
INTRODUCTION
Cutaneous T-cell lymphomas (CTCL) are a wide group of heterogeneous diseases representing approximately 2.6% of all non-Hodgkin lymphomas worldwide. Global genomic approaches recently revealed the existence of recurrent alterations, some having potential diagnosis or prognosis value [1, 2, 3, 4, 5, 6, 7, 8]. Recurrent monoallelic rearrangements of the 6p25.3 locus were identified in peripheral T cell lymphomas (PTCL) [9, 10, 11, 12]. Feldman et al., then our team, showed that these 6p25.3 rearrangements are quite specific of CD30-positive/ALK-negative anaplastic large T-cell lymphomas (ALCL), mainly of cutaneous origin (cALCL) [9, 10, 11, 12], but also occur in transformed/tumor stage mycosis fungoides (T-MF) [11] and in rare lymphomatoid papulosis variants [13]. Systemic ALCL with 6p25.3 rearrangements seem to have better clinical outcomes than cases without rearrangements [14].
Among the 3 genes located in this region, IRF4, an oncogene activated in various types of hematological [15, 16, 17, 18, 19, 20] and skin [21, 22] cancers, was initially considered the strongest candidate [10, 11]. Then, in one ALCL case, next generation sequencing identified a t(6;7)(p25.3;q32.3) translocation interrupting the DUSP22 (Dual Specificity Phosphatase 22) gene, encoding a tyrosine/serine/threonine phosphatase [23], within intron 1 [12]. Fluorescence in situ hybridization (FISH) showed that 7q32.3 was the partner locus in about 30% of PTCL with 6p25.3 rearrangements. Regardless of the partner (7q32.3, other or unknown), tested PTCL cases with monoallelic 6p25.3 alterations exhibited DUSP22 down-regulation [12], making it a candidate tumor-suppressor at this locus.
Beside its inhibitory effects on various signaling pathways, such as T-cell receptor and STAT3, and on cell migration [23, 24, 25, 26, 27, 28, 29], little is known about the physiopathological roles of DUSP22. Notably, its implication in oncogenesis was never functionally addressed so far. The status of the second allele of DUSP22 in PTCL with monoallelic 6p25.3 breakpoints-associated silencing was also not yet studied. Finally, sequence databases indicated the existence of alternative DUSP22 transcripts predicted to encode carboxy-terminally truncated proteins, but their expression levels and functions were never studied. Our goal was thus to investigate the expression status of DUSP22 isoforms in normal lymphocytes and PTCL, the mechanism of second allele silencing in PTCL with 6p25.3 rearrangements, and whether DUSP22 exerted tumor-suppressor properties.
RESULTS
The DUSP22 gene encodes various transcripts which are silenced in cutaneous T-cell lymphomas with monoallelic 6p25.3 rearrangements
Four alternatively spliced DUSP22 gene transcripts were already deposited in databases (Figure 1A, Table 1, Supplementary Figures S1, S2). The transcript encoding the known 184 amino-acids protein [23, 24, 25, 26, 27, 28, 29] comprises 8 exons. An alternative transcript with unspliced intron 7 is predicted to generate a 205 amino-acids protein diverging from the former in the carboxy-terminal region. Exon 4 can be spliced from these two transcripts (Δ exon 4), leading to frameshift-associated premature STOP codon, and a predicted 54 amino-acids carboxy-terminally truncated protein. Expression of these transcripts and their putative proteins had not yet been evaluated in any physiopathological condition.
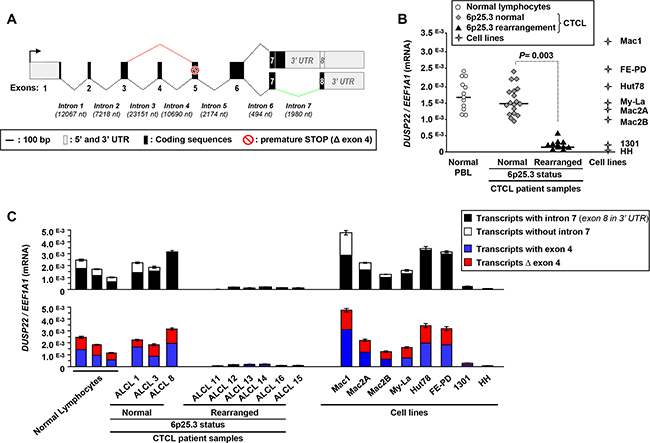
Figure 1: Silencing of DUSP22 alternative transcripts in cutaneous T-cell lymphomas with monoallelic 6p25.3 breakpoints. A. Schematic representation of DUSP22 alternative transcripts. Numbered boxes indicate the exons, with 5’ and 3’ untranslated (UTR) regions in grey and coding region in black. Nt: nucleotides. The black arrow indicates the position of the transcription initiation site. Dotted lines indicate the regular and alternative splicing events (in red, without exon 4; in green, without intron 7). The red symbol highlights the presence of a premature STOP codon which is in frame in Δ exon 4 transcripts. B. Quantitative RT-PCR analysis of DUSP22 transcript levels (forward primer within exon 1 and reverse primer overlapping the junction between exons 6 and 7), normalized by EEF1A1 expression, in normal peripheral blood leukocytes (PBL), cutaneous T-cell lymphomas cases (CTCL) with or without 6p25.3 rearrangements, and lymphoid T-cell lines. Mean from independent measurements are shown. C. DUSP22 alternatively spliced transcipts levels were analyzed by quantitative RT-PCR, normalized by EEF1A1 expression, in normal PBL, CTCL cases and lymphoid T-cell lines. Mean ± SEM from independent measurements are shown. For transcripts with and without intron 7 (Top panel), the common forward primer was within exon 1 and isoform-specific reverse primers were either at the beginning of intron 7, or at the beginning of exon 8. For transcripts with and without (Δ) exon 4 (Bottom panel), isoform-specific forward primers were overlapping either exons 3 and 4 or exons 3 and 5, respectively, and the common reverse primer was overlapping the junction between exons 6 and 7.
Table 1: SNP haplotypes, methylation and expression/splicing status of DUSP22 and its paralog
DUSP22 (6p25.3) |
16p11.2 paralog |
|
|---|---|---|
SNPs alleles |
||
rs11242812 |
G |
A |
rs1129085 |
G and/or A |
G |
rs1046656 |
C |
T |
Methylation status (5’ CpG island) |
No methylation$($ in normal tissues & tested PTCL cases) |
Methylated‡ (‡ if not deleted in the 5’ region) |
Expression |
Expressed in normal tissues& PTCL without 6p25.3 rearrangement Silenced in PTCL with 6p25.3 break |
Completely silent or hypomorphic |
Isoforms |
Alternative mRNA splicing |
Not detected (expression absent or very weak) |
Regular splicing, 8 coding exons Protein: 184 a.a. Expressed at the RNA level in PBL Silenced in PTCL with 6p25.3 break |
Not detected |
|
Intron 7 not spliced, leading to alternative reading frame, exon 8 then being in the 3’ UTR Predicted protein: 205 a.a., p.A170fs37* Predominant in normal PBL Silenced in PTCL with 6p25.3 break |
Not detected |
|
Exon 4 spliced, leading to frameshift Predicted protein: 54 a.a., p.G47fs9* Expressed at the RNA level in PBL Silenced in PTCL with 6p25.3 break Not detected at the protein level |
Not detected |
Legends: The SNP haplotypes, methylation and expression status of DUSP22 and its 16p11.2 are summarized here. For isoforms, the splicing events and the predicted consequence at the protein levels (number of amino acids -a. a.- and when there is a alternative reading frame, position of the first amino acid affected by frameshift –fs-, and number of alternative amino acids before the STOP codon -*-). UTR, untranslated region; PTCL, peripheral T-cell lymphomas; PBL, normal peripheral blood lymphocytes.
Here we showed that normal peripheral blood lymphocytes (PBL) predominantly expressed transcripts with unspliced intron 7, over those with intron 7-splicing (Supplementary Figure S1B, Figure 1C, Table 1). Δ exon 4 transcripts, predicted as candidates to nonsense-mediated decay, were readily detected in normal (PBL) and actually expressed equivalently than transcripts with exon 4 (Supplementary Figure S1B, Figure 1C, Table 1).
Other deposited transcripts originating from alternative initiation sites were not detected in the tested normal PBL, cutaneous T-cell lymphomas (CTCL) and cell lines, and not studied further.
Consistent with observations of Feldman et al. [12], quantitative RT-PCR showed that, as compared with normal PBL, all cALCL and T-MF cases (Supplementary Table S1) with monoallelic 6p25.3 breakpoints [11] exhibited DUSP22 silencing (Figure 1B). Alternatively spliced DUSP22 transcripts were all silenced in CTCL with 6p25.3 alterations (Figure 1C, Table 1, Supplementary Tables S1 and S2), while there was no significant effect on the expression of the neighboring IRF4 and EXOC2/SEC5 genes (Supplementary Figure S3A).
Lack of DUSP22 deletion or methylation in CTCL with monoallelic 6p25.3 rearrangements
We next looked for inactivating mutations, deletions or methylation-mediated silencing [30] of DUSP22 in PTCL with 6p25.3 rearrangements. Such rearrangements were previously found monoallelic and there was no deletion of the second allele [10, 11, 12]. As FISH probes used then were mainly flanking the gene [10, 11, 12], we looked for interstitial deletions using probes encompassing DUSP22 (Supplementary Figure S4A).
Consistent with bioinformatics predicting the existence of a paralog of DUSP22 on 16p11.2 [31, 32], these probes gave, apart from the 6p25.3 locus, additional hybridization signals on 16p11.2 (Figure 2A, Supplementary Figures S4C and S5A).
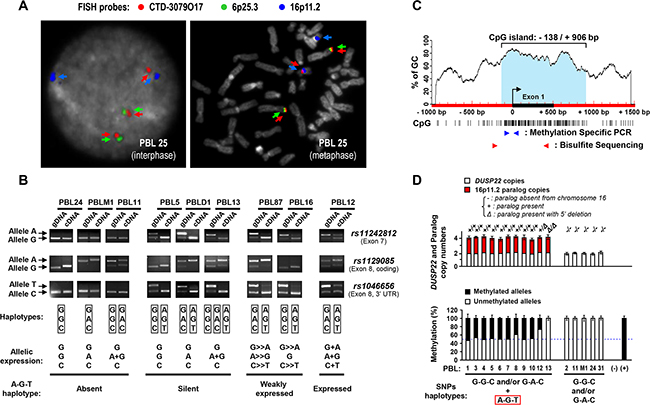
Figure 2: Mapping and haplotype identification of a transcriptionally inactive paralog of the DUSP22 gene on 16p11.2. A. Metaphase and interphase FISH analysis of normal peripheral blood lymphocytes (PBL) with a 6p25.3-specific probe (RP11-164H16, Spectrum Green labeled), a probe encompassing the DUSP22 gene (CTD-3079O17, Spectrum Red labeled), and a 16p11.2-specific probe (RP11-488I20, Spectrum Gold labeled, here displayed in blue (pseudocolor) as colocalization of signals from the former probes may appear in yellow). DAPI (4’,6-diamidino-2-phenylindole dihydrochloride), here visualized in grey (pseudocolor), was used to stain nuclear DNA and chromosomes. Cases exhibiting, in addition to 6p25.3, 1 or 2 additional CTD-3079O17 probe signals, illustrate the existence of a DUSP22-related sequence subjected to copy number variations on the 16p11.2 locus (See also Supplementary S5A, B). B. Genotype (gDNA, genomic DNA) and allelic expression status (cDNA, complementary DNA) of SNPs rs11242812, rs1129085 and rs1046656 in representative cases of normal PBL, analyzed by PCR and restriction enzyme digestion (MspI, MspI and BssSI, respectively). Arrows indicate each SNP allele. SNPs genotypes and allelic expression status are summarized for each case under the electrophoresis profiles. C. Schematic representation of the CpG island (vertical bars indicate CpG sites) encompassing the 5’ region of the DUSP22 gene. The black box and curved arrow indicate exon 1 and transcription initiation site, and blue and red arrowheads the primers used for methylation specific PCR (MSP) and bisulfite genomic DNA sequencing (BGS) analyses. D. Top panel: DUSP22 and paralog copy numbers, determined by quantitative analysis of SNP rs1046656 (allele C= DUSP22; allele T= paralog), in normal PBL. KLK3 (19q13.41) was used as a control gene for normalization. Data were matched with results from3 color FISH (discriminating DUSP22 -6p25.3- from its paralog -16p11.2-, Figure 2A, Supplementary Table S6). The occurrence of deletion in the 5’ region of the paralog was evidenced by the observation of altered qPCR signal ratios between exon 1 and exon 6. Bottom panel: Quantitative MSP analysis of DUSP22/paralog 5’ CpG island in normal PBL according to the absence or presence of paralog alleles, determined as described above. No DNA (-) and in vitro methylated DNA (+) were used as negative and positive controls. Percentages of unmethylated and methylated alleles (Mean ± SEM from independent measurements) are shown.
The presumed high sequence similarity between DUSP22 and its paralog [31, 32] might alter the interpretation of genetic and epigenetic analyses of DUSP22. We thus developed a 3 color FISH approach combined with molecular analyses enabling to distinguish DUSP22 and its paralog.
Using FISH, we showed that the 16p11.2 paralog was present in most individuals (on one or both chromosomes 16) and absent in ≈10% of cases (Figure 2A, Supplementary Figure S5A, Supplementary Table S6 and Supplementary text file).
Genotyping SNPs rs11242812, rs1129085 and rs1046656, we identified specific haplotypes discriminating DUSP22 (G-G-C and G-A-C alleles) from the 16p11.2 paralog (A-G-T allele) (Figure 2B, Table 1, Supplementary Figure S2, Supplementary Table S6 and Supplementary text file).
Analyzing these SNPs, we next found that, in normal cells, the 16p11.2 paralog was silent, while DUSP22 was always bi-allelically expressed (Figure 2B, Table 1, Supplementary Figures S6A, S6B, Supplementary Table S6 and Supplementary text file). The paralog was also not expressed in tested CTCL samples.
Finally, consistent with its lack of transcriptional activity, we showed that, while the 5’ CpG island of the DUSP22 gene was never methylated, the 16p11.2 paralog was mainly methylated (≈80% of alleles) or deleted in that region (Figures 2C, 2D, Table 1, Supplementary Figures S5 and S6, Supplementary Table S6 and Supplementary text file).
Knowledge of the paralog status and identification of DUSP22-specific haplotypes allowed comprehensive analysis of DUSP22 copy number and methylation status in normal PBL and CTCL. Except cases 15 (without paralog) and 11 (six methylated paralog copies), all tested cALCL carried two copies of the methylated and inactive paralog (Figure 3A). In our series of cALCL cases, we found neither DUSP22 gene deletion (Figure 3A, Top panel) nor DUSP22 5’CpG island methylation (Figure 3A, Bottom panel). When detected in tumors, methylation was always attributable to the presence of the paralog (Figure 2D). Indeed, methylation levels were identical in tumors and matching constitutional DNA and proportional to paralog copy numbers (Figure 3A).
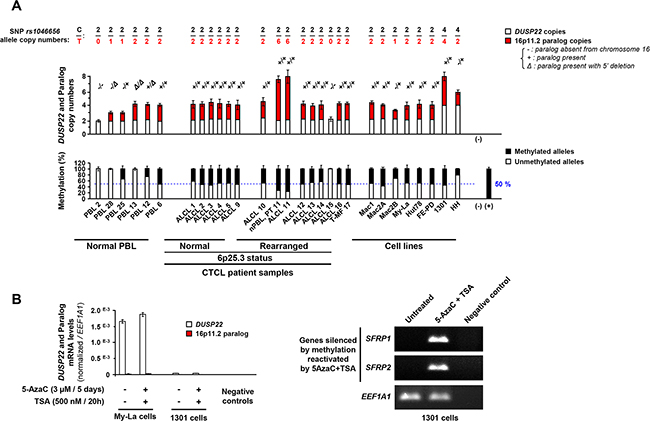
Figure 3: Copy number and methylation status of DUSP22 and its 16p11.2 paralog in anaplastic large T-cell lymphomas. A. Top panel: DUSP22 and paralog copy numbers, determined by quantitative analysis of SNP rs1046656 (allele C= DUSP22; allele T= paralog), in normal lymphocytes (PBL) and cutaneous T-cell lymphomas cases (CTCL) and lymphoid T-cell lines. KLK3 (19q13.41) was used as a control gene for normalization. The occurrence of deletion in the 5’ region of the paralog was evidenced by the observation of altered qPCR signal ratios between exon 1 and exon 6. PBL cases with different 16p11.2 paralog status (2: no copy; 28: one copy with 5’ deletion; 25: one copy; 13: two copies with 5’ deletion; 12: two copies, one having 5’ deletion; 6: two copies) were used as controls. For ALCL case 10, found to carry 6 copies of the 16p11.2 paralog, analysis of the patient’s normal PBL (Normal, PT 10) showed that this was a constitutional feature. A. Bottom panel: Quantitative methylation specific PCR (MSP) analysis of 5’ CpG island present in DUSP22 and most 16p11.2 paralog alleles, in normal PBL and CTCL samples and cell lines. No DNA (-) and in vitro methylated DNA (+) were used as negative and positive controls. Percentages of unmethylated and methylated alleles (Mean ± SEM from independent measurements) are shown. For ALCL case 10, found to constitutionally carry 6 copies of the 16p11.2 paralog (Top panel), comparative analysis with the patient’s normal PBL (Normal, PT 10) showed that the high level of methylation observed in that tumor is attributed to the paralog copies. B. Left panel: DUSP22 and paralog transcript levels were analyzed by allele specific (SNP rs1046656: C = DUSP22; T = paralog) quantitative RT-PCR, normalized by EEF1A1 expression, in lymphoid T-cell lines treated or not with 5-Aza-2’-deoxycytidine (DNA methyltransferase inhibitor) trichostatin A (histone deacetylase inhibitor). Mean ± SEM from independent measurements are shown. Such treatment did not significantly alter DUSP22 expression and notably did not upregulated expression in the 1301 cell line with low levels. Right panel: as a control of drug efficiency, SFRP1 and SFRP2 genes, known to be silenced by promoter methylation in various cancers, were reactivated by combined treatment with 5-Aza-2’-deoxycytidine and trichostatin A.
Identification of DUSP22 somatic mutations in cALCL cases
Further searching for a second hit, we analyzed tumor DNA samples for DUSP22 mutations in the promixal promoter, exons and splice sites. This screening was performed on 20 cALCL and 19 T-MF, another type of CTCL exhibiting DUSP22 rearrangements [11] (Supplementary Table S1).
We identified somatic mutations (absent in the constitutional DNA from the same patient) in 2 out of 20 tested cALCL cases (10%), but in none of the T-MF cases.
The first case (#12, Supplementary Table S1) harbored a missense c.4G>A nucleotide change immediately after the ATG translation initiation codon, predicted to result in a p.Gly2Arg (p.G2R) amino-acid substitution. It concomitantly exhibited, with minor allele frequency, a c.-28G>A nucleotide variation in the 5’UTR, 28 base-pairs upstream of the initiating codon (Figure 4A). Given the very high estimated homology (99.9%) between DUSP22 and its paralog [31, 32], present as two methylated copies in this case (Figure 3A, case 12), the PCR primers amplified exon 1 from both DUSP22 and the paralog. We then digested the tumor DNA with the methylation-sensitive HpaII restriction enzyme, thus fragmenting unmethylated DUSP22 alleles but not restriction-resistant methylated paralog alleles. Subsequent PCR amplification and sequencing showed only wild-type sequences (Figure 4A), indicating that the mutations affected DUSP22 and not its paralog. As this case also harbored monoallelic 6p25.3 rearrangement with nearly complete DUSP22 silencing (Supplementary Table S1 and Figure 1), one or both of these mutations may inactivate the second allele. Sequencing of the weakly expressed tumor DUSP22 cDNA revealed only wild-type sequences (Figure 4A) attributed to normal cells, indicating that mutant alleles were not expressed, consistent with a silencing effect of these mutations.
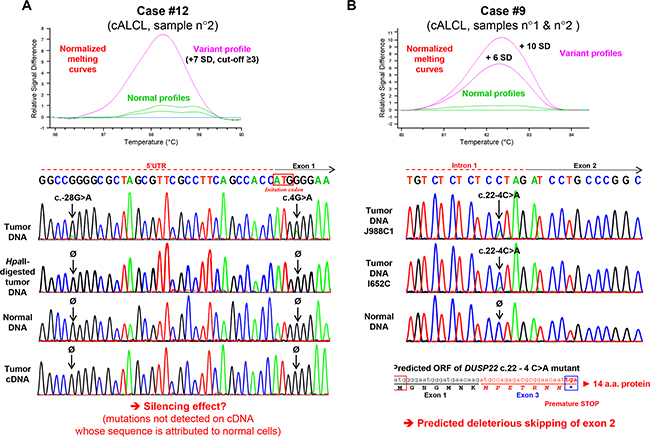
Figure 4: Identification of somatic DUSP22 mutations in cutaneous anaplastic large T-cell lymphomas. A. Top panel: High resolution melting (HRM) profile of exon 1 genomic DNA PCR products from normal samples and cutaneous anaplastic large T-cell lymphoma (cALCL) case 12, known to harbor monoallelic 6p25.3 breakpoint and DUSP22 gene silencing. Bottom panel: Sanger sequencing analysis of case 12 constitutional and tumor genomic DNA (the latter being digested or not with the HpaII methylation-sensitive restriction endonuclease), as well as tumor complementary DNA (cDNA, obtained from RNA by reverse transcription-PCR). B. Top panel: High resolution melting (HRM) profile of genomic DNA PCR products encompassing exon 2 and flanking splice sites from normal samples and cutaneous anaplastic large T-cell lymphomas (cALCL) case 9. Bottom panel: Sanger sequencing analysis of case 9 constitutional and tumor genomic DNA, with predicted effect of the identified mutation on messenger RNA splicing.
Another case (#9, Supplementary Table S1) harbored a c.22-4C>A somatic intronic mutation, 4 base-pairs upstream of exon 2, predicted to lead to exon 2 skipping, with deleterious frameshift effect (Figure 4A). Mutant allele frequency and quantitative SNP analysis were consistent with a heterozygous mutation and the second allele of DUSP22 was neither rearranged nor deleted (Supplementary Table S1). For this case, frozen material was not available and RNA levels and exon skipping could not be evaluated.
Characterization of DUSP22 genetic and expression status in lymphoid T-cell lines
To identify relevant models to study the role of DUSP22 in oncogenesis, we investigated CTCL cell lines and two other lymphoid T-cell lines (Supplementary Table S1) [33, 34, 35, 36, 37, 38, 39]. Unlike CTCL tumors, none of these 8 cell lines harbored a break-apart FISH pattern with probes flanking DUSP22 (Supplementary Figure S7A). FISH and high resolution array-comparative genomic hybridization [5] indicated that, in the FE-PD cell line, a 13 Mb region downstream of DUSP22 was duplicated on 9qter (Supplementary Figure S7A, S7B). FE-PD and most cell lines expressed DUSP22 as normal PBL, except HH and 1301 which exhibited very weak DUSP22 transcripts levels (Figure 1B, 1C, Supplementary Figure S1B), though not harboring 6p25.3 rearrangements (Supplementary Figure S7A). The 1301 and HH cell lines, both nearly tetraploid, carried 4 copies of DUSP22 and respectively 4 and 2 copies of the 16p11.2 paralog, all paralog alleles being methylated in both cell lines (Figure 3, Supplementary Figure S7C). Neither deletion, nor mutations nor 5’ CpG island hypermethylation of DUSP22 could account for the weak transcript levels observed in the 1301 and HH cell lines, methylation being attributable only to the paralog and proportional to its copy number (Figure 3, Supplementary Figure S7C). Further proving that methylation was not involved in DUSP22 silencing in these cell lines, there was no reactivation of its expression upon combined 5-aza-2’-deoxycytidine (inhibitor of DNA methyltransferases)/Trichostatin A (inhibitor of histone deacetylases) treatment (Figure 3B, left panel). As a control, such treatment reactivated expression of SFRP1 and SFRP2 genes known to be silenced by methylation in various types of cancers (Figure 3B, right panel).
DUSP22 exerts tumor suppressive functions
The 205 amino-acids isoform is encoded by the transcript with unspliced intron 7, which is predominant in normal PBL and silenced in PTCL with 6p25.3 rearrangements (Figure 1A, 1C, Table 1, Supplementary Figure S1). This was the only isoform detected at the protein level in normal PBL and cell lines expressing endogenous DUSP22 transcripts (Supplementary Figure S8A).
Western blot analysis of cells transduced with DUSP22 lentiviral expression vectors revealed bands with estimated molecular weight corresponding to the ectopically expressed 205 and 184 amino-acids isoforms (Figure 5C, Table 1, Supplementary Figures S1 and S8D). The Δ exon 4 isoform was never detected, even using an antibody directed against an N-terminal epitope conserved in the DUSP22 isoforms (Bottom panels in Figure 5C and Supplementary Figure S8D), suggesting that this isoform is weakly stable.
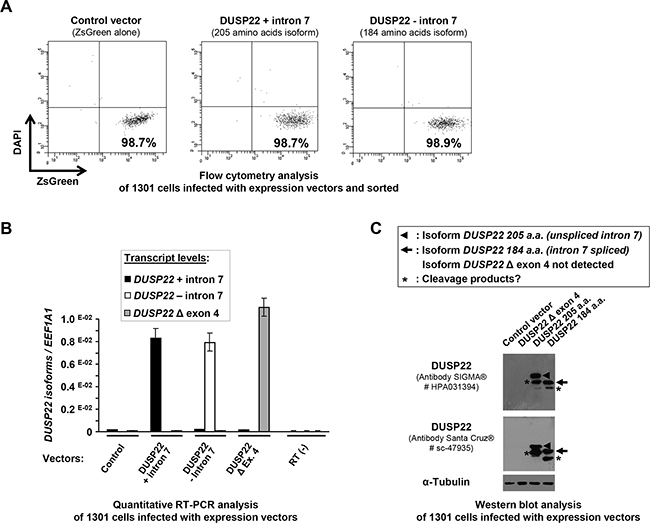
Figure 5: Cell sorting after lentiviral infection and analysis of ectopic expression of DUSP22 isoforms in 1301 lymphoid T cells. A. 1301 cells were transduced with lentiviral vectors (Control -encoding the ZsGreen reporter alone- and bicistronic vectors encoding DUSP22 isoforms together with ZsGreen). Alive (DAPI-) and transduced (ZsGreen+) cells were sorted by flow cytometry using ARIA II cell sorter. Dot plots show cell fraction purity after sorting. B. DUSP22 isoforms transcript levels were analyzed by quantitative real-time reverse transcription-PCR (qRT-PCR) on RNA isolated from the same cells transduced with either control or DUSP22 isoforms expression vectors and sorted. DUSP22 isoform-specific primers were used (Figure 1C and Supplementary Table S2) and transcript levels were normalized for EEF1A1 gene expression and plotted at the y axis. Mean ± SEM from independent measurements are shown. C. Western blot analysis of DUSP22 expression in 1301 cells infected with the control and DUSP22 isoforms vectors and sorted. The anti-DUSP22 antibodies used were from SIGMA® (# HPA031394, as in (A)) and Santa Cruz® (# sc-47935), the latter being directed against an N-terminal peptide common to the 3 isoforms. Alpha-tubulin (α-Tubulin) was used as a control for protein loading.
Functional studies were thus performed with the 205 amino-acids isoform to investigate the potential tumor suppressor role of DUSP22 in T-cells. The HH and 1301 cell lines were selected as they exhibited a very low expression of DUSP22 transcripts and undetectable protein levels (Figure 1B, 1C and Supplementary Figure S8A). Conversely, FE-PD cells were chosen as a control given their high endogenous expression of DUSP22 protein (Supplementary Figure S8A).
FE-PD and 1301 cell lines were very efficiently infected by our lentiviral vectors (95% and 80%, respectively), whereas HH cells could not (as reflected by a percentage of ZsGreen positive cells below 5%) and were thus not further studied. FE-PD and 1301 living infected cells were sorted by flow cytometry prior to functional analyses and checked for DUSP22 expression at the RNA and protein levels (Figure 5, Supplementary Figure S8).
Ectopic expression of the DUSP22 205 amino-acids isoform had no effect on proliferation of DUSP22-proficient FE-PD cells, and only weakly decreased their viability by slightly enhancing apoptosis (Supplementary Figures S8, S9). In 1301 cells, showing low endogenous DUSP22 levels, lentivirus-mediated expression of this DUSP22 isoform significantly impaired the cell population increase. There was no significant inhibition of cell proliferation but a 5-fold enhancement of apoptosis (Figure 6A–6C).
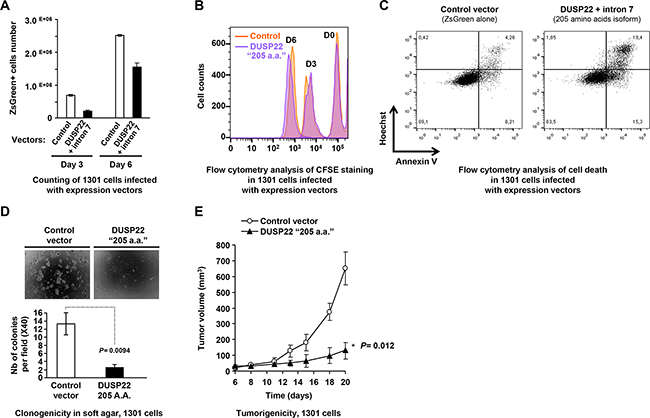
Figure 6: Tumor suppressive effects of DUSP22 in lymphoid T cells. A. Effect of ectopic expression of the main DUSP22 isoform detected in lymphoid cells on proliferation and viability of 1301 (T cell-acute lymphoblastic leukemia-T-ALL-) cells. Cells infected with either the control (empty) or the DUSP22 205 amino acids (a.a.) isoform (Figure 1A and Supplementary Figure S1) lentiviral vectors and sorted were diluted in equal numbers and grown in the presence of 10% fetal calf serum and counted at different time points. Mean ± SEM from a representative experiment are shown. B. Proliferation assay of 1301 cells transduced with either the control (empty) or the DUSP22 205 amino acids (a.a.) isoform lentiviral vectors and sorted. Cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) at day-0 (D0) and its dilution was followed at day-3 (D3) and day-6 (D6) of culture. C. Apoptosis and necrosis analyses were performed on infected and sorted 1301 cells by Annexin V/Hoescht 333542 staining 3 days after replating. Single Hoescht 333542-positivity indicates necrotic cells, while double Annexin V/Hoescht positivity hallmarks apoptotic cells. D. 1301 T-ALL cells transduced with either the control or DUSP22 (205 a.a. isoform) vectors were assayed for clonogenicity in soft agar. Upper panels: Visualization of colonies using phase contrast microscopy. Bottom panels: Colonies counting plotted as mean ± SEM from independent measurements. E. Tumorigenicity of 1301 T-ALL cells, transduced with either the control or DUSP22 (205 a.a isoform) vectors and grafted in immunodeficient mice. Tumor growth is shown for a representative experiment (mean ± SEM from 5 different mice for each condition at different time points).
FE-PD cells neither produced colonies in soft agar, as reported previously [34], nor formed tumors after subcutaneous injection in immunodeficient mice, preventing their use in further functional studies. Remarkably, lentivirus-mediated expression of the physiological DUSP22 205 amino-acids isoform strongly inhibited the clonogenicity of 1301 cells (Figure 6D) and also impaired their tumorigenic properties in a subcutaneous xenograft model (Figure 6E). Altogether, our study shows that inactivation of DUSP22 contributes to T cell transformation and supports a tumor suppressor function of DUSP22.
DISCUSSION
The silencing of DUSP22 in PTCL with 6p25.3 rearrangements [12] pointed out this gene as a candidate tumor suppressor whose inactivation may contribute to the pathogenesis of PTCL subtypes, notably ALCL (essentially cutaneous) and T-MF [9, 10, 11, 12, 13, 14]. However, in our cALCL and T-MF cases, 6p25.3 chromosomal alterations were always found to be monoallelic. We thus looked for interstitial deletions or methylation that could account for silencing of the second allele of DUSP22. During the course of this analysis, we cytogenetically and molecularly characterized a paralog, mapped on 16p11.2, recently predicted by bio-informatics analyses [31, 32].
This paralog presumably exhibited 99.9% sequence similarity with DUSP22 [31, 32]. Nonetheless, we could design a strategy combining 3 colors FISH, SNP genotyping and methylation analyses enabling to distinguish DUSP22 (6p25.3) from its paralog (16p11.2) and allowing comprehensive genetic and epigenetic analyses of DUSP22 in normal cells, tumors and cell lines.
We showed that this paralog is present in 90% of individuals, on one (30%) or both (60%) 16p11.2 alleles. We also identified DUSP22-specific SNP haplotypes and showed that this paralog is either methylated or deleted in the 5’ region and transcriptionally inactive.
CTCL cases with mono-allelic 6p25.3 rearrangements exhibit neither deletion nor methylation of the second allele of DUSP22. Methylation of the 5’ CpG island was indeed always linked to the presence of the inactive paralog, except when deleted. The paralog was silent in tested CTCL tumors and cell lines indicating that it is not involved in CTCL pathogenesis.
The above-described strategy can avoid potential misinterpretation of DUSP22 status. This was first illustrated in 2 CTCL cases (number 15 and 18, Supplementary Table S1) initially thought to exhibit focal heterozygous deletions of DUSP22 based on previous high resolution comparative genomic hybridization (CGH) data [5]. FISH and quantitative SNP analysis of tumor and matching normal cells and DNA from these patients revealed that these tumors constitutionally lacked paralog sequences but did not exhibit DUSP22 deletions. Initial misinterpretation of DUSP22 gene dosage was due to comparison to a commercial pool of normal DNAs, as usual in array CGH analysis [5], most of them carrying paralog alleles. Besides, Kadin, Feldman and colleagues reported 3 cases of cALCL with abnormal FISH patterns interpreted as bi-allelic DUSP22 rearrangements [40]. Given the existence of the highly prevalent 16p11.2 paralog, the complex FISH profiles of such cases are hardly conclusive without concomitant analysis of matching normal cells and hybridization with 16p11.2 probes, as used here. Finally, two studies recently reported DUSP22 5’ region hypomethylation upon fire smoke exposure [41] or hypermethylation in Alzheimer’s disease [42]. As we observed that the DUSP22 gene 5’ region is not methylated in normal tissues, the methylation detected in these studies is likely to result from the presence of the paralog. Therefore, a comprehensive assessment of DUSP22 status cannot be conducted without knowledge of its paralog copy number and 5’ methylation or deletion status.
Two out of the 20 studied cALCL cases harbored predicted inactivating somatic mutations, one of which also exhibiting monoallelic rearrangement and DUSP22 silencing, while the second case had neither rearrangement nor deletion of the second allele. Most of our CTCL cases with monoallelic breakpoints thus did not exhibit a second hit that could explain silencing of DUSP22. A growing number of tumor suppressor genes are reported to be haploinsufficient, as heterozygous alterations leading to a 50% reduction in gene function are enough to promote oncogenesis [43]. Here, this hypothesis is however unlikely as complete silencing was always observed in tumors with monoallelic DUSP22 rearrangements. As we also found that DUSP22 is biallelically expressed in normal tissues, the second allele of 6p25.3 rearranged tumors is therefore somatically inactivated by an unknown transcriptional or post-transcriptional mechanism.
We then confirmed the existence of distinct DUSP22 transcripts, produced by alternative splicing of exon 4 or intron 7 and predicting the production of 3 protein isoforms, only one of which being naturally detected in lymphoid T-cells. We showed that these DUSP22 transcripts were all silenced in CTCL with 6p25.3 rearrangements. As anti-DUSP22 antibodies are not reliable for immuno-histochemical analyses [44], quantitative RT-PCR or RNA sequencing analyses are necessary to assess DUSP22 expression levels.
Restoring expression of the physiological DUSP22 isoform in the 1301 DUSP22-deficient T cell leukemia cell line enhanced apoptosis and strongly impaired both clonogenicity in soft agar and tumorigenicity in immuno-deficient mice. Altogether, our study supports a tumor suppressor function of DUSP22 and that its inactivation contributes to the development of PTCL. However, lack of spontaneous tumorigenesis in Dusp22 knock-out mice [29] clearly indicates that DUSP22 is not a gatekeeper tumor suppressor whose inactivation is sufficient to initiate tumor development. DUSP22 rearrangements are moreover correlated with better clinical outcomes in systemic ALCL [14], but not in our series of cutaneous ALCL [11, 44] which however have an overall excellent prognosis. As for other tumor suppressors whose inactivation is a secondary event, loss of DUSP22 function may thus only contribute to transformation in cells already carrying other initiating oncogenic events. Global copy number analysis [5] did not reveal associations between DUSP22 and other genomic alterations in our cALCL cases. DUSP22 rearrangements are found mostly in ALK negative-ALCL [10, 11, 12, 14, 45], recently shown to harbor various molecular alterations leading to constitutive activation of the JAK/STAT3 pathway [46, 47]. Among other pathways [23, 24, 25, 26, 27, 28, 29], DUSP22 was previously shown to inhibit STAT3 signalling [26]. Mutations in this pathway may cooperate with DUSP22 rearrangements in ALCL oncogenesis. Genome-wide massive parallel sequencing approaches will determine which molecular alterations coexist with DUSP22 inactivation in PTCL and may account for the favorable outcome of 6p25.3 rearranged tumors.
Besides, several types of cancers exhibit 6p25-pter allelic losses [48, 49, 50, 51, 52, 53, 54], suggesting the inactivation of a tumor suppressor gene in this region. Given its tumor suppressive behavior in PTCL, DUSP22 is a candidate to be tested in tumors with 6p25 deletions, using a comprehensive approach such as described herein.
MATERIALS AND METHODS
Patients’ tissues and normal peripheral blood lymphocytes
Normal peripheral blood lymphocytes (PBL) and cutaneous T cell lymphomas (Supplementary Table S1) samples were collected and stored frozen at -140°C until nucleic acids and proteins extraction. Tumor samples presented at least 70% of cancer cells, based on histopathological review. Normal PBL were obtained upon Ficoll gradient centrifugation of blood samples from healthy donors at the établissement Français du Sang (Bordeaux, France). Other normal tissues (skin, lung, stomach, colon, kidney, bladder) were obtained from the Biobank of the University Hospital (Bordeaux, France). The study was performed after obtaining informed written consent from patients and healthy donors according to the Declaration of Helsinki Principles and was approved by the medical ethical committee of Aquitaine, France.
Cell culture
The HH cell line was purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany) and MAC cell lines [33] were a generous gift from Pr Marshall Kadin (Providence, USA). Other cell lines (My-La, FE-PD, HUT78, 1301; Supplementary Table S1) were obtained from sources previously described [34, 35, 36, 37, 38, 39]. HH and 1301 cell lines were grown in RPMI 1640 supplemented with 10% fetal bovine serum (Life Technologies). FE-PD cells were grown in IMDM supplemented with 20% fetal bovine serum (Life Technologies).
Fluorescent in situ hybridization (FISH)
Samples preparation, hybridizations and 6p25.3 locus FISH analyses were carried out following standard protocols, as previously described [11]. Probes are listed in Figure 2 and Supplementary Figures S4, S5 and S7.
DNA and RNA extraction
High-molecular-weight genomic DNA was extracted from fresh or fresh-frozen biopsy samples, normal peripheral blood lymphocytes or cell lines using a standard protocol with proteinase K digestion, phenol/chloroform extraction and ethanol precipitation, as previously described [34]. Total RNA was extracted using TRIzol Reagent (Invitrogen) and treated with DNAse I (Invitrogen) to avoid contamination with genomic DNA.
Gene expression analysis
Gene expression levels were determined by reverse transcription/real-time quantitative PCR (qRT-PCR), as previously described [34], using the comparative MNE (Mean Normalized Expression) method [55]. The primers used for qRT-PCR analyses are listed in Supplementary Table S2. Melting curves, gel electrophoresis and sequencing analyses showed that these primers amplified only the specific fragments.
SNP genotyping
The genotype and allelic expression status of DUSP22 and its paralog was initially determined by genotyping of SNPs rs11242812 and rs1046656, performed by Sanger sequencing of PCR products obtained from genomic DNA and complementary DNA (cDNA) of lymphocytes from healthy donors. Sequencing reactions were carried out on a 3130xl Genetic Analyzer (Applied Biosystems), using the Big Dye Terminator kit (Applied Biosystems). Further SNPs genotyping and allelic expression status analyses of DUSP22 and its paralog, IRF4 and EXOC2 genes were performed by restriction enzyme digestion of PCR products obtained from genomic DNA and cDNA of lymphocytes, normal tissues or cell lines. PCR products encompassing SNPs rs11242812 (DUSP22, exon 7), rs1129085 (DUSP22, exon 8, coding region), rs1046656 (DUSP22, exon 8, 3’ UTR), rs2316515 (IRF4, exon 11) and rs11242914 (EXOC2, exon 28) were respectively digested by MspI, MspI, BssSI, HpyCH4V and HaeIII restriction endonucleases. Alleles of each SNP, digested or not by the respective endonucleases, were evaluated after 4% low melting agarose electrophoresis.
Quantitative SNP (qSNP) analysis was performed to determine allele-specific copy numbers for rs1046656 (alleles C and T). These analyses were performed on genomic DNA isolated from normal PBL, peripheral T-cell lymphomas (PTCL) cases and lymphoid T-cell lines. We first designed allele-specific primers to analyze the allelic status of this SNP by real-time quantitative PCR, as previously described [34]. Optimal PCR amplification conditions were 95°C for 3 minutes followed by 45 cycles of 95°C for 20 seconds and 62°C for 20 seconds. Quantification of each SNP allele was performed according to the MNE (Mean Normalized Expression) method [55], here adapted to DNA copy numbers. Data were normalized using KLK3 (located on 19q13.41) as a control gene, but also ALB (4q13.3), CDH1 (16q22.1) or CECR1 (22q11.2) to ensure that there was no alteration of KLK3 copy numbers. Data from tumor DNA were compared with DNA from matching normal tissue from the same patient. Data from cell line DNA, DNA from a series of normal PBL was used as a reference. To validate this real time PCR, we also developed a restriction fragment length polymorphism-PCR assay, performed with fluorescently labeled primers, with numbers of cycles adjusted to fall within the linear range of the amplification. PCR products encompassing SNP rs1046656 were digested by the BssSI restriction endonuclease and quantified using capillary electrophoresis. Allele-specific copy numbers, determined by quantifying the area under curve of each SNP allele’s peak, were normalized using KLK3 as a control gene. The primers used for amplification of genomic DNA and cDNA fragments for SNP genotyping by sequencing, restriction enzyme digestion and gel electrophoresis, quantitative real time PCR or capillary electrophoresis are listed in Supplementary Table S3. Amplicons specificity was verified by gel electrophoresis and sequencing analyses.
DNA methylation analysis
Genomic DNA was subjected to sodium bisulfite treatment using the EpiTect Bisulfite Kit, according to the manufacturer’s instructions (QIAGEN, Hilden, Germany). Methylation was then analyzed using real-time quantitative methylation-specific PCR (qMSP) and confirmed for selected representative samples using bisulfite genomic sequencing (BGS), using standard protocols [56]. As for gene expression and gene copy number determination, qMSP was quantified using the using the comparative MNE (Mean Normalized Expression) method [55]. Data were displayed as percentage of unmethylated and methylated alleles. The primers used for qMSP and BGS analyses were designed using the Primer Express v 1.0 software (Applied Biosystems) and are listed in Supplementary Table S4. Specificity was verified by melting curves, gel electrophoresis and sequencing analyses.
DUSP22 mutation analysis
Genomic DNA from normal and tumor tissues were screened for the presence of mutations by quantitative PCR amplification followed by high resolution melting (HRM) analysis on a LC480 device (Roche Diagnostics), using the manufacturer’s kit, as previously described [57]. Amplicons exhibiting a melting profile (± 3 Standard Deviations) or temperature (Tm) differing from control DNA were subjected to Sanger DNA sequencing, as described in the SNP genotyping section. The primers, encompassing the 8 DUSP22 gene exons and splice sites, used for HRM analysis and Sanger sequencing are listed in Supplementary Table S5.
Lentiviral vectors and transduction
To generate expression vectors, the coding sequences of DUSP22 isoforms were amplified by polymerase chain reaction (PCR) using complementary DNA (cDNA) from normal lymphocytes as a template. The following primers were used: exon 1 forward primer (5’-aaactgcagAGCCACCATGGGGAATGGGAT-3’, low capitals italic characters being an overhang and engineered PstI site) with either intron 7 reverse primer (5’-aaaacgcgtTTAGGTCTCCGTCGTATAATTATCG-3’, low capitals italic characters being an overhang and engineered MluI site) or exon 8 reverse primer (aaaacgcgtCATTACAGTCTTCTGAGAAAGG, low capitals italic characters being an overhang and engineered MluI site). PCR products were cloned in a bicistronic self-inactivating (SIN) lentiviral vector, under the control of the myeloproliferative sarcoma virus enhancer, upstream of an internal ribosomal entry site (IRES2) followed by the ZsGreen fluorescent protein coding sequence. A similar lentiviral vector containing only the ZsGreen coding sequence under the control of the myeloproliferative sarcoma virus enhancer was used as a control. Constructs were verified by sequencing. Lentiviral vectors were produced by triple-transient transfection of 293T cells, the viral titer was determined and cell lines were transduced at an optimal multiplicity of infection (MOI) of 30 viral particles per cell, as previously described [58].
Western blot analysis
DUSP22 protein levels were assessed by Western blot, following standard protocols. The antibodies used were DUSP22 (HPA031394, Sigma-Aldrich, St. St. Louis, MO 63103, USA, 16514-1-AP, Proteintech, Chicago, IL 60612, USA, and sc-47935, Santa Cruz Biotechnology, Dallas, TX 75220, USA) and α-Tubulin (Sigma-Aldrich).
Flow cytometry, cell sorting, cell proliferation and viability assays
Cell sorting
The lentiviral infection efficiency of the tested cells was monitored by flow cytometry evaluating ZsGreen expression on a FACS Calibur and BD FACS Canto II (Becton Dickinson, San Jose, CA). Transduced 1301 and FE-PD cells were stained with DAPI (1μg/ml) for 15 min. Viable (DAPI negative)-transduced (ZsGreen positive) cells were sorted using a BD ARIA II device (BD Biosciences). Cell fraction purity was checked and selected cell populations were used for qRT-PCR, western blotting and phenotypic assays. Cell proliferation was evaluated by FACS at different time points (0, 3 and 6 days after plating) after staining with Cell trace™ Violet, according to manufacturer recommendations (Molecular Probes®, Eugene, OR, USA). Apoptosis/necrosis was evaluated by FACS after staining with an anti-annexin V-PE conjugated antibody according to manufacturer recommendations (BD Biosciences, Le Pont-de Claix, France) and Hoechst 33342 was added 5 minutes before sample acquisition. Acquisition and analysis were performed using a BD FACS Canto II (BD Biosciences) and the FlowJo software (Tree Star Inc.) respectively.
Soft agar assays and tumorigenicity in immunodeficient mice
Clonogenicity and tumorigenicity assays with cells infected with control or DUSP22 ectopic expression vectors were performed as described [34].
Statistical analysis
The statistical significance of differences in DUSP22 expression levels and proliferation, viability, clonogenicity, or tumorigenicity of cells infected with control and DUSP22 expression vectors was evaluated by the Student’s t-test, using the MedCalc software. All graphs represent mean values ± s.e.m. P values are indicated on the figures, P ≤ 0.05 being considered significant.
ACKNOWLEDGMENTS
This study was supported a grant from the Ligue Contre le Cancer (Comités Aquitaine-Charentes), France, and by the TENOMIC-PAIR INca program (Institut National du Cancer, Paris, France). We thank Nathalie Carrère and Séverine Verdon (Service de Biologie des Tumeurs - Tumorothèque et Centre de Ressources Biologiques Cancer, Centre Hospitalier Universitaire de Bordeaux, France), for assistance with normal and pathological samples collection and processing, Jackie Ferrer and Chiara Barili (Université de Bordeaux, France) for technical assistance with DNA and RNA extractions, reverse transcriptions, methylation specific PCR analysis and SNP genotyping, Marshall Kadin for providing the MAC cell lines, Keld Kaltoft for providing the My-La cell line and Laurence Lamant (Inserm UMR1037, Centre de Recherches en Cancérologie de Toulouse, France) for providing the FE-PD cell line.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Scarisbrick JJ, Woolford AJ, Russell-Jones R, Whittaker SJ. Loss of heterozygosity on 10q and microsatellite instability in advanced stages of primary cutaneous T-cell lymphoma and possible association with homozygous deletion of PTEN. Blood. 2000; 95:2937-2942.
2. Vermeer MH, van Doorn R, Dijkman R, Mao X, Whittaker S, van Voorst Vader PC, Gerritsen MJ, Geerts ML, Gellrich S, Söderberg O, Leuchowius KJ, Landegren U, Out-Luiting JJ et al. Novel and highly recurrent chromosomal alterations in Sézary syndrome. Cancer Res. 2008; 68:2689-2698.
3. van Doorn R, van Kester MS, Dijkman R, Vermeer MH, Mulder AA, Szuhai K Knijnenburg J, Boer JM, Willemze R, Tensen CP. Oncogenomic analysis of mycosis fungoides reveals major differences with Sezary syndrome. Blood. 2009; 113:127-136.
4. Laharanne E, Chevret E, Idrissi Y, Gentil C, Longy M, Ferrer J Dubus P, Jouary T, Vergier B, Beylot-Barry M, Merlio JP. CDKN2A-CDKN2B deletion defines an aggressive subset of cutaneous T-cell lymphoma. Mod Pathol. 2010; 23:547-558.
5. Laharanne E, Oumouhou N., Bonnet F, Carlotti M, Gentil C, Chevret E Jouary T, Longy M, Vergier B, Beylot-Barry M, Merlio JP. Genome-wide analysis of cutaneous T-cell lymphomas identifies three clinically relevant classes. J Invest Dermatol. 2010; 130:1707-1718.
6. Cristofoletti C, Picchio MC, Lazzeri C, Tocco V, Pagani E, Bresin A, Mancini B, Passarelli F, Facchiano A, Scala E, Lombardo GA, Cantonetti M, Caprini E et al. Comprehensive analysis of PTEN status in Sezary syndrome. Blood. 2013; 122:3511-3520.
7. McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA Zwerner JP, Hucks D, Dave U, Zhao Z, Eischen CM. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood. 2015; 126:508-519.
8. Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, Bjornson RD, Maman Y, Wang T, Tordoff J, Carlson K, Overton JD, Liu KJ et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015; 47:1011-1019.
9. Chevret E, Prochazkova M, Beylot-Barry M, Merlio JP. A suggested protocol for obtaining high-quality skin metaphases from primary cutaneous T-cell lymphoma. Cancer Genet Cytogenet. 2006; 167:89-91.
10. Feldman AL, Law M, Remstein ED, Macon WR, Erickson LA, Grogg KL, Kurtin PJ, Dogan A. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia. 2009; 23:574-580.
11. Pham-Ledard A, Prochazkova-Carlotti M, Laharanne E, Vergier B, Jouary T, Beylot-Barry M, Merlio JP. IRF4 Gene Rearrangements Define a Subgroup of CD30+ Cutaneous T-Cell Lymphoma: A Study of 54 Cases. J. Invest. Dermatol. 2010; 130:1707-1718.
12. Feldman AL, Dogan A, Smith DI, Law ME, Ansell SM, Johnson SH, Porcher JC, Ozsan N, Wieben ED, Eckloff BW, Vasmatzis G. Discovery of recurrent t(6;7)(p25.3;q32.3) translocations in ALK-negative anaplastic large cell lymphomas by massively parallel genomic sequencing. Blood. 2011; 117:915-919.
13. Karai LJ, Kadin ME, Hsi ED, Sluzevich JC, Ketterling RP, Knudson RA, Feldman AL. Chromosomal rearrangements of 6p25.3 define a new subtype of lymphomatoid papulosis. Am J Surg Pathol. 2013; 37:1173-1181.
14. Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, Sidhu JS, Hsi ED, Karikehalli S, Jiang L, Vasmatzis G, Gibson SE, Ondrejka S et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood. 2014; 124:1473-1480.
15. Iida S, Rao PH, Butler M, Corradini P, Boccadoro M, Klein B, Chaganti RS, Dalla-Favera R. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat Genet. 1997; 17:226-230.
16. Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, Zeng Y, Chen B, Epstein J et al. IRF4 addiction in multiple myeloma. Nature. 2008; 454:226-231.
17. Michaux L, Wlodarska I, Rack K, Stul M, Criel A, Maerevoet M, Marichal S, Demuynck H, Mineur P, Kargar Samani K, Van Hoof A, Ferrant A, Marynen P et al. Translocation t(1;6)(p35.3;p25.2): a new recurrent aberration in “unmutated” B-CLL. Leukemia. 2005; 19:77-82.
18. Pratt G, Fenton JA, Allsup D, Fegan C, Morgan GJ, Jackson G, Sunter NJ, Hall AG, Irving JA, Allan JM. A polymorphism in the 3’ UTR of IRF4 linked to susceptibility and pathogenesis in chronic lymphocytic leukaemia and Hodgkin lymphoma has limited impact in multiple myeloma. Br J Haematol. 2010; 150:371-373.
19. Havelange V, Pekarsky Y, Nakamura T, Palamarchuk A, Alder H, Rassenti L, Kipps T, Croce CM. IRF4 mutations in chronic lymphocytic leukemia. Blood. 2011; 118:2827-2829.
20. Salaverria I, Philipp C, Oschlies I, Kohler CW, Kreuz M, Szczepanowski M, Burkhardt B, Trautmann H, Gesk S, Andrusiewicz M, Berger H, Fey M, Harder L. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood. 2011; 118:139-147.
21. Duffy DL, Iles MM, Glass D, Zhu G, Barrett JH, Höiom V, Zhao ZZ, Sturm RA, Soranzo N, Hammond C, Kvaskoff M, Whiteman DC, Mangino M. IRF4 variants have age-specific effects on nevus count and predispose to melanoma. Am J Hum Genet. 2010; 87:6-16.
22. Han J, Qureshi AA, Nan H, Zhang J, Song Y, Guo Q, Hunter DJ. A germline variant in the interferon regulatory factor 4 gene as a novel skin cancer risk locus. Cancer Res. 2011; 71:1533-1539.
23. Patterson KI, Brummer T, O’Brien PM, Daly RJ. Dual-specificity phosphatases: critical regulators with diverse cellular targets. Biochem J. 2009; 418:475-489.
24. Chen AJ, Zhou G, Juan T, Colicos SM, Cannon JP, Cabriera-Hansen M, Meyer CF, Jurecic R, Copeland NG, Gilbert DJ, Jenkins NA, Fletcher F, Tan TH et al. The dual specificity JKAP specifically activates the c-Jun N-terminal kinase pathway. J Biol Chem. 2002; 277:36592-36601.
25. Alonso A, Merlo JJ, Na S, Kholod N, Jaroszewski L, Kharitonenkov A, Williams S, Godzik A, Posada JD, Mustelin T. Inhibition of T cell antigen receptor signaling by VHR-related MKPX (VHX), a new dual specificity phosphatase related to VH1 related (VHR). J Biol Chem. 2002; 277:5524-5528.
26. Sekine Y, Tsuji S, Ikeda O, Sato N, Aoki N, Aoyama K, Sugiyama K, Matsuda T. Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene. 2006; 25:5801-5806.
27. Sekine Y, Ikeda O, Hayakawa Y, Tsuji S, Imoto S, Aoki N, Sugiyama K, Matsuda T. DUSP22/LMW-DSP2 regulates estrogen receptor-alpha-mediated signaling through dephosphorylation of Ser-118. Oncogene. 2007; 26:6038-49.
28. Li JP, Fu YN, Chen YR, Tan TH. JNK pathway-associated phosphatase dephosphorylates focal adhesion kinase and suppresses cell migration. J Biol Chem. 2010; 285:5472-5478.
29. Li JP, Yang CY, Chuang HC, Lan JL, Chen DY, Chen YM, Wang X, Chen AJ, Belmont JW, Tan TH. The phosphatase JKAP/DUSP22 inhibits T-cell receptor signalling and autoimmunity by inactivating Lck. Nat Commun. 2014; 5:3618.
30. Ting AH, McGarvey KM, Baylin SB. The cancer epigenome--components and functional correlates. Genes Dev. 2006; 20:3215-3231.
31. Genovese G, Handsaker RE, Li H, Altemose N, Lindgren AM, Chambert K, Pasaniuc B, Price AL, Reich D, Morton CC, Pollak MR, Wilson JG, McCarroll SA. Using population admixture to help complete maps of the human genome. Nat Genet. 2013; 45:406-414.
32. Genovese G, Handsaker RE, Li H, Kenny EE, McCarroll SA. Mapping the human reference genome’s missing sequence by three-way admixture in Latino genomes. Am J Hum Genet. 2013; 93:411-421.
33. Kadin ME, Cavaille-Coll MW, Gertz R, Massagué J, Cheifetz S, George D. Loss of receptors for transforming growth factor beta in human T-cell malignancies. Proc Natl Acad Sci U S A. 1994; 91:6002-6006.
34. Chevret E, Andrique L, Prochazkova-Carlotti M, Ferrer J, Cappellen D, Laharanne E Idrissi Y, Boettiger A, Sahraoui W, Ruiz F, Pham-Ledard A, Vergier B, Belloc F et al. Telomerase functions beyond telomere maintenance in primary cutaneous T-cell lymphoma. Blood. 2014; 123:1850-1859.
35. Kaltoft K, Bisballe S, Dyrberg T, Boel E, Rasmussen PB, Thestrup-Pedersen K. Establishment of two continuous T-cell strains from a single plaque of a patient with mycosis fungoides. In Vitro Cell Dev Biol. 1992; 28A:161-167
36. del Mistro A, Leszl A, Bertorelle R, Calabro ML, Panozzo M, Menin C, D’Andrea E, Chieco-Bianchi L. A CD30-positive T cell line established from an aggressive anaplastic large cell lymphoma, originally diagnosed as Hodgkin’s disease. Leukemia. 1994; 8:1214-1219.
37. Dirks WG, Zaborski M, Jäger K, Challier C, Shiota M, Quentmeier H, Drexler HG. The (2;5)(p23;q35) translocation in cell lines derived from malignant lymphomas: absence of t(2;5) in Hodgkin-analogous cell lines. Leukemia. 1996 ; 10:142-149.
38. Bunn PA Jr, Foss FM. T-cell lymphoma cell lines (HUT102 and HUT78) established at the National Cancer Institute: history and importance to understanding the biology, clinical features, and therapy of cutaneous T-cell lymphomas (CTCL) and adult T-cell leukemia-lymphomas (ATLL). J Cell Biochem Suppl. 1996; 24:12-23.
39. Starkebaum G, Loughran TP Jr, Waters CA, Ruscetti FW. Establishment of an IL-2 independent, human T-cell line possessing only the p70 IL-2 receptor. Int J Cancer. 1991; 49:246-253.
40. Csikesz CR, Knudson RA, Greipp PT, Feldman AL, Kadin M. Primary cutaneous CD30-positive T-cell lymphoproliferative disorders with biallelic rearrangements of DUSP22. J Invest Dermatol. 2013; 133:1680-1682.
41. Ouyang B, Baxter CS, Lam HM, Yeramaneni S, Levin L, Haynes E, Ho SM. Hypomethylation of dual specificity phosphatase 22 promoter correlates with duration of service in firefighters and is inducible by low-dose benzo[a]pyrene. J Occup Environ Med. 2012; 54:774-780.
42. Sanchez-Mut JV, Aso E, Heyn H, Matsuda T, Bock C, Ferrer I, Esteller M. Promoter hypermethylation of the phosphatase DUSP22 mediates PKA-dependent TAU phosphorylation and CREB activation in Alzheimer’s disease. Hippocampus. 2014; 24:363-368.
43. Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, & Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell, 2013; 155: 948-962
44. Fauconneau A, Pham-Ledard A, Cappellen D, Frison E, Prochazkova-Carlotti M, Parrens M, Dalle S, Joly P, Viraben R, Franck F, Ingen-Housz-Oro S, Giacchero D, Jullié ML et al. Assessment of diagnostic criteria between primary cutaneous anaplastic large-cell lymphoma and CD30-rich transformed mycosis fungoides; a study of 66 cases. Br J Dermatol. 2015; 172:1547-1554.
45. Hapgood G, Savage KJ. The biology and management of systemic anaplastic large cell lymphoma. Blood. 2015; 126:17-25.
46. Crescenzo R, Abate F, Lasorsa E, Tabbo’ F, Gaudiano M, Chiesa N, Di Giacomo F, Spaccarotella E, Barbarossa L, Ercole E, Todaro M, Boi M, Acquaviva A et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell. 2015; 27:516-532.
47. Merkel O, Hamacher F, Griessl R, Grabner L, Schiefer A, Prutsch N, Baer C, Egger G, Schlederer M, Krenn PW, Hartmann TN, Simonitsch-Klupp I, Plass C. Oncogenic role of miR-155 in anaplastic large cell lymphoma lacking the t(2;5) translocation. J Pathol. 2015; 236:445-456.
48. Foulkes WD, Ragoussis J, Stamp GW, Allan GJ, Trowsdale J. Frequent loss of heterozygosity on chromosome 6 in human ovarian carcinoma. Br J Cancer. 1993; 67:551-559.
49. Honchel R, McDonnell S, Schaid DJ, Thibodeau SN. Tumor necrosis factor-alpha allelic frequency and chromosome 6 allelic imbalance in patients with colorectal cancer. Cancer Res. 1996; 56:145-149.
50. Chatterjee A, Pulido HA, Koul S, Beleño N, Perilla A, Posso H, Manusukhani M, Murty VV. Mapping the sites of putative tumor suppressor genes at 6p25 and 6p21.3 in cervical carcinoma: occurrence of allelic deletions in precancerous lesions. Cancer Res. 2001; 61:2119-2123.
51. Dumur CI, Dechsukhum C, Ware JL, Cofield SS, Best AM, Wilkinson DS, Garrett CT, Ferreira-Gonzalez A. Genome-wide detection of LOH in prostate cancer using human SNP microarray technology. Genomics. 2003; 81:260-269.
52. McEvoy CR, Morley AA, Firgaira FA. Evidence for whole chromosome 6 loss and duplication of the remaining chromosome in acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2003; 37:321-325.
53. Hurst CD, Fiegler H, Carr P, Williams S, Carter NP, Knowles MA. High-resolution analysis of genomic copy number alterations in bladder cancer by microarray-based comparative genomic hybridization. Oncogene. 2004; 23:2250-2263.
54. Aldaz CM, Chen T, Sahin A, Cunningham J, Bondy M. Comparative allelotype of in situ and invasive human breast cancer: high frequency of microsatellite instability in lobular breast carcinomas. Cancer Res. 1995; 55:3976-3981.
55. Simon P. Q-Gene: processing quantitative real-time RT-PCR data. Bioinformatics. 2003; 19: 1439-1440.
56. Lo YM, Wong IH, Zhang J, Tein MS, Ng MH, Hjelm NM. Quantitative analysis of aberrant p16 methylation using real-time quantitative methylation-specific polymerase chain reaction. Cancer Res. 1999; 59:3899-3903.
57. Caumont C, Gros A, Boucher C, Mélard P, Prochazkova-Carlotti M, Laharanne E, Pham-Ledard A, Vergier B, Chevret E, Beylot-Barry M, Merlio JP, Cappellen D. PLCG1 Gene Mutations Are Uncommon in Cutaneous T-Cell Lymphomas. J Invest Dermatol. 2015; 135:2334-2337.
58. Robert-Richard E, Moreau-Gaudry F, Lalanne M, Lamrissi-Garcia I, Cario-André M, Guyonnet-Dupérat V, Taine L, Ged C, de Verneuil H. Effective gene therapy of mice with congenital erythropoietic porphyria is facilitated by a survival advantage of corrected erythroid cells. Am J Hum Genet. 2008; 82:113-124.