
Introduction
Gastric cancer is the fifth most common neoplasm and the third leading cause of cancer death worldwide. [1] Most cases of gastric cancer are attributable to persistent chronic infection by Helicobacter pylori (H. pylori), including almost 90% of cases of non-cardia gastric cancer. [2] Since H. pylori was designated a definite gastric carcinogen in 1994, [3] the pathogenesis of H. pylori infection has been extensively studied, but exactly how this bacterium and/or the associated gastric inflammatory response contributes to gastric carcinogenesis remains poorly defined.
Gastric malignancy usually develops after a multi-step, decades-long process, as proposed by Correa in 1975. [4] In this “conventional model”, the epithelial cells and glands of the gastric mucosa undergo progressive histological change, as confirmed by numerous murine studies. [5-7] Additional steps to this model were proposed by Houghton et al in 2004, [8] based upon H. felis infection in wild type C57BL/6 mice. In that model, chronic Helicobacter infection triggered the migration of bone marrow-derived cells (BMDC) to the stomach, where they gave rise to neoplastic gastric epithelial cells and progressed to gastric adenocarcinoma via the classic changes described by Dr. Correa. [8] H. pylori infection in mice induces less severe gastric mucosal alteration, and rarely if ever causes adenocarcinoma. Nevertheless, support for the “BMDC model” has come from similar experiments by Varon and colleagues using mice infected by H. pylori Sydney Strain 1 (SS1), [9] in which BMDC were identified in approximately 25% of dysplastic gastric lesions, which are thought to be precursors of gastric adenocarcinoma.
Since wild type C57BL/6 mice rarely develop gastric cancer, even after 80 weeks H. pylori infection, [10] our goal was to evaluate the contribution of BMDC to gastric cancer in a mouse model that is susceptible to H. pylori-induced gastric carcinogenesis. [11] For the current study, we chose p27Kip1 deficient (p27 KO) mice, since our laboratory has shown that these mice develop gastric cancer about a year after H. pylori SS1 infection, after slowly progressing through a sequence of pathological changes, recapitulating human H. pylori-associated gastric carcinogenesis. [12]
We used the p27-deficient gastric cancer prone model to evaluate the relative contributions of BMDC to epithelial cells in H. pylori-induced gastric cancer. BMDC are critical for the development of dysplasia and neoplasia in the Helicobacter models, however, the relative contribution of genetic defects in the epithelial and BM cell compartments have not been studied. We hypothesized that if BMDC were critical for H. pylori-induced gastric carcinogenesis in p27-deficient mice, then the p27-deficient bone marrow-derived, rather than the p27-deficient gastric epithelial cells would be responsible for gastric tumorigenesis following H. pylori infection. To determine whether the loss of p27 expression in bone marrow-derived cells (in a background of wild type epithelium) or loss of p27 expression in gastric epithelial cells (with wild type BM cells) contributes to in H. pylori-infected gastric mucosa, we generated chimeric mice by transplantation of BM via lateral tail vein injection of p27 knock-out or wild type BMDC into wild type and p27-deficient recipients, respectively, using wild type to wild type autologous mice to control for the effects of radiation and bone marrow transplantation. After bone marrow transplantation and long term H. pylori infection, we evaluated cell lineages in the context of gastric pathological changes to address the cellular origins of H. pylori-associated gastric carcinoma.
Results
There were three groups of mice in the present study, all on a C57BL/6 background, as shown in Table 1. All donors were males and recipients were females.
Table 1: Experimental Design
Donors |
Recipients |
|
Group 1 (Control) |
♂ Wild Type (GFP-expressing) |
♀ Wild Type |
Group 2 |
♂ p27 -/- Mice |
♀ Wild Type |
Group 3 |
♂ Wild Type (GFP-expressing) |
♀ p27-/- Mice |
Donor and recipient mice in three experimental groups. In this study, all of the mice have C57BL/6 background and all donors are males, recipients are females. ♂ : Male, ♀ : Female, GFP: Green Fluorescent Protein.
Successful bone marrow transplantation
For the mice that recovered from sublethal irradiation, bone marrow transplantation was evaluated 52 weeks after H. pylori infection as shown in Figure 1. Since the bone marrow donor mice in experimental groups 1 and 3 expressed enhanced green fluorescent protein (GFP), immunofluorescence staining was performed to detect these bone marrow-derived cells (stained green) in the stomachs of recipient mice (Figure 1A). In the mice in experimental group 2, the presence of donor bone marrow-derived cells in the stomachs of the recipients was instead confirmed by staining Y-chromosomes from the male donors by FISH. Donor derived Y-chromosomes were labeled with a Cy3-conjugated probe (red), located within the DAPI stained nucleus (blue) in merged image (Figure 1B). The presence of bone marrow-derived GFP-positive cells (group 3) or Y-chromosome stained cells (group 2) could be frequently detected in the dysplastic gastric glands in these two experimental groups of mice (Figure 1).
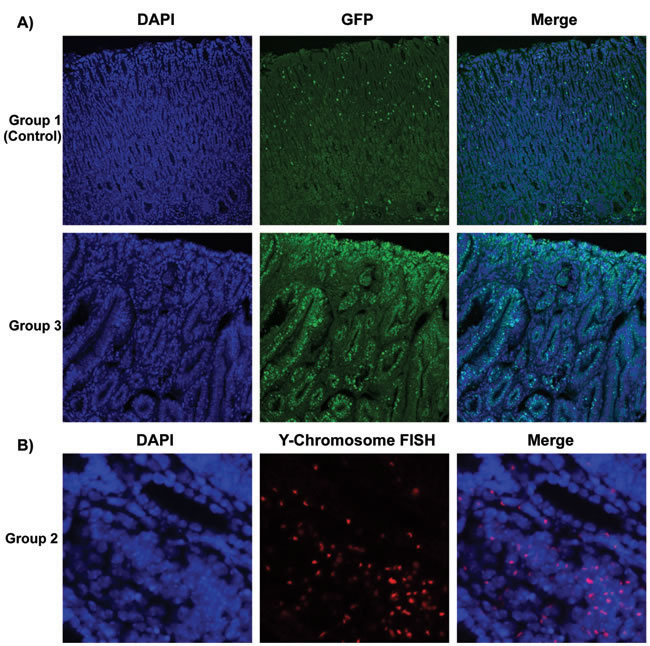
Figure 1: Determination of Bone Marrow-derived GFP-expressing Cells And Male Donor-derived Y-chromosome in Recipient Mice infected by H. pylori. A. Immunofluorescence staining detected marrow-derived cells, which were stained green in group 1 and group 3. Nuclei labeled blue by DAPI in left panels. B. In group 2, Y-chromosomes from male donor mice were detected by performing Fluorescence in situ hybridization (FISH). Donor derived Y-chromosomes were labeled with Cy3-conjugated probe (red), which were located within the DAPI stained nucleus (blue) in merged images. Many of the donor cells were incorporated in gastric glands.
Histological evaluation
Mice were euthanized 52 weeks after H. pylori infection by gavage (all groups), and the gastric histology was evaluated by hematoxylin and eosin (H&E) staining. Representative H&E stained histological images from the three groups at 52 weeks post infection are shown in Figure 2, with higher magnification images in the left lower corner of each panel. In the control group (Group 1), all the mice survived until the end of the study. Although our aim was to determine the origin of gastric cancer cells, we did notice differences in survival curves during the study. In Kaplan-Meier analysis, group 3 p27 KO mice which received wild type bone marrow had significantly poorer overall survival compared with group 1 control mice and group 2 wild type mice which received p27 KO marrow cells (data not shown). The cause of premature death in our group 3 mice was likely from multiple endocrine and/or hematological tumors rather than their stomach pathology. [13-15]
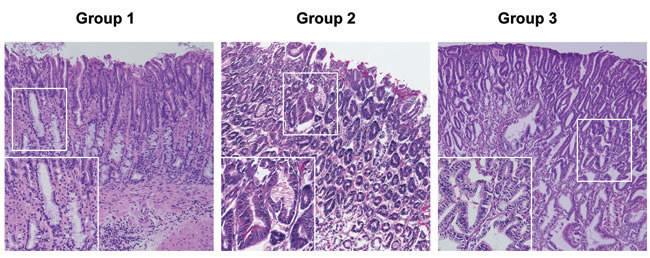
Figure 2: Representative Gastric Histopathology in Recipient Mice. Representative histopathology from the three groups of mice at 52 weeks post H. pylori infection (Hematoxylin and eosin staining). Mice in the control group (Group 1) developed mild gastric epithelial hyperplasia and mild pseudopyloric metaplasia but, unlike the mice in groups 2 and 3, there was no progression to dysplasia. In contrast there was evidence of more advanced gastric pathological abnormality in mice in groups 2 and 3. Original magnification x 100, higher power (insets) x 200.
Although group 1 mice have intensive gastric inflammatory cell infiltration, they retained normal gastric epithelial architecture, and only developed mild hyperplasia and pseudopyloric metaplasia (higher magnification image), but no dysplasia. In contrast, mice in groups 2 and 3 exhibited more complex gastric mucosal architecture with dysplastic features including atypical and heterogeneous gastric glands, coupled with variability in epithelial cell size and enlarged and hyperchromatic nuclei (see higher magnification image). These histopathological findings were quantified for each animal (Figure 3) by scoring inflammation, epithelial defects, oxyntic atrophy, hyperplasia, pyloric metaplasia, and dysplasia separately on a scale of 0 – 4 and comparing differences among groups (Figure 3A). [11] The total Histologic Activity Index (Figure 3B), a combination of the above individual criteria scores, was also calculated. In general, wild type (WT) mice in the control group showed a milder gastric phenotype for all parameters, whereas mice in groups 2 and 3 developed statistically significantly more severe lesions across criteria. The statistical analysis showed no significant differences among the scores for group 2 and group 3 mice. None of the mice developed invasive cancer.
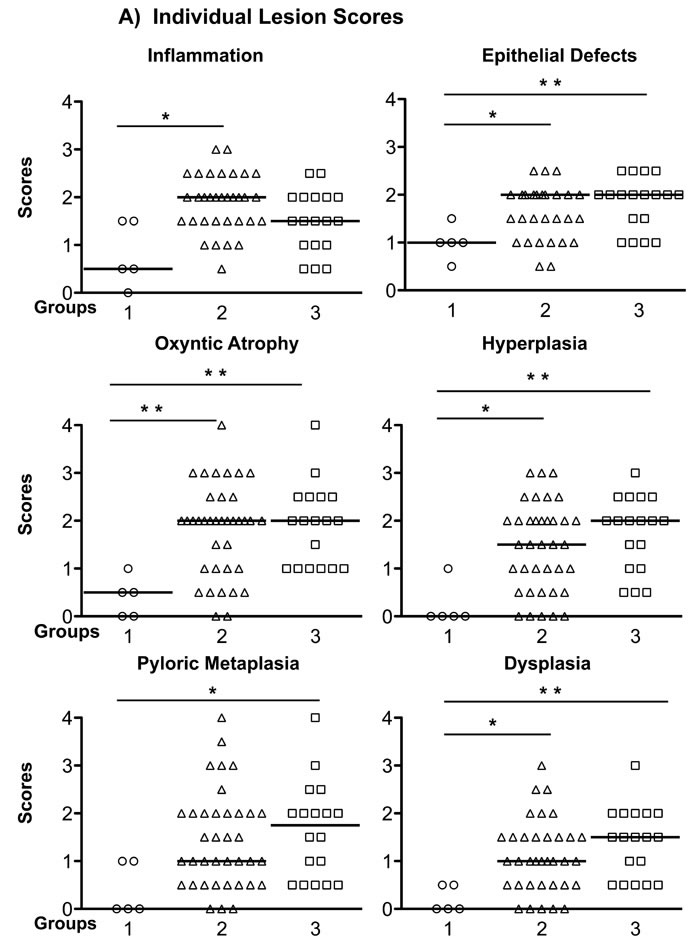
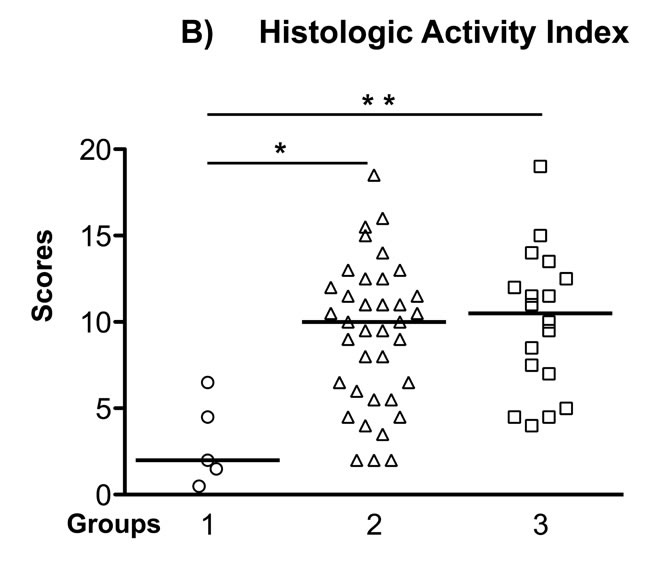
Figure 3: Gastric Histology Scores in H. pylori-infected Recipient Mice. A. The Individual gastric lesion scores of every mouse in each group, including inflammation, epithelial defects, oxyntic atrophy, hyperplasia, pyloric metaplasia, and dysplasia, were graded on a scale of 0 - 4. B. Total Histological Activity Index is a combination of all the six individual criteria scores (scored from 0 - 24). In general, wild type mice in the control group showed a milder gastric pathology, while the two other groups developed advanced pathological lesions. Each empty circle, triangle and square represents one mouse in each group, respectively. The bar represents the median score in each group. N = 5, 37, 18 in groups 1, 2, 3, respectively. *P < 0.05, **P < 0.01 vs group 1 (control). There was no statistically significant difference between group 2 and group 3 mice.
Bone marrow-derived cells express E-cadherin and incorporate into gastric glands
Cadherins are important calcium-dependent cell transmembrane adhesion proteins present on gastric epithelial cells; downregulation of E-cadherin has been implicated in tumorigenesis. [16] As group 2 mice received BMDC from p27 deficient mice, no GFP-expressing cells were detectable. To detect whether bone marrow-derived cells in mice express E-cadherin and incorporate into gastric glands, we performed triple-color staining in each group. We conducted E-Cadherin (red) immunofluorescence staining and combined it with either Y-Chromosome FISH in groups 1 and 2, or immunofluorescence staining for GFP (green) in groups 1 and 3. Subsequently, nuclei were stained with DAPI (blue) to provide composite images (Figure 4A and 4B). The staining clearly showed male donor-derived BMDC incorporated into gastric glands. Gastric epithelial cells were marked with E-Cadherin. Bright, intact E-Cadherin staining was seen in the control group whereas there was much weaker and discontinuous E-Cadherin expression in dysplastic areas in group 2 and 3 mice. Y-chromosome positive or GFP-expressing donor mouse derived BMDCs were abundant in H. pylori-infected gastric mucosa and in the dysplastic gastric glands of group 2 and 3, while only very few BMDCs in group 1 (control) were detected. In group 3 mice, there were areas of numerous GFP-positive cells in some irregular dysplastic glands, with very few non-donor-derived cells. And within the glands, these donor cells also expressed E-cadherin (Figure 4A and 4B).
In comparison with control mice, donor-derived Y-chromosome positive cells were more frequent in group 2 mice (Figure 4C, left panel, 17 ± 1 vs 68 ± 16 per high power field, *P < 0.05). More GFP-expressing BMDCs were engrafted in the inflamed gastric mucosa and incorporated into the gastric glands, especially in dysplastic glands, in group 3 mice compared with the control (group 1 mice) (Figure 4C, right panel, 15 ± 2 vs 62 ± 14 per high power field, *P < 0.05). Data were expressed as mean ± standard error of the mean (SEM).
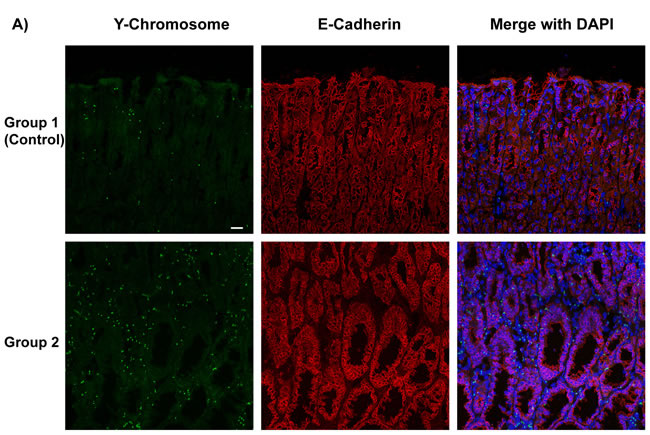
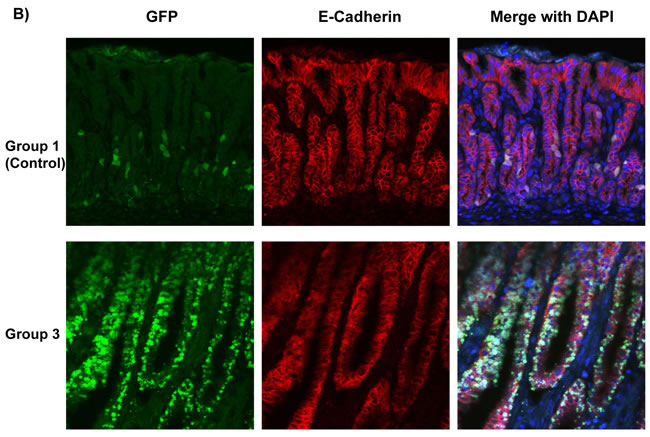
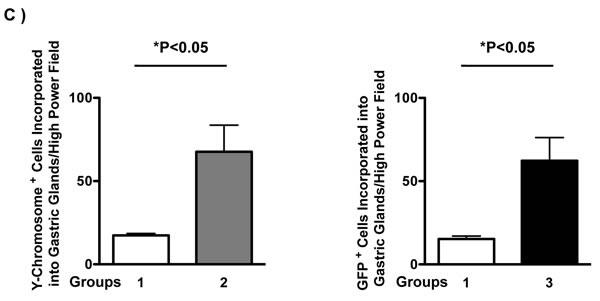
Figure 4: Bone Marrow-Derived Cells (BMDCs) Incorporated into Gastric Glands. A. Y-Chromosome detection by FISH (green) was combined with E-Cadherin (red) immunofluorescence staining, then merged with DAPI (blue) in groups 1, 2. Bar: 20 μm. B. GFP (green) and E-Cadherin (red) immunofluorescence staining were merged with DAPI (blue) in groups 1 and 3. These images clearly showed that BMDCs incorporated into gastric glands. BMDCs were frequently observed in dysplastic gastric glands in group 2 and 3. C. Left panel: Quantification of Y-Chromosome positive BMDCs incorporated into gastric glands per high power field. *P < 0.05. Right panel: Quantification of GFP positive BMDCs incorporated into gastric glands per high power field. *P < 0.05. GFP: Green Fluorescent Protein.
Expression of proinflammatory cytokines and chemokines in gastric mucosa
To evaluate the nature of the inflammatory milieu we assessed the gastric mucosal levels of three proinflammatory cytokines and six chemokines using a customized multiplexed protein quantification assay, according to the manufacturer’s instructions (EMD Millipore, MA, USA). Data were expressed as mean ± SEM. There were no significant differences in protein levels of MCP-1 and MIP-1 α among the three groups. However, in comparison with controls, TNF-α levels were reduced in group 3 (5.75 ± 0.85 pg/mg protein, P < 0.05 vs control 12.61 ± 1.93 pg/mg protein), and there was lower IL-1β expression in groups 2 and 3 (23.45 ± 3.48 pg/mg protein; 23.75 ± 3.18 pg/mg protein, respectively, P < 0.05 vs control 44.37 ± 5.23 pg/mg protein). When compared with group 3 mice, IFN-γ (43.62 ± 9.36 pg/mg protein) and MIP-1 β expression (42.39 ± 6.19 pg/mg protein) in group 2 were markedly elevated (P < 0.05 vs 16.91 ± 2.27 pg/mg protein; 24.83 ± 2.26 pg/mg protein in group 3, respectively). In addition, IP-10, RANTES and MIG levels were significantly increased in group 2 (64.38 ± 9.94; 81.79 ±15.82; 286.1 ± 75.39 pg/mg protein, when compared with group 3 (20.84 ± 4.1; 23.21 ± 6.79; 55.31 ± 29.4 pg/mg protein, respectively, P < 0.05 vs group 3 for each comparison). N = 5, 22, 18 in groups 1, 2, 3, respectively (Figure 5).
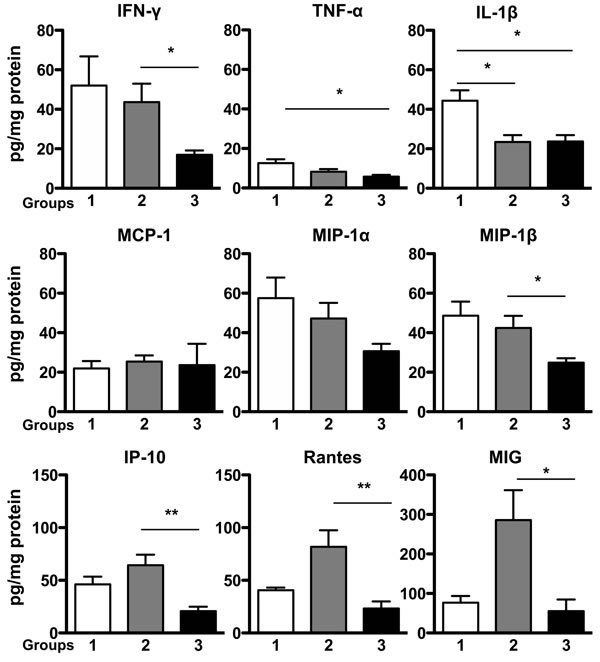
Figure 5: Expression of Proinflammatory Cytokines and Chemokines in Gastric Mucosa. Protein levels of specific proinflammatory cytokines and chemokines in mice stomach tissue were analyzed in a multiplex assay. There were no significant differences in protein levels of MCP-1 and MIP-1 α among the three groups, while IFN-γ, MIP-1 β, IP-10, RANTES and MIG levels were significantly elevated in group 2 when compared with group 3. TNF-α levels in group 3, and IL-1β expression in group 2 and group 3 were significantly lower than those protein levels in control group, respectively. Data were expressed as mean ± SEM (pg/mg protein). SEM: the standard error of the mean. N = 5, 22, 18 in groups 1, 2, 3, respectively. *P < 0.05, **P < 0.01 vs control.
Discussion
Chronic H. pylori infection is a major risk factor for the development of distal gastric carcinoma. [17] Despite decades of research in this field, how H. pylori and its associated gastric inflammatory response promote gastric neoplasia remains unclear. Studies in mouse models using H. felis, a murine-adapted Helicobacter, supports the notion that the cancer stem cell in gastric cancer originates from bone marrow-derived cells drawn to an inflamed gastric milieu by chronic bacterial colonization. [8, 9] Similar findings have been reported using H. pylori, lending more human relevance to the findings. However, H. pylori infection in the mouse model progresses to dysplasia and not to frank carcinoma. Therefore, it remains uncertain whether gastric epithelial cells or bone marrow-derived stem cells initiate gastric cancer development in the H. pylori-infected mouse model.
In the present study, we utilized cancer-prone p27-deficient mice that develop gastric cancer about one year after experimental H. pylori infection [12] to explore whether their increased cancer susceptibility was due to p27 deficiency in their bone marrow-derived cells or in their gastric epithelial cells. Wild type mice receiving autologous bone marrow followed by H. pylori infection served as an experimental control to evaluate the effects of radiation, bone marrow transplantation, and to compare with the other two groups in terms of pathological changes and cytokine expression. Using GFP and Y-chromosome markers to track bone marrow-derived cells, we observed that very few GFP+ bone marrow-derived cells were incorporated into the normal gastric glands of the control mice whereas many cells derived from the bone marrow appeared to be incorporated into the dysplastic gastric glands of mice deficient in p27. Image quantification analysis further confirmed that compared with group 1 control mice, there was a higher frequency of Y-chromosome positive or GFP-expressing donor mouse derived BMDCs incorporated into the recipient’s gastric glands, especially within the dysplastic areas of group 2 and 3 mice. These results indicate that the irradiation protocol does not cause significant structural damage in gastric mucosa, and that engrafted bone marrow cells participate in the development of gastric dysplasia related to H. pylori stimulation. These results therefore support previous findings in different Helicobacter-infected mouse models. [8, 9]
Since our previous reports demonstrated that H. pylori-infected p27-deficient mice are susceptible to advanced gastric preneoplastic and neoplastic lesions, [12, 18] we predicted that if gastric cancer originated in bone marrow-derived cells, then wild type mice that received p27-deficent bone marrow cells (group 2) should have the highest cancer incidence of the three experimental mouse groups. However, our results indicated that the increased susceptibility of these mice to H. pylori-induced gastric pathology was equal to that of the mice in group 3 that lacked p27 in all cells except the bone marrow.
Group 2 mice, which received p27-deficient bone marrow, showed significantly higher inflammation scores compared with control mice. Although p27 deficiency does not increase the number of stem cells in the bone marrow, mice lacking p27 have increased cytokine-stimulated T cell proliferation in the peripheral lymphoid organs, [19-21] which may lead to increased cytokine secretion and enhanced inflammatory responses in the stomachs of the wild type H. pylori-infected mice that received p27-deficient bone marrow cells. Mice in group 3 that lacked p27 in all cells except their bone marrow cells also displayed increased inflammation compared with autologous wild type controls, though the difference was not statistically significant, probably because of a small sample size in the controls. Nevertheless, this indicates that gastric epithelial cell p27 may normally play a role in down-regulating inflammatory and pro-neoplastic responses to H. pylori.
Cytokines and chemokines are important mediators that mediate and coordinate host immune responses to pathogen infection and invasion. Previous studies have demonstrated that H. pylori infection induces the expression of multiple inflammatory cytokines and chemokines. [22-24] We investigated the nature of gastric inflammatory during chronic H. pylori infection in all groups of mice in our study through sampling the expression of several proinflammatory cytokines (IFN-γ, TNF-α and IL-1β) and chemotactic chemokines (MCP-1, MIP-1α, MIP-1β, IP-10, RANTES and MIG) by a protein multiplex array. At 52 weeks after H. pylori infection, there was very low expression of the T helper 1 cytokine TNF-α, and no significant differences in MCP-1 and MIP-1α among the three groups. Unexpectedly, in contrast to other studies, IL-1β expression in groups 2 and 3 that developed gastric dysplasia was significantly lower than that of control group. [25, 26]
Interestingly, although the severity of mucosal lesions and the degree of inflammation assessed by standard pathological criteria were similar in group 2 and group 3 mice, they displayed quite different cytokine/chemokine protein expression, with significantly increased protein levels of IFN-γ, MIP-1β, IP-10, RANTES and MIG in group 2 mice. MIP-1β and RANTES are members of the CC chemokine family, and are homologous to their human CCL4 and CCL5 counterparts, respectively. Both these CC cytokines can bind to CCR5 receptor and act as potent chemoattractants. CCL5 is also involved in wound healing [27]. Recent studies have showed that RANTES is associated with gastric cancer progression, [28, 29] and increased expressions of IP-10 and MIG have also been implicated in gastric cancer. [30, 31] IP-10 and MIG, binding to the same CXCR3 receptor, are IFN-γ related chemokines, and they usually increase in parallel during inflammation. [32-34] Our prior experiments in p27-deficient mice support a role for markedly elevated IP-10 and MIG protein levels in the dysplastic gastric mucosa consequent to persistent H. pylori infection. [18] These elevated cytokines and chemokines in group 2 mice that received p27 deficient bone marrow cells support the idea that BMDC are recruited to the gastric mucosa by increased production of chemoattractants locally that may serve to perpetuate ongoing inflammation. Control and group 3 mice that received wild type bone marrow showed a similar and relatively low level of IP-10, RANTES and MIG, compared with mice in group 2. In contrast to prior experimental bone marrow transplantation in the context of H. pylori infection, [8, 9] group 3 mice that were lacking p27 in all cells except in their bone marrow displayed a high frequency of bone marrow-derived inflammatory cells incorporated into their dysplastic glands. However, since the gastric cytokine and chemokine profiles of these mice were generally similar to that observed in the control mice, which suggests that the mechanisms driving bone marrow cell recruitment to the gastric mucosa and incorporation of BMDC into dysplastic glands must be distinct from the CC/IP-10/MIG-associated processes underlying the carcinogenesis in group 2 mice.
A possible limitation of this study as it applies to human H. pylori-induced gastric carcinogenesis is the use of a single mouse-adapted H. pylori strain, SS1, that is known to lose cag pathogenicity island functionality in vivo [35] We used SS1 because it is the standard H. pylori strain for mouse models [36] and because it is known to induce high-grade dysplasia in p27 knock out mice in our laboratory. [12] However, we appreciate that additional effects related to the translocation of cagA, an important virulence factor for human gastric cancer development, may also be clinically relevant.
In conclusion, our results demonstrate that both bone marrow-derived and gastric epithelial cells contribute to the increased gastric cancer susceptibility of p27-deficient H. pylori-infected mice. Based on the results from three experimental groups in the present study, we conclude that both BMDC and gastric epithelial cells are involved in the development of gastric neoplasia following H. pylori infection, acting in concert but through two distinct pathways. Clinical and translational studies to address the relevance of these findings to human gastric carcinogenesis should now be considered.
Materials and Methods
Mice, bone marrow transplantation and H. pylori infection
Male and female homozygous p27kip1-deficient mice were bred in our laboratory from six to eight week-old heterozygous p27-deficient founders purchased from The Jackson Laboratory (Bar Harbor, ME, USA) as described previously. [18] Male C57BL/6-Tg (CAG-EGFP) 131 Osb/LeySopJ mice (Stock number: 006567) were used as bone marrow donors in group one and group 3; female C57BL/6 wild type mice were used as recipients in group one and group 2. All mice were housed in micro-isolator cages under positive air pressure and fed autoclaved laboratory chow and water ad libitum throughout. The protocol was approved by Rhode Island Hospital’s Animal Care and Use Committee.
At 6 weeks of age, recipient mice were sub-lethally exposed to two doses of 450 centigray (cGy) on the same day, two hours apart, using a Gammacell® 40 Exactor Irradiator (Best Theratronics, Ottawa, Ontario K2K 0E4, Canada) and reconstituted with 3 x106 bone marrow (BM) cells from GFP transgenic or p27-deficient mice by lateral tail vein injection as previously described. [8]
Four weeks after bone marrow transplantation, after allowing the recipient mice to recover from irradiation, all mice were gavaged with H. pylori Sydney Strain (SS1) of approximately 109 bacterial colony forming units (CFU) in (200 μl) volume on three occasions over five days as described previously. [18]
All mice were euthanized and their stomachs were removed 52 weeks after H. pylori infection, or sooner if their condition met Rhode Island Hospital’s Animal Care and Use Committee criteria for signs of distress. Stomachs were opened along the greater curvature and cut into several linear strips for DNA isolation, protein extraction and histologic analysis. Mouse serum samples were collected for anti-H. pylori IgG detection. H. pylori infection status was determined by a combination of positive findings on polymerase chain reaction (PCR) of DNA isolated from mouse gastric tissue, and anti-H. pylori IgG ELISA, as described previously. [37] Only infected mice were included in the final analyses.
Y-chromosome detection by fluorescence in situ hybridization (FISH), immunofluorescent histochemical staining, and image quantification
Y-chromosomes from male donor mice in group 2 were detected by performing Fluorescence in situ hybridization (FISH). Cyanine (Cy™3) labeled Y-chromosome was detected by following the kit’s two-day protocol (Cambio Ltd, Cambridge, UK). In brief, slides were immersed in pre-warmed denaturation buffer (70% formamide in 2x saline sodium citrate buffer, Invitrogen, Carlsbad, CA, USA) for 2-5 minutes at 65 °C and the probe was denatured at 65 °C for 10 minutes. A mixture of probe (2 μl) and hybridization buffer (8 μl) were carefully added to the tissue on each slide. Then the slides were placed in a sealed humidified chamber, and hybridized at 37 °C overnight. After washing, the tissue section was mounted with Vectashield anti-fade mounting medium with 4’, 6-diamidino-2-phenylindole (DAPI) to counterstain nuclei (Vector Laboratories, Burlingame, CA).
Y-chromosome detection by FISH and anti-E-cadherin immunofluorescence staining was also performed on frozen tissue sections of group 1 and 2 mice. The mouse Y-chromosome probe used for this experiment was labeled with Green 5-Fluorescein dUTP (Empire Genomics, Buffalo, NY, USA). Hybridization was carried out according to the manufacture’s protocol. Anti-GFP and anti-E-cadherin immunofluorescence staining were performed on paraffin-embedded tissue sections after antigen retrieval and permeabilization of cell membranes. Polyclonal chicken anti-mouse GFP antibody was applied at a dilution of 1:200 (Invitrogen Corporation, Carlsbad, CA, USA), and monoclonal anti-E-cadherin antibody was used at 1:400 (Cell Signaling Technology, Danvers, MA, USA). The slides were incubated overnight at 4°C. FITC-conjugated donkey anti-chicken and Cy™3-conjugated donkey anti-rabbit secondary antibodies (Jackson Immunoresearch Laboratories Inc., West Grove, PA, USA) were applied at 1:400 dilutions for 1 hour at room temperature. All slides were mounted with Vectashield anti-fade mounting medium with DAPI (Vector Laboratories, Burlingame, CA, USA). Images were obtained with either a Nikon C1si confocal microscope or a Zeiss LSM 710 confocal laser-scanning microscope. Identification of Y-chromosome positive cells was based on the presence of bright green spots inside DAPI-stained blue nuclei. Positive GFP cells were detected as bright green staining localized in the cytoplasm.
To quantify the number of Y-chromosome positive BMDCs that were incorporated into gastric glands, we counted such cells in tissue sections from 3 representative mice in each group. Five 8-bit grayscale consecutive high power fields per slide were acquired with a Nikon E800 microscope (Nikon Inc. Mellville, NY, USA) using a 40x Plan Apo objective. Quantitative data were expressed as the mean of donor-derived cells incorporated into the gastric glands ± SEM per high power field for each experimental group.
Histologic evaluation
Each mouse stomach was opened along the greater curvature and quickly cut into linear strips. Longitudinal gastric strips (including two from the lesser curvature) were fixed in 10% neutral-buffered formalin, embedded in paraffin, and stained with H&E. Gastric mucosal lesions in each mouse were evaluated by a veterinary pathologist who was blinded to the experimental details using the scoring criteria (0 - 4.0) described previously. [11] As mucous metaplasia and hyalinosis may develop spontaneously in mice, [6] these sub-scores were excluded from the calculation of the histological activity index (HAI).
Proinflammatory cytokines and chemokines measurement
We evaluated a panel of three proinflammatory cytokines and three chemokines at the protein level by a customized multiplex magnetic beads array (EMD Millipore, Billerica, MA, USA). The panel comprised interferon-gamma (IFN-γ), tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), monocyte chemoattractant protein-1 (MCP-1), macrophage inflammatory protein 1-alpha (MIP-1α) and macrophage inflammatory protein 1-beta (MIP-1β), interferon gamma-induced protein 10 (IP-10) regulated upon activation, normal T cells-expressed and secreted chemokine (RANTES), and Monokine induced by gamma interferon (MIG). For these analyses a strip of stomach tissue from each mouse was homogenized and total protein concentration was measured by BCA protein assay kit (Pierce Biotechnology, Rockford, IL). Aliquots of the protein samples were then applied to a customized multiplex magnetic bead array (EMD Millipore, Billerica, MA, USA) according to the manufacturer’s instructions. Data were normalized to total protein concentration in each sample and expressed as mean ± SEM pg/mg protein.
Statistics
All statistical analysis was performed using Graphpad Prism 5.0 software (San Diego, CA) with a P level < 0.05 considered as statistically significant. Histopathological scores were compared by Kruskal-Wallis one-way analysis of variance (ANOVA) followed by Dunn’s multiple comparison tests. Y-chromosome positive and GFP positive donor-derived cells were compared by student’s t-test. Cytokine and chemokine expression were compared with ANOVA followed by Bonferroni’s multiple comparison method. All analyses were confined to those mice that were confirmed to be successfully infected by H. pylori.
Acknowledgments
The authors appreciate the help of Mr. Mark Dooner for advice on mouse irradiation and bone marrow transplantation, the help of Dr. Mark T. Whary for mice serum anti-H. pylori IgG detection, and the assistance of Ms. Virginia Hovanesian for digital imaging.
Conflicts of Interest
The authors have no conflicts to disclose.
Grant Support
The research was funded by the National Institutes of Health (CA 125126) to SFM, and the American Gastroenterology Association (Student Research Fellowship Award) to WK.
References
1.http://globocan.iarc.fr/old/FactSheets/cancers/stomach-new.asp. Assessed on Dec. 15, 2015.
2. Plummer M, Franceschi S, Vignat J, Forman D, de Martel C. Global burden of gastric cancer attributable to Helicobacter pylori. Int J Cancer. 2015; 136, 487-490. doi: 10.1002/ijc.28999.
3. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994; 61: 1-241. PMID: 7715068.
4. Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet. 1975; 306: 58-60. doi: 10.1016/S0140-6736(75)90498-5.
5. Fox JG, Rogers AB, Ihrig M, Taylor NS, Whary MT, Dockray G, Varro A, Wang TC. Helicobacter pylori-associated gastric cancer in INS-GAS mice is gender specific. Cancer Res. 2003; 63: 942-950. PMID: 12615707.
6. Lee CW, Rickman B, Rogers AB, Ge Z, Wang TC, Fox JG. Helicobacter pylori eradication prevents progression of gastric cancer in hypergastrinemic INS-GAS mice. Cancer Res. 2008; 68: 3540-3548. doi: 10.1158/0008-5472.CAN-07-6786.
7. Cai X, Carlson J, Stoicov C, Li H, Wang TC, Houghton J. Helicobacter felis eradication restores normal architecture and inhibits gastric cancer progression in C57BL/6 mice. Gastroenterology. 2005; 128: 1937-1952. doi:http://dx.doi.org/10.1053/j.gastro.2005.02.066.
8. Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004; 306: 1568-1571. doi: 10.1126/science.1099513.
9. Varon C, Dubus P, Mazurier F, Asencio C, Chambonnier L, Ferrand J, Giese A, Senant-Dugot N, Carlotti M, Megraud F. Helicobacter pylori infection recruits bone marrow-derived cells that participate in gastric preneoplasia in mice. Gastroenterology. 2012; 142: 281-291. doi: 10.1053/j.gastro.2011.10.036.
10. Kim DH, Kim SW, Song YJ, Oh TY, Han SU, Kim YB, Joo HJ, Cho YK, Dim DY, Cho SW, Kim MW, Kim JH, Hahm KB. Long-term evaluation of mice model infected with Helicobacter pylori: focus on gastric pathology including gastric cancer. Aliment Pharmacol Ther. 2003; 18 Suppl 1: 14-23. doi: 10.1046/j.1365-2036.18.s1.4.x.
11. Rogers AB, Houghton J. Helicobacter-based mouse models of digestive system carcinogenesis. Methods Mol Biol. 2009; 511: 267-295. doi: 10.1007/978-1-59745-447-6_11.
12. Kuzushita N, Rogers AB, Monti NA, Whary MT, Park MJ, Aswad Bl, Shirin H, Koff A, Eguchi G, Moss SF. p27kip1 deficiency confers susceptibility to gastric carcinogenesis in Helicobacter pylori-infected mice. Gastroenterology. 2005; 129: 1544-1556. doi: 10.1053/j.gastro.2005.07.056.
13. Nakayama K, Ishia N, Shirane M, Inomata A, Inoue T, Shishido N, Horii l, Loh DY, Nakayama K. Mice lacking p27 (Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996; 85:707-20. doi: http://dx.doi.org/10.1016/S0092-8674(00)81237-4.
14. Kiyokawa H, Hineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27 (Kip1). Cell. 1996; 85: 721–732. doi: 10.1016/S0092-8674(00)81238-6.
15. Fero ML, Rivkin M, Tasch M, Porter P, Carrow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perimutter RM, Kaushansky K, Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27 (Kip1)-deficient mice. Cell. 1996; 85: 733-44. doi: 10.1016/S0092-8674(00)81239-8.
16. Van Roy F, Berx G. The cell-cell adhesion molecule E-cadherin. Cell Mol Life Sci. 2008; 65: 3756–3788. doi: 10.1007/s00018-008-8281-1.
17. Wroblewski LE, Peek RM. Helicobacter pylori in gastric carcinogenesis: mechanisms. Gastroenterol Clin North Am. 2013; 42: 285-298. doi: 10.1016/j.gtc.2013.01.006.
18. Zhang S, Lee DS, Morrissey R, Aponte-Pieras JR, Rogers AB, Moss SF. Early or late antibiotic intervention prevents Helicobacter pylori-induced gastric cancer in a mouse model. Cancer Lett. 2015; 359: 345-351. Erratum in: Cancer Lett. 2015; 359: 344. doi: 10.1016/j.canlet.2014.09.010.
19. Cheng T, Rodrigues N, Dombowski D, Stier S, Scadden DT. Stem cell repopulation efficiency but not pool size is governed by p27 (kip1). Nat Med. 2000; 6:1235-1240. doi: 10.1038/81335.
20. Shen R, Kaplan MH. The homeostasis but not the differentiation of T cells is regulated by p27 (Kip1). J Immunol. 2002; 169: 714-21. doi: 10.4049/jimmunol.169.2.714.
21. Zhang S, Lawless VA, Kaplan MH. Cytokine-stimulated T lymphocyte proliferation is regulated by p27Kip1. J Immunol. 2000; 165: 6270-6277. doi: 10.4049/jimmunol.165.11.6270.
22. Yamaoka Y, Kita M, Kodama T, Sawai N, Tanahashi T, Kashima K, Imanishi J. Chemokines in the gastric mucosa in Helicobacter pylori infection. Gut. 1998; 42: 609-17. doi: 10.1136/gut.42.5.609.
23. Eck M, Schmausser B, Scheller K, Toksoy A, Kraus M, Menzel T, Muller-Hermelink HK, Gillitzer R. CXC chemokines Gro (alpha)/IL-8 and IP-10/MIG in Helicobacter pylori gastritis. Clin Exp Immunol. 2000; 122: 192-199. doi: 10.1046/j.1365-2249.2000.01374.x.
24. Yamaoka Y, Yamauchi K, Ota H, Sugiyama A, Ishizone S, Graham DY, Maruta F, Murakami M, Katsuyama T. Natural history of gastric mucosal cytokine expression in Helicobacter pylori gastritis in Mongolian gerbils. Infect Immun. 2005; 73: 2205-2212. doi: 10.1128/IAI.73.4.2205-2212.2005.
25. Bartchewsky W, Martini MR, Masiero M, Squassoni AC, Alvarez MC, Ladeira MS, Salvatore D, Trevisan M, Pedrazzoli J, Ribeiro ML. Effect of Helicobacter pylori infection on IL-8, IL-1beta and COX-2 expression in patients with chronic gastritis and gastric cancer. Scand J Gastroenterol. 2009; 44: 153-161. doi: 10.1080/00365520802530853.
26. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Richman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl O, Fox JG, Wang TC. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell. 2008; 14: 408-419. doi: 10.1016/j.ccr.2008.10.011.
27. Ishida Y, Kimura A, Kuninaka Y, Inui M, Matsushima K, Mukaida N, Konodo T. Pivotal role of the CCL5/CCR5 interaction for recruitment of endothelial progenitor cells in mouse wound healing. J Clin Invest. 2012; 122: 711-721. doi: 10.1172/JCI43027.
28. Okita K, Furuhata T, Kimura Y, Kawakami M, Yamaguchi K, Tsuruma T, Zembutsu H, Hirata K. The interplay between gastric cancer cell lines and PBMCs mediated by the CC chemokine RANTES plays an important role in tumor progression. J Exp Clin Cancer Res. 2005; 24: 439-446. PMID: 16270531.
29. Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014; 2014: 292376. doi: 10.1155/2014/292376.
30. Lee HJ, Song IC, Yun HJ, Jo DY, Kim S. CXC chemokines and chemokine receptors in gastric cancer: From basic findings towards therapeutic targeting. World J Gastroenterol. 2014; 20:1681-1693. doi: 10.3748/wjg.v20.i7.1681.
31. Rajkumar T, Vijayalakshmi N, Gopal G, Sabitha K, Shirley S, Raja UM, Ramakrishnan SA. Identification and validation of genes involved in gastric tumorigenesis. Cancer Cell Int. 2010; 10: 45. doi: 10.1186/1475-2867-10-45.
32. Colvin RA, Campanella GS, Sun J, Luster AD. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J Biol Chem. 2004; 279: 30219-30227. doi: 10.1074/jbc.M403595200.
33. Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011; 89: 207-215. doi: 10.1038/icb.2010.158.
34. Kraft M, Riedel S, Maaser C, Kucharzik T, Steinbuechel A, Domschke W, Luegering N. IFN-gamma synergizes with TNF-alpha but not with viable H. pylori in up-regulating CXC chemokine secretion in gastric epithelial cells. Clin Exp Immunol. 2001; 126: 474-81. doi: 10.1046/j.1365-2249.2001.01634.x.
35. Crabtree JE, Ferrero RL, Kusters JG. The mouse colonizing Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter. 2002; 7: 139-40. doi: 10.1046/j.1083-4389.2002.00071.x.
36. Poh AR, O’Donoghue RJ, Ernst M, Putoczki TL. Mouse models for gastric cancer: Matching models to biological questions. J Gastroenterol Hepatol. 2016; 31:1257-72. doi: 10.1111/jgh.13297.
37. Whary MT, Ge Z, Fox JG. Verifying and quantifying Helicobacter pylori infection status of research mice. Methods Mol Biol. 2012; 921:143-56. doi: 10.1007/978-1-62703-005-2_19.