
Introduction
Autophagy is a highly conserved cellular degradation pathway that mediates the degradation of macromolecules and organelles by the lysosome [1]. Serving as an intracellular “recycling” pathway, autophagy ensures the continued availability of building blocks for macromolecular synthesis during periods of cellular stress and nutrient deprivation. Autophagy also exerts a quality control function by removing misfolded or unfolded proteins and damaged organelles. In light of these many important functions, it is not surprising that autophagy plays an essential role in normal physiology and protection against diseases, including neurodegenerative diseases, infection and aging [2, 3].
The role of autophagy in cancer remains controversial, with studies suggesting both pro- and anti-tumor effects of autophagy genes [4]. Supporting a tumor-suppressive effect of autophagy, several studies indicate that autophagy prevents tumor initiation. BECLIN 1 (BECN1) is frequently monoallelically deleted in human breast and ovarian cancers [5, 6]; Becn1 heterozygous-deficient mice have an increased frequency of spontaneous malignancies including lymphomas, liver, lung and breast tumors [7-9]; and decreased BECN1 expression is associated with poor outcomes in human breast cancer [10]. Mice deficient in Atg4C have increased chemically-induced fibrosarcomas [11], and Bif-1 deletion in mice results in an increased frequency of spontaneous lymphomas and solid tumors [12]. The anticancer effects of tumor-suppressor genes such as PTEN, LKB1 and TSC1/TSC2 derive, at least in part, from their regulation of PI3K-mTOR signaling, which normally opposes the autophagy pathway. Consistent with this idea, oncogenes such as AKT, EGFR and BCL2 may promote tumorigenesis by inhibiting autophagy, either through a positive effect on mTOR signaling (reviewed in [13]) or directly via effects on Beclin 1 [14-16].
On the other hand, several studies suggest that the autophagy pathway can promote the survival of cancer cells under conditions of environmental stress. Inhibition of autophagy by deletion FIP200 (also known as RB1CC1) suppresses the development of mammary tumors in a polyoma Middle T-driven breast cancer model [17]. Autophagy promotes survival and increases tumorigenesis in mutant Kras-expressing cells [18-20], and Atg7 deletion in mice suppresses progression of Kras and BrafV600E induced lung cancers and Kras driven pancreatic carcinoma [21-23]. Beclin 1 appears to be required for maintenance of a cancer stem cell population in breast cancers [24]. Taken together, these studies suggest a model whereby autophagy plays different roles in different stages of tumorigenesis. Autophagy may oppose the initiation of tumors by removing damaged organelles and proteins from the cytoplasm, exerting quality control and avoiding the generation of reactive oxygen species (ROS) by damaged mitochondria. Once a tumor is established however, autophagy may promote cancer cell survival in the face of stresses caused by rapid growth and the metabolic effects of oncogenes. In addition to different roles in different stages of tumorigenesis, autophagy may have different functions in the context of different tumor mutational status; for example, although loss of autophagy delays tumorigenesis in Kras-driven pancreatic tumors, it accelerates tumorigenesis in Kras-driven pancreatic tumors with concurrent p53 mutation [22].
To further address the role of autophagy in tumor development in an in vivo model system and to probe the interrelationship of autophagy and p53 deficiency, we inhibited autophagy in a tissue-specific fashion in zebrafish by expressing a dominant-negative form of the autophagy gene atg5 (atg5K130R) from the well-characterized microphthalmia-associated transcription factor a (mitfa) promoter. mitfa is expressed in the developing neural crest and in melanocytes [25], and a previous study showed that transgenic expression of mutant BRAFV600E kinase under the control of the mitfa promoter leads to melanoma in p53-mutant fish [26]. Fish stably expressing the mitfa:atg5K130R transgene in the p53 mutant background developed several types of tumors including small round cell tumors, neuroendocrine tumors and malignant peripheral nerve sheath tumors (MPNSTs). Compared to control heterozygous tp53M214K/+ heterozygotes, Tg(mitfa:atg5K130R); tp53M214K/+ fish displayed decreased latency and increased incidence of MPNSTs. Coexpression of a sox10-eGFP transgene demonstrated the neural crest origin of the tumors. Moreover, expression of Atg5K130R caused increased DNA damage and accelerated loss-of-heterozygosity (LOH) of p53 in the p53M214K/+ background. Taken together, these results suggest that autophagy acts as a barrier to tumor formation, and that defective autophagy may contribute to genome instability and tumorigenesis.
Results
Expression of dominant-negative autophagy gene, atg5K130R impairs autophagy in zebrafish
Previous studies have established that expression of Atg5 in which lysine 130 is mutated to arginine inhibits autophagy via a dominant-negative mechanism [27-30]. To confirm that the expression of Atg5K130R inhibits autophagy in zebrafish in vivo, we took advantage of a Tg(cmv:GFP-lc3) autophagy reporter line [31]. During autophagy induction, Lc3-I (diffusely distributed in the cytoplasm) is conjugated with phosphatidylethanolamine to form Lc3-II, which stably associates with the autophagosomal membrane. GFP-tagged Lc3-II accumulates in punctate structures (autophagosomes), serving as a useful in vivo marker of autophagy [32]. We injected mRNA encoding mCherry, zebrafish atg5 or dominant-negative atg5K130R mRNA, or a morpholino directed against atg5 that we previously validated [33] into one-cell stage Tg(CMV:GFP-Lc3) embryos. We prepared primary gastrula-stage cultures from the injected embryos and used fluorescence microscopy to quantify the number of GFP-Lc3 puncta. Compared to mCherry or atg5, atg5K130R mRNA significantly reduced the number of GFP-LC3 puncta, similar to the effect of the atg5 morpholino, indicating inhibition of autophagosome formation by expression of Atg5K130R in vivo (Figure 1A, 1B).
We further confirmed that the expression of Atg5K130R inhibits autophagy by immunoblot analysis of Lc3 in lysates prepared from embryos injected with the mRNAs. atg5K130R mRNA- injected embryo lysates showed reduced levels of Lc3-II compared to control atg5 or mcherry mRNA-injected embryos, suggesting that autophagy is inhibited by the expression of Atg5K130R (Figure 1C, 1D). In summary, the overexpression of atg5K130R RNA efficiently inhibited autophagy in zebrafish in vivo.
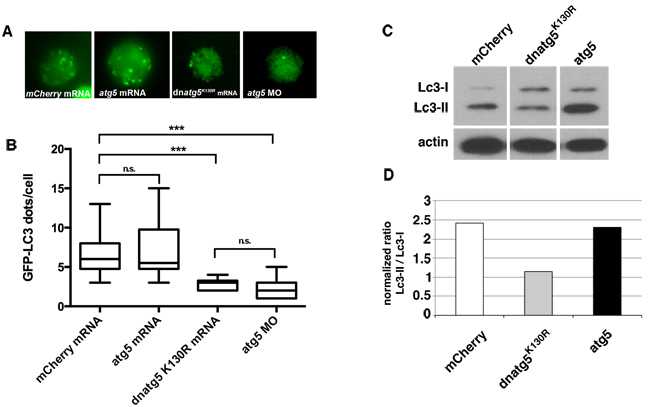
Figure 1: Autophagy is inhibited by expression of atg5K130R mRNA. A.-B. Representative images A. and quantification B. of GFP-Lc3 puncta in primary cells prepared from blastula-stage Tg(cmv:GFP-LC3) embryos injected with mRNA encoding mCherry control, wildtype atg5 or dominant-negative atg5K130R, or with a morpholino targeting atg5. At least 15 cells/group were scored. Bars represent mean ± SEM of 50 to 100 cells per group. Similar results were observed in three independent experiments. ***P < 0.001 for atg5K130R mRNA- or morpholino-injected embryos versus mCherry mRNA-injected embryos; two-tailed t-test. C. Immunoblot analysis of Lc3 in primary cell lysates from embryos injected at the one-cell stage with in vitro transcribed mCherry, atg5K130R or atg5 mRNA. Actin is shown as a loading control. D. Quantification of immunoblot.
Stable expression of mitfa: atg5K130R results in tumors in a tp53M214K/+ background
Having shown that expression of Atg5K130R inhibits autophagy in zebrafish embryos, we used this approach to study the role of autophagy in tumor suppression. To investigate the role of autophagy in tumor suppression, we expressed atg5K130R from the mitfa promoter. During zebrafish development, Mitf is expressed in neural crest and melanocytes and has been implicated as a key regulator in pigment cell development [25]. A previous report showed that transgenic zebrafish expressing mutant BRAFV600E driven by the mitfa promoter formed fish nevi and developed melanomas in a p53-mutant background [26].
We used the Tol2 transposon system [34] to generate genomic insertions of the mitfa:atg5K130R transgene, and identified founders by PCR analysis of F1 progeny (Supplementary Figure 1). Stable transgenics were raised to adulthood and monitored for tumor onset. Previous reports showed that chimeric knockout of the autophagy gene Atg5 resulted in hepatomas in mice [35]. However, transgenic fish expressing Atg5K130R did not develop tumors. We suspected that inhibition of autophagy alone might be insufficient to cause tumors, and therefore tested the effect of the transgene in the sensitized tp53-mutant background. Homozygous mutant tp53M214K fish were previously shown to have defects in the apoptotic response to γ-irradiation, as well as increased tumor susceptibility. The most common tumors in these fish are MPNSTs [36]. In our cohort, 55% of tp53M214K/M214K homozygotes developed MPNSTs beginning at 7 months of age (median age of onset 16.5 months); tp53M214K/+ heterozygotes exhibited lower incidence (17%) and longer latency (median age of onset 20 months) (Figure 2), consistent with previous reports [36].
Transgenic expression of the dominant-negative autophagy construct increased tumor incidence and decreased latency in p53-deficient fish (Figure 2). In the p53-heterozygous background; nearly 40% of Tg(mitfa:atg5K130R);tp53M214K/+ heterozygotes developed tumors, with a median age of onset of 17 months. (p < 0.0001 for comparison to non-transgenic tp53M214K/+ fish by log-rank test). Expression of Atg5K130R in a tp53M214K/M214K background caused slightly earlier and higher tumor incidence compared to tp53M214K/M214K homozygotes (60% incidence: median age of onset 16 months), but the difference did not reach statistical significance (Figure 2). To verify that the tumors expressed the atg5K130R transgene, we prepared short-term primary cell cultures of the tumors (to enrich for tumor cells and allow for depletion of nonproliferating normal cells), purified RNA from the cells and performed RT-PCR with primers specific for atg5. This assay validated expression of the atg5K130R transgene in the tumors (Supplementary Figure 1).
For consistency, in these experiments all Tg(mitfa:atg5K130R) animals were derived from a single founder. To confirm these results, and to ensure that the effect of autophagy inhibition on tumor incidence was not due to unintended consequences of transgene genomic insertion, we generated an independent transgenic line in which the mitfa promoter drives expression of a dominant-negative form of the autophagy protein Atg4b [Tg(mitfa:atg4bC74A)]. Mutation of this conserved cysteine residue to alanine in mouse Atg4b was previously shown to inhibit autophagy [28]. Using the GFP-Lc3 assay, we showed that expression of zebrafish Atg4bC74A significantly impairs GFP-Lc3 puncta formation (Supplementary Figure 2). We crossed Tg(mitfa:atg4bC74A) to tp53M214K/M214K homozygotes to generate Tg(mitfa:atg4bC74A); tp53M214K/+ heterozygotes and non-transgenic tp53M214K/+ heterozygous siblings. 5/48 (10.4%) of Tg(mitfa:atg4bC74A); tp53M214K/+ heterozygotes developed tumors with a median tumor onset of 12.7 months. In contrast, none of the tp53M214K/+ heterozygous siblings (n = 48) had developed tumors by 18 months of age at which point the experiment was stopped (not shown). Thus, these data confirm that inhibition of autophagy decreases the latency of tumor onset in tp53M214K/+ heterozygotes.
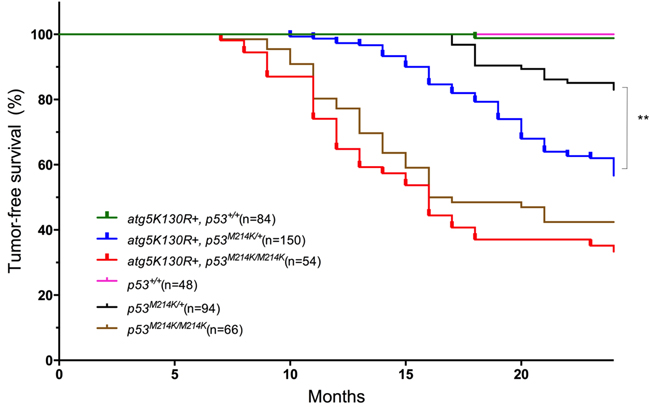
Figure 2: Atg5K130R accelerates tumor development in the p53-mutant background. Tumor-free survival for each indicated fish genotype is shown. Numbers in parentheses indicate total number of animals observed per genotype. **p < 0.001 for Tg(mitfa:atg5K130R); p53M214K/+ vs non-transgenic tp53M214K/+ (Log-rank (Mantel-Cox) test).
Tumor spectrum of zebrafish expressing Tg(mitfa:atg5K130R) in the tp53 mutant background
Macroscopically, tumors predominantly occurred in the abdominal region (Figure 3A) and were characterized by the presence of prominent blood vessels on the ventral surface. A small number of tumors also arose behind the eyes and in the jaw and the gills (data not shown). The most common tumors were MPNSTs, characterized by wavy fascicles of spindle cells with elongate nuclei showing tapered ends. The next most common tumors were characterized by cords, nests, and gland-like structures composed of epithelioid cells with high nuclear to cytoplasmic ratio, hyperchromatic nuclei, and scant eosinophilic cytoplasm, morphologically resembling human poorly differentiated neuroendocrine carcinomas. Five of the Tg(mitfa:atg5K130R);tp53M214K/+ fish and one Tg(mitfa:atg5K130R);tp53M214K/M214K fish developed malignant small cell tumors, which contained sheets of small round cells with finely dispersed chromatin, high nuclear-cytoplasmic ratio and scant eosinophilic cytoplasm. Small cell tumors were not found in non-transgenic tp53M214K/+ or tp53M214K/M214K controls (Figure 3A, 3B, and Table 1).
Table 1: Tumor histology in different genetic backgrounds
p53 Genotype |
+/+ |
M214K/+ |
M214K/M214K |
|||
mitfa:atg5K130R transgene |
- |
+ |
- |
+ |
- |
+ |
Total # per genotype |
48 |
84 |
94 |
150 |
66 |
54 |
Total # with tumor (%) |
0 |
0 |
14 (15%) |
61 (40%) |
39 (58%) |
36 (67%) |
MPNST |
0 |
0 |
11 |
43 |
26 |
26 |
Neuroendocrine cell tumor |
0 |
0 |
3 |
5 |
3 |
2 |
Small round cell tumor |
0 |
0 |
0 |
5 |
0 |
1 |
Vascular tumor |
0 |
0 |
0 |
0 |
2 |
0 |
Not determined |
0 |
0 |
0 |
4 |
8 |
7 |
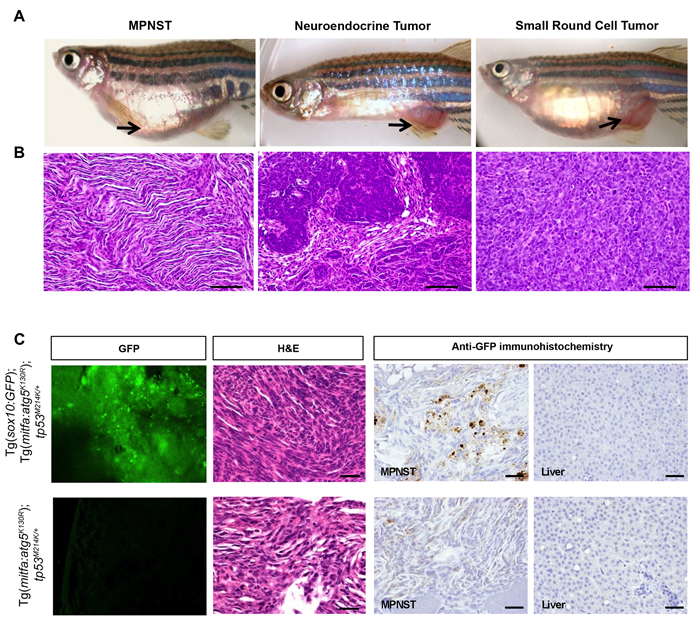
Figure 3: MPNSTs arising in Tg(mitfa:atg5K130R); tp53M214K/+ fish express sox10. A.-B. Representative photomicrographs of gross macroscopic appearance of tumors A. and hematoxylin and eosin (H&E) stained microscopic sections of corresponding tumors B. in Tg(mitfa:atg5K130R); tp53M214K/+ fish. MPNST, malignant peripheral nerve sheath tumor. Scale bars, 50 μm. See Table 1 for details of numbers of fish with each tumor type. C. Representative confocal images to detect GFP expression, H&E staining, and anti-GFP immunoperoxidase staining of an MPNST from a Tg(mitfa:atg5K130R); Tg(sox10:EGFP); tp53M214K/+ fish (top) and an MPNST from a GFP-negative control Tg(mitfa:atg5K130R); tp53M214K/+ (bottom). Liver is shown for the anti-GFP immunoperoxidase staining as a representative non-tumor tissue (right panels). Scale bars, 100 µm.
Tg(mitfa:atg5K130R) MPNSTs express atg5K130R and sox10
Tg(mitfa:atg5K130R);tp53M214K/+ fish showed a significantly higher tumor incidence than tp53M214K/+ heterozygous mutant fish. However, both fish lines developed MPNSTs (Table 1), which was somewhat surprising for the Tg(mitfa:atg5K130R) fish, because expression of activated mutant BRAFV600E from the mitfa promoter was previously shown to cause melanomas in zebrafish [26]. To confirm that the atg5K130R transgene was expressed in tumors arising in Tg(mitfa:atg5K130R); tp53M214K/+ fish, we isolated tumors, disaggregated the cells and monitored cell growth in primary cell cultures for up to 3 passages to obtain homogeneous cells. We isolated RNA from the cells and performed RT-PCR analysis to detect atg5 mRNA. PCR products were confirmed by sequence analysis and showed the expression of atg5K130R RNA in tumor tissue (Supplementary Figure 2). We then sought further confirmation that these tumors indeed arose from a neural lineage. Specifically, we asked whether the tumors expressed markers of neural crest, which gives rise to Schwann cells [37-39], the cell of origin of MPNSTs [40, 41]. Expression of mitfa is regulated by sox10, a transcription factor expressed in neural crest cells [42] and re-expressed during melanoma initiation [43]. We crossed Tg(mitfa:atg5K130R);tp53M214K/+ fish to the neural crest cell reporter fish line, Tg(−4.9sox10:GFP) [44] and monitored the fish for tumor development. A representative tumor arising in this line is shown in Figure 3C. The tumor expressed GFP as detected by confocal microscopy (Figure 3C, first column). Histologically, the tumor was identified as MPNST (Figure 3C, second column) and GFP expression in tumor tissue was confirmed by immunostaining (Figure 3C, third column). GFP expression was variable; some areas of the tumor showed strong expression of GFP, but spindle-like tumor cells showed very weak or almost negative expression of GFP, likely caused by loss of sox10 promoter activity in fully differentiated cells [45]. GFP expression was not detected in non-tumorigenic liver (Figure 3C, fourth column). These results confirm that MPNSTs in Tg(mitfa:atg5K130R);tp53M214K/+ fish arise from neural crest cells and suggest that impairment of autophagy in this lineage increases tumor susceptibility.
Cell proliferation rates are equivalent in MPNSTs from atg5K130R transgenic and non-transgenic tp53M214K/+ heterozygotes
MPNSTs arising in Tg(mitfa:atg5K130R);tp53M214K/+ fish were grossly evident much earlier than similar tumors occurring in tp53M214K/+ heterozygous mutant fish. This raised the possibility that tumors in atg5K130R transgenics might undergo more rapid cell proliferation than those in non-transgenic tp53M214K/+ heterozygotes, making the tumors detectable earlier. To test this possibility, we prepared histologic sections of MPNSTs from atg5K130R transgenic and non-transgenic tp53M214K/+ heterozygotes and performed immunohistochemistry for the Serine 10-phosphorylated form of Histone H3, a marker of mitotic cells [46]. MPNSTs from both atg5K130R-transgenic and non-transgenic animals exhibited similar levels of cell proliferation (Figure 4), suggesting that the early appearance of MPNSTs in atg5K130R-transgenic p53 heterozygotes is not due to intrinsically higher rates of cell proliferation in these tumors.
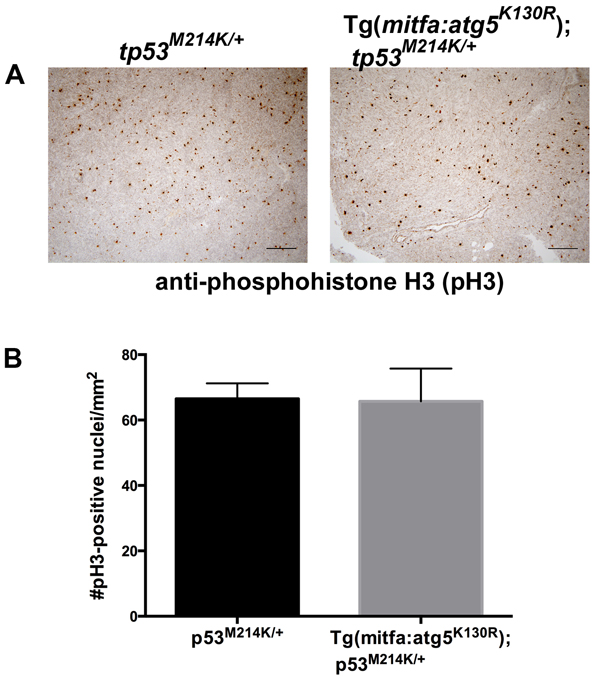
Figure 4: Cell proliferation rates are equivalent in MPNSTs from atg5K130R transgenic and non-transgenic p53-deficient fish. A. Anti-phosphohistone H3 immunohistochemistry of tp53M214K/+ and Tg(mitfa:atg5K130R); tp53M214K/+ fish. B. Quantification of staining. Mean +/- SEM of 5 tumors per genotype is shown.
MPSNTs arising in Tg(mitfa:atg5K130R) fish have increased double-stranded DNA breaks.
To further probe the mechanism of increased tumor incidence in Tg(mitfa:atg5K130R);tp53M214K/+ fish, we considered whether inhibition of autophagy in neural crest lineage cells caused genome instability. Previous studies have shown that cells deficient in autophagy exhibit chromosome instability manifested by increased DNA damage, gene amplification, and aneuploidy [47, 48]. Irradiated glioblastoma cells in which autophagy is inhibited displayed more pronounced and prolonged foci of phosphohistone H2AX, a marker of DNA double-strand breaks [49]. To test whether inhibition of autophagy caused elevated levels of DNA damage in the transgenic fish, we stained MPNSTs from Tg(mitfa:atg5K130R);tp53M214K/+ and tp53M214K/+ heterozygous mutants with a validated antibody specific for the phosphorylated form of Histone H2AX (pH2AX) [50]. Tumors arising in fish with the mitfa:atg5K130R transgene exhibited significantly higher numbers of pH2AX foci (Figure 5). Thus, inhibition of autophagy was associated with increased generation and/or persistence of double-strand DNA breaks in Tg(mitfa:atg5K130R);tp53M214K/+ tumors.
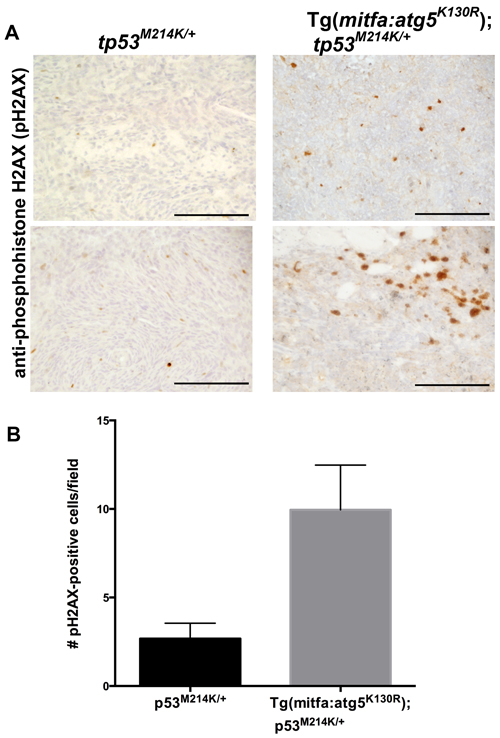
Figure 5: MPSNTs arising in atg5K130R transgenics have increased double-stranded DNA breaks. A. Anti-phosphohistone H2AX immunohistochemistry of tp53M214K/+ and Tg(mitfa:atg5K130R); tp53M214K/+ fish. Scale bar: 100 µm. B. Quantification of staining. Mean +/- SEM of 40 fields from each of 5 tumors per genotype is shown.
Inhibition of autophagy promotes tp53 loss-of-heterozygosity and tumorigenesis
We did not observe tumors in fish that stably express Atg5K130R in a tp53 wild-type background. Since increased tumor incidence due to Atg5K130R expression was only detected in a tp53 heterozygous mutant background, we hypothesized that expression of Atg5K130R might affect the function or expression of tp53. tp53 functions as a homotetramer in cells, and the defective apoptosis phenotype of heterozygous tp53M214K/+ mutant fish was found to be intermediate compared to that of the homozygous tp53M214K/M214K mutant fish, implying that the total cellular level of p53 protein is functionally important [36]. We also noted that the tumor incidence in Tg(mitfa:atg5K130R);tp53M214K/+ fish resembles that of p53 homozygous tp53M214K/M214K mutant (non-transgenic) fish, with a slightly later onset. TP53 loss of heterozygosity (LOH) frequently occurs during the development of human cancers, including MPNSTs [51-55]. The elevated incidence of DNA damage in Tg(mitfa:atg5K130R);tp53M214K/+ MPNSTs prompted us to test the incidence of tp53 LOH in these tumors (Figure 6). Pair-matched tumor and normal tissue genomic DNA was prepared from the individual tumor-bearing fish, and a fragment of the tp53 gene containing the M214K mutation was PCR-amplified and sequenced. LOH of tp53 occurred in > 70% of tumors from both Tg(mitfa:atg5K130R);tp53M214K/+ and non-transgenic tp53M214K/+ fish [22/30 (73.3%) of tumors from Tg(mitfa:atg5K130R);tp53M214K/+ and 7/10 (70%) of tumors in heterozygous tp53M214K/+ fish (70%)] (representative sequences are shown in Figure 6). Given the significantly shortened latency of tumorigenesis in the Tg(mitfa:atg5K130R); tp53M214K fish, these data suggest that inhibition of autophagy by the expression of Atg5K130R may accelerate LOH of p53. These results further suggest a possible role for autophagy in tumor suppression by regulating LOH of tumor suppressor genes.
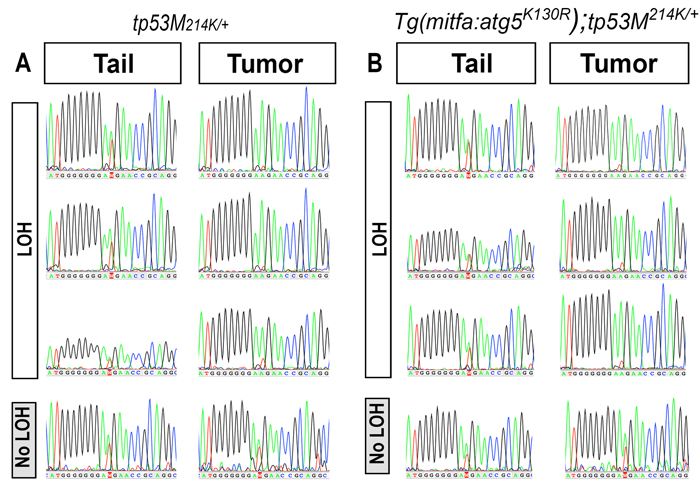
Figure 6: p53 loss of heterozygosity in MPNSTs arising in atg5K130R transgenic and non-transgenic tp53M214K/+ heterozygotes.Genomic DNA was amplified from MPNST tumor and from non-tumor tail tissue from tp53M214K/+ and Tg(mitfa:atg5K130R); tp53M214K/+ fish. Representative sequence traces of a fragment of the tp53 coding sequence harboring the M214K mutation are shown.
Discussion
There is ongoing controversy regarding the role of autophagy in tumorigenesis (reviewed in [4, 13]). Autophagy may play a role in tumor suppression, as suggested by studies involving genetic deletion of autophagy proteins in mouse models. However, autophagy is also known to play a critical role in the survival of cells under stress, and therefore autophagy may promote the survival and growth of cancer cells. The data we present here suggest that inhibition of autophagy in a specific developmental lineage increases the incidence of cancer in that lineage, not through a direct oncogenic mechanism but rather by modulating a pre-existing cancer susceptibility, in this case due to tp53 deficiency.
Expression of dominant-negative proteins as a strategy to inhibit autophagy has not previously been reported in zebrafish; therefore, we took several steps to validate this approach. We showed by immunoblot analysis and using a live autophagy reporter line that expression of atg5K130R inhibits autophagy in vivo. We further showed that the transgene is expressed in tumors arising in transgenic fish.
Previous studies employing the zebrafish mitfa promoter to drive mutated human BRAFV600E or NRASQ61K have documented a high incidence of melanomas in the transgenic fish [26, 56]. Notably, in both these cases, melanomas only arose in the context of tp53 deficiency. One study reported that ATG5 is often downregulated in human melanomas compared to preneoplastic nevi, and that low ATG5 expression is an adverse prognostic indicator in melanoma [57]. The same study demonstrated that the autophagy pathway appears to serve as a barrier to oncogene-induced senescence in melanocytes expressing mutant BRAF. In our study, however, Tg(mitfa:atg5K130R) zebrafish displayed normal pigmentation and failed to develop nevi or melanomas, in either wildtype and p53-deficient backgrounds. Taken together with our data, these results suggest that the tumor-promoting effects of deficient autophagy depend on specific cellular contexts. In melanocytes, deficient autophagy may contribute to the development or progression of melanoma, but is likely not sufficient to cause melanoma in the absence of a RAS or BRAF activating mutation.
While we did not detect melanomas, Tg(mitfa:atg5K130R);tp53M214K/+ and Tg(mitfa:atg5K130R); tp53M214K/M214K lines developed several different types of tumors, most commonly MPNSTs and neuroendocrine tumors. This tumor spectrum is consistent with the developmental origins of mitfa-expressing cells. In zebrafish, as in other vertebrates, neural crest cells give rise to peripheral neurons, glia, Schwann cells and melanocytes during migration from the dorsal neural tube into target tissues [58, 59]. More recently, a second possible cell of origin for melanocytes has been identified, namely Schwann Cell Precursors (SCPs) in peripheral nerves [60]. While we cannot rule out the possibility that dominant-negative autophagy protein expression driven by the mitfa promoter alters cell fate determination of cranial neural crest cells, resulting in aberrant Schwann cell development and MPNSTs, it is more likely that inhibition of autophagy affects the development of SCPs and promotes tumorigenesis. In this setting, expression of atg5K130R and inhibition of autophagy may prevent normal development of melanocytes from migrating SCPs. Indeed, Adameyko et al. showed that Sox10+/Krox20- SCPs acquire Mitf expression as cells migrate away from peripheral nerves [60]. A recent study from Kaufman and Zon and colleagues showed that expression of the embryonic marker crestin in neural crest progenitors is driven by Sox10 together with the Mitf and Tfap2 transcription factors. In a model expressing activated mutant BRAFV600E, re-expression of crestin marked the earliest events in melanoma formation, and overexpression of Sox10 accelerated the formation of melanomas [43]. Interestingly, 49% of human MPNSTs express SOX10 [61]. However, we note that MPNSTs also arise in tp53-deficient animals in the absence of the atg5K130R transgene, and thus these results do not preclude haploinsufficiency of tp53 itself, as opposed to a direct effect of impaired autophagy in Schwann cell precursors, may be the main driving force in the development of MPNSTs.
Another possibility is that the mitfa promoter fragment used in these studies (~5 kb of genomic DNA) does not perfectly recapitulate the activity of the endogenous mitfa locus, leading to ectopic transgene expression in peripheral nerve tissues. However, the development of non-MPNST neural tumors such as neuroendocrine and small-cell tumors in the transgenic line suggests that autophagy inhibition acts directly on a neural precursor cell and mediates or facilitates tumorigenesis. In this regard, it is interesting to note that the PI3-Kinase mTOR pathway, which acts to inhibit the autophagy pathway [62, 63], has very recently been shown to contribute to the pathogenesis of both MPNSTs and neuroendocrine tumors, the two most common tumor types manifested by Tg(mitfa:atg5K130R);tp53M214K/+ fish [64, 65]. Our studies, however, do not directly test the possible role of the PI-3 kinase pathway in zebrafish MPNSTs, and further experiments will be required to distinguish these possibilities.
Several zebrafish cancer models demonstrate a propensity to develops MPNSTs, including those harboring mutations in tp53, ribosomal proteins or mismatch repair proteins, or mutations causing genomic instability [36, 66-69]. These mutant lines share the common characteristic of genomic instability. In this regard, it is particularly interesting that inhibition of autophagy has also been reported to cause genomic instability [47]. We demonstrated increased levels of DNA double-strand breaks in MPNSTs in the Tg(mitfa:atg5K130R);tp53M214K/+ background. Thus the mechanism of accelerated MPNST development in fish with impaired autophagy may be increased genomic instability. However, our experiments do not definitively establish that impaired autophagy, and not another cause, is responsible for the excess DNA damage in the MPNSTs arising in Tg(mitfa:atg5K130R);tp53M214K/+ fish.
Consistent with this hypothesis, expression of the atg5K130R transgene greatly accelerated the development of tumors in tp53M214K+ heterozygous mutants. Berghmans et al. reported tumor incidence only in homozygous mutant tp53M214K fish [36], whereas in our studies, p53 heterozygous mutant fish also developed MPNSTs after 16 months of age. The inconsistency in tumor phenotypes may due to the late onset of tumors in the heterozygous tp53 mutant fish line, arising after 16 months of age, beyond the age range reported by Berghmans et al. The relatively long latency of tumor development in tp53M214K/+ heterozygotes suggested that biallelic mutation of p53 is necessary for tumor development. To address this hypothesis, each tumor bearing-fish was analyzed for its p53 status. Strikingly, more than 70% of tumors showed LOH of tp53, both in the Tg(mitfa:atg5K130R) heterozygous tp53 M214K mutant background or the non-transgenic heterozygous tp53 M214K/+ mutant line, suggesting LOH may be the central route whereby tumors arise in a tp53M214K/+ background. In zebrafish cancer models of tp53 deficiency, LOH of tp53 has not been reported except in the tp53I166T mutant fish line [70]. Here, we showed that expression of Atg5K130R did not increase the frequency of LOH but clearly accelerated LOH of p53 in a tp53M214K/+ background and led to enhanced tumor development. The precise mechanisms of increased tumor susceptibility are not yet clear, but may relate to genomic instability caused by impaired autophagy. Of relevance, a recent study demonstrated that the contribution of autophagy to tumor susceptibility in a Kras mouse model of pancreatic cancer depends on the p53 status [22]. In the presence of wildtype p53 levels, loss of autophagy impaired tumor progression. In contrast, in the absence of p53 function, loss of autophagy greatly accelerated the onset of pancreatic carcinoma. This raises the possibility that impaired autophagy may contribute to tumorigenesis in at least two ways: first by promoting genomic instability and LOH of tp53, and then by providing a selective advantage to the growth of tp53-deficient cells.
Materials and Methods
Zebrafish strains and maintenance
Fish were raised and maintained under standard conditions [71]. Embryos from the wild type AB strain were used for injection to generate the Tg(mitfa:atg5K130R) line. All animals crossed with wild type AB, the autophagy reporter line Tg(cmv:GFP-lc3) [31], or the zebrafish p53 mutant line tp53M214K [36] and maintained. Autophagy reporter fish Tg(cmv:GFP-lc3) was were obtained from Dr. Daniel Klionsky (University of Michigan). Tg (-4.9sox10:eGFP) zebrafish were kindly provided by Dr. Tom Schilling (UC-Irvine). Tp53M214K mutant zebrafish were a gift of Dr. A. Thomas Look (Dana-Farber Cancer Institute). All animal protocols were approved by the UT Southwestern Medical Center Institutional Animal Care and Use Committee.
In vitro transcription and RNA injection
To construct a dominant-negative version of zebrafish atg5, zebrafish cDNA was prepared from 24 hpf embryos. Amplified atg5 cDNA was cloned into the pGEM-T Easy vector (Promega) and mutated using the Quick Change XL Site-Directed mutagenesis kit (Stratagene) to generate the K130R mutation. mCherry cDNA was amplified using pME-mCherry (kindly provided by Dr. Nathan Lawson, University of Massachusetts Medical Center) and cloned into pGEM-T Easy vector. Constructs were verified for their sequences before in vitro transcription. Capped mRNAs used for injection were prepared by in vitro transcription using mMessage mMachine kit (Ambion, AM1340). In vitro transcribed RNAs were injected into one-cell stage embryos.
Zebrafish embryonic cell culture
Approximately 50 embryos injected with atg5, atg5K130R or mCherry mRNA or with atg5 morpholino were used for primary cell culture. Injected embryos at the shield stage were sterilized with 70% ethanol, and dissociated in trypsin/EDTA solution. Primary cells were plated on a multi-chamber slide (Nunc) in LDF (Leibovitz 50%, DMEM 35%, F-12 15%) medium containing antibiotics and incubated at 25 ᵒC for 18 hours. Cells were imaged with epifluorescence microscopy and the number of GFP-LC3 dots/cell was quantified.
Western blot analysis
Single cell-stage embryos were injected with 200 pg mRNA encoding mCherry (control), zebrafish atg5, or dominant-negative atg5K130R and primary cell cultures were prepared and used for immunoblotting. Cells were harvested and incubated for 30 min on ice in lysis buffer containing 150mM NaCl, 10mM Tris pH7.4, 0.2% Triton X-100, 0.3% NP-40, 0.2mM Na3VO4 and protease inhibitors (Roche). After centrifugation at 13,000 x g, supernatants were used for imunoblotting. Atg5 antibody (Novus, NB110-53818), LC3 antibody (Novus, NB100-2220) and β-actin antibody (Millipore, MAB1501R) were used for immunoblotting.
Microscopic imaging
Images of GFP-Lc3 in primary cells were acquired on a Zeiss LSM 510 META confocal microscope. Primary cells were fixed in 4% PFA for 30 min and mounted using ProLong Gold antifade reagent (Invitrogen, P36935). Images of whole-mount zebrafish were taken with a Nikon Coolpix 4500 camera mounted on a Leica MZ125 stereo dissecting microscope.
Cloning of expression constructs for transgenic line generation
Transgenesis constructs were assembled using Gateway recombinational cloning (Invitrogen) as previously described [72]. Transgenic zebrafish lines were generated using the Tol2-system as described [34]. Supplementary Table 1 lists the primers used. All expression constructs were verified for their sequences before use.
Transgenic fish generation, screening and genotyping
Expression constructs (50 ng/μL) with transposase mRNA (100 ng/μL) were injected into one cell stage-embryos. Injected embryos were raised to adulthood and in-crossed for transgenic screening. Pools of embryos were screened for transgene incorporation by PCR reactions. Putative founders were out-crossed for further screening and transgenic F1 adults were identified by tail clipping and genomic PCR reactions.
Zebrafish RNA preparation and RT-PCR
To confirm the expression of transgenes in transgenic fish lines, RNA was extracted from Tg(mitfa:atg5K130R) tumor tissue or cultured primary tumor cells using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. cDNAs were synthesized using 5 μg of purified RNAs by RT2 HT first-strand kit (Qiagen). PCR reactions and sequencing analysis were performed to detect endogenous or transgenic atg5 transcripts.
Histological analysis
Zebrafish were euthanized with 50% Tricaine solution, fixed in 4% PFA for 48 h and decalcified for 5 days in 0.5M EDTA. Tumor sections were stained with H&E for tissue pathological diagnosis.
Primary cell culture of zebrafish MPNSTs
Tumors were dissected from euthanized zebrafish. Tissues were washed twice in PBS containing antibiotics (Gibco), and digested for 30 min at 37˚C using dispase (Becton Dickinson). Digested tissues were washed twice in PBS, plated on a multi-chamber slide (Nunc) in LDF (Leibovitz 50%, DMEM 35%, F-12 15%) medium containing antibiotics, and incubated at 25ᵒC. After 3 passages, cells were harvested and used for RT-PCR.
Assessment of tumor incidence
To generate cohorts of animals for the tumor study, Tg(mitfa:atg5K130R) heterozygotes (all derived from the same founder) were crossed to tp53M214K/+ heterozygotes to generate Tg(mitfa:atg5K130R); tp53M214K/+ animals. These were backcrossed to tp53M214K/+ heterozygotes to generate the following genotypes: 1) non-transgenic, tp53+/+; 2) non-transgenic, tp53M214K/+; 3) non-transgenic, tp53M214K/M214K; 4) Tg(mitfa:atg5K130R), tp53+/+; 5) Tg(mitfa:atg5K130R), tp53M214K/+; 6) Tg(mitfa:atg5K130R), tp53M214K/M214K. Animals were raised in groups of 40 fish per 9L tank on a single aquarium system and monitored daily. The end-point for all animals was the first appearance of tumor. Once a tumor was detected, animals were sacrificed. Tail tissue was taken for genotyping for the atg5K130R transgene and the p53 M214K mutation. Tumor tissue was divided; part of the tumor was fixed and submitted for histologic analysis and the remainder was flash-frozen and preserved for molecular analysis. At 24 months of age all remaining tumor-free animals were sacrificed and genotyped. Tumor incidence was analyzed statistically using the log-rank test.
Immunohistochemistry
For immunostaining, slides were deparaffinized and antigen retrieval was performed for 15 min in Trilogy reagent (Cell Marque). After quenching peroxidase activity and blocking nonspecific binding, slides were incubated for 1 h with anti-GFP antibody (MBL), anti-phosphohistoneH3 Serine 10 antibody (Santa Cruz Biotechnology) or anti-phosphohistone H2AX [50] followed by incubation with HRP-conjugated anti-rabbit antibody (Immpress kit, Vector Labs) for 30 min in a wet chamber. Slides were developed with DAB solution, counterstained with hematoxylin (Invitrogen), dehydrated, and mounted with Permount mounting media (Fisher).
Acknowledgments
We thank Kathryn Tucker and John Robertson for zebrafish care. We thank Daniel Klionsky, Tom Schilling and Tom Look for providing critical reagents and Haley Smith for providing assistance with manuscript preparation.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported by grants from the National Institutes of Health (R01CA135731 to J.F.A. and R01CA109618 to B.L.), the Amon G. Carter Foundation to J.F.A; and the Cancer Prevention and Research Institute of Texas RP120718-P1 to B.L.
Author Contributions
E.L., B.L., and J.F.A. conceived and designed experiments; E.L., Y.W. and Z.Z. performed experiments; E.L., D.R., B.L., and J.F.A. analyzed data; E.L., B.L, and J.F.A. wrote the manuscript. All authors approved the final version of the manuscript.
References
1. Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol. 2011; 76:397-402.
2. Levine B and Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132(1):27-42.
3. Mizushima N, Levine B, Cuervo AM and Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008; 451(7182):1069-1075.
4. Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, Kimmelman A, Kumar S, Levine B, Maiuri MC, Martin SJ, Penninger J, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015; 34(7):856-880.
5. Aita VM, Liang XH, Murty VV, Pincus DL, Yu W, Cayanis E, Kalachikov S, Gilliam TC and Levine B. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999; 59(1):59-65.
6. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H and Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999; 402(6762):672-676.
7. Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G and Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. The Journal of clinical investigation. 2003; 112(12):1809-1820.
8. Yue Z, Jin S, Yang C, Levine AJ and Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003; 100(25):15077-15082.
9. Cicchini M, Chakrabarti R, Kongara S, Price S, Nahar R, Lozy F, Zhong H, Vazquez A, Kang Y and Karantza V. Autophagy regulator BECN1 suppresses mammary tumorigenesis driven by WNT1 activation and following parity. Autophagy. 2014; 10(11):2036-2052.
10. Tang H, Sebti S, Titone R, Zhou Y, Isidoro C, Ross TS, Hibshoosh H, Xiao G, Packer M, Xie Y and Levine B. Decreased BECN1 mRNA Expression in Human Breast Cancer is Associated with Estrogen Receptor-Negative Subtypes and Poor Prognosis. EBioMedicine. 2015; 2(3):255-263.
11. Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N and Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2007; 282(25):18573-18583.
12. Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ and Wang HG. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007; 9(10):1142-1151.
13. Rubinsztein DC, Codogno P and Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov. 2012; 11(9):709-730.
14. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD and Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005; 122(6):927-939.
15. Wang RC, Wei Y, An Z, Zou Z, Xiao G, Bhagat G, White M, Reichelt J and Levine B. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012; 338(6109):956-959.
16. Wei Y, Zou Z, Becker N, Anderson M, Sumpter R, Xiao G, Kinch L, Koduru P, Christudass CS, Veltri RW, Grishin NV, Peyton M, Minna J, Bhagat G and Levine B. EGFR-mediated Beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013; 154(6):1269-1284.
17. Wei H, Wei S, Gan B, Peng X, Zou W and Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes & development. 2011; 25(14):1510-1527.
18. Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, Coller HA, Dipaola RS, Gelinas C, Rabinowitz JD and White E. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011; 25(5):460-470.
19. Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G and Lee SJ. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011; 286(15):12924-12932.
20. Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM and Debnath J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011; 22(2):165-178.
21. Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, Snyder E, Santanam U, Dipaola RS, Jacks T, Rabinowitz JD and White E. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes & development. 2013; 27(13):1447-1461.
22. Rosenfeldt MT, O’Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ and Ryan KM. p53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013; 504(7479):296-300.
23. Strohecker AM and White E. Autophagy promotes -driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy. 2013; 10(2).
24. Gong C, Bauvy C, Tonelli G, Yue W, Delomenie C, Nicolas V, Zhu Y, Domergue V, Marin-Esteban V, Tharinger H, Delbos L, Gary-Gouy H, Morel AP, Ghavami S, Song E, Codogno P, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013; 32(18):2261-2272, 2272e 2261-2211.
25. Widlund HR and Fisher DE. Microphthalamia-associated transcription factor: a critical regulator of pigment cell development and survival. Oncogene. 2003; 22(20):3035-3041.
26. Patton EE, Widlund HR, Kutok JL, Kopani KR, Amatruda JF, Murphey RD, Berghmans S, Mayhall EA, Traver D, Fletcher CD, Aster JC, Granter SR, Look AT, Lee C, Fisher DE and Zon LI. BRAF mutations are sufficient to promote nevi formation and cooperate with p53 in the genesis of melanoma. Curr Biol. 2005; 15(3):249-254.
27. Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun JI, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y and Jung YK. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. The Journal of biological chemistry. 2005; 280(21):20722-20729.
28. Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T and Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008; 19(11):4651-4659.
29. Orvedahl A, MacPherson S, Sumpter R, Jr., Talloczy Z, Zou Z and Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell host & microbe. 2010; 7(2):115-127.
30. Hamacher-Brady A, Brady NR and Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. The Journal of biological chemistry. 2006; 281(40):29776-29787.
31. He C, Bartholomew CR, Zhou W and Klionsky DJ. Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy. 2009; 5(4):520-526.
32. Mizushima N, Yoshimori T and Levine B. Methods in mammalian autophagy research. Cell. 2010; 140(3):313-326.
33. Lee E, Koo Y, Ng A, Wei Y, Luby-Phelps K, Juraszek A, Xavier RJ, Cleaver O, Levine B and Amatruda JF. Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 2014; 10(4).
34. Kawakami K, Takeda H, Kawakami N, Kobayashi M, Matsuda N and Mishina M. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Developmental cell. 2004; 7(1):133-144.
35. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K and Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011; 25(8):795-800.
36. Berghmans S, Murphey RD, Wienholds E, Neuberg D, Kutok JL, Fletcher CD, Morris JP, Liu TX, Schulte-Merker S, Kanki JP, Plasterk R, Zon LI and Look AT. tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(2):407-412.
37. Smith-Thomas LC and Fawcett JW. Expression of Schwann cell markers by mammalian neural crest cells in vitro. Development. 1989; 105(2):251-262.
38. Stemple DL and Anderson DJ. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 1992; 71(6):973-985.
39. Woodhoo A and Sommer L. Development of the Schwann cell lineage: from the neural crest to the myelinated nerve. Glia. 2008; 56(14):1481-1490.
40. Yasuda T, Sobue G, Ito T, Doyu M, Sugiura I, Hashizume Y and Kato K. Human peripheral nerve sheath neoplasm: expression of Schwann cell-related markers and their relation to malignant transformation. Muscle & nerve. 1991; 14(9):812-819.
41. Gupta G, Mammis A and Maniker A. Malignant peripheral nerve sheath tumors. Neurosurgery clinics of North America. 2008; 19(4):533-543, v.
42. Elworthy S, Lister JA, Carney TJ, Raible DW and Kelsh RN. Transcriptional regulation of mitfa accounts for the sox10 requirement in zebrafish melanophore development. Development. 2003; 130(12):2809-2818.
43. Kaufman CK, Mosimann C, Fan ZP, Yang S, Thomas AJ, Ablain J, Tan JL, Fogley RD, van Rooijen E, Hagedorn EJ, Ciarlo C, White RM, Matos DA, Puller AC, Santoriello C, Liao EC, et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science. 2016; 351(6272):aad2197.
44. Wada N, Javidan Y, Nelson S, Carney TJ, Kelsh RN and Schilling TF. Hedgehog signaling is required for cranial neural crest morphogenesis and chondrogenesis at the midline in the zebrafish skull. Development. 2005; 132(17):3977-3988.
45. Greenhill ER, Rocco A, Vibert L, Nikaido M and Kelsh RN. An iterative genetic and dynamical modelling approach identifies novel features of the gene regulatory network underlying melanocyte development. PLoS Genet. 2011; 7(9):e1002265.
46. Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP and Allis CD. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997; 106(6):348-360.
47. Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S and White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007; 21(11):1367-1381.
48. Bae H and Guan JL. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol Cancer Res. 2011; 9(9):1232-1241.
49. Ito H, Daido S, Kanzawa T, Kondo S and Kondo Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int J Oncol. 2005; 26(5):1401-1410.
50. Sidi S, Sanda T, Kennedy RD, Hagen AT, Jette CA, Hoffmans R, Pascual J, Imamura S, Kishi S, Amatruda JF, Kanki JP, Green DR, D’Andrea AA and Look AT. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell. 2008; 133(5):864-877.
51. Campo E, de la Calle-Martin O, Miquel R, Palacin A, Romero M, Fabregat V, Vives J, Cardesa A and Yague J. Loss of heterozygosity of p53 gene and p53 protein expression in human colorectal carcinomas. Cancer research. 1991; 51(16):4436-4442.
52. Eccles DM, Brett L, Lessells A, Gruber L, Lane D, Steel CM and Leonard RC. Overexpression of the p53 protein and allele loss at 17p13 in ovarian carcinoma. British journal of cancer. 1992; 65(1):40-44.
53. Lynn M, Wang Y, Slater J, Shah N, Conroy J, Ennis S, Morris T, Betts DR, Fletcher JA and O’Sullivan MJ. High-resolution genome-wide copy-number analyses identify localized copy-number alterations in Ewing sarcoma. Diagnostic molecular pathology. 2013; 22(2):76-84.
54. Studniak E, Maloney E, Ociepa T, Urasinski T, Skonieczka K, Haus O, Poluha A, Kowalczyk J and Zajaczek S. Allelic loss of selected tumor suppressor genes in acute lymphoblastic leukemia in children. Polish journal of pathology. 2013; 64(2):121-128.
55. Thomas L, Mautner VF, Cooper DN and Upadhyaya M. Molecular heterogeneity in malignant peripheral nerve sheath tumors associated with neurofibromatosis type 1. Human genomics. 2012; 6:18.
56. Dovey M, White RM and Zon LI. Oncogenic NRAS cooperates with p53 loss to generate melanoma in zebrafish. Zebrafish. 2009; 6(4):397-404.
57. Liu H, He Z, von Rutte T, Yousefi S, Hunger RE and Simon HU. Down-regulation of autophagy-related protein 5 (ATG5) contributes to the pathogenesis of early-stage cutaneous melanoma. Sci Transl Med. 2013; 5(202):202ra123.
58. Kelsh RN and Eisen JS. The zebrafish colourless gene regulates development of non-ectomesenchymal neural crest derivatives. Development. 2000; 127(3):515-525.
59. Mellgren EM and Johnson SL. The evolution of morphological complexity in zebrafish stripes. Trends Genet. 2002; 18(3):128-134.
60. Adameyko I and Lallemend F. Glial versus melanocyte cell fate choice: Schwann cell precursors as a cellular origin of melanocytes. Cellular and molecular life sciences. 2010; 67(18):3037-3055.
61. Nonaka D, Chiriboga L and Rubin BP. Sox10: a pan-schwannian and melanocytic marker. Am J Surg Pathol. 2008; 32(9):1291-1298.
62. Strozyk E and Kulms D. The role of AKT/mTOR pathway in stress response to UV-irradiation: implication in skin carcinogenesis by regulation of apoptosis, autophagy and senescence. Int J Mol Sci. 2013; 14(8):15260-15285.
63. Desantis A, Bruno T, Catena V, De Nicola F, Goeman F, Iezzi S, Sorino C, Ponzoni M, Bossi G, Federico V, La Rosa F, Ricciardi MR, Lesma E, De Meo PD, Castrignano T, Petrucci MT, et al. Che-1-induced inhibition of mTOR pathway enables stress-induced autophagy. EMBO J. 2015; 34(9):1214-1230.
64. Rahrmann EP, Watson AL, Keng VW, Choi K, Moriarity BS, Beckmann DA, Wolf NK, Sarver A, Collins MH, Moertel CL, Wallace MR, Gel B, Serra E, Ratner N and Largaespada DA. Forward genetic screen for malignant peripheral nerve sheath tumor formation identifies new genes and pathways driving tumorigenesis. Nature genetics. 2013; 45(7):756-766.
65. Qian ZR, Ter-Minassian M, Chan JA, Imamura Y, Hooshmand SM, Kuchiba A, Morikawa T, Brais LK, Daskalova A, Heafield R, Lin X, Christiani DC, Fuchs CS, Ogino S and Kulke MH. Prognostic significance of MTOR pathway component expression in neuroendocrine tumors. Journal of clinical oncology. 2013; 31(27):3418-3425.
66. Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA and Hopkins N. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004; 2(5):E139.
67. Moore JL, Rush LM, Breneman C, Mohideen MA and Cheng KC. Zebrafish genomic instability mutants and cancer susceptibility. Genetics. 2006; 174(2):585-600.
68. Feitsma H, Kuiper RV, Korving J, Nijman IJ and Cuppen E. Zebrafish with mutations in mismatch repair genes develop neurofibromas and other tumors. Cancer Res. 2008; 68(13):5059-5066.
69. Rhodes J, Amsterdam A, Sanda T, Moreau LA, McKenna K, Heinrichs S, Ganem NJ, Ho KW, Neuberg DS, Johnston A, Ahn Y, Kutok JL, Hromas R, Wray J, Lee C, Murphy C, et al. Emi1 maintains genomic integrity during zebrafish embryogenesis and cooperates with p53 in tumor suppression. Mol Cell Biol. 2009; 29(21):5911-5922.
70. Parant JM, George SA, Holden JA and Yost HJ. Genetic modeling of Li-Fraumeni syndrome in zebrafish. Dis Model Mech. 2010; 3(1-2):45-56.
71. Westerfield M. (2000). The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). (Eugene, Oregon: University of Oregon Press).
72. Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP and Chien CB. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn. 2007; 236(11):3088-3099.