was quantified in Image J. All experiments were repeated thrice.
RG7112 suppresses HIF-1alpha in hypoxic conditions and reduces tumor vascularity
Finally, we examined whether RG7112 affects HIF-1alpha expression and tumor vascularity in clear cell carcinomas. Exposure to RG7112 (Figure 5A) and MDM2 siRNA (Supplementary Figure 7A) suppressed hypoxia (1% O2)-induced expression of HIF-1alpha in two cell lines with wild type TP53. In addition, large vessels stained with the endothelial marker CD31 were observed in tumor sections from control mice, whereas only small vessels were observed in RG7112-treated mice (Figure 5B and Supplementary Figure 7B). Indeed, the number of microvessels in RMG-I and OVISE tumors was significantly reduced in RG7112-treated mice (P = 0.011 and P = 0.016, respectively), indicating that RG7112 inhibits angiogenesis (Figure 5C and Supplementary Figure 7C).
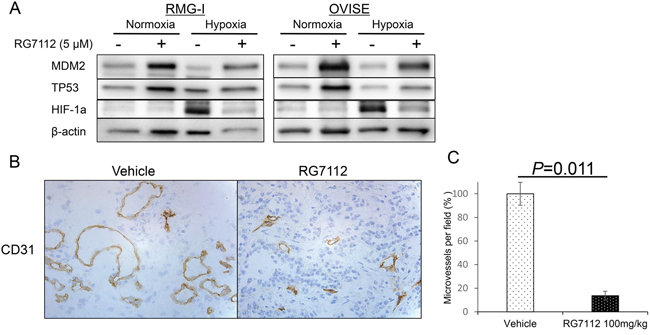
Figure 5: RG7112-induced suppression of HIF-1alpha during hypoxia and reduction of tumor vascularity. A. Normoxic and hypoxic (under 1% O2) expression of MDM2, p53, and HIF-1alpha in RMG-I and OVISE cells treated with or without 5 μM RG7112. B. Immunohistochemical staining of CD31 to detect microvessels in RMG-I tumors from control and RG7112-treated mice. C. Microvessels per field was calculated in RMG-I tumors from control and RG7112-treated mice, and compared by t-test.
DISCUSSION
In this study, we found that abundant expression of MDM2 was significantly associated with poor prognosis (PFS and OS) in ovarian clear cell carcinomas, and that the MDM2 inhibitor RG7112 has in vitro and in vivo antitumor and proapoptotic activities in clear cell carcinomas without TP53 mutations. MDM2 is a major ubiquitin ligase that cooperates with MDM4 to degrade TP53 via the ubiquitin-proteasome system [21, 32, 33]. MDM2 is negatively regulated by specific microRNAs such as miR-1827 and miR-340 [34–36], and overexpression via multiple mechanisms is observed in various types of human malignancies, including sarcomas, gliomas, hematological malignancies, and breast cancer [34, 37]. In particular, MDM2 overexpression is more likely to occur in cancers without TP53 mutations [37], as MDM2 also contains TP53-binding sites in the first intron, and is thus directly inducible by wild type TP53 [34]. Accordingly, MDM2 expression was significantly lower in high-grade serous and clear cell carcinomas with mutated TP53. Thus, the prognostic value of MDM2 has attracted much attention. For instance, MDM2 overexpression was associated with poor prognosis in renal cell carcinomas [38]. In line with this result, we found that MDM2 overexpression is an independent marker of poor prognosis in clear cell carcinomas. Importantly, MDM2 overexpression and subsequent inactivation of TP53 may be associated with chemoresistance [39, 40]. Taken together, the data provide a strong rationale to target MDM2 in clear cell carcinoma.
RG7112, Nutlin-3a, and RG7337 are potent and selective MDM2 antagonists, which inhibit the interaction between TP53 and MDM2, and thereby stabilizes TP53 [27, 41, 42]. We found that RG7112 has antitumor activity exclusively in clear cell tumors with wild type TP53. In vitro, the inhibitor increased the number of cells in sub-G1 phase. In vivo, the compound induced apoptosis by stimulating phosphorylation of TP53 at Ser-46, inducing expression of the proapoptotic TP53 target genes PUMA and BAX, and suppressing the antiapoptotic protein survivin. Although both cell-cycle arrest and apoptotic cell death can be induced by RG7112, the mechanism underlying this “switch” (from cytostatic to cytotoxic effect) has not been elucidated [27, 43]. We previously demonstrated that phosphorylation at Ser-46 is required for TP53-dependent apoptosis, and is induced by either overexpression of TP53 or by severe DNA damage [16]. Recently, inhibition of MDM2 was reported to induce p53-induced senescence or quiescence in cells with activation of the mTOR (mammalian target of rapamycin) pathway [44, 45]. Thus, depending on TP53 status, the cytostatic or cytotoxic effects of RG7122 may vary among tumor types and even among cells in cancers with heterogeneous cell populations.
RG7112 antitumor activity was also observed in xenograft mouse models. This activity might be partly independent of apoptosis, as induction of TP21 suggests that cell cycle arrest is an underlying mechanism, as is suppression of tumor vascularization. In this study, RG7112 inhibited tumor vascularization by stabilization of TP53, which is known to prevent angiogenesis via suppression of HIF-1alpha [46]. We note that HIF-1alpha expression is significantly elevated in clear cell carcinomas compared to other types of ovarian cancers [47, 48]. Taken together, the data suggest that RG7112 may play multiple roles in inhibiting the progression of clear cell tumors.
Overexpression of MDM4 has also been reported in various cancers, and MDM4 inhibitors were recently developed along with dual inhibitors against MDM2 and MDM4 [49, 50]. However, we found that MDM4 is expressed only in low levels in clear cell carcinomas. Moreover, knockdown of MDM2 by siRNA clearly suppressed cell viability and induced apoptosis. Therefore, MDM2 might be a more suitable target than MDM4 in clear cell carcinomas. RG7112 is currently in phase I/II clinical trials against several cancers, but not against clear cell carcinomas. RG7112 might prove to be particularly promising against clear cell carcinoma.
This study has several limitations. For instance, the mechanisms of MDM2 overexpression are yet to be elucidated. As MDM2 is also expressed in normal cells, the feasibility, pharmacokinetics, and pharmacodynamics of RG7112 should be carefully considered in any potential clinical application. In addition, it is unclear whether the presence or absence of TP53 mutations is a sufficient biomarker by itself to predict sensitivity to MDM2 inhibitors. Finally, further studies are needed to establish whether MDM2 abundance is associated with sensitivity to RG7112.
In conclusion, we demonstrated that MDM2 is frequently overexpressed in clear cell carcinomas, and that MDM2 overexpression is associated with poor prognosis. An MDM2 inhibitor, RG7112, significantly suppressed the growth of clear cell tumors with wild type TP53 both in vitro and in vivo. Furthermore, RG7112 elicited apoptotic cell death via phosphorylation of TP53 at Ser-46, and via induction of proapoptotic TP53 target genes. These findings suggest that MDM2 inhibitors such as RG7112 may be potent, targeted therapies against clear cell carcinomas.
MATERIALS AND METHODS
Tumor samples
Surgical samples were obtained with written informed consent from 91 patients at the University of Tokyo Hospital and Saitama Medical University International Medical Center, of whom 75 had clear cell carcinoma and 16 had high-grade serous carcinoma. The high proportion of carcinoma cells (>50%) in each sample was confirmed by a pathologist as described previously [51]. All 91 patients received primary surgery followed by 6-8 cycles of chemotherapy with paclitaxel and carboplatin. RNA was extracted and hybridized as described previously to HG-U133 Plus 2.0 Arrays (Affymetrix) containing 54,675 probes for human genes [51]. Microarray data for 41 patients (25 clear cell and 16 high-grade serous carcinomas) are available from Gene Expression Omnibus under accession number GSE65986, and 50 clear cell carcinomas were added in this study. We observed no significant biases between previous and additional microarray datasets. Patient characteristics are listed in Supplementary Table 1. Genomic DNA was extracted from 72 of 75 clear cell carcinomas, and TP53 mutations (all exons) were determined by Sanger sequencing [52]. Total RNA from 13 normal tissues were purchased from Clontech (Mountain View, CA, USA), Ambion (Waltham, MA, USA), and Stratagene (Cedar Creek, TX, USA).
RG7112 and cell lines
RG7112 was purchased from MedChem Express (Monmouth Junction, NJ, USA) and APEXBIO (Houston, TX, USA). OVISE, OVTOKO, and RMG-I cell lines were purchased from the Japanese Collection of Research Bioresources Cell Bank (Ibaraki, Osaka, Japan), while cell lines JHOC-7 and JHOC-9 were purchased from RIKEN Cell Bank (Ibaraki, Tsukuba, Japan). ES-2 and SKOV3 cell lines were purchased from American Type Culture Collection (ATCC) (Manassas, VA, USA). All these 7 cell lines are classified histologically as clear cell carcinoma, were cultured as described previously [48]. An immortalized cell line derived from ovarian surface epithelium was a generous gift from Dr. Hidetaka Katabuchi [53], and COS-7, and 293T cell lines were purchased from ATCC. The cell lines were authenticated by short tandem repeats.
Proliferation assays
Cell proliferation was measured using Cell Counting Kit-8, which is based on the tetrazolium salt WST-8 (Dojindo; Minatoku, Tokyo, Japan) derived from methyl thiazolyl tetrazolium (MTT). Briefly, cells were seeded in 96-well plates at 2,000 cells/well. After 24 h, cells were incubated with 0-10 μM RG7112 for 72 h. Proliferation was quantified by monitoring the absorbance at 450 nm using a microplate reader (Bio Tek; Winooski, VT, USA). Cell proliferation was normalized relative to cell cultures treated with DMSO. Experiments were repeated at least three times.
Immunoblotting
Cells were treated with RG7112 for the indicated time at indicated concentrations, and then lysed as described previously [54]. Lysates were probed with antibodies specific for MDM2, TP21, BAX, TP53 (Santa Cruz; Dallas, TX, USA), HIF-1alpha (Becton, Dickinson and Company; Franklin Lakes, NJ, USA), phosphorylated TP53 (Ser46), phosphorylated TP53 (Ser15), cleaved PARP, PUMA (Cell Signaling Technology; Danvers, MA, USA), survivin (Novus Biologicals; Littleton, CO, USA), and beta-actin (Sigma-Aldrich; St. Louis, MO, USA).
Cell cycle analysis
Cells (5 × 105) were seeded in 60-mm dishes and treated with RG7112 for 48 h. Floating and adherent cells were collected by trypsinization, washed twice with phosphate-buffered saline (PBS), resuspended in cold 70% ethanol, and incubated at 4 °C overnight. Cells were then washed another two times with PBS, treated with 0.25 mg/mL RNase A (Sigma-Aldrich) for 30 min at 37 °C, stained with 50 μg/mL propidium iodide (Sigma-Aldrich) at 4 °C for 30 min in the dark, and analyzed on FACS Calibur HG (Becton, Dickinson and Company). Data were analyzed in CELL Quest pro ver. 3.1. (Becton, Dickinson and Company), and experiments were repeated three times.
Detection of apoptosis
Cells (5 × 105) were seeded in 60-mm plates for 24 h, and exposed to either DMSO or RG7112 for 72 h. Cells were then trypsinized, washed twice, doubly stained with annexin V-fluorescein isothiocynate and propidium iodide according to the manufacturer’s instructions (Annexin V-FITC Apoptosis Detection Kit; BD Biosciences), and analyzed by flow cytometry. Experiments were repeated thrice.
Gene silencing
Small interfering RNAs (siRNAs) specific for MDM2 (Stealth siRNAs HSS142909 and HSS142911) were purchased from Invitrogen (Carlsbad, CA, USA). Cells were plated in 100-mm plates at approximately 30% confluence, incubated for 24 h, and transfected for 72 h with 20 nM siRNA duplexes in Lipofectamine RNAiMAX (Invitrogen) and Opti-MEM medium (Life Technologies; Carlsbad, CA, USA). A negative control kit (Invitrogen) was used for comparison.
Tumor xenografts in nude mice
Specific pathogen-free female nude mice (BALB/cAJc1-nu/nu) were purchased from CLEA Japan, Inc. (Meguro, Tokyo, Japan). At 5-6 weeks of age, mice were injected subcutaneously into the flank with 107 RMG-I or OVISE cells in 200 μL PBS. Tumors were harvested after exponential growth, minced into 3-mm pieces, and transplanted subcutaneously into the right flank of other mice. After tumors had grown to 5 mm in diameter, mice were randomly assigned into two groups of 5-6 animals. One group received daily oral injections (100 mg/kg) of RG7112 suspended in 0.5 w/v% methyl cellulose 400 (Wako Pure Chemical Industries; Osaka, Osaka, Japan), while the other received similar doses of methyl cellulose. Tumor size was measured with a digital caliper three times weekly to estimate tumor volume according to the formula ([major axis] × [minor axis]2)/2. Mice were sacrificed humanely after 20 days of treatments, and tumors were preserved in 4% paraformaldehyde for immunoblotting analysis.
Immunohistochemistry
To immunostain CD31, tumor sections were fixed at 4 °C for 10 min in 4% paraformaldehyde, immersed in 1% H2O2 at room temperature for 30 min to quench endogenous peroxidases, and blocked at room temperature for 30 min with Blocking One (Nacalai tesque; Kyoto, Japan). Sections were then probed at 4 °C overnight with 1:500 anti-CD31 (PECAM-1; BD Biosciences; Franklin Lakes, NJ), washed in Tris-buffered saline, and labeled at room temperature for 45 min with 1:400 biotinylated rabbit anti-rat (DAKO), and then at room temperature for 45 min with LSAB (DAKO). Finally, sections were stained with 3,3-diaminobenzidine (DAKO) and hematoxylin (Wako).
Quantification of microvascularization
Harvested subcutaneous tumors were immunostained with anti-mouse CD31 (PECAM-1; BD Biosciences). The number of microvessels stained was counted at 400× in four random fields in each tumor.
Statistical analysis
Survival was analyzed by Kaplan-Meier method and log rank test. Univariate and multivariate analyses were performed using the Cox proportional hazard model. Groups were compared by t-test. Data were analyzed in JMP v11 (Cary, NC, USA) and GraphPad Prism 6 (La Jolla, CA, USA), with P < 0.05 considered to be significant.
ACKNOWLEDGMENTS
We thank Drs. Masako Ikemura and Daichi Maeda for insightful comments, Prof. Hidetaka Katabuchi for generously providing OSE cells, and Ms. Kaori Tomita for technical assistance. We thank Editage (www.editage.com) for English language editing.
This work was financially supported by Grants-in-Aid for Scientific Research (C), 26462515 (to K Oda), and by Grants-in-Aid for Young Scientific Research (B), 25893229 (to Y Ikeda), 15K20128 (to K Sone), 16K20176 (to M Mori-Uchino), and a Grant-in-Aid for Research Activity Start-up (15H06173 to T Kashiyama) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan. This study was also a research program of the Project for Development of Innovative Research on Cancer Therapeutics (P-Direct) (to T Yano) and the Project for Cancer Research And Therapeutic Evolution (P-CREATE) (to H Aburatani and K Oda) from the Japan Agency for Medical Research and development, AMED.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Kurman RJ, Shih IeM. The Dualistic Model of Ovarian Carcinogenesis: Revisited, Revised, and Expanded. Am J Pathol 2016; 186:733-74.
2. Okamoto A, Glasspool RM, Mabuchi S, Matsumura N, Nomura H, Itamochi H, Takano M, Takano T, Susumu N, Aoki D, Konishi I, Covens A, Ledermann J, et al. Gynecologic Cancer InterGroup (GCIG) consensus review for clear cell carcinoma of the ovary. Int J Gynecol Cancer 2014; 24:S20-25.
3. Heintz AP, Odicino F, Maisonneuve P, Quinn MA, Benedet JL, Creasman WT, Ngan HY, Pecorelli S, Beller U. Carcinoma of the ovary. FIGUREO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet 2006; 95:S161-192.
4. Kurman RJ, Shih IeM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum Pathol 2011; 42:918-931.
5. Lim A, Mesher D, Gentry-Maharaj A, Balogun N, Widschwendter M, Jacobs I, Sasieni P, Menon U. Time to diagnosis of Type I or II invasive epithelial ovarian cancers: a multicentre observational study using patient questionnaire and primary care records. BJOG 2015.
6. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474:609-615.
7. Berns EM, Bowtell DD. The changing view of high-grade serous ovarian cancer. Cancer Res 2012; 72:2701-2704.
8. Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, Kassahn KS, Newell F, Quinn MC, et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015; 521:489-494.
9. Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, Suzuki M, Sato I, Taguchi K. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer 2000; 88:2584-2589.
10. Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, Glas R, Slamon D, Velculescu VE, Kuman RJ, Shih IeM. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol 2009; 174:1597-160.
11. Ho ES, Lai CR, Hsieh YT, Chen JT, Lin AJ, Hung MH, Liu FS. p53 mutation is infrequent in clear cell carcinoma of the ovary. Gynecol Oncol 2001; 80:189-193.
12. Jones S, Wang TL, Shih IeM, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, Kinzler KW, Velculescu VE, Papadopoulos N. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2016; 330:228-231
13. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000; 408:307-310.
14. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 2014; 25:304-317.
15. Powell E, Piwnica-Worms D, Piwnica-Worms H. Contribution of p53 to metastasis. Cancer Discov 2014; 4:405-414.
16. Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 2000; 102:849-862.
17. Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 2000; 14:34-44.
18. Hong B, van den Heuvel AP, Prabhu VV, Zhang S, El-Deiry WS. Targeting tumor suppressor p53 for cancer therapy: strategies, challenges and opportunities. Curr Drug Targets 2014; 15:80-89.
19. Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol 2010; 80:724-730.
20. Micel LN, Tentler JJ, Smith PG, Eckhardt GS. Role of ubiquitin ligases and the proteasome in oncogenesis: novel targets for anticancer therapies. J Clin Oncol 2013; 31:1231-1238.
21. Pei D, Zhang Y, Zheng J. Regulation of p53: a collaboration between Mdm2 and Mdmx. Oncotarget 2012; 3:228-235. doi: 10.18632/oncotarget.443.
22. Wang X, Jiang X. Mdm2 and MdmX partner to regulate p53. FEBS Lett 2012; 586:1390-6.
23. Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucleic Acids Res 1998; 26:3453-3459.
24. Yu Q, Li Y, Mu K, Li Z, Meng Q, Wu X, Wang Y, Li L. Amplification of Mdmx and overexpression of MDM2 contribute to mammary carcinogenesis by substituting for p53 mutations. Diagn Pathol 2014; 9:71.
25. Crane EK, Kwan SY, Izaguirre DI, Tsang YT, Mullany LK, Zu Z, Richards JS, Gershenson DM, Wong KK. Nutlin-3a: a potential therapeutic opportunity for TP53 wild-type ovarian carcinomas. PLoS One 2015; 10:e0135101.
26. Ray-Coquard I, Blay JY, Italiano A, Le Cesne A, Penel N, Zhi J, Heil F, Rueger R, Graves B, Ding M, Geho D, Middleton SA, Vassilev LT, et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 2012; 13:1133-1140.
27. Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, Tardell C, Garrido R, Lee E, Kolinsky K, To KH, Linn M, Podlaski F, et al. MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res 2013; 73:2587-2597.
28. Verreault M, Schmitt C, Goldwirt L, Pelton K, Haidar S, Levasseur C, Guehennec J, Knoff D, Labussière M, Marie Y, Ligon AH, Mokhtari K, Hoang-Xuan K, et al. Preclinical efficacy of the MDM2 inhibitor RG7112 in MDM2-amplified and TP53 wild-type glioblastomas. Clin Cancer Res 2016; 22:1185-1196.
29. Millard M, Pathania D, Grande F, Xu S, Neamati N. Small-molecule inhibitors of p53-MDM2 interaction: the 2006-2010 update. Curr Pharm Des 2011; 17:536-559.
30. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov 2014; 13:217-236.
31. Drummond MW, Vyas P, Kirschbaum M, Iyer SP, Ruvolo V, et al. Results of the phase I trial of RG7112, a small-Molecule MDM2 antagonist in leukemia. Clin Cancer Res 2016; 22:868-876.
32. Lagrange M, Charbonnier S, Orfanoudakis G, Robinson P, Zanier K, Masson M, Lutz Y, Trave G, Weiss E, Deryckere F. Binding of human papillomavirus 16 E6 to p53 and E6AP is impaired by monoclonal antibodies directed against the second zinc-binding domain of E6. J Gen Virol 2005; 86:1001-1007.
33. Devine T, Dai MS. Targeting the ubiquitin-mediated proteasome degradation of p53 for cancer therapy. Curr Pharm Des 2013; 19:3248-3262.
34. Onel K, Cordon-Cardo C. MDM2 and prognosis. Mol Cancer Res 2004; 2:1-8.
35. Leach FS, Tokino T, Meltzer P, Burrell M, Oliner JD, Smith S, Hill DE, Sidransky D, Kinzler KW, Vogelstein B. p53 mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res 1993; 53:2231-2234.
36. Huang K, Tang Y, He L, Dai Y. MicroRNA-340 inhibits prostate cancer cell proliferation and metastasis by targeting the MDM2-p53 pathway. Oncol Rep 2016; 35:887-895.
37. Li Q, Lozano G. Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res 2013; 19:34-41.
38. Noon AP, Vlatković N, Polański R, Maguire M, Shawki H, Parsons K, Boyd MT. p53 and MDM2 in renal cell carcinoma: biomarkers for disease progression and future therapeutic targets? Cancer 2010; 116:780-790.
39. Knappskog S, Lonning PE. P53 and its molecular basis to chemoresistance in breast cancer. Expert Opin Ther Targets 2012; 16:S23-30.
40. Yang-Hartwich Y, Soteras MG, Lin ZP, Holmberg J, Sumi N, Craveiro V, Liang M, Romanoff E, Bingham J, Garofalo F, Alvero A, Mor G. p53 protein aggregation promotes platinum resistance in ovarian cancer. Oncogene. 2015, 34:3605-3616.
41. Zhang X, Cheng L, Minn K, Madan R, Godwin AK, Shridhar V, Chien J. Targeting of mutant p53-induced FoxM1 with thiostrepton induces cytotoxicity and enhances carboplatin sensitivity in cancer cells. Oncotarget. 2014, 5:11365-11380. doi: 10.18632/oncotarget.2497.
42. Zanjirband M, Edmondson RJ, Lunec J. Pre-clinical efficacy and synergistic potential of the MDM2-p53 antagonists, Nutlin-3 and RG7388, as single agents and in combined treatment with cisplatin in ovarian cancer. Oncotarget. 2016; 7:40115-40134. doi: 10.18632/oncotarget.9499.
43. Richmond J, Carol H, Evans K, High L, Mendomo A, Robbins A, Meyer C, Venn NC, Marschalek R, Henderson M, Sutton R, Kurmasheva RT, Kees UR, et al. Effective targeting of the P53-MDM2 axis in preclinical models of infant MLL-rearranged acute lymphoblastic leukemia. Clin Cancer Res 2015; 21:1395-1405.
44. Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344-352. doi: 10.18632/aging.100160.
45. Lane DP, Verma C, Fang CC. The p53 inducing drug dosage may determine quiescence or senescence. Aging (Albany NY) 2010; 2:748. doi: 10.18632/aging.100229.
46. Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev 2000; 14:34-44.
47. Mabuchi S, Kawase C, Altomare DA, Morishige K, Hayashi M, Sawada K, Ito K, Terai Y, Nishio Y, Klein-Szanto AJ, Burger RA, Ohmichi M, Testa JR, et al. Vascular endothelial growth factor is a promising therapeutic target for the treatment of clear cell carcinoma of the ovary. Mol Cancer Ther 2010; 9:2411-2422.
48. Lee S, Garner EI, Welch WR, Berkowitz RS, Mok SC. Over-expression of hypoxia-inducible factor 1 alpha in ovarian clear cell carcinoma. Gynecol Oncol 2007; 106:311-317.
49. Wang H, Ma X, Ren S, Buolamwini JK, Yan C. A small-molecule inhibitor of MDMX activates p53 and induces apoptosis. Mol Cancer Ther 2011; 10:69-79.
50. Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To KH, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, et al. Stapled α-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A. 2013; 110:E3445-3454.
51. Uehara Y, Oda K, Ikeda Y, Koso T, Tsuji S, Yamamoto S, Asada K, Sone K, Kurikawa R, Makii C, Hagiwara O, Tanikawa M, Maeda D, Hasegawa K, Nakagawa S, Wada-Hiraike O, Kawana K, Fukayama M, Fujiwara K, Yano T, et al. Integrated copy number and expression analysis identifies profiles of whole-arm chromosomal alterations and subgroups with favorable outcome in ovarian clear cell carcinomas. PLoS One 2015; 10:e0128066.
52. Kashiyama T, Oda K, Ikeda Y, Shiose Y, Hirota Y, Inaba K, Makii C, Kurikawa R, Miyasaka A, Koso T, Fukuda T, Tanikawa M, Shoji K, et al. Antitumor activity and induction of TP53-dependent apoptosis toward ovarian clear cell adenocarcinoma by the dual PI3K/mTOR inhibitor DS-7423. PLoS One 2014; 9:e87220.
53. Maeda T, Tashiro H, Katabuchi H, Begum M, Ohtake H, Kiyono T, Okamura H. Establishment of an immortalised human ovarian surface epithelial cell line without chromosomal instability. Br J Cancer. 2005; 93:116-123.
54. Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res 2005; 65:10669-10673.