
INTRODUCTION
Thyroid cancer is the most common endocrine malignancy worldwide. Thyroid cancers consist of papillary (75–85%), follicular (10–20%), medullary (~5%) and anaplastic carcinomas (< 5%) [1, 2]. Follicular thyroid carcinoma (FTC) has relatively worse clinical courses than papillary thyroid carcinoma (PTC) with frequent hematogenous spread and recurrence [1]. Ten to 15% of patients with FTC develop metastatic diseases, mostly involving lung, bone and liver by hematogenous spread [3]. Approximately 11–39% of patients with FTC develop recurrence of the cancers [4, 5]. Among the four types of thyroid cancers, only FTC has a benign counterpart (follicular thyroid adenoma, FTA). FTA and FTC fall within a biologic continuum and capsular invasion and/or vascular invasion status are gold standards for distinction between FTA and FTC [1, 6]. In this scenario, FTA originated from the thyroid follicle penetrates the tumor capsule and eventually progresses to FTC. Like other tumors, FTC is considered to arise from a single clone as a result of accumulation of mutations in driver genes and subsequent clonal selection of the mutant progeny with increasingly aggressive behaviors [7]. Thus, genomic comparison of FTA and FTC genomes may provide new insights regarding genetic origins of FTC and also potential genetic determinants of the progression from FTA to FTC.
In FTC, through gene-to-gene analysis, point mutations in RAS (KRAS, HRAS and NRAS) and PIK3CA, and paired box gene 8/peroxisome proliferator-activated receptor gamma (PAX8-PPARγ) gene fusion have been well-documented [2]. These alterations are also found in FTA, but less common than FTC [3], further suggesting that FTA is a genetic precursor of FTC. For example, 30% to 40% of FTCs harbor RAS mutations, while approximately 20% of FTAs harbor them [8]. Also, PAX8-PPARγ fusion transcript is found in 4% to 13% of FTA, but 30% to 60% of FTC [9]. These genetic alterations are surely drivers for follicular tumor development, but previous genome analyses in other cancers [10, 11] suggest there should be more mutations in the FTA and FTC besides them. For a comprehensive elucidation of genetic alterations in cancers, whole-exome (WES), whole-genome or whole-transcriptome sequencing (WTS) analyses would be ideal. Recently, large-scale genome analyses for PTC and anaplastic thyroid carcinoma using next-generation sequencing have been reported [12, 13]. However, to date, there have been no such genomic studies on FTC.
In this study, to further characterize FTA and FTC genomes and extend the knowledge on genetic progression from FTA to FTC, the following questions were investigated: (i) whether FTA and FTC genomes have differences in their somatic mutation, copy numbers and gene fusion profiles and (ii) whether there are any recurrent genetic alterations that may drive FTA progression to FTC.
RESULTS
Whole-exome sequencing of FTA and FTC genomes
To explore genomic profiles of FTA and FTC, a total of 27 thyroid follicular tumor genomes (14 FTAs and 13 FTCs) were analyzed in this study (Table 1). Coverage of depth was median of 82X (63–177X) for tumor samples and 74X (56–196X) for matched normal samples (Table S1). Using the MuTect [14] and the SomaticIndelDetector [15], we identified 8–40 point mutations and indels per sample (median of 20 somatic variants) (Figure 1A, Table S2). Two FTA patients were found to have previous history of other tumors (multiple myeloma in FTA03 and breast cancer in FTA04) (Table 1). Their mutation profiles, however, were not significantly different from other FTAs including number and sequence composition of mutations (P > 0.05). In the germline data of these two patients, we were not able to find any known cancer predisposition mutations. Somatic mutation density (average of 0.3 nonsynonymous mutation per Mb) of the FTCs was not significantly different with that of PTCs (P = 0.244) [12], but much lower than lung cancer (P < 0.001) or colon cancer (P < 0.001) [10, 16]. No significant difference in the numbers of nonsynonymous mutations was observed between FTA (7–25; median of 12 mutations) and FTC (6–30; median of 16 mutations) genomes (P = 0.675) (Figure 1B, Table 2). In addition, there was no significant difference between conventional and Hürthle tumor genomes (P = 0.327), between FTA and minimally invasive FTC genomes (P = 0.544), and between minimally and widely invasive FTC genomes (P = 0.692). Regarding the mutation spectra, no significant difference was observed between FTA and FTC genomes, either (Figure 1C). There was no correlation of the genomic features with other clinicopathologic features (Table S3).
Table 1: Clinocopathologic features of the patients and tumors
Case |
Age/sex |
Diagnosis |
subtype |
Size (diameter) |
TNM |
Extent of carcinoma |
Other cancers |
|---|---|---|---|---|---|---|---|
FTA01 |
48/Female |
Follicular adenoma |
Hürthle cell |
2.0 cm |
N/A |
N/A |
None |
FTA02 |
35/Female |
Follicular adenoma |
Conventional |
1.3 cm |
N/A |
N/A |
None |
FTA03 |
57/Male |
Follicular adenoma |
Hürthle cell |
1.6 cm |
N/A |
N/A |
Multiple myeloma |
FTA04 |
71/Female |
Follicular adenoma |
Hürthle cell |
5.4 cm |
N/A |
N/A |
Breast cancer |
FTA05 |
70/Female |
Follicular adenoma |
Conventional |
3.8 cm |
N/A |
N/A |
None |
FTA06 |
47/Female |
Follicular adenoma |
Conventional |
2.5 cm |
N/A |
N/A |
None |
FTA07 |
56/Male |
Follicular adenoma |
Hürthle cell |
1.0 cm |
N/A |
N/A |
None |
FTA08 |
47/Female |
Follicular adenoma |
Hürthle cell |
1.6 cm |
N/A |
N/A |
None |
FTA09 |
27/Female |
Follicular adenoma |
Conventional |
N/A |
N/A |
N/A |
None |
FTA10 |
58/Female |
Follicular adenoma |
Conventional |
1.8 cm |
N/A |
N/A |
None |
FTA11 |
59/Female |
Follicular adenoma |
Hürthle cell |
1.2 cm |
N/A |
N/A |
None |
FTA12 |
65/Male |
Follicular adenoma |
Hürthle cell |
0.4 cm |
N/A |
N/A |
None |
FTA13 |
51/Female |
Follicular adenoma |
Conventional |
3.8 cm |
N/A |
N/A |
None |
FTA14 |
61/Female |
Follicular adenoma |
Hürthle cell |
5.0 cm |
N/A |
N/A |
None |
FTC01 |
35/Female |
Follicular carcinoma |
Hürthle cell |
2.2 cm |
T2N1M0 |
Widely invasive |
None |
FTC02 |
60/Female |
Follicular carcinoma |
Hürthle cell |
7.0 cm |
T3N0M0 |
Minimally invasive |
None |
FTC03 |
29/Female |
Follicular carcinoma |
Conventional |
6.0 cm |
T3N0M0 |
Minimally invasive |
None |
FTC04 |
25/Female |
Follicular carcinoma |
Conventional |
2.8 cm |
T2N0M0 |
Minimally invasive |
None |
FTC05 |
39/Female |
Follicular carcinoma |
Conventional |
3.2 cm |
T2N0M0 |
Minimally invasive |
None |
FTC06 |
44/Female |
Follicular carcinoma |
Hürthle cell |
4.5 cm |
T3N0M0 |
Minimally invasive |
None |
FTC07 |
64/Male |
Follicular carcinoma |
Conventional |
3.4 cm |
T2N0M0 |
Minimally invasive |
None |
FTC08 |
34/Female |
Follicular carcinoma |
Conventional |
1.9 cm |
T1N0M0 |
Minimally invasive |
None |
FTC09 |
70/Male |
Follicular carcinoma |
Conventional |
8.0 cm |
T3N0M0 |
Minimally invasive |
None |
FTC10 |
73/Female |
Follicular carcinoma |
Conventional |
N/A |
N/A |
Minimally invasive |
None |
FTC11 |
74/Male |
Follicular carcinoma |
Conventional |
4.3 cm |
T3N0M0 |
Minimally invasive |
None |
FTC12 |
59/Male |
Follicular carcinoma |
Hürthle cell |
1.8 cm |
T1N0M0 |
Widely invasive |
None |
FTC13 |
55/Female |
Follicular carcinoma |
Conventional |
5.2 cm |
T3N0M0 |
Minimally invasive |
None |
FTA: follicular thyroid adenoma, FTC: follicular thyroid carcinoma, N/A: not available.
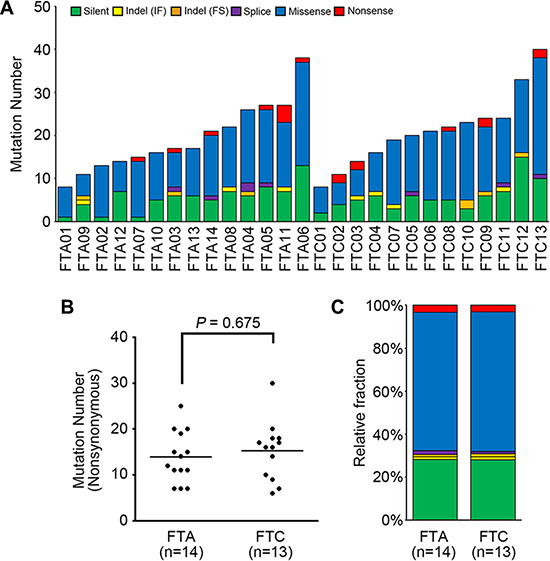
Figure 1: The mutational features of 27 thyroid follicular tumor genomes. (A) The numbers of somatic mutations are shown for six functional categories. (B) Frequencies of nonsynonimous mutations in FTA (follicular thyroid adenoma) and FTC (follicular thyroid carcinoma) genomes. Horizontal black bars represent the mean values. No significant difference in the numbers of nonsynonymous mutations was observed between FTA (7–25; median of 12 mutations) and FTC (6–30; median of 16 mutations) genomes (P = 0.675). (C) Relative fractions of the mutations for FTA and FTC genomes are shown for the six functional categories. IF: in-frame, FS: frameshift.
Table 2: Summary of comparison data between FTA and FTC genomes
FTA vs. FTC |
|
|---|---|
Somatic mutation number |
No significant difference |
Mutation allele frequency |
FTC > FTA (P < 0.001) |
Inferred evolutionary age |
No significant difference |
Driver mutation number |
No significant difference |
Number of CNA |
FTC > FTA (P = 0.004) |
FTA: follicular thyroid adenoma, FTC: follicular thyroid carcinoma, CNA: copy number alteration.
As for variant allele frequencies (VAFs) of the point mutations, mutations of the FTC had significantly higher allele frequencies than those of the FTA (mean VAF 0.25 in FTCs and mean VAF 0.20 in FTAs, P < 0.001). Based on these VAFs, we inferred evolutionary ages of the 27 follicular thyroid tumor genomes. We adopted an evolutionary model that used somatic mutations as molecular clocks. In this model, the relative timing between the birth of a founder cell and the emergence of the last common ancestor before the last cycle of clonal amplification was estimated. The numbers of clonal mutations in the FTA genomes were 5 to 27 that gave conservative estimates of evolutionary ages of 200 to 1080 cell cycles. The FTC genomes showed 5 to 23 clonal mutations corresponding to 200 to 920 cell cycles of evolutionary ages. The evolutionary ages estimated from the FTC genomes were not significantly different with those estimated from the FTA genomes (P = 0.085, Table 2). Details of the estimated evolutionary ages are available in Figure S1.
Copy number alterations and their distributions in FTA and FTC genomes
We next analyzed CNAs for the same 27 thyroid follicular tumor genomes with their matched normal genomes as references by array-CGH. A total of 37 CNAs were identified in the 13 samples (Table S4). The FTC genomes harbored significantly higher numbers of CNAs than those of the FTA genomes (mean of 0.4 (range, 0–2) for FTAs and 2.5 (range, 0–11) for FTCs, P = 0.004) (Figure 2A, Table 2).
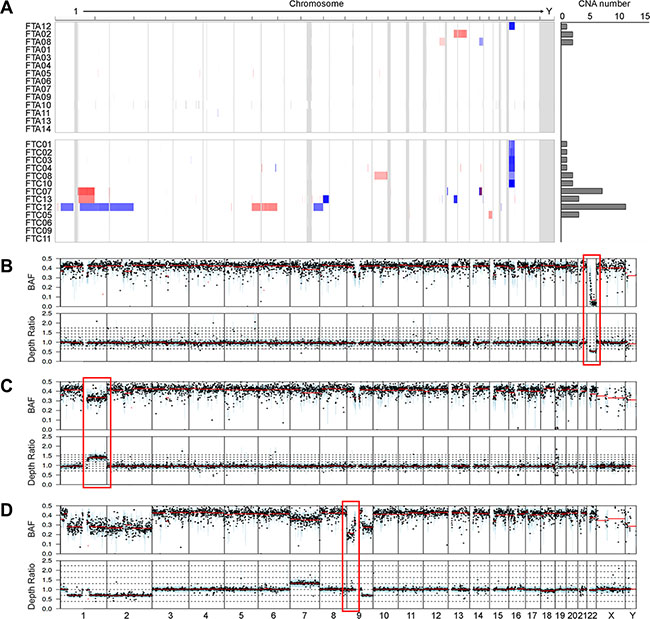
Figure 2: Copy number profiles and copy-neutral loss of heterozygosity (LOH). (A) Heatmap shows the chromosomal copy gains (red) and lesses (blue) in each sample; rows represent samples classified into FTAs (follicular thyroid adenomas) and FTCs (follicular thyroid carcinomas). Boundaries of individual chromosomes are indicated by vertical bars. On the right of heatmap, the numbers of copy number alterations (CNAs) are shown for each sample. (B) An example of 22q deletion in the case FTC02. The red box represents the copy number loss on chromosome 22, where NF2 gene is located. (C) An example of 1q gain in the case FTC07. The red box represents the copy number gain on chromosome 1, where NTRK1 gene is located. (D) The red box represents the copy-neutral LOH on chromosome 9 in the case FTC12. X-axis represnts chromosomes. BAF, B-allele frequency; Depth ratio is scaled on log2.
Of the 37 CNAs detected, two CNA regions (gain on 1q and loss on 22q) were identified recurrently (> 2 cases) (Figure 2A). The most recurrent CNA was 22q11.1-q13.33 deletion, a 35 Mb-sized region that encompasses NF2, EP300, MKL1 and CHEK2 genes. Six of 13 FTCs (46%) harbored the 22q11.1-q13.33 deletion, while one of 14 FTAs (7%) harbored this deletion (P = 0.033) (Figure 2A). Recurrent copy number losses in chromosome 22 have been reported not only in FTCs [17, 18] but also in other human neoplasms, such as meningiomas and mesotheliomas [19]. When we examined the B allele profiles using WES data, all of 22q deletions were also shown (Figure 2B). Recurrent copy gains on 1q, where NTRK1, PBX1 and ABL2 oncogenes are located, were detected in the two FTCs (15% of FTCs) by both array-CGH and B allele analysis (Figure 2A and 2C), but none in FTAs. Recurrent copy number gains in chromosome 1q have been reported in FTCs but not in FTAs [20]. In addition to CNAs, we found that one FTC (case FTC12) harbored copy-neutral loss of heterozygosity (LOH) event on 9p24.3-p13.1 (Figure 2D), which had been reported in FTC [21].
Driver mutations and pathways of thyroid follicular tumor genomes
A total of 17 candidate driver genes with nonsilent mutations were identified based on the COSMIC (NRAS, EZH1, HRAS, BRAF, EIF1AX, KDM5C, BRIP1, SPOP, TSHR and KMT2C), recurrence (NRAS, n = 6; EZH1, n = 6; TG, n = 3; IGF2BP3, n = 3, HRAS, n = 2; KMT2C, n = 2; EIF1AX, n = 2) and the CHASM (NRAS, HRAS, BRAF, NF1, SPOP, TSHR, TCF12, CNOT1, BRIP1, KDM5C, STAG2 and MAP4K3). In the CHASM analysis, mutations that significantly predicted as driver (FDR < 0.2) and overlapped either with the cancer Gene Census or the Cancer Driver Database were considered potential driver mutations. TG gene encodes thyroglobulin that is produced predominantly in the thyroid gland and plays essential roles for synthesis and storage of thyroid hormones [22]. Somatic TG mutations have been reported in autonomous thyroid adenoma and PTC [23, 24] and all TG mutations identified in this study were validated as somatic using Sanger sequencing (Figure S2). Functionally, however, it remains uncertain whether TG mutation is causally implicated in thyroid tumorigenesis. Also, TG is one of the largest genes in human genome, many of which can be easy targets for somatic mutation as in the case of TTN gene [25], suggesting that TG mutations are likely to be passengers. NRAS, HRAS, EIF1AX and KMT2C mutations were detected in both FTA and FTC genomes. BRAF, BRIP1, TCF12, CNOT1, STAG2, MAP4K3 and IGF2BP3 mutations were observed only in FTC while EZH1, TSHR, SPOP, KDM5C and NF1 mutations were detected only in FTA. Overall, 16 genes with 31 nonsilent mutations were identified as potential driver mutations; NRAS, HRAS, EIF1AX, KDM2C, EZH1, TSHR, SPOP, KDM5C, NF1, BRAF, BRIP1, TCF12, CNOT1, STAG2, MAP4K3 and IGF2BP3 (Figure 3A, Table S2). Twelve of 14 FTAs and 10 of 13 FTCs harbored one or more potential driver mutations (Figure 3A). There was no statistical difference in the number of potential driver genes between FTAs and FTCs (P = 0.822, Table 2).
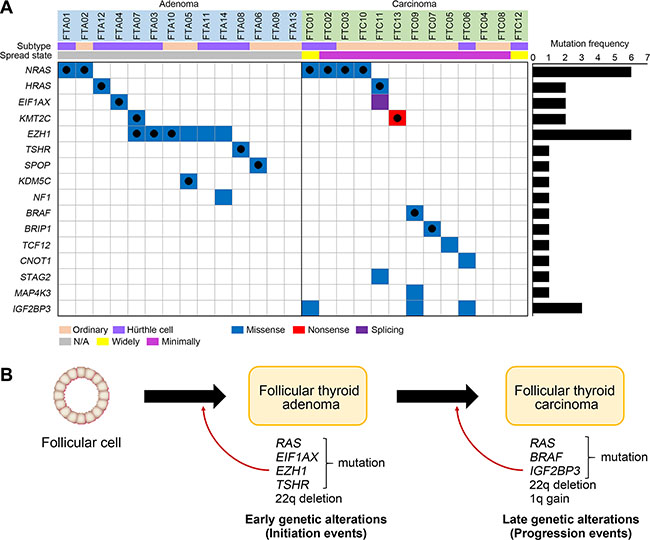
Figure 3: Driver mutations and pathway analyses. (A) 16 genes with 31 nonsilent mutations are shown. On the right, the numbers of non-silent mutations are shown for each gene. Black filled circles represent the reported variants in the COSMIC database. (B) Schematic representation of early and late genetic alterations in thyroid follicular tumor. Development of FTA requires early genetic alteration (initiation events) such as RAS and EZH1 mutations. Additional genetic alterations (progression events) such as RAS and BRAF mutations and copy loss on chromosome 22q may contribute to progression of FTA to FTC.
Next, to investigate pathway-level relationship of the individual mutations, we performed the DAVID analysis (http://david.abcc.ncifcrf.gov) and found that mutated genes in the FTCs were significantly associated with tumorigenesis-related gene functions, including ‘cell adhesion’ (P = 0.004), ‘MAPKinase signaling’ (P = 0.026) and ‘Thyroid cancer pathway (P = 0.048). According to the DAVID analysis, only one cancer-relate functional gene set (‘Ras protein signal transduction’, P = 0.011) was significantly enriched in the FTA genomes. Further details of the DAVID analysis are available in Table S5.
Gene fusions
We explored the fusion transcripts for 10 thyroid follicular tumors (nine FTCs and one FTA) available for WTS. A total of three fusion transcripts (MAST3-COL5A3, FAM168A-RAB6A and UPF3A-CDC16) were identified from the 10 tumors (Table S6). The MAST3-COL5A3 identified in one FTC (FTC07) is a novel fusion never reported in any database. FTC07 showed complex recombination events on chromosome 19p, where MAST3 and COL5A3 resides, and both genes are located at the breakpoints of the recombination, suggesting that recombination on chromosome 19p might contribute this fusion event (Figure S3). The other two fusion events (FAM168A-RAB6A and UPF3A-CDC16) have been reported in solid cancers (FAM168A-RAB6A in lung cancer and UPF3A-CDC16 in thyroid cancer), both of which have the same junction breakpoints as the previous reports [26]. All three fusions were validated by reverse transcription-polymerase chain reaction (RT-PCR) followed by Sanger sequencing (Figure S4). The PAX8-PPARγ, the most common fusion transcript in FTC, was not detected in our samples.
DISCUSSION
As tumors progress, they in general become aggressive at morphologic, genetic and clinicopathologic levels. However, it remains unclear whether the clinocopathologic manifestations accompany genetic changes. Clinically, distinction between FTA and FTC is critical for proper selection of therapeutic modalities. With respect to the spatial progression of FTA to FTC, FTA is likely to precede FTC by definition. However, whether FTA progression to FTC is accompanied by genomic alterations remains unknown. To our knowledge, genome-wide mutational landscapes of neither FTA nor FTC are reported. In this study, we attempted to find whether FTA and FTC have differences in their somatic mutations and CNAs. We found that not only the quantity but also the quality of the somatic mutations was not significantly different between FTA and FTC. Only CNA numbers were significantly higher in FTC than that in FTA. Of note, FTC genomes harbored significantly higher numbers of the 22q11.1-q13.33 deletion than those of FTA genomes, suggesting this deletion would be FTC-specific.
Genomic studies on thyroid cancers have focused on PTC, the most common thyroid cancer, and to date those on FTC, the second most common, is lacking. We found that FTC genomes had similar amount of genetic alterations to the PTC genome [12] but harbored lower frequencies of somatic mutations and CNAs than other solid cancers such as lung and colon cancers that showed mutation densities around 10 per megabase [10, 16]. The earlier TCGA report on PTC and thyroid medullary carcinoma [27] genomes also showed lower frequencies of genetic alterations than those in other solid cancers [26, 28]. Only anaplastic carcinoma of thyroid, the most aggressive type, exhibited a high density of somatic mutations (~2.5 per Mb) [13]. These data suggest that thyroid cancers except for anaplastic carcinoma may need lower number of mutational events for the development than other solid cancers.
Among the 16 potential driver genes identified in the present study, RAS is the most recurrent gene with NRAS (n = 6) and HRAS (n = 2) identified in p.Q61K/R, the hotspot sites. The reported RAS mutations lies 18% to 30% in FTA and 24% to 57% in FTC [29, 30], which are in agreement with our data (21% in FTA and 38% in FTC). EZH1 mutation was the second most recurrent mutation (n = 6), which had been described in 27% of FTAs [24]. Of note, all EZH1 mutations identified in our study were found in the FTAs (43% of FTAs) along with two mutational hotspots (p.Y642F (n = 3) and p.Q571R (n = 3)), but none in FTCs. The EZH1 p.Q571R mutation was the same variant identified in the previous study and it caused increased histone H3 trimethylation and increased proliferation of thyroid cells [24]. The other mutation p.Y642F was reported in PTC (the COSMIC database). Taken together, missense mutations of EZH1 may play a genomic feature of FTA.
Somatic mutations of NRAS, HRAS, BRAF, TSHR and EIF1AX have been reported in follicular tumors while those of EZH1, SPOP, NF1, TCF12, IGF2BP3, KMT2C, CNOT1, BRIP1, KDM5C, STAG2 and MAP4K3 have not been reported in FTC (the COSMIC database). EZH1, SPOP, NF1, CNOT1, BRIP1, KMT2C and STAG2 missense mutations have been described in PTC as well [12, 31]. By contrast, somatic mutations of TCF12, KDM5C, IGF2BP3 and MAP4K3 have not been reported in any types of thyroid cancers. At variant level, somatic mutations of KDM5C (p.L756I) have been identified in other cancers including lung and uterus cancers (the COSMIC database). Collectively, we confirmed that NRAS, HRAS, EIF1AX, EZH1, TSHR and BRAF mutations previously identified in follicular tumor in our FTC or FTA. Of these, EZH1, TSHR and EIF1AX mutations were exclusively detected in FTAs, while BRAF mutation was exclusively detected in FTCs. These data indicate that EZH1, TSHR, EIF1AX and BRAF mutations may play roles in the initial development of follicular tumors and progression of follicular tumor, respectively (Figure 3B). RAS mutation was detected in both FTAs and FTCs, but more common in FTCs, suggesting that it may play a role in both development and progression of follicular tumors. TCF12, KDM5C, IGF2BP3 and MAP4K3 mutations newly identified in our study might possibly be follicular tumor-specific thyroid mutations. SPOP, NF1, BRIP, CNOT1, KMT2C and STAG2 mutations previously identified in PTC might be involved in thyroid tumorigenesis in a type-nonspecific manner. However, since these mutations in six genes are singleton mutations, further studies in a larger cohort are necessary to confirm our hypothesis.
Until now, there exist few mutation data on preneoplastic conditions or early cancers at whole-genome or whole-exome level. Of note, in breast (ductal carcinoma in situ vs. invasive ductal carcinoma) [32] and gastric (early vs. advanced gastric cancers) cancers [33], even the early primary tumors (ductal carcinoma in situ and early gastric cancer) are already matured in terms of the quantity and quality of the mutations. Also, microsatellite-unstable colon adenomas are known to be nearly as old as invasive colon cancers [34]. By contrast, uterine cervix and prostate cancers showed far different genetic progression pattern. For examples, carcinoma in situ of uterine cervix and high-grade prostate intraepithelial tumor exhibit significantly lower numbers of mutation than their corresponding invasive cancers [35, 36]. Our mutation-based evolutionary analyses revealed that the time intervals required from the initiation of a tumor to the emergence of the last common ancestor were similar between FTA and FTC genomes. The evolutionary analysis data also support our WES data that showed no significant differences between FTA and FTC genomes. Together, like breast and gastric cancers, these data indicate that FTA genomes may be as old as FTC genomes, thus suggesting that follicular thyroid tumor genomes during the transition from FTA to FTC may stand stable at genomic levels in contrast to the discernable changes at pathologic and clinical levels [6, 37]. However, because we included only two cases of widely invasive FTC, it might be better to say that the genomic difference between FTA and minimally invasive FTC might be insignificant, supporting the findings that minimally invasive FTCs can have very few pathologic differences from FTAs and act very benignly with different treatment recommendations when compared to widely invasive FTC’s [38]. As for the Hürthle cell FTCs, they have some differences in biological behavior and molecular mechanisms as compared to the conventional FTC [38]. However, our study suggests that such differences are not evident in mutational profiles at least of FTA and minimally invasive FTCs.
The CNA profiles in the present study were largely consistent with those in the earlier studies [18, 39]. Unlike mutations, the FTC genomes harbored significantly higher numbers of CNAs than those of the FTA genomes. Of note, copy number loss on 22q (46% of FTCs) and copy gain on 1q (15% of FTCs) were detected recurrently in FTCs, while only one FTA (FTA12) harbored copy number loss on 22q. Earlier study showed that 22q loss and 1q gain were detected in FTAs albeit less common than FTCs [17], supporting our findings. Together, our data and the previous data indicate that 22q loss and 1q gain play roles in both development and progression of follicular tumors. In the WES data, two FTAs (case FTA09 and 13) and three FTCs (cases FTC04, 08 and 12) did not harbor any driver mutations. FTC04 and 08 harbored 22q loss and FTC12 harbored the greatest number of CNAs (Figure 2A), suggesting a possibility that CNAs might precede somatic mutations in FTCs. The FTA without any driver mutation or CNA but with several non-driver mutations could be a naïve tumor that is too young to harbor clonal driver mutations.
In the present study, we couldn’t find any PAX8-PPARγ fusion transcript by either WTS or RT-PCR. PAX8 gene is essential for the genesis of the thyroid follicular cell lineage [40], and PAX8-PPARγ fusion transcript is frequently detected in FTC [9, 41]. However, frequency of PAX8-PPARγ fusion transcript is very low (0%~4%) in Asia [42–44], which is in agreement with our data. Rather than this, we detected three fusion transcripts (Table S6). However, all the three fusion (MAST3-COL5A3, FAM168A-RAB6A and UPF3A-CDC16 fusions) were predicted to be out-of-frame and actually did not retain any intact open reading frame. Moreover, cancer-related functions in any of the 6 fusion partner genes have not been known. Our observations suggest that gene fusion transcripts besides PAX8-PPARγ may not play a major and recurrent role in FTC development.
In summary, our data for the first time analyzed genomic profiles of thyroid follicular tumors and found previously unreported eleven somatic mutations in FTAs or FTCs. Our data suggest that the time course from FTA to FTC progression at genomic levels may be different from that at pathologic levels (capsular invasion and/or vascular invasion). It is theoretically possible that the uncoupling between genomic and pathologic findings could arise from non-genetic factors such as tissue microenvironment. Finally, the early genomic maturation or aging of FTA might emphasize strategies for the genomic diagnosis of follicular thyroid tumors.
MATERIALS AND METHODS
Thyroid follicular adenoma and carcinoma tissues
Tumor and matched normal thyroid tissues obtained by surgeries from 14 patients with FTA (six conventional and eight Hürthle cell variants) and 13 patients with FTC (nine conventional and four Hürthle cell variants) were obtained from the Tissue Banks of Seoul St. Mary Hospital of Catholic University (Seoul, Korea), Guro Hospital of Korea University (Seoul, Korea), Pusan National University Hospital (Pusan, Korea) and Keimyung University Dongsan Hospital (Daegu, Korea). All 27 patients were Korean, and none had evidence for hereditary thyroid tumors or other cancers except two cases (multiple myeloma in FTA03 and breast cancer in FTA04). Approval for this study was obtained from the institutional review board at the Catholic University of Korea, College of Medicine. Clinicopathologic features and histology of the patients are summarized in Table 1 and Figure S5, respectively. Initially, frozen tissues from the tissue banks were cut, stained with the hematoxylin/eosin and examined under microscope by a pathologist, who identified areas rich in FTA cells or FTC cells in the frozen tissues. In order to have matched normal tissues from each patient, we used thyroid tissues that were confirmed to be free of tumor cells by examination under the microscope. The purities of FTA and FTC cells were approximately 90% and 90%, respectively. For genomic DNA and RNA extraction from the frozen tissues, we used the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) and mirVana miRNA Isolation Kit (Invitrogen, Carlsbad, CA, USA), respectively.
Whole-exome sequencing
WES was performed for the genomic DNA obtained from tumors and matched normal tissues using the Agilent SureSelect Human All Exome 50 Mb kit (Agilent Technologies, Santa Clara, CA) and Illumina HiSeq2000 platform according to the manufacturer’s instructions. Acquisition and processing of the sequencing data were performed as previously described [33]. A Burrows-Wheeler aligner was used to align the sequencing reads onto the human reference genome (UCSC hg19). The aligned sequencing reads were evaluated using Qualimap [45], and the sequences were deposited in the SRA database (Project ID: PRJNA320003).
Identification of somatic variants and driver mutation
Somatic variants were identified using MuTect [14] and SomaticIndelDetector [15] for point mutations and indels, respectively. The ANNOVAR package [46] was used to select somatic variants located in the exonic sequences and to predict their functional consequences. To obtain reliable and robust mutation calling, the following somatic variants were eliminated: (i) read depth fewer than 20 in either tumor or matched normal; and (ii) polymorphisms referenced in either 1000 Genomes Project or Exome Aggregation Consortium or Exome Sequencing Project with a minor allele frequency greater than 2% in Asian. Among the filtered variants, mutations overlapped with COSMIC database were manually rescued. We used the CHASM analysis with ‘thyroid’ option for the cancer tissue type in order to identify putative cancer-related mutations [47]. In addition, we looked up the cancer Gene Census [48] and Cancer Drivers Database [49]. Mutations that significantly predicted in the CHASM analysis (FDR < 0.2) and overlapped with the cancer Gene Census (http://cancer.sanger.ac.uk/census) or Cancer Driver Database (https://www.intogen.org/) were considered as potential driver mutations.
DNA copy number and LOH analysis
DNA copy number profiling was performed using Agilent Sure Print G3 Human comparative genomic hybridization (CGH) Microarray 180K (Agilent Technologies). The genomic DNA from thyroid tumor genomes were hybridized onto the array with genomic DNA from matched normal genomes as described elsewhere [36]. Background correction and normalization form array images were done using the Agilent Feature Extraction Software v10.7.3.1. The RankSegmentation statistical algorithm in NEXUS software v7.5 (Biodiscovery Inc., El Segundo, CA) was used to define copy number alterations (CNAs) of each sample. CNAs identified by array-CGH were cross validated using WES data. For copy number analysis from the exome sequencing data, ngCGH and RankSegmentation statistical algorithm in NEXUS software v7.5 were used. We inferred the loss of heterozygosity (LOH) events using the Sequenza algorithm [50]. All of the identified CNAs and LOH events were manually curated in terms of depth ratio and B allele frequency.
Gene set analyses
To investigate the gene ontology of individual mutations, we performed the DAVID analysis (http://david.abcc.ncifcrf.gov/) [51]. The gene ontologies including ‘biological process’, ‘cellular components’ and ‘molecular function’ category and pathways including ‘KEGG pathway’, ‘PANTHER pathway’ and ‘BIOCARTA pathway’ were sorted by significance.
Whole-transcriptome sequencing for gene fusion analyses
The mRNA of ten thyroid tumors (one FTA and nine FTCs) was converted into cDNA library using TruSeq RNA Library Preparation Kit (Illumina). WTS was performed using an Illumina HiSeq2000 platform. Sequencing reads were mapped onto the human reference genome (GRCh37, hg19). Acquisition and processing of the sequencing data were performed as previously described [52]. Fusion transcripts were identified by searching for the discordant reads and junction spanning reads by using the pyPRADA program [53].
Evolutionary models using somatic mutations
Estimation of evolutionary ages was performed as described elsewhere [33]. In brief, we adopted an evolutionary model for colorectal cancer genome which had 5 × 10–10 mutations per base pair per generation [54]. Using this, we calculated the mutation rate of r = 50.0 × 106 × 5 × 10–10 ≈ 0.025 per generation or cell cycle. The number of the cell divisions required to obtain N mutations follows a distribution with a mean of N/r. Clonally fixed somatic mutations obtained by a founder cell since its first cell division until the emergence of the last common ancestor in a single lineage were used in this analysis.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This study was supported by grants from National Research Foundation of Korea (2012 R1A5A2047939, NRF-2015R1C1A1A01051525 and NRF-2015M3C9A4053389).
REFERENCES
1. Rosai J, Carcangiu M, DeLellis R. Tumors of the thyroid gland. In: Atlas of tumor pathology, 3rd series, fas 5. Washington, DC: Armed Forces Institute of Pathology. 1992:21–48.
2. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013; 13:184–199.
3. D’Avanzo A, Treseler P, Ituarte PH, Wong M, Streja L, Greenspan FS, Siperstein AE, Duh QY, Clark OH. Follicular thyroid carcinoma: histology and prognosis. Cancer. 2004; 100:1123–1129.
4. DeGroot LJ, Kaplan EL, Shukla MS, Salti G, Straus FH. Morbidity and mortality in follicular thyroid cancer. J Clin Endocrinol Metab. 1995; 80:2946–2953.
5. Chow SM, Law SC, Mendenhall WM, Au SK, Yau S, Yuen KT, Law CC, Lau WH. Follicular thyroid carcinoma: prognostic factors and the role of radioiodine. Cancer. 2002; 95:488–498.
6. McHenry CR, Phitayakorn R. Follicular adenoma and carcinoma of the thyroid gland. Oncologist. 2011; 16:585–593.
7. Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006; 6:292–306.
8. Motoi N, Sakamoto A, Yamochi T, Horiuchi H, Motoi T, Machinami R. Role of ras mutation in the progression of thyroid carcinoma of follicular epithelial origin. Pathol Res Pract. 2000; 196:1–7.
9. Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW, 2nd, Tallini G, Kroll TG, Nikiforov YE. RAS point mutations and PAX8-PPAR gamma rearrangement in thyroid tumors: evidence for distinct molecular pathways in thyroid follicular carcinoma. J Clin Endocrinol Metab. 2003; 88:2318–2326.
10. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337.
11. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–550.
12. Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014; 159:676–690.
13. Kunstman JW, Juhlin CC, Goh G, Brown TC, Stenman A, Healy JM, Rubinstein JC, Choi M, Kiss N, Nelson-Williams C, Mane S, Rimm DL, Prasad ML, et al. Characterization of the mutational landscape of anaplastic thyroid cancer via whole-exome sequencing. Hum Mol Genet. 2015; 24:2318–2329.
14. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013; 31:213–219.
15. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011; 43:491–498.
16. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008; 455:1069–1075.
17. Hemmer S, Wasenius VM, Knuutila S, Franssila K, Joensuu H. DNA copy number changes in thyroid carcinoma. Am J Pathol. 1999; 154:1539–1547.
18. Roque L, Rodrigues R, Pinto A, Moura-Nunes V, Soares J. Chromosome imbalances in thyroid follicular neoplasms: a comparison between follicular adenomas and carcinomas. Genes Chromosomes Cancer. 2003; 36:292–302.
19. Bjorkqvist AM, Tammilehto L, Anttila S, Mattson K, Knuutila S. Recurrent DNA copy number changes in 1q, 4q, 6q, 9p, 13q, 14q and 22q detected by comparative genomic hybridization in malignant mesothelioma. Br J Cancer. 1997; 75:523–527.
20. Qureshi AA, Collins VP, Jani P. Genomic differences in benign and malignant follicular thyroid tumours using 1-Mb array-comparative genomic hybridisation. Eur Arch Otorhinolaryngol. 2013; 270:325–335.
21. Tung WS, Shevlin DW, Bartsch D, Norton JA, Wells SA, Jr, Goodfellow PJ. Infrequent CDKN2 mutation in human differentiated thyroid cancers. Mol Carcinog. 1996; 15:5–10.
22. Mazzaferri EL, Robbins RJ, Spencer CA, Braverman LE, Pacini F, Wartofsky L, Haugen BR, Sherman SI, Cooper DS, Braunstein GD, Lee S, Davies TF, Arafah BM, et al. A consensus report of the role of serum thyroglobulin as a monitoring method for low-risk patients with papillary thyroid carcinoma. J Clin Endocrinol Metab. 2003; 88:1433–1441.
23. Siraj AK, Masoodi T, Bu R, Beg S, Al-Sobhi SS, Al-Dayel F, Al-Dawish M, Alkuraya FS, Al-Kuraya KS. Genomic Profiling of Thyroid Cancer Reveals a Role for Thyroglobulin in Metastasis. Am J Hum Genet. 2016; 98:1170–1180.
24. Calebiro D, Grassi ES, Eszlinger M, Ronchi CL, Godbole A, Bathon K, Guizzardi F, de Filippis T, Krohn K, Jaeschke H, Schwarzmayr T, Bircan R, Gozu HI, et al. Recurrent EZH1 mutations are a second hit in autonomous thyroid adenomas. J Clin Invest. 2016; doi: 10.1172/JCI84894.
25. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446:153–158.
26. Yoshihara K, Wang Q, Torres-Garcia W, Zheng S, Vegesna R, Kim H, Verhaak RG. The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene. 2015; 34:4845–4854.
27. Agrawal N, Jiao Y, Sausen M, Leary R, Bettegowda C, Roberts NJ, Bhan S, Ho AS, Khan Z, Bishop J, Westra WH, Wood LD, Hruban RH, et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J Clin Endocrinol Metab. 2013; 98:E364–369.
28. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–218.
29. Bae JS, Choi SK, Jeon S, Kim Y, Lee S, Lee YS, Jung CK. Impact of NRAS Mutations on the Diagnosis of Follicular Neoplasm of the Thyroid. Int J Endocrinol. 2014; 2014:289834.
30. Fukahori M, Yoshida A, Hayashi H, Yoshihara M, Matsukuma S, Sakuma Y, Koizume S, Okamoto N, Kondo T, Masuda M, Miyagi Y. The associations between RAS mutations and clinical characteristics in follicular thyroid tumors: new insights from a single center and a large patient cohort. Thyroid. 2012; 22:683–689.
31. Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho JC, Krishnamoorthy GP, Xu B, Schultz N, Berger MF, Sander C, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest. 2016; 126:1052–1066.
32. Kim SY, Jung SH, Kim MS, Baek IP, Lee SH, Kim TM, Chung YJ, Lee SH. Genomic differences between pure ductal carcinoma in situ and synchronous ductal carcinoma in situ with invasive breast cancer. Oncotarget. 2015; 6:7597–7607. doi: 10.18632/oncotarget.3162.
33. Kim TM, Jung SH, Kim MS, Baek IP, Park SW, Lee SH, Lee HH, Kim SS, Chung YJ, Lee SH. The mutational burdens and evolutionary ages of early gastric cancers are comparable to those of advanced gastric cancers. J Pathol. 2014; 234:365–374.
34. Kim TM, An CH, Rhee JK, Jung SH, Lee SH, Baek IP, Kim MS, Lee SH, Chung YJ. Clonal origins and parallel evolution of regionally synchronous colorectal adenoma and carcinoma. Oncotarget. 2015; 6:27725–27735. doi: 10.18632/oncotarget.4834.
35. Jung SH, Choi YJ, Kim MS, Baek IP, Lee SH, Lee AW, Hur SY, Kim TM, Lee SH, Chung YJ. Progression of naive intraepithelial neoplasia genome to aggressive squamous cell carcinoma genome of uterine cervix. Oncotarget. 2015; 6:4385–4393. doi: 10.18632/oncotarget.2981.
36. Jung SH, Shin S, Kim MS, Baek IP, Lee JY, Lee SH, Kim TM, Lee SH, Chung YJ. Genetic Progression of High Grade Prostatic Intraepithelial Neoplasia to Prostate Cancer. Eur Urol. 2016; 69:823–830.
37. Min HS, Kim JH, Ryoo I, Jung SL, Jung CK. The role of core needle biopsy in the preoperative diagnosis of follicular neoplasm of the thyroid. APMIS. 2014; 122:993–1000.
38. Haugen BR, Alexander EK, Bible KC, Doherty GM, Mandel SJ, Nikiforov YE, Pacini F, Randolph GW, Sawka AM, Schlumberger M, Schuff KG, Sherman SI, Sosa JA, et al. 2015 American Thyroid Association Management Guidelines for Adult Patients with Thyroid Nodules and Differentiated Thyroid Cancer: The American Thyroid Association Guidelines Task Force on Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2016; 26:1–133.
39. Hemmer S, Wasenius VM, Knuutila S, Joensuu H, Franssila K. Comparison of benign and malignant follicular thyroid tumours by comparative genomic hybridization. Br J Cancer. 1998; 78:1012–1017.
40. Esposito C, Miccadei S, Saiardi A, Civitareale D. PAX 8 activates the enhancer of the human thyroperoxidase gene. Biochem J. 1998; 331:37–40.
41. Kroll TG, Sarraf P, Pecciarini L, Chen CJ, Mueller E, Spiegelman BM, Fletcher JA. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science. 2000; 289:1357–1360.
42. Hibi Y, Nagaya T, Kambe F, Imai T, Funahashi H, Nakao A, Seo H. Is thyroid follicular cancer in Japanese caused by a specific t(2; 3)(q13; p25) translocation generating Pax8-PPAR gamma fusion mRNA? Endocr J. 2004; 51:361–366.
43. Mochizuki K, Kondo T, Oishi N, Tahara I, Inoue T, Kasai K, Nakazawa T, Okamoto T, Shibata N, Katoh R. Low frequency of PAX8-PPARgamma rearrangement in follicular thyroid carcinomas in Japanese patients. Pathol Int. 2015; 65:250–253.
44. Jeong SH, Hong HS, Kwak JJ, Lee EH. Analysis of RAS mutation and PAX8/PPARgamma rearrangements in follicular-derived thyroid neoplasms in a Korean population: frequency and ultrasound findings. J Endocrinol Invest. 2015; 38:849–857.
45. Garcia-Alcalde F, Okonechnikov K, Carbonell J, Cruz LM, Gotz S, Tarazona S, Dopazo J, Meyer TF, Conesa A. Qualimap: evaluating next-generation sequencing alignment data. Bioinformatics. 2012; 28:2678–2679.
46. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164.
47. Carter H, Samayoa J, Hruban RH, Karchin R. Prioritization of driver mutations in pancreatic cancer using cancer-specific high-throughput annotation of somatic mutations (CHASM). Cancer Biol Ther. 2010; 10:582–587.
48. Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004; 4:177–183.
49. Rubio-Perez C, Tamborero D, Schroeder MP, Antolin AA, Deu-Pons J, Perez-Llamas C, Mestres J, Gonzalez-Perez A, Lopez-Bigas N. In silico prescription of anticancer drugs to cohorts of 28 tumor types reveals targeting opportunities. Cancer Cell. 2015; 27:382–396.
50. Favero F, Joshi T, Marquard AM, Birkbak NJ, Krzystanek M, Li Q, Szallasi Z, Eklund AC. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Ann Oncol. 2015; 26:64–70.
51. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44–57.
52. Choi YJ, Jung SH, Kim MS, Baek IP, Rhee JK, Lee SH, Hur SY, Kim TM, Chung YJ, Lee SH. Genomic landscape of endometrial stromal sarcoma of uterus. Oncotarget. 2015; 6:33319–33328. doi: 10.18632/oncotarget.5384.
53. Torres-Garcia W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, Verhaak RG. PRADA: pipeline for RNA sequencing data analysis. Bioinformatics. 2014; 30:2224–2226.
54. Jones S, Chen WD, Parmigiani G, Diehl F, Beerenwinkel N, Antal T, Traulsen A, Nowak MA, Siegel C, Velculescu VE, Kinzler KW, Vogelstein B, Willis J, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008; 105:4283–4288.