
INTRODUCTION
Translocations involving MLL are associated with poor prognosis in AML. The MLL-AF9 rearrangement, in particular, was found in 30.4% of all AML patients [1]. The abnormal transcriptional program imposed by the MLL-AF9 fusion protein causes a blockade to myeloid differentiation and maintains a state of self-renewal that is similar to that of embryonic stem cells [2]. In recent years, several epigenetic regulators have emerged that play critical roles in MLL-induced leukemia, such as the methyltransferase Dot1L, the Polycomb repressive complex 2 (PRC2) or the chromatin remodeling complex SWI/SNF [3-6]. In a study that characterized the cellular programs underlying oncogene addiction in AML, MLL-AF9 was shown to be required for expression of the Smyd2 gene, as well as of the oncogenic transcription factors Myb and Myc, albeit the mechanisms underlying Smyd2 regulation remained unclear [7]. As will be shown in more detail in this work, genome-wide datasets produced in our laboratory [8, 9] pointed to a direct regulation of Smyd2 by Myc. We thus decided to address the role of Smyd2 in MLL-AF9- and Myc-induced malignancies.
Over 50 human genes encode SET-domain methyltransferases: five of these cluster into the Smyd subfamily, in which the SET domain is split by an intervening MYND domain that can mediate protein-protein interactions [10, 11]. Three family members, Smyd1, -2 and -3, share a high degree of sequence homology and were proposed to control gene expression through histone methylation [12-14]. Smyd-family members have been involved both in development and cancer. Deletion of the Smyd1 (or Bop) gene resulted in defective cardiac maturation and embryonic lethality [11]. Smyd3 is over-expressed in hepatocellular and colorectal carcinomas [12, 15]; recent work showed that it is required for the development of those tumors through the up-regulation of a set of cancer promoting genes [16], and also enhances the tumorigenic capacity of esophageal squamous cell carcinoma [17].
Smyd2, the focus of the present work, is expressed in embryos and in a wide range of normal tissues [14]. The human SMYD2 gene maps to the chromosomal region 1q32, which is amplified in diverse human solid tumors. Its over-expression was associated with poor prognosis in esophageal squamous cell carcinoma (ESCC) [18, 19], childhood acute lymphoblastic leukemia (ALL) [20] and gastric cancer [21]. Recent studies showed that Smyd2 over-expression may be critical in different tumor types, including HPV-unrelated head-and-neck carcinoma [22], pancreatic ductal adenocarcinoma (PDAC) [23], as well as CLL, where together with SMYD3 it may be associated with the acquisition of complex karyotypic alterations [24]. In a mouse model of PDAC, in particular, genetic ablation of Smyd2 significantly delayed tumor progression [23].
The involvement of Smyd2 in gene regulation via histone methylation remains unclear. Smyd2 was first proposed to methylate H3 Lys36 and to associate with the Sin3A histone deacetylase complex to repress gene expression [14]. A subsequent study reported that H3 Lys4 methylation by Smyd2 correlated with up-regulation of a set of genes [25]. On the other hand, several reports indicated that SMYD2 methylates a series of non-histone proteins that may also impact gene expression. First, SMYD2 was reported to methylate p53 on lysine 370, repressing its activity [26]. Biochemical characterization revealed that SMYD2 preferentially binds and methylates p53 rather than histones in-vitro [27, 28]. Another non-histone substrate is the tumor suppressor RB, which can be methylated by SMYD2 at lysine 860, an event regulated both through the cell cycle and in response to DNA damage [29]. SMYD2 also methylates RB on lysine 810, leading to increased serine 807/811 phosphorylation and release of the E2F transcription factor, thus favoring E2F activity and cell growth [30]. Hence, SMYD2 appears to antagonize both of the major tumor suppressors, p53 and RB. SMYD2 also methylates the estrogen receptor α (ERα), antagonizing its function as a transactivator [31], as well as PARP1, favoring its poly(ADP-ribosyl)ation activity [32].
The substrate specificity of Smyd2 might be more complex than anticipated [33] and might extend beyond nuclear activities, as SMYD2 appears to lack a nuclear localization signal (NLS) and predominantly localizes to the cytoplasm [34]. One of the cytoplasmic substrates of SMYD2 is Hsp90, methylation of which may play a significant role in muscle myofilament organization [34]. It is noteworthy here that, unlike Smyd1, Smyd2 has not been found to play a role in cardiac development in the mouse [35, 36]. In PDAC, finally, Smyd2 was proposed to coordinate growth and stress signals in part through the methylation of the protein kinase MAPKAPK3 [23].
In this study, we report that germ-line deletion of Smyd2 has no impact on normal embryonic development. Smyd2 knockout mice were born healthy, grew to adulthood with no observable defects, and showed a lifespan comparable to that of control animals. Moreover, combined loss of Smyd2 and of the closest family member, Smyd3, had no effect on survival. Our analyses showed that normal hematopoiesis was not significantly affected by Smyd2 loss. On the other hand, Smyd2 deletion from HSCs significantly delayed the progression of MLL-AF9 induced leukemia, Smyd2-deleted leukemic cells showing a substantial competitive disadvantage relative to control cells. Finally, our data indicated that Smyd2 expression is controlled by Myc. However, despite the prominent phenotype observed in AML, Smyd2 deletion did not affect Myc-induced lymphomagenesis. We propose that Smyd2 may have a specific role in a Myc-dependent leukemogenesis program.
RESULTS
Smyd2 knockout and Smyd2; Smyd3 double-knockout mice are viable and healthy
In order to assess the physiological importance of mammalian Smyd2 in survival and development, we used mice carrying a conditional knockout allele (Smyd2lox) and derived germ-line deleted Smyd2+/- animals (see Methods), which were intercrossed to generate Smyd2-/- mice. Smyd2-/- mice were viable and born at Mendelian frequency, with 99 live-born pups yielding 28 Smyd2+/+, 41 Smyd2+/-, and 30 Smyd2-/- animals. Over an 18-month observation period, Smyd2-/- mice showed a lifespan comparable to that of heterozygous and wild-type controls, were fertile (Supplementary Table 1) and showed no obvious tumor predisposition. Thus, Smyd2 is a non-essential gene in the mouse.
Comparison of protein sequences among Smyd family members showed that Smyd2 and Smyd3 are the closest members of the family (Table 1). We hypothesized that these proteins may functionally compensate each other, thereby masking their potential roles in survival and development. In order to test this hypothesis, we derived Smyd3 knockout mice (see Methods) and crossed the Smyd2 and Smyd3 mutant strains (Table 2): accounting for stochastic variation, the data showed that single and double-mutant mice were born at near-Mendelian ratios, reached adulthood without evident pathologies, were fertile, and showed weights and life-spans comparable to those of control littermates (Figure 1, Supplementary Table 1). Altogether, our data show that Smyd2 and Smyd3 are dispensable for development and survival.
Table 1: Percent identities of the amino acid sequences of Smyd1, 3, 4 and 5 relative to Smyd2
Smyd1 |
Smyd3 |
Smyd4 |
Smyd5 |
|
Full-length |
26.3 |
30.4 |
11.9 |
11.2 |
SET-domain |
32.5 |
33 |
21.9 |
12.3 |
MYND-domain |
56.4 |
46 |
35 |
30 |
Table 2: Breeding strategy for the derivation of Smyd2/3 double knockout mice and observed frequencies of each genotype.
i) Breeding: Smyd2+/-; Smyd3+/- with Smyd2+/-; Smyd3-/- mice
Genotype of pups (Smyd2; Smyd3) |
Observed Frequency (nr.) |
Expected Frequency (nr.) |
+/+ +/- |
0% (0) |
12.5% (2) |
+/+ -/- |
31.25% (5) |
12.5% (2) |
+/- +/- |
25% (4) |
25% (4) |
+/- -/- |
12.5% (2) |
25% (4) |
-/- +/- |
25% (4) |
12.5% (2) |
-/- -/- |
6.25% (1) |
12.5% (2) |
Total |
100% (16) |
ii) Breeding: Smyd2+/-; Smyd3-/- with Smyd2-/-; Smyd3+/- mice
Genotype of pups (Smyd2/Smyd3) |
Observed Frequency (nr.) |
Expected Frequency (nr.) |
+/- +/- |
34.78 % (24) |
25% (17-18) |
+/- -/- |
20.28 % (14) |
25% (17-18) |
-/- +/- |
26.08% (18) |
25% (17-18) |
-/- -/- |
18.84% (13) |
25% (17-18) |
Total |
100% (69) |
iii) Breeding: Smyd2-/-; Smyd3-/+ with Smyd2-/-; Smyd3-/+ mice
Genotype of pups (Smyd2/Smyd3) |
Observed Frequency (nr.) |
Expected Frequency (nr.) |
-/- +/+ |
15.625% (5) |
25% (8) |
-/- +/- |
56.25% (18) |
50% (16) |
-/- -/- |
28.125% (9) |
25% (8) |
Total |
100% (32) |
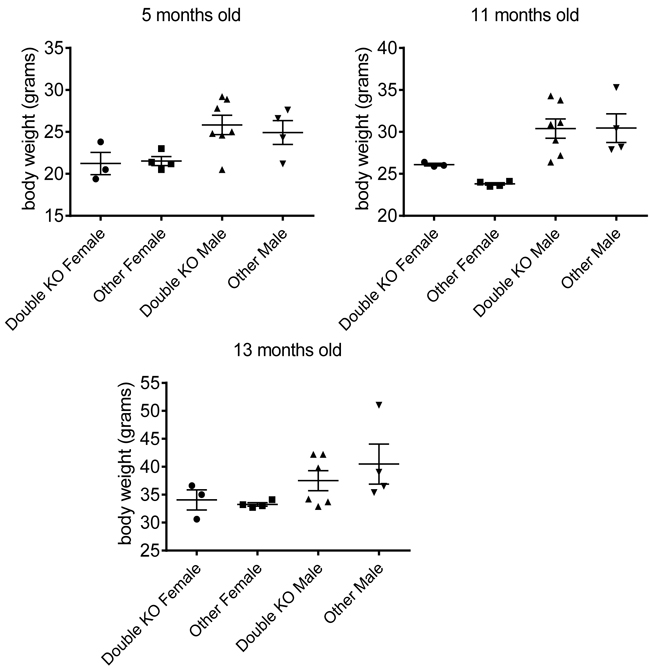
Figure 1: Smyd2/Smyd3 double knock-out does not affect body weight. Weight of male and female mice of different genotypes at the indicated ages (5, 11, and 13 months old). The mice studied here were born from breedings between Smyd2+/-; Smyd3+/- and Smyd2+/-; Smyd3+/- parents. Double-deleted mice lacking both Smyd2 and Smyd3 are compared with littermates of all other genotypes.
Smyd2 deletion has no effect on blood or bone marrow cell numbers
As a premise to address the effect of Smyd2 deficiency in AML, we first assessed its effect on hematopoiesis. No significant differences were observed in the numbers of either total or Lineage-negative (Lin-) cells in the bone marrow of Smyd2-/- relative to wild type mice (Figure 2A). Peripheral blood analysis revealed no significant difference between the percentages of mature blood cell populations (neutrophils, lymphocytes, eosinophils and basophils) in Smyd2-/- and Smyd2+/+ mice, at either 2 or 11 months of age (Figure 2B). We noted a slight increase in eosinophils in old Smyd2-/- mice, the relevance of which remains to be addressed. Thus, Smyd2 deficient-mice showed largely normal hematopoiesis at steady state.
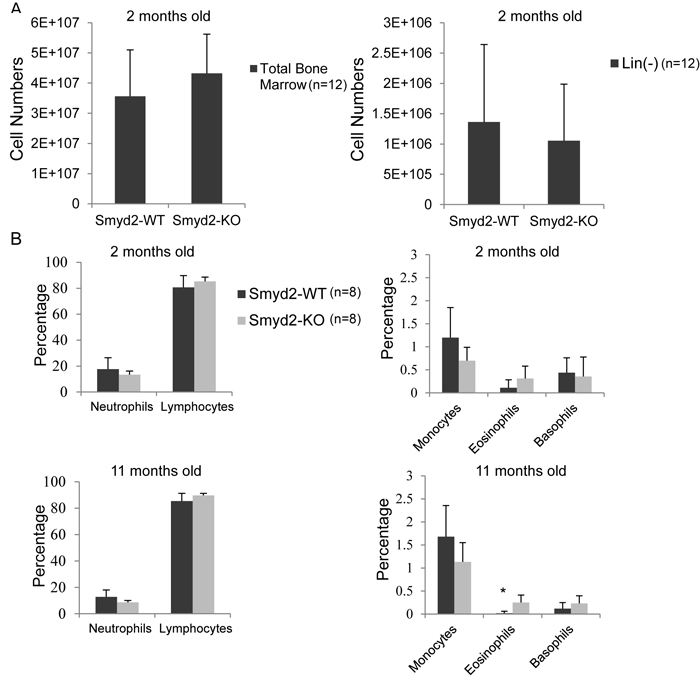
Figure 2: Smyd2 deletion affects neither bone marrow, nor peripheral blood cell counts. A. Total and Lineage-depleted bone marrow cell counts in Smyd2+/+ and Smyd2-/- mice (n = 12 each). P-values are 0.2 and 0.5, respectively. B. Peripheral blood samples were collected from the tail vein of 8 Smyd2+/+ and 8 Smyd2-/- mice at 2 and 11 months of age, as indicated, and directly analyzed in an automated blood cell counter (AcT 5 diff, Beckman Coulter) to determine the numbers of neutrophils, lymphocytes, monocytes, eosinophils and basophils. P-values for the difference in counts between WT and KO mice were 0.23, 0.22, 0.08, 0.1 and 0.68 respectively at 2 months, and 0.17, 0.18, 0.16, 0.02 (marked by the asterisk) and 0.23 respectively at 11 months (calculated using 2-tailed Student’s t-test).
Smyd2 deletion delays MLL-AF9-induced leukemogenesis
In order to test the role of Smyd2 in AML, we transduced Smyd2-/- and Smyd2+/+ hematopoietic stem and progenitor cells (HSPCs) with retroviruses expressing the MLL-AF9 and NRasG12D oncogenes, and transplanted the infected cells into sub-lethally irradiated syngeneic wild-type recipients. Smyd2 deficiency caused a significant delay and reduced penetrance of AML-associated death (Figure 3A). Upon disease development, however, Smyd2-/- and Smyd2+/+ HSPCs gave rise to undistinguishable AML phenotypes, as judged by either leukemic blast counts in peripheral blood (Figure 3B), spleen size (Figure 4A) or the Gr-1+CD3- immunophenotype of MLL-AF9-positive tumor cells (tracked by the associated Venus fluorescent marker), confirming their myeloid identity (Figure 4B).
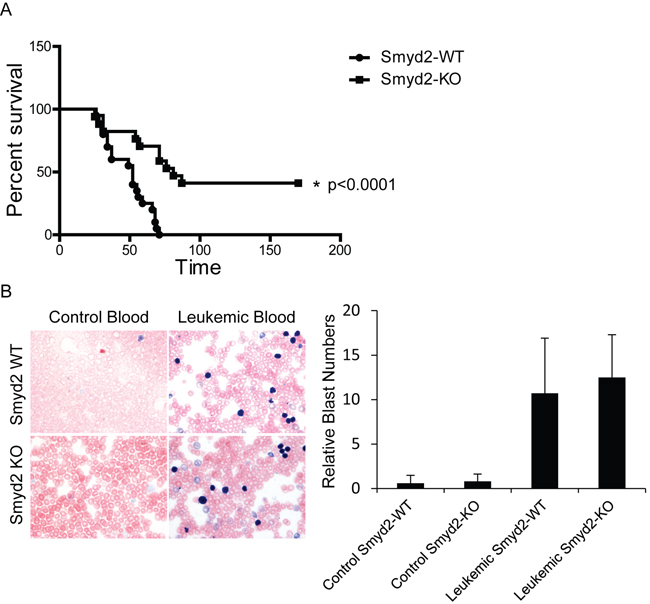
Figure 3: Smyd2 deletion impairs MLL-AF9/NRasG12D-driven leukemogenesis. A. Kaplan Meier curve representing disease-free survival of recipient mice transplanted with MLL-AF9/NRasG12D-infected HSPCs derived from Smyd2+/+ and Smyd2-/- donors, as indicated. HSPCs from 6 Smyd2+/+ and 5 Smyd2-/- donors were transferred into 20 and 17 recipients, respectively. Log rank test shows a statistically significant survival advantage in Smyd2-/- HSPC reconstituted AMLs (p < 0.0001). B. Representative peripheral blood smears from control and leukemic recipient mice of Smyd2+/+ and Smyd2-/- genotypes (60X objective). Leukemic recipients have significantly high amount of circulating immature blasts. The samples were stained with May-Grünwald-Giemsa (Left panel). Counting the number of blast cells of 10 regions under the microscope using 60X objective revealed that no significant difference in the numbers of circulating blast cells between the leukemic recipients of Smyd2+/+ and Smyd2-/- progenitor cells (Right panel); p = 0.62 (Student’s t-test).
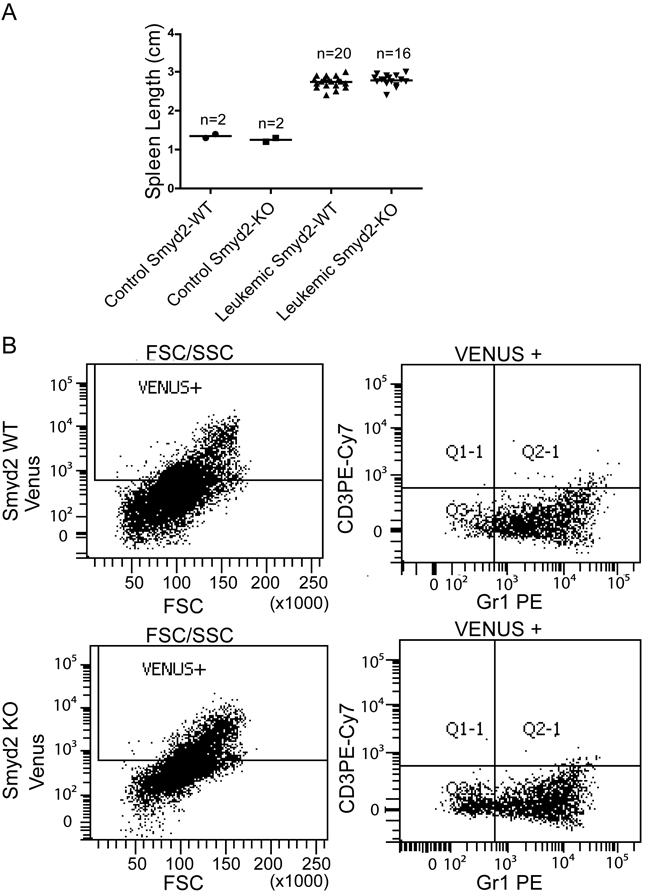
Figure 4: Spleen size and immunophenotypic analysis of MLL-AF9/NRasG12D induced leukemic recipients. A. Spleen length in control Smyd2+/+ and Smyd2-/- mice (n = 2 each) and recipients with leukemia derived from either Smyd2+/+ or Smyd2-/- HSPCs (n = 20 and n = 16, respectively). No significant difference was observed between the two leukemic genotypes (p=0.4; Student’s t-test). B. Representative immunophenotypic analysis of Venus-positive peripheral blood cells from Smyd2+/+ and Smyd2-/- leukemic recipients: detection of Venus positivity, Forward scatter (FSC), GR1 and CD3 staining were assessed by flow-cytometry, as indicated.
Lack of Smyd2 does not impair clonogenic potential but causes competitive disadvantage in leukemic cells in vitro
We then compared the growth and colony forming capacity of Smyd2+/+ and Smyd2-/- leukemic cells in semi-solid medium. Although clonogenic capacity was variable among biological samples of the same phenotype, the range was comparable between the two groups (Figure 5A), with comparable colony shape and size (Figure 5B).
To address the competitive fitness of Smyd2+/+ relative to Smyd2-/- leukemic cells, we mixed these cells in 1:1 proportions, and serially passaged the resulting mixtures in liquid culture. The relative amounts of cells of each genotype were assessed by monitoring the proportion of wild type and knockout Smyd2 alleles by PCR analysis (Figure 6A): Smyd2-deleted cells were reproducibly eliminated, wild-type leukemic cells taking over the cultures at the third passage. As the same result was obtained in all the mixtures, the loss of the mutant allele was thus unlikely to stem from stochastic clonal variations, but rather from a selective disadvantage of Smyd2-/- leukemic cells upon serial replating. Quantitative RT-PCR analysis of the wild-type Smyd2 mRNA in 2 mixed populations showed a progressive increase with ultimate doubling at passage 3, consistent with the full takeover of the cultures by wild-type cells (Figure 6B). Finally, wild-type leukemic cells infected with a pMSCV-GFP vector were mixed with Smyd2-/- leukemic cells infected with pMSCV-Cherry and serially passaged, revealing a gradual decrease in Cherry- and increase in GFP-positive cells (Figure 6C).
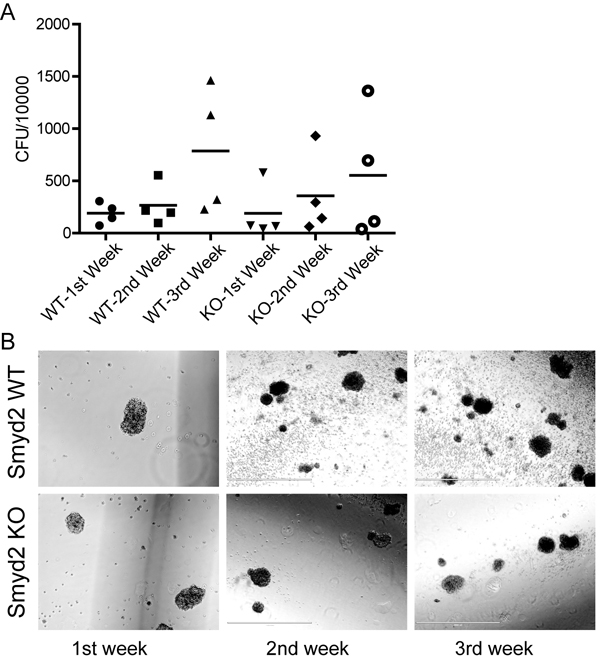
Figure 5: The colony-forming capacity of MLL-AF9/ NRasG12D induced leukemic stem cells is unaffected by the loss of Smyd2. A. Colony forming units (CFU) per 10,000 primary leukemic cells sorted from the spleen of leukemic recipients. The cells were plated in methylcellulose supplemented with HSPC cytokines, and serially passaged three times. B. Morphology and size of representative blast colonies from each genotype (10X objective, scale bar 1000 µm).
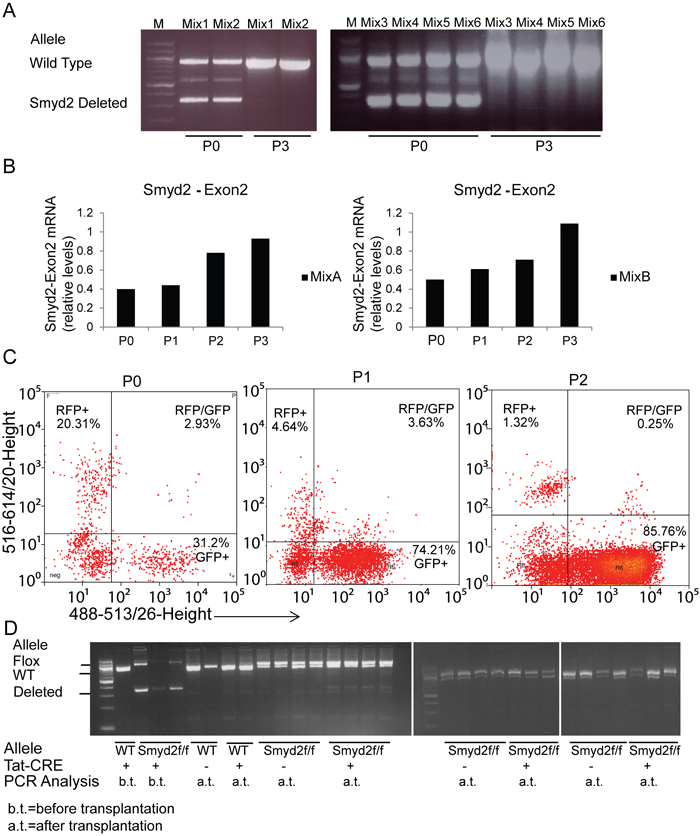
Figure 6: Competitive disadvantage of Smyd2-deleted leukemic cells. A. In vitro competition assay: PCR analysis of wild type and deleted Smyd2 alleles in cell mixtures. Smyd2+/+ and Smyd2-/- primary leukemic cells were mixed (1:1 ratio) and analyzed at passages 0 and 3. Six independent mixes using 4 Smyd2+/+ and Smyd2-/- biological replicates were used in this assay. Primers that flank Smyd2 exon2 (the deleted exon in Smyd2-null cells) were used for the PCR, allowing simultaneous detection of the wild type and deleted alleles. B. Quantitative RT-PCR analysis of Smyd2 exon 2 in two representative mixtures at the indicated passages. The Tbp gene was used as a normalizer. The bars show the average of two experimental replicates. C. Flow cytometric analysis of the selective disadvantage of Smyd2-/- leukemic cells. Wild type and Smyd2-/- leukemic cells were infected with pMSCV-GFP and pMSCV-cherry retroviruses, respectively. The experiment was repeated 3 times with different biological replicates, with very similar results: a representative experiment is shown here. D. In vivo competition assay: primary Smyd2flox/flox leukemic cells were incubated with either Tat-CRE (+) or vehicle (-), and transplanted into secondary recipients. PCR analysis: DNA samples were analyzed either before transplantation (b.t) or after transplantation and development of secondary leukemia (a.t.) with the same primers as in A. Loss of the Smyd2-deleted relative to the non-deleted (Flox) allele is clearly visible in the secondary leukemia derived from Tat-CRE incubated cells, indicating loss of the deleted cells.
Smyd2-null leukemic cells show competitive disadvantage in vivo
We next aimed to test the maintenance of Smyd2-deleted leukemic cells in vivo. Primary leukemia were generated by introducing MLL-AF9 and NRasG12D in either Smyd2flox/flox or Smyd2wt/wt HSPCs. Leukemic cells collected from the spleens of recipient mice were incubated in vitro with a recombinant Tat-CRE recombinase [37]. PCR amplification, performed to test the level of Smyd2 deletion, revealed incomplete deletion of the Smyd2flox allele (the percentage of the deleted allele was 61, 82, and 75 respectively for the three Smyd2flox/flox donors), resulting in heterogeneous populations containing both Smyd2 deleted and undeleted cells. Each Tat-CRE- and vehicle-treated leukemia sample was transplanted into 4 recipients. PCR analysis of secondary leukemia samples showed elimination of the mutant allele (Figure 6D), indicating a competitive disadvantage of Smyd2-deleted cells in vivo.
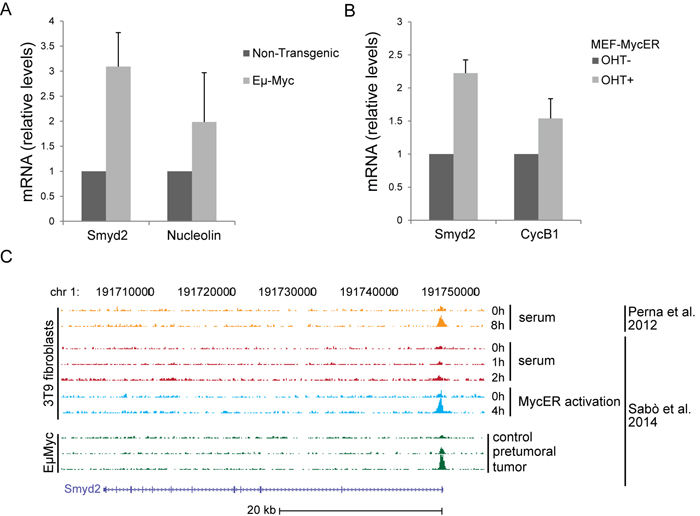
Figure 7: Smyd2 is a direct Myc target. Quantitative RT-PCR analysis of the Smyd2 mRNA in splenic B-cells of Eµ-myc and non-transgenic mice A. and upon hydroxy-tamoxifen (OHT)-induced MycER activation in mouse embryo fibroblasts (MEF-MycER) B. Nucleolin and Cyclin B1 (CycB1) are known Myc targets, used as positive controls. Averages values and error bars are from three biological replicates. Tbp was used as the house-keeping gene to calculate relative mRNA levels. C. Myc ChIP-Seq profiles at the Smyd2 locus in 3T9 fibroblasts and B-cell samples, as indicated [8, 9].
The Smyd2 gene is a direct Myc target
Transcription factors, such as Myb and c-Myc, mediate the regulatory network exerted by MLL fusion proteins. In the same leukemia model used here, in particular, MLL-AF9 bound to the c-myc promoter and induced expression of the gene [7]. In our previous RNA profiling datasets, Smyd2 classified as a Myc-dependent serum response (MDSR) gene in mouse fibroblasts [8] and was induced during lymphoma development in Eµ-myc mice [9], the latter validated here by RT-PCR (Figure 7A). Albeit not classified as significant in our RNA-seq data [9], Smyd2 also showed activation following ectopic MycER activation in fibroblasts (Figure 7B). In all of the above models, ChIP-Seq profiles showed that Myc bound to the Smyd2 promoter, indicating that Smyd2 activation by Myc is direct (Figure 7C). Chip-PCR data confirmed binding of Myc to the Smyd2 promoter in MEF-MycER and Eµ-myc B-cells (Figure 8A, 8B). Using the same assay, we showed that the Smyd2 promoter was directly targeted by Myc in MLL-AF9 leukemic cells from three independent tumors (Figure 8C). Altogether, our results indicate that Smyd2 is activated by Myc downstream of MLL-AF9 and is a critical mediator of the oncogenic signal.
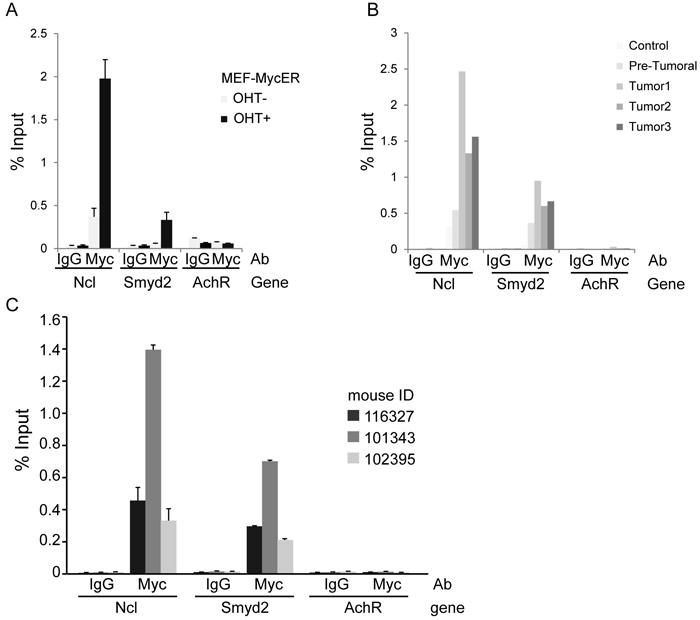
Figure 8: ChIP-qPCR analysis confirms binding of Myc to the Smyd2 promoter. A. Myc-ER fibroblasts; B. Non-transgenic, pre-tumoral and tumoral Eµ-myc transgenic B-cells. Ncl (nucleolin) and AchR (acetylcholine receptor) were used as positive and negative controls, respectively. C. ChIP-qPCR analysis of Myc enrichment on the Smyd2 promoter in splenic cell samples obtained from three terminally sick MLL-AF9 leukemic recipients. The bars represent the average of three experimental replicates in Fig. 8A and C, and two experimental replicates in Fig. 8B.
Smyd2 is dispensable for Myc-induced lymphomagenesis
To address the role of Smyd2 in Myc-induced lymphoma formation, Eμ-myc transgenic mice [38] were bred with Smyd2flox/flox CD19-CRE mice. Conditional deletion of Smyd2 was tested by quantitative RT-PCR on RNA samples isolated from the pre-tumoral B cells of 6-8 weeks old animals (Figure 9A). Flow-cytometry with B220 and Ki67 staining showed that the cell cycle distribution of B cells in the blood of young (pre-tumoral) mice was not affected by the loss of Smyd2 (Figure 9B). Consistent with this result, Eµ-myc Smyd2flox/flox mice with or without CD19-CRE showed no significant difference in disease onset (Figure 10A). The absence of Smyd2 in lymphomas arising with CD19-CRE was confirmed at the mRNA level (Figure 10B): it is noteworthy here that lymphomas are generally monoclonal, explaining the full loss of Smyd2, while mixed pre-tumoral populations still showed residual expression (Figure 9A). Finally, germ-line deleted Eμ-myc Smyd2-/- animals and Eμ-myc Smyd2+/+ controls showed comparable lymphoma onset (Figure 10C). Thus, Smyd2 deletion did not affect Myc-induced lymphomagenesis. We note that, albeit not significantly deregulated during lymphomagenesis, Smyd3 is expressed in mouse B-cells [9]: whether combined loss of Smyd2 and Smyd3 may affect lymphomagenesis - or may further impair AML progression - remains to be addressed.
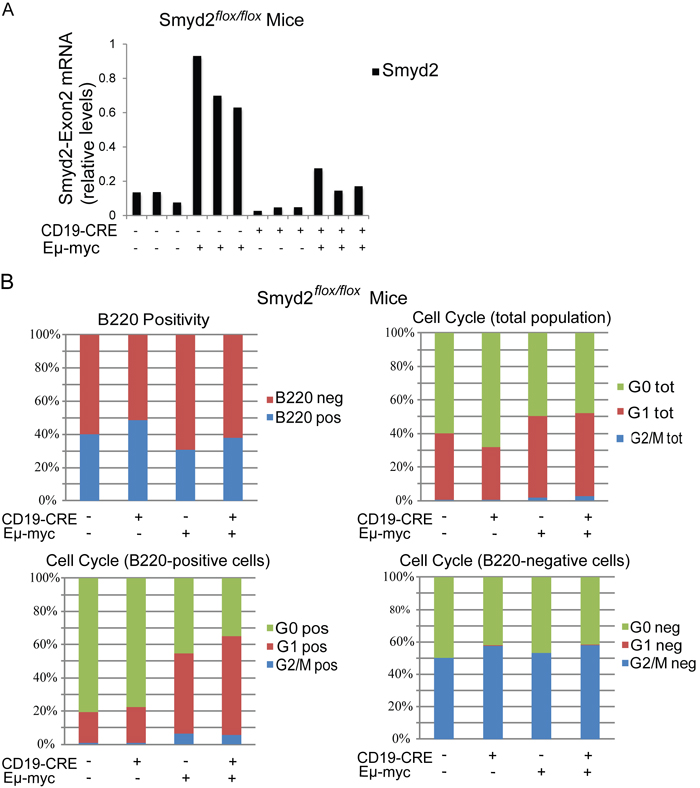
Figure 9: Smyd2 deletion does not affect pre-tumoral B cell expansion and cell cycle distribution in Eµ-myc transgenic mice. A. Quantitative RT-PCR analysis of Smyd2 exon2 in the B cells isolated from mice of the indicated genotypes (n = 3 each). Relative Smyd2 mRNA levels were calculated using Tbp as the housekeeper gene. The bars represent the average of two experimental replicates. B. Multiparameter FACS analysis of peripheral blood cells. Cells were stained with B220-PE or B220-PE and Ki67-Alexa 488 just before FACS acquisition. Cell Cycle distributions based on Ki67 staining are shown for the total, the B220-positive and the B220-negative cell populations. Six biological replicates were included in the analysis for each genotype, with negligible standard deviations (not included).
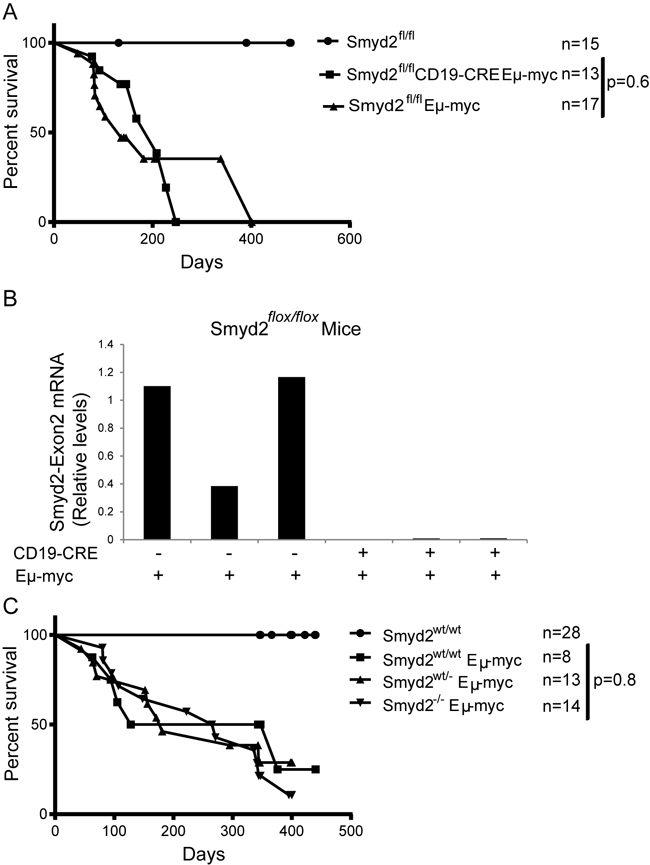
Figure 10: Eµ-myc induced lymphoma formation was not affected by Smyd2 deletion. A. Disease-free survival curves for control (Smyd2flox/flox), Eµ-myc Smyd2flox/flox and Eµ-myc Smyd2flox/flox CD19-CRE mice. B. Quantitative RT-PCR analysis of Smyd2 exon2 shows complete elimination of Smyd2 by CD19-CRE in lymphoma samples. The bars represent the average of two experimental replicates. C. Survival curves for control (Smyd2wt/wt) and Eµ-myc transgenic mice of the indicated Smyd2 genotypes. P-values were calculated using Log-rank (Mantel-Cox) test.
DISCUSSION
Our study addressed the role of Smyd2 methyltransferase in normal mammalian development, hematopoiesis, and MLL-AF9 induced leukemic transformation. Smyd2 is one of the five MYND domain containing SET-proteins, with demonstrated methyltransferase activity. Unlike Smyd1, which is expressed in heart and muscle in mouse and human [11, 39, 40], Smyd2 and -3 are expressed in a broad range of tissues [14]. While Smyd1 is required for mouse development, knockout animals dying at embryonic day 10.5 [11], we show here that germ-line deletion of either Smyd2 or Smyd3, or the two together, has no obvious effect on mouse development, fertility or lifespan.
A series of studies pointed to the potential involvement of Smyd2 and Smyd3 in various human cancers [16, 17, 22-24, 41-44]. In our hands, mining of the Oncomine database revealed that SMYD2 transcription is significantly elevated in patients with bladder cancer, colon adenoma and colorectal carcinoma, hepatocellular carcinoma, prostate carcinoma, hereditary clear cell renal cell carcinoma, and chronic lymphocytic leukemia (Oncomine™; Compendia Bioscience, Ann Arbor, MI). SMYD2 was also reported to methylate several cancer-associated substrates, including p53, RB and ERα [26, 30, 31]. Nevertheless, the involvement of Smyd2 in different types of tumors remained to be evaluated experimentally. Moreover, the role of Smyd2 in normal development had never been genetically addressed.
As enforced Smyd2 expression has been associated with elevated self-renewal of hematopoietic progenitors [7], we investigated the number of total bone marrow, lineage-depleted cells, and differentiated circulating blood cells in Smyd2 KO mice. According to our results, loss of Smyd2 did not appreciably alter normal hematopoiesis, although more detailed analyses will be required to address its possible roles under stress conditions.
Our data show that Smyd2 is critical for leukemogenesis induced by the MLL-AF9 oncogene, Smyd2 knockout primary leukemic cells showing marked competitive disadvantages over their wild-type counterparts either in vitro or in vivo. We demonstrate that the Smyd2 gene is a direct target of Myc, which itself was proposed to be important for stemness in leukemic cells [45]. Moreover, Smyd2 is directly targeted by Myc in fibroblasts and B-cells, including normal and Myc-transformed cells. The c-myc gene itself is a direct transcription target of MLL-AF9, as well as of the Myb transcription factor, which may mediate oncogene addiction in AML [7]. Altogether, these observations point to a transcriptional regulatory cascade in leukemic cells, with MLL-AF9 and Myb activating c-myc, leading to Myc accumulation and activation of Smyd2.
The role of Smyd2 downstream of Myc must be dependent on cellular context, as Smyd2 deletion did not affect lymphomagenesis in Eµ-myc transgenic mice. Interestingly, while MLL-AF9-induced leukemias depend on a subset of self-renewing stem cells [46], this does not appear to be the case in Eµ-myc lymphomas [47]. We speculate that Smyd2 is part of a Myc-controlled self-renewal network downstream of MLL-AF9.
MATERIALS AND METHODS
Mouse breedings and handling
C57Bl/6J mouse strains carrying the Smyd2tm1a(KOMP)Wtsi and Smyd3tm1a(KOMP)Wtsi alleles, respectively, were obtained from the European Conditional Mouse Mutagenesis Consortium (Eucomm). These alleles include LoxP elements flanking exon 2 of the Smyd2 and Smyd3, respectively, and an FRT-flanked selection cassette containing the Engrailed-1 splice acceptor (sA), β-Gal and neomycin selection elements. Animals from both strains were crossed with FlpE recombinase transgenic mice [48] in order to eliminate the selection cassette, resulting in the Smyd2flox or Smyd3flox allele. Homozygous Smyd2fl/fl and Smyd3fl/fl mice were viable and fertile, and showed no abnormalities compared to fl/wt or wt/wt counterparts. In order to delete Smyd2, or Smyd3, from the germ line, homozygous flox/flox mice were crossed with the Deleter strain [49]. Heterozygous Smyd2 or Smyd3-targeted mice, obtained from the Smyd2fl/fl-Deleter or Smyd3fl/fl- Deleter crosses, were then bred with wild type mice in order to obtain Smyd2+/- or Smyd3+/- free of the Deleter-Cre allele. The heterozygous mice were then intercrossed to generate Smyd2-/- and Smyd3-/- mice, which were subsequently used to obtain double knockout animals. Mice were maintained on a C57Bl/6 background. To target Smyd2 in lymphomas, Smyd2fl/fl mice were first bred with C57Bl/6J CD19-CRE transgenic mice [50], and Smyd2fl/+ CD19-CRE mice then crossed to obtain Smyd2fl/fl CD19-CRE animals. The latter were bred with C57Bl/6J Eμ-myc transgenic mice. The resulting Eμ-myc Smyd2fl/fl and Eμ-myc Smyd2fl/fl, CD19-CRE animals were monitored twice a week for lymphoma development by lymph node palpation. Alternatively, C57Bl/6J Eμ-myc transgenic mice were serially bred with germ line deleted Smyd2-/- mice. The resulting Smyd2 wild type, heterozygous and knock-out Eμ-myc transgenic littermates were monitored for lymphoma development as described before. The PCR primers used for genotyping all strains are listed in supplementary Table 2.
Experiments involving animals have been done in accordance with the Italian Laws (D.lgs. 26/2014), which enforces Dir. 2010/63/EU (Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes).
Generation of AML
AML was modeled as described [7]. Lineage-depleted hematopoietic stem and progenitor cells (HSPCs), were isolated from total bone marrow of Smyd2-/- and Smyd2+/+ mice with the Lineage Cell Depletion Kit, mouse (cat. no. 130-090-858, Milteny Biotech). The cells were cultured overnight in HSC specific medium (RPMI, 10% HSC qualified FBS, 50 µg/ml SCF, 10 ng/ml IL3, 10 ng/ml IL6) before being infected with a 1:1 mix of pMSCV-MLL-AF9-IRES-Venus and p-MSCV-Luci-IRES-mNRasG12D retroviruses [7]. Two days after infection, HSPCs were transplanted by tail vein injection into sub-lethally irradiated (7 grays) syngeneic recipients (1.5x105 cells/mouse). Leukemia onset was diagnosed by the presence of the myeloblastoid cells in blood smears detected by May-Grünwald-Giemsa staining, peripheral blood cell counts in a Beckman-Coulter automated blood cell counter, and observation of enlarged spleen by palpation. The percentages of leukemic cells were determined by flow cytometric detection of Venus positivity. Flow-cytometric analysis of Gr-1-PE (BD Biosciences Pharmingen, cat. no. 561084, 1/100 dilution) and CD3- PE Cy7 (BD Biosciences Pharmingen, cat. no. 561100, 1/200 dilution) staining was used to confirm myeloid origin of the Venus positive tumor cells. Statistical evaluation of survival was performed by the log-rank (Mantel-Cox) test for comparison of the Kaplan-Meier event time format.
Colony forming assay
Leukemic spleen cells were sorted for Venus (MLL-AF9 oncogene) positivity, and 10.000 cells from 4 biological replicates of each genotype, obtained from recipients transformed with different HSPC donors, were seeded in methyl cellulose (MethoCult M3234, StemCell Technologies) supplemented with HSPC cytokines (50 µg/ml SCF, 10 ng/ml IL3, 10 ng/ml IL6). The colonies were observed and counted for three weeks, with weekly passages.
Competition assays
For in vitro competition assays, Smyd2+/+ and Smyd2-/- leukemic cells, obtained from sorting the Venus positive spleen cells from terminally sick mice, were mixed in 1:1 proportion and seeded in HSC medium. The DNA preparations obtained from each passage were subjected to a PCR analysis (28 cycles) using a set of primers which amplify wild type, flox and deleted alleles (Smyd2_flox-F and Smyd2_flox-R) (Supplementary Table 2). Smyd2 RNA levels in wild type/Smyd2-null leukemia mixtures were monitored by quantitative RT-PCR with a primer set that amplifies exon 2 (Supplementary Table 2).
For in vivo competition assays, MLL-AF9 leukemias were generated from the HSPCs of 3 Smyd2fl/fl and 2 Smyd2w/w donors. Leukemic spleen cells were isolated as above and incubated with custom made recombinant Tat-CRE recombinase (100 µM, for 3 hours) [37]. Tat-CRE and vehicle treated leukemia samples were separately transplanted into 4 recipients each, as described above (0.9x106 cells per recipient). Recipient animals were irradiated (5 gray) 24 hours before tail vein injection. Secondary leukemia occurrence was observed from 17 to 37 days after transplantation, monitoring general physical health, blood cell counts and enlarged spleen, as described above. Smyd2 alleles were analyzed by PCR, using the primers described above (Smyd2_flox-F and Smyd2_flox-R) on the DNA samples of Venus-sorted spleen cells.
Primary cells and cell lines
Bone marrow cells were isolated from tibias and femurs of the mice, stained with trypan blue, and counted in a haemocytometer. Lineage-depleted cells (lin-) were obtained from total bone marrow cells using the Lineage Cell Depletion Kit, mouse (cat. no. 130-090-858, Milteny Biotech) and counted as above.
For pre-tumoral analysis, blood samples from 6-8 weeks old Eµ-myc transgenic mice with no infiltration of peripheral lymph nodes were used. Isolation and processing of control, pretumoral and tumor primary B-cells for ChIP assays were performed as described [9]. The 3T9-MycER fibroblast cells used in ChIP assay were described [9]. Fibroblasts were grown in DMEM medium supplemented with 10% serum, penicillin/streptomycin, 2 mM L-Gln and 1% β-mercaptoethanol in low-oxygen conditions (3%).
The antibodies used in multiparameter flow cytometry to characterize pre-tumoral B cells are; B220-PE (1/200 dilution, cat. no. 553081, BD Pharmingen), and Ki67-Alexa 488 (1/25 dilution, cat. no. 561165, BD Pharmingen). For Ki67 staining; the B220-PE stained cells were fixed in formaldehyde and permeabilized using the permeabilization solution from the Ki67 staining kit (BD Biosciences Pharmingen, cat. no. 558616) for 30 minutes, and incubated with the Ki67 antibody for 1 hour. 1/10000 diluted Hoechst solution was used to counter-stain the cells (cat. no. 33342, Life Technologies).
Chromatin immunoprecipitation
ChIP assay was performed as described [9]. For the leukemia ChIP analysis, 30 million spleen cells from three wild type leukemic recipients from different donors were seeded in HSC medium supplemented with cytokines 48 hours before formaldehyde fixation. Myc N262 (Santa Cruz, sc-764) and rabbit IgG (Santa Cruz, sc-2027) antibodies were used for ChIP. The primers used for Q-PCR analysis following ChIP are listed in Supplementary Table2.
Analysis of protein identity
Uniprot Alignment Tool was used to determine the percent identities of full-length proteins, or the SET and MYND domains separately [51], as shown in Table 1. The accession numbers used were Q8R5A0 (mouse Smyd1), P97443 (mouse Smyd2), Q9CWR2 (mouse Smyd3), Q8BTK5 (mouse Smyd4), Q3TYX3 (mouse Smyd5).
ACKNOWLEDGMENTS
We thank Dr. Johannes Zuber for kindly providing the pMCSV-MLL-AF9-Venus and pMCSV-NRasG12D plasmids. We thank Andrea Piontini and Manuella Capillo for their help with the management of mouse colonies, Simona Ranzoni for assistance with cell sorting and flow cytometry, Mattia Pellizola, Heiko Muller and Giovanni Faga for bioinformatic analysis, Matteo Del Molin for assistance in cell culture, Massimiliano Mazza for critical discussion about AML. SB thanks Michael Bremang, Leo Bremang and Maya Bremang for support and inspiration.
CONFLICTS OF INTEREST
The Authors declare no conflicts of interest.
GRANT SUPPORT
S.B. was supported by the Structured International Postdoc Program of the European School of Molecular Medicine, and by the Umberto Veronesi Foundation. This work was supported by grants from the European Research Council, the Italian Health Ministry and the Italian Association for Cancer Research (AIRC) to BA.
REFERENCES
1. Zhang Y, Chen A, Yan XM and Huang G. Disordered epigenetic regulation in MLL-related leukemia. Int J Hematol. 2012; 96:428-437.
2. Somervaille TC, Matheny CJ, Spencer GJ, Iwasaki M, Rinn JL, Witten DM, Chang HY, Shurtleff SA, Downing JR and Cleary ML. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell. 2009; 4:129-140.
3. Bernt KM, Zhu N, Sinha AU, Vempati S, Faber J, Krivtsov AV, Feng Z, Punt N, Daigle A, Bullinger L, Pollock RM, Richon VM, Kung AL and Armstrong SA. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011; 20:66-78.
4. Deshpande AJ, Chen L, Fazio M, Sinha AU, Bernt KM, Banka D, Dias S, Chang J, Olhava EJ, Daigle SR, Richon VM, Pollock RM and Armstrong SA. Leukemic transformation by the MLL-AF6 fusion oncogene requires the H3K79 methyltransferase Dot1l. Blood. 2013; 121:2533-2541.
5. Neff T, Sinha AU, Kluk MJ, Zhu N, Khattab MH, Stein L, Xie H, Orkin SH and Armstrong SA. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci U S A. 2012; 109:5028-5033.
6. Shi J, Whyte WA, Zepeda-Mendoza CJ, Milazzo JP, Shen C, Roe JS, Minder JL, Mercan F, Wang E, Eckersley-Maslin MA, Campbell AE, Kawaoka S, Shareef S, et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013; 27:2648-2662.
7. Zuber J, Rappaport AR, Luo W, Wang E, Chen C, Vaseva AV, Shi J, Weissmueller S, Fellmann C, Taylor MJ, Weissenboeck M, Graeber TG, Kogan SC, Vakoc CR and Lowe SW. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011; 25:1628-1640.
8. Perna D, Faga G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung WK, Sanges R, Stupka E, Oskarsson T, Trumpp A, Wei CL, Muller H and Amati B. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene. 2012; 31:1695-1709.
9. Sabò A, Kress TR, Pelizzola M, de Pretis S, Gorski MM, Tesi A, Morelli MJ, Bora P, Doni M, Verrecchia A, Tonelli C, Faga G, Bianchi V, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature. 2014; 511:488-492.
10. Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD and Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000; 406:593-599.
11. Gottlieb PD, Pierce SA, Sims RJ, Yamagishi H, Weihe EK, Harriss JV, Maika SD, Kuziel WA, King HL, Olson EN, Nakagawa O and Srivastava D. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat Genet. 2002; 31:25-32.
12. Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, Yagyu R and Nakamura Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. 2004; 6:731-740.
13. Tan X, Rotllant J, Li H, De Deyne P and Du SJ. SmyD1, a histone methyltransferase, is required for myofibril organization and muscle contraction in zebrafish embryos. Proc Natl Acad Sci U S A. 2006; 103:2713-2718.
14. Brown MA, Sims RJ, 3rd, Gottlieb PD and Tucker PW. Identification and characterization of Smyd2: a split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol Cancer. 2006; 5:26.
15. Lin YM, Furukawa Y, Tsunoda T, Yue CT, Yang KC and Nakamura Y. Molecular diagnosis of colorectal tumors by expression profiles of 50 genes expressed differentially in adenomas and carcinomas. Oncogene. 2002; 21:4120-4128.
16. Sarris ME, Moulos P, Haroniti A, Giakountis A and Talianidis I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell. 2016; 29:354-366.
17. Zhu Y, Zhu MX, Zhang XD, Xu XE, Wu ZY, Liao LD, Li LY, Xie YM, Wu JY, Zou HY, Xie JJ, Li EM and Xu LY. SMYD3 stimulates EZR and LOXL2 transcription to enhance proliferation, migration, and invasion in esophageal squamous cell carcinoma. Hum Pathol. 2016; 52:153-163.
18. Rooney PH, Murray GI, Stevenson DA, Haites NE, Cassidy J and McLeod HL. Comparative genomic hybridization and chromosomal instability in solid tumours. Br J Cancer. 1999; 80:862-873.
19. Komatsu S, Imoto I, Tsuda H, Kozaki KI, Muramatsu T, Shimada Y, Aiko S, Yoshizumi Y, Ichikawa D, Otsuji E and Inazawa J. Overexpression of SMYD2 relates to tumor cell proliferation and malignant outcome of esophageal squamous cell carcinoma. Carcinogenesis. 2009; 30:1139-1146.
20. Sakamoto LH, Andrade RV, Felipe MS, Motoyama AB and Pittella Silva F. SMYD2 is highly expressed in pediatric acute lymphoblastic leukemia and constitutes a bad prognostic factor. Leuk Res. 2014; 38:496-502.
21. Komatsu S, Ichikawa D, Hirajima S, Nagata H, Nishimura Y, Kawaguchi T, Miyamae M, Okajima W, Ohashi T, Konishi H, Shiozaki A, Fujiwara H, Okamoto K, et al. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br J Cancer. 2015.
22. Ohtomo-Oda R, Komatsu S, Mori T, Sekine S, Hirajima S, Yoshimoto S, Kanai Y, Otsuji E, Ikeda E and Tsuda H. SMYD2 overexpression is associated with tumor cell proliferation and a worse outcome in human papillomavirus-unrelated nonmultiple head and neck carcinomas. Hum Pathol. 2016; 49:145-155.
23. Reynoird N, Mazur PK, Stellfeld T, Flores NM, Lofgren SM, Carlson SM, Brambilla E, Hainaut P, Kaznowska EB, Arrowsmith CH, Khatri P, Stresemann C, Gozani O and Sage J. Coordination of stress signals by the lysine methyltransferase SMYD2 promotes pancreatic cancer. Genes Dev. 2016; 30:772-785.
24. Oliveira-Santos W, Rabello DA, Lucena-Araujo AR, de Oliveira FM, Rego EM, Pittella Silva F and Saldanha-Araujo F. Residual expression of SMYD2 and SMYD3 is associated with the acquisition of complex karyotype in chronic lymphocytic leukemia. Tumour Biol. 2016.
25. Abu-Farha M, Lambert JP, Al-Madhoun AS, Elisma F, Skerjanc IS and Figeys D. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics. 2008; 7:560-572.
26. Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T and Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006; 444:629-632.
27. Wang L, Li L, Zhang H, Luo X, Dai J, Zhou S, Gu J, Zhu J, Atadja P, Lu C, Li E and Zhao K. Structure of human SMYD2 protein reveals the basis of p53 tumor suppressor methylation. J Biol Chem. 2011; 286:38725-38737.
28. Wu J, Cheung T, Grande C, Ferguson AD, Zhu X, Theriault K, Code E, Birr C, Keen N and Chen H. Biochemical characterization of human SET and MYND domain-containing protein 2 methyltransferase. Biochemistry. 2011; 50:6488-6497.
29. Saddic LA, West LE, Aslanian A, Yates JR, 3rd, Rubin SM, Gozani O and Sage J. Methylation of the retinoblastoma tumor suppressor by SMYD2. J Biol Chem. 2010; 285:37733-37740.
30. Cho HS, Hayami S, Toyokawa G, Maejima K, Yamane Y, Suzuki T, Dohmae N, Kogure M, Kang D, Neal DE, Ponder BA, Yamaue H, Nakamura Y and Hamamoto R. RB1 methylation by SMYD2 enhances cell cycle progression through an increase of RB1 phosphorylation. Neoplasia. 2012; 14:476-486.
31. Zhang X, Tanaka K, Yan J, Li J, Peng D, Jiang Y, Yang Z, Barton MC, Wen H and Shi X. Regulation of estrogen receptor alpha by histone methyltransferase SMYD2-mediated protein methylation. Proc Natl Acad Sci U S A. 2013; 110:17284-17289.
32. Piao L, Kang D, Suzuki T, Masuda A, Dohmae N, Nakamura Y and Hamamoto R. The histone methyltransferase SMYD2 methylates PARP1 and promotes poly(ADP-ribosyl)ation activity in cancer cells. Neoplasia. 2014; 16:257-264, 264 e252.
33. Olsen JB, Cao XJ, Han B, Chen LH, Horvath A, Richardson TI, Campbell RM, Garcia BA and Nguyen H. Quantitative Profiling of the Activity of Protein Lysine Methyltransferase SMYD2 Using SILAC-Based Proteomics. Mol Cell Proteomics. 2016; 15:892-905.
34. Donlin LT, Andresen C, Just S, Rudensky E, Pappas CT, Kruger M, Jacobs EY, Unger A, Zieseniss A, Dobenecker MW, Voelkel T, Chait BT, Gregorio CC, Rottbauer W, Tarakhovsky A and Linke WA. Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev. 2012; 26:114-119.
35. Diehl F, Brown MA, van Amerongen MJ, Novoyatleva T, Wietelmann A, Harriss J, Ferrazzi F, Bottger T, Harvey RP, Tucker PW and Engel FB. Cardiac deletion of Smyd2 is dispensable for mouse heart development. PLoS One. 2010; 5:e9748.
36. Voelkel T, Andresen C, Unger A, Just S, Rottbauer W and Linke WA. Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim Biophys Acta. 2013; 1833:812-822.
37. Peitz M, Pfannkuche K, Rajewsky K and Edenhofer F. Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: a tool for efficient genetic engineering of mammalian genomes. Proc Natl Acad Sci U S A. 2002; 99:4489-4494.
38. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD and Brinster RL. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985; 318:533-538.
39. Hwang I and Gottlieb PD. The Bop gene adjacent to the mouse CD8b gene encodes distinct zinc-finger proteins expressed in CTLs and in muscle. J Immunol. 1997; 158:1165-1174.
40. Li D, Niu Z, Yu W, Qian Y, Wang Q, Li Q, Yi Z, Luo J, Wu X, Wang Y, Schwartz RJ and Liu M. SMYD1, the myogenic activator, is a direct target of serum response factor and myogenin. Nucleic Acids Res. 2009; 37:7059-7071.
41. Mazur PK, Reynoird N, Khatri P, Jansen PW, Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak D, Tummino PJ, Kruger RG, Garcia BA, et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014; 510:283-287.
42. Zeng B, Li Z, Chen R, Guo N, Zhou J, Zhou Q, Lin Q, Cheng D, Liao Q, Zheng L and Gong Y. Epigenetic regulation of miR-124 by hepatitis C virus core protein promotes migration and invasion of intrahepatic cholangiocarcinoma cells by targeting SMYD3. FEBS Lett. 2012; 586:3271-3278.
43. Cock-Rada AM, Medjkane S, Janski N, Yousfi N, Perichon M, Chaussepied M, Chluba J, Langsley G and Weitzman JB. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 2012; 72:810-820.
44. Tsuge M, Hamamoto R, Silva FP, Ohnishi Y, Chayama K, Kamatani N, Furukawa Y and Nakamura Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5’ flanking region of SMYD3 is a risk factor for human cancers. Nat Genet. 2005; 37:1104-1107.
45. Kim J, Woo AJ, Chu J, Snow JW, Fujiwara Y, Kim CG, Cantor AB and Orkin SH. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010; 143:313-324.
46. Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, Golub TR and Armstrong SA. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006; 442:818-822.
47. Kelly PN, Dakic A, Adams JM, Nutt SL and Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science. 2007; 317:337.
48. Fiering S, Kim CG, Epner EM and Groudine M. An “in-out” strategy using gene targeting and FLP recombinase for the functional dissection of complex DNA regulatory elements: analysis of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1993; 90:8469-8473.
49. Schwenk F, Baron U and Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995; 23:5080-5081.
50. Rickert RC, Roes J and Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997; 25:1317-1318.
51. Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S, Gasteiger E, Huang H, Lopez R, Magrane M, Martin MJ, Natale DA, O’Donovan C, Redaschi N and Yeh LS. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004; 32:D115-119.