
INTRODUCTION
Translation initiation plays a critical role in both general and gene-specific regulation of gene expression and thereby in the control of cell proliferation, differentiation, and survival. Unrestricted translation initiation causes malignant transformation in vitro and likely plays a causative role in the genesis of some human cancers [1-3]. Consistently, translation initiation factors are over-expressed or otherwise activated in breast, lung, cervical, and hematologic cancers and predict poor prognosis [4-9]. In contrast, restricting translation initiation reverses transformed phenotypes in vitro and in vivo [10-13]. This experimental and clinical evidence indicates that translation initiation factors are potential targets for the development of mechanism-specific anti-cancer therapies. Indeed, an anti-sense oligonucleotide-based therapy targeting eukaryotic translation initiation factor (eIF) 4E inhibits tumor growth in animal models of cancer and has recently been evaluated in a phase-I-II human clinical trial[11]. However, whether small molecules can effectively target translation initiation in vivo has not been demonstrated. The studies reported here were designed to determine whether the eIF2.GTP.Met-tRNAi translation initiation (ternary) complex is a bona fide target for the development of anti-cancer therapeutic agents.
Formation of the eIF2.GTP.Met-tRNAi ternary complex is the first critical step in the translation initiation cascade. Upon assembly of the 80S ribosome at the initiation codon, GTP in the ternary complex is hydrolyzed, releasing the eIF2.GDP binary complex [14]. To initiate a new round of translation, eIF2.GDP must be converted to eIF2.GTP by eIF2B, the eIF2 guanine nucleotide exchange factor. Phosphorylation of the alpha subunit of eIF2 (eIF2α) on S51 increases the affinity of eIF2 for eIF2B and functionally converts eIF2 into a competitive inhibitor of eIF2B[15]. Because of the low eIF2B/eIF2 ratio, even partial phosphorylation of eIF2α limits the amount of the eIF2.GTP.Met-tRNAi ternary complex and thereby inhibits translation initiation [16].
Restriction of the ternary complex by phosphorylation of eIF2α plays a critical role in the translational regulation of specific genes or gene clusters [17-22]. For example, the translation of mRNAs containing excessive and stable secondary structures, like those encoding for most oncogenic proteins, is preferentially down-regulated by phosphorylation of eIF2α at levels that would minimally affect translation of most housekeeping proteins encoded by mRNAs with simple 5’UTRs [23-29]. Paradoxically, the translation of a subset of mRNAs with particular features in their 5’UTRs, like the one coding for activating transcription factor-4 (ATF-4), is more efficient under conditions that limit the amount of the ternary complex [30, 31]. Increased translation of ATF-4 results in the transcriptional activation of its target genes including the chaperone BiP and the pro-apoptotic transcription factor CHOP [32].
We previously identified three chemically distinct small molecular weight compounds, Clotrimazole (CLT), eicosapentaenoic acid (EPA), and troglitazone (TRO), that cause phosphorylation of eIF2α in non-transformed cell lines [18, 20, 22, 33]. We used these compounds as molecular probes to determine whether restriction of the ternary complex via phosphorylation of eIF2α can be achieved pharmacologically for cancer treatment.
We report here that CLT, EPA, and TRO cause phosphorylation of eIF2α, restrict the amount of the ternary complex, and thereby inhibit translation initiation in a wide variety of cancer cell lines. In experimental cancer models, the compounds abrogate xenograft tumor growth and increase the mean survival time of p53-/- mice. Abrogation of tumor growth is associated with phosphorylation of eIF2α, reduced availability of the ternary complex, down-regulation of oncogenes, and up-regulation of ATF-4-dependent genes. Importantly, we show that administration of TRO to liposarcoma patients caused phosphorylation of eIF2α and increased expression of the ATF-4-dependent protein BiP. These findings provide the first direct in vivo evidence that the ternary complex can be pharmacologically targeted for cancer therapy.
RESULTS and DISCUSSION
CLT, EPA, and TRO inhibit cancer cell proliferation.
We determined the effect of CLT, EPA, and TRO on the proliferation and/or survival of cancer cell lines by sulforhodamine B cell proliferation assay (SRB). Table 1 presents the IC50 of each compound for the cancer cell lines tested. The three molecular probes inhibited in a dose-dependent manner the proliferation of a wide variety of cancer cell lines, including several multiple drug-resistant ones.
Table 1: Effect of CLT, EPA, and TRO on Ca++ metabolism and proliferation in cancer cells.
CLT |
TRO |
EPA |
|||||||
Cell line |
IC501 |
Ca++ release2 |
SOC close2 |
IC50* |
Ca++ release2 |
SOC close2 |
IC50* |
Ca++ release2 |
SOC close2 |
KLN HeLa Calu-6 MCF-7 ACHN DU-145 U118MG HCT 15 SK-OV3 SK-MEL 28 HTB 174 CRL 1933 SV480 HCT 116 HT 29 CRL 231 A549 MMRU HEP-G2 |
5 3 1 2 7 7 5 2 12 3 1.5 10 3.4 3 7 2 3 2 5 |
Y Y Y Y Y Y Y Y N Y Y Y ND ND Y ND Y Y Y |
Y Y Y Y Y Y Y Y N Y Y N ND ND ND ND Y Y Y |
8 12 15 10 18 17 34 12 17 30 20 15 14 8 20 26 8 ND 23 |
Y Y ND Y Y Y Y ND N ND Y Y ND ND Y ND Y ND Y |
Y Y ND Y Y Y N ND N ND N ND ND ND N ND Y ND ND |
20 10 40 43 62 36 36 47 96 46 79 ND ND ND 72 ND 54 36 ND |
Y Y Y Y Y Y Y ND N ND Y ND ND ND Y ND Y Y ND |
Y Y Y Y ND Y Y ND N ND N ND ND ND ND ND Y Y ND |
1Concentration of drug that inhibits cell growth by 50%.
2 Y = yes, N = no, ND = not done
CLT, EPA, and TRO deplete internal Ca++ stores in cancer cells.
We have previously reported that in NIH 3T3 cells, CLT, EPA, and TRO release Ca++ from internal stores and simultaneously inhibit their refilling through store-operated Ca++ channels (SOC) [18, 20, 22, 33]. This causes phosphorylation of eIF2α and inhibition of translation initiation [16, 20, 22]. To determine whether inhibition of cancer cell proliferation by CLT, EPA, and TRO is mediated by their effect on intracellular Ca++ homeostasis, we measured cytosolic Ca++ in Fura-2–loaded cancer cells. Table 1 shows that in most cancer cells, particularly those sensitive to their anti-proliferative effect, CLT, EPA, and TRO released Ca++ from internal stores – as indicated by an increased cytosolic Ca++ in Ca++-free media. To determine whether CLT, EPA, and TRO inhibit SOC in cancer cells, we first challenged Fura-2–loaded cancer cells with thapsigargin (TG), a specific inhibitor of SERCA-ATPase [34, 35]. Treatment with TG in media containing Ca++ results in sustained elevation of cytosolic Ca++, a functional expression of capacitative Ca++ influx through SOC. Addition of CLT, EPA, or TRO after TG closes SOC in most cancer cell lines, as indicated by a sharp reduction of cytosolic Ca++ towards the baseline levels (Table 1). To document directly whether the compounds release Ca++ from the ER stores, as suggested by the FURA-2 experiments, we generated stable cell lines that express calcium-indicator cameleon proteins targeted to the ER[36]. These experiments formally demonstrate that CLT, EPA, and TRO partially deplete ER Ca++ stores (see Supplemental Results and in Supplemental Figure 1A for details).
Taken together, the data presented in Table 1 and Supplemental Figure 1A demonstrate that CLT, EPA, and TRO partially deplete ER- Ca++ stores in a variety of human cancer cell lines. Because the effect on intracellular Ca++ was similar in all cancer cell lines sensitive to the anti-proliferative effect of the compounds, further mechanistic and in vivo studies were conducted in selected cancer cell lines.
CLT, EPA, and TRO induce phosphorylation of eIF2α and inhibit translation initiation in cancer cells.
Extensive experimental evidence shows that partial depletion of ER Ca++ stores induces phosphorylation of eIF2α [18, 20]. As expected from their effect on ER Ca++, CLT, EPA, and TRO caused strong phosphorylation of eIF2α in KLN squamous cell carcinoma cells (Figure 1A), as well as in MCF-7 breast cells and DU145 prostate cancer cells (data not shown).
To determine the functional consequences of eIF2α phosphorylation, we took advantage of the well-documented translational up-regulation of ATF-4 and consequent induction of ATF-4-responsive genes that result from eIF2α phosphorylation, which reduces the amount of eIF2.GTP.Met-tRNAi ternary complex [32]. As shown in Figure 1B, CLT, EPA, and TRO strongly induce the expression of CHOP, a transcriptional target of ATF-4. These data suggest that CLT, EPA, and TRO reduce the amount of the ternary complex and thereby inhibit translation initiation in cancer cells.
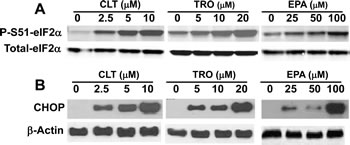
Figure 1: CLT, EPA, and TRO cause eIF2α phosphorylation and induce CHOP expression in KLN cells. KLN cells were incubated with indicated concentrations of CLT, EPA, and TRO for 2 (A) or 8 (B) hours, the cells were lysed, and equal amounts of protein were separated by SDS-PAGE and blotted with anti-PS51-eIF2α, anti-total eIF2α (2 hours treated samples, A) or anti-CHOP and anti-β-actin (8 hours treated samples, B) specific antibodies.
CLT, EPA, and TRO restrict formation of the ternary complex in vitro and in vivo.
To confirm that CLT, EPA, and TRO restrict the abundance of the eIF2.GTP.Met-tRNAi ternary complex, we developed a quantitative cell-based assay whose read-out depends on the availability of this complex. The assay takes advantage of the striking differential effect that restriction of the ternary complex exerts on the translation of mRNAs. In particular, translation of a subset of mRNAs characterized by the presence of multiple upstream open reading frames (uORF) in their 5’UTR increases when the ternary complex is restricted, as characteristically represented by the mRNA encoding for ATF-4 and GCN2 [32, 37, 38]. Fusion of the 5’ UTR of either mRNA to a reporter gene confers enhanced translation when the ternary complex is scarce [32, 38].
Assay development has been described elsewhere [39, 40]. Briefly, we constructed a bi-directional plasmid in which a common promoter/enhancer complex drives the transcription of firefly luciferase (F-luc) ORF fused to the 5’UTR of ATF-4, and of the renilla luciferase (R-luc) ORF fused to a simple 90-nucleotide 5’UTR derived from the plasmid (Figure 2A). The relative expression of each luciferase was established by the F-luc to R-luc ratio determined with a dual luciferase assay. In stably transfected KLN cells, CLT, EPA, or TRO increased the F-luc to R-luc ratio in a dose-dependent manner without affecting the ratio of the respective mRNAs (Figures 2B and 2C). These data indicate that CLT, EPA, and TRO increase the translation of the reporter F-luc mRNA fused to the 5’UTR of ATF-4. To establish the cause-effect relationship between phosphorylation of eIF2α and the increased translation of F-luc mRNA shown in Figure 1, we transiently co-transfected KLN cells with the bi-directional construct described in Figure 2A and an expression vector coding for either wild-type eIF2α (eIF2α-WT) or a constitutively active non-phosphorylatable eIF2α mutant (eIF2α-S51A). Figure 2D shows that co-transfection with the eIF2α-S51A mutant abrogated the increase in the F-luc to R-luc ratio observed in response to CLT, EPA, or TRO, while co-transfection with eIF2α-WT had no effect. Furthermore, a single nucleotide insertion that removes the second uORF in the reporter construct abrogated the translational up-regulation of F-luc induced by these agents (Figure 2E). Abrogation of F-Luc translational up-regulation by either non-phosphorylatable eIF2α or by deletion of one uORF demonstrates conclusively that CLT, EPA, or TRO reduces the availability of the ternary complex by inducing phosphorylation of eIF2α. Consistently, anti-cancer agents that do not cause eIF2α phosphorylation failed to up-regulate F-luc (see supplementary Table 1).
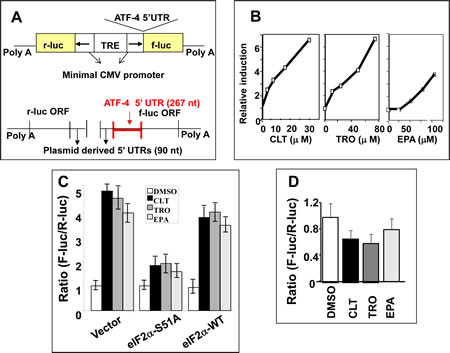
Figure 2: CLT, EPA, and TRO limit formation of the ternary complex. A) Schematic depiction of expression vector and its mRNA products utilized for establishment of a ternary complex availability assay. B) KLN cells stably transfected with this vector were incubated with CLT, EPA, or TRO for 24 hours, and reporter activities were determined by DLR (Promega, Inc.). Changes in the F-luc/R-luc ratio were expressed as multiples of the same ratio in vehicle-treated cells. C) Poly A plus mRNA was isolated from KLN cells in B and hybridized with probes specific to F-luc or R-luc mRNA. D) KLN cells were co-transfected with the bi-directional vector shown in Figure 2A and the eIF2α-WT or eIF2α-51A expression vectors. The F-luc/R-luc ratio in response to CLT, EPA, or TRO was measured by DLR assay as in B. E) A single nucleotide was inserted into the second uORF of the ATF-4 5’UTR, which eliminated the ORF2. Cells transfected with this mutant construct were treated with CLT, EPA, or TRO, as in 2B.
CLT, EPA, and TRO inhibit translation initiation.
The results depicted in Figures 1 and 2 strongly indicate that CLT, EPA, and TRO inhibit translation initiation in cancer cells, a conclusion that can be tested experimentally by analyzing polysome profiles of cell lysates. When translation initiation is inhibited, polysome profiles shift from heavy toward lighter polysomes. As shown in Figure 3A-C, the polysome profiles of CLT, EPA, and TRO-treated KLN cells shifted from heavy to light polysomes and free ribosomal subunits, demonstrating that the three agents inhibit translation initiation in cancer cells. This conclusion was confirmed in pulse-labeling experiments demonstrating that CLT, EPA, and TRO inhibit protein synthesis in different cancer cell lines (Supplemental Figure 1B).
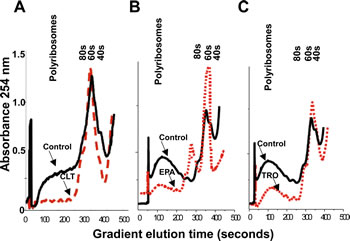
Figure 3: CLT, EPA, and TRO inhibit translation initiation in cancer cells. KLN cells were incubated with CLT (A, 15 µM), EPA (B, 50 µM), or TRO (C, 30 µM) for 2 hours, lysed, and an equal amount of OD 260 material was separated by sucrose density gradient centrifugation. The gradient was eluted from the bottom with constant monitoring at 254 nm.
CLT-, EPA-, and TRO-induced cell cycle arrest in cancer cells is mediated by phosphorylation of eIF2α.
To determine the effect of CLT, EPA, and TRO on cell cycle progression in cancer cells, we treated exponentially growing KLN cancer cells with these three agents, stained them with propidium iodide, and analyzed them by FACS for cell cycle distribution and apoptosis. CLT, EPA, and TRO blocked the KLN cancer cell cycle in G1 with no apparent effect on the proportion of apoptotic cells (Supplemental Figure 2). This cell cycle arrest is mediated by phosphorylation of eIF2α on S51, because it was abrogated in cells transiently transfected with the non-phosphorylatable eIF2α-S51A mutant (Table 2).
Table 2: Effect of CLT, EPA, and TRO on cell cycle progression of eIFα-51A/GFP or eIF2α-WT/GFP transfected KLN-mouse carcinoma cells* (the percentage of cells in each cell cycle stage)
Transfected Vector |
Treatment |
|||
DMSO |
CLT |
EPA |
TRO |
|
eIF2α-WT/GFP |
G1: 47 S: 25 G2: 28 |
G1: 65 S: 19 G2: 16 |
G1: 61 S: 22 G2: 17 |
G1: 67 S: 21 G2: 12 |
eIF2α-51A/GFP |
G1: 45 S: 26 G2: 29 |
G1: 52 S: 27 G2: 21 |
G1: 49 S: 29 G2: 22 |
G1: 53 S: 24 G2: 23 |
* KLN cells transiently transfected with either GFP-eIF2α-51A (A, B) or GFP-eIF2α-WT (C, D) were treated with CLT, EPA, or TRO, sequentially fixed with 3% paraformaldhyde and 70% ethanol, stained with propidium iodide, and cell cycle distribution of GFP-expressing cells was determined by FACS analysis.
CLT, EPA, and TRO cause phosphorylation of eIF2α, induce expression of BiP, and down-regulate expression of cyclin D1 in tumors.
To determine directly whether phosphorylation of eIF2α in tumors can be achieved pharmacologically, we implanted KLN cells intradermally in mice to form orthotopic-syngenic tumors. Mice carrying small (2-4 mm in diameter) KLN tumors were treated for either one or seven days orally with CLT (120 mg/kg), EPA (2.5 g/kg), TRO (350 mg/kg), or the respective vehicles. Staining of excised tumors with anti-PS51-eIF2α−specific antibodies showed that all three agents induced significant and sustained phosphorylation of eIF2α, providing the first experimental evidence that phosphorylation of eIF2α in tumors can be achieved pharmacologically (Figure 4). Consistently, in tumors from treated animals, all three agents increased the expression of BiP, another ATF-4 dependent surrogate marker for depletion of the ternary complex that is induced by phosphorylation of eIF2α[30-32, 41]. CLT, EPA, and TRO also down-regulated the expression of cyclin D1 in the tumors excised from treated mice (Figure 4). This finding is consistent with depletion of the ternary complex, which preferentially affects translation of mRNAs containing highly structured 5’UTRs, including those encoding for most oncogenic proteins such as cyclin D1[18, 20, 22, 42]. To confirm restriction of the ternary complex in the tumors, we injected mice intradermically with the engineered KLN cells shown in Figure 2B and treated tumor-bearing mice with CLT (120 mg/kg/day) or vehicle for two days. Supplemental Figure 3 shows that CLT significantly increased the F-luc to R-luc ratio in the tumors from CLT-treated mice. Taken together, the experiments described above demonstrate that restriction of the ternary complex and inhibition of translation initiation in tumors can be pharmacologically achieved.
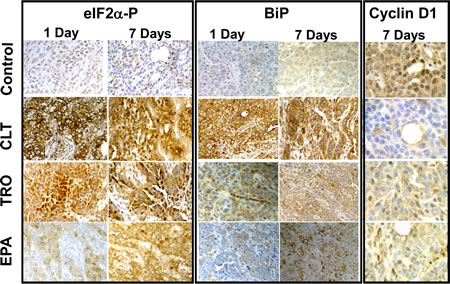
Figure 4: CLT, EPA, and TRO cause phosphorylation of eIF2α and induction of BiP and suppress expression of cyclin D1 in KLN tumors. Mice with small KLN tumors were treated with CLT (120 mg/kg/day), EPA (2.5.g/kg/day), TRO (350 mg/kg/day), or vehicle for one day (left panels) or seven days (right panels). The tumors were excised and stained with antibodies specific to PS51-eIF2α, BiP, or cyclin D1 and counter-stained with hematoxyline.
CLT, EPA, and TRO inhibit tumor growth in animal models.
To determine the functional consequence of reducing the amount of the ternary complex in tumors, we treated KLN-cell tumor-bearing mice orally with CLT (120 mg/kg), EPA (2.5 g/kg), or TRO (350 mg/kg) daily for 5 weeks. Control mice for CLT or TRO received gum arabica (the vehicle used to dissolve the drugs), while control mice for EPA received equicaloric amounts of corn oil. As shown in Figure 5A, CLT, EPA, and TRO significantly reduced the growth of KLN cell tumors, demonstrating that phosphorylation of eIF2α and restriction of the ternary complex is associated with inhibition of tumor growth.
The observation that EPA induces phosphorylation of eIF2α and inhibition of KLN-tumor growth in vivo is remarkable and has major public health implications, because EPA, the main n-3 fatty acid in marine fish oil, is a component of the human diet. Indeed, some human population studies suggest that consumption of diets rich in EPA may reduce cancer risk [43-45]. For this reason, we probed further the effect of EPA on tumor growth and progression in a DU-145 human prostate cancer xenograft and in a genetically engineered p53-/- mouse cancer model. Treatment with EPA significantly reduced the growth of DU-145 human prostate cancer xenograft tumors (Figure 5B). In p53-/- mice, oral administration of EPA significantly extended the mean survival time from 185 to 360 days (p<0.005, Figure 5C). This experiment is remarkable, because p53 is the most commonly mutated or deleted gene in human cancers, and p53-/- mice develop various cancers with 100% penetrance and all die before 40 weeks of age.
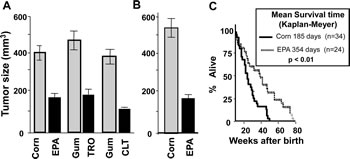
Figure 5: CLT, EPA, and TRO inhibit the growth of tumors and EPA extends the life expectancy of p53-/- mice. A) DBA/2J mice were injected with 2.5 x 106 KLN cells, the animals were randomly assigned to the treatment and control groups after establishment of tumors and treated with 120 mg/kg/day CLT, or 350 mg/kg/day TRO, both in gum arabica, or EPA (2.5 g/kg, administered as fish oil concentrate). Gum arabica-treated mice were used as a control for CLT and TRO, and corn oil-treated mice were used as a control for EPA treatment. The experiments were carried out in pairs (CLT vs. gum arabica, TRO vs. gum arabica, EPA vs. corn oil) with 15 mice per group. Tumor dimensions were determined with electronic calipers, tumor volume was calculated, and the results were analyzed by Student’s t-test. B). Male nude mice were injected with 1x106 human DU145 prostate cancer cells, and mice with visible tumors were fed fish oil- or corn oil-enriched diets for five weeks. C) p53-/- mice were treated with either EPA (2.5 g/kg administered as fish oil concentrate) or an equicaloric amount of corn oil starting at four weeks of age; the time of death was recorded. The data were analyzed by Kaplan-Mayer test (p<0.01).
Troglitazone causes phosphorylation of eIF2α and induction of BiP expression in human cancers.
While this work was in progress, we became aware of a clinical trial in which TRO was administered to liposarcoma patients between a diagnostic biopsy and the surgical excision of the tumor [46]. We obtained paraffin-embedded pathology samples from these patients and stained them with anti-PS51-eIF2α- and BiP-specific antibodies. Figure 6 shows that treatment with TRO caused a remarkable increase in eIF2α phosphorylation and BiP expression in the excised human liposarcoma tumors compared with the pre-treatment biopsy.
Clinical and experimental studies indicate that dysregulation of translation initiation plays a role in malignant transformation and tumor progression, highlighting the potential of therapeutics that exert antitumor and/or chemopreventive effects by selectively targeting the translation initiation cascade[1, 47-49]. The findings reported in this manuscript provide conclusive evidence that translation initiation can be pharmacologically targeted in vivo with small molecules, a critical prerequisite for the development of translation initiation-specific anti-cancer agents. Specifically, we demonstrate that 1) pharmacological agents that induce phosphorylation of eIF2α and reduce the ternary complex in vitro similarly phosphorylate eIF2α and reduce the ternary complex in animal and human tumors, and 2) phosphorylation of eIF2α in vivo is associated with suppression of tumor growth and extension of life expectancy in various animal cancer models. Direct in vivo demonstration of the cause-effect relationship studies between induction of eIF2α phosphorylation and tumor by replacing endogenous eIF2α with recombinant eIF2α-S51A mutant could not be carried out despite our attempts because tumors formed by these cells failed to grow well after an initial phase. Similar results have been reported by others [50].
Together, the in vivo experiments depicted in Figure 5 and 6 indicate that pharmacological inhibition of translation initiation can be used for cancer treatment and perhaps prevention in high-risk populations. Second generation of translation initiation inhibitors being developed as well as progress in resolving the structure of translation initiation factors and their regulators should accelerate development of translation initiation inhibitors for cancer therapy [39, 51, 52]. Furthermore, since EPA is the main component of marine fish oils and can be readily administered to humans with minimal or no side effects, our studies provide a strong favorable rationale and the analytical tools for human clinical trials aimed at evaluating whether translation initiation inhibitors that induce phosphorylation of eIF2α would reduce either recurrence in cancer patients who are in remission or the incidence of cancer in high-risk populations.
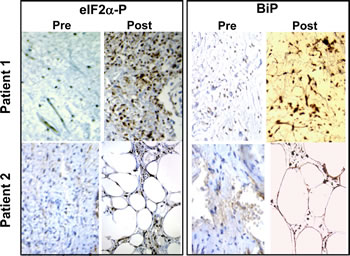
Figure 6: TRO induces phosphorylation of eIF2α in human liposarcoma tumors. Biopsy samples of liposarcomas obtained before treatment (left panels) and tumor samples of the same patients after treatment with TRO (right panels) in a clinical trial conducted independently of our work[46] were stained with antibodies specific to PS51-eIF2α or to BiP.
METHODS
Ethics Statement.
All animal studies in this report are carried out per Harvard Medical School Standing Committee on Animals, IACCUC, that oversees protocols involving animals per approved protocol #03151. The end point in studies with p53-/- and p53+/+ was lifespan. Animals were observed daily and weighed twice weekly. They were sacrificed in they had locomotion or breathing difficulties or lost more than 15% of body weight or appeared cachectic. In all other studies, animals were maintained for total of four weeks after the start of treatment. They were observed at least three times a week, any animal that had tumor volume greater than 15% of body weight or lost 15% body weight or had breathing or locomotion difficulties or other signs of pain or suffering per approved protocol were sacrificed. All animals were given analgesic (bprunex) as needed.
Human samples are retrospective discarded pathological materials with no identifiers. Partners HealthCare Human Research Committee, the Partners Internal Review Board for Human Studies approved the collection of samples as de-identified discarded pathological materials not requiring consent.
Cell growth assay.
Cell growth was measured by the SRB assay as described in [22].
Measurements of free cytosolic and ERCa++.
Free intracellular Ca++ was measured using Fura-2, AM (Molecular Probes, Eugene, OR) as described by Clementi et al. [53]. Fluorescence was monitored with a Photon Technology International dual-wavelength spectrofluorometer (PTI, Monmouth Junction, NJ). Excitation was at 340/380 nm, and emission at 510 nm. For measurement of ER Ca++, we established stable cell lines expressing ER-targeted cameleon proteins [36, 54] and monitored ER Ca++ using the PTI dual-wavelength spectrofluorometer.
Polysome profiles.
Polysome profiles were obtained by sucrose density gradient centrifugation as described in[55].
Stable and transient transfection.
Cells were seeded at the density of 105 in 60-mm (stable transfection) or 106 cells per 150-mm (transient transfection) plates and transfected one day later using the Qiagen transfectamine transfection kit. For selection of stable cell lines, transfected cells were transferred to 100-mm plates and selected with appropriate antibiotics.
Cell cycle analysis.
Untransfected KLN cells were fixed with ethanol (70%), stained with propidium iodide (50 µg/ml PI with 20 µg/ml RNAse A) for 30 minutes, and analyzed by FACS[56]. Cells transiently transfected with GFP-eIF2α-51A or eIF2α-WT were fixed with 3% paraformaldhyde, washed twice with PBS, fixed again with 70% methanol, and stained with PI as described above. Cell cycle distribution of GFP-expressing cells was analyzed by FACS.
Western blotting.
Cell extracts were separated by SDS-PAGE and probed with anti-phosphoserine-51-eIF2α (PS51-eIF2α), anti-total eIF2α-specific antibodies (PS51-eIF2α) (Biosource International, Hopkinton, MA), anti-CHOP, or anti β-actin as described [56].
Animal studies.
KLN squamous cell carcinoma cells were injected intradermically (2.5 x 105 cells in 0.1 ml of PBS) into 6-week-old female DBA/2J mice (Jackson Laboratories, Bar Harbor, ME). Four days after implantation, tumor-bearing mice were randomized into control and treatment groups and then treated with the vehicle or CLT (120 mg/kg in gum arabica), TRO (400 mg/kg in gum arabica), or EPA (2.5 g/kg, lipid concentrate) daily by gavage. Control mice were treated with the respective vehicle alone. Tumor volumes were calculated as in [20] and results analyzed by Student’s t-test. DU145 human prostate cancer cells were subcutaneously injected to nude mice that were fed a diet containing either 20% corn oil or 1% corn oil and 19% menhaden oil, starting five days after inoculation of tumors. Tumor volumes were calculated[20] and results analyzed by Student’s t-test. p53-/- mice were administered either EPA (2.5 g/kg in fish oil refined for high content of EPA) or equicaloric corn oil daily by gavage until the death of the last animal. Life expectancies of control and EPA-treated mice were compared by Kaplan-Meyer test.
The end point in studies with p53-/- and p53+/+ was lifespan. Animals were observed daily and weighed twice weekly. They were sacrificed in they had locomotion or breathing difficulties or lost more than 15% of body weight or appeared cachectic. In all other studies, animals were maintained for total of four weeks after the start of treatment. They were observed at least three times a week, any animal that had tumor volume greater than 15% of body weight or lost 15% body weight or had breathing or locomotion difficulties or other signs of pain or suffering per approved protocol were sacrificed. All animals were given analgesic (bprunex) as needed.
Immunocytochemistry.
Formalin-fixed, paraffin-embedded tumor sections were immunostained with anti-PS51-eIF2α, anti-BiP, and anti-cyclin D1 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), and counter-stained with hematoxylin.
Dual luciferase assay.
Cells or minced tumors expressing F-luc and R-luc were lysed and the extracts assayed with a glow-type dual luciferase assay kit, (Promega, Inc., Madison, WI).
ACKNOWLEDGMENTS
The authors are grateful to Dr. D.C. Tosteson and Dr. M.T. Tosteson for critical comments and continuous support, to C. Fletcher for valuable help scoring the human liposarcoma samples, and R. Tsien for providing ER-targeted cameleons expression plasmids. This work was supported in part by NIH grants CA78411, CA101034 and U19 CA87427 (NCDDG) to JAH and Department of Defense grant W81xWH-05-1-096 and Susan B. Komen Breast Cancer Research Foundation grant BCTR0402565 to HA.
References
1. De Benedetti A and Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004; 23(18):3189-3199.
2. Cohen N, Sharma M, Kentsis A, Perez JM, Strudwick S and Borden KL. PML RING suppresses oncogenic transformation by reducing the affinity of eIF4E for mRNA. Embo J. 2001; 20(16):4547-4559.
3. Graff JR and Zimmer SG. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metastasis. 2003; 20(3):265-273.
4. Nathan CA, Liu L, Li BD, Abreo FW, Nandy I and De Benedetti A. Detection of the proto-oncogene eIF4E in surgical margins may predict recurrence in head and neck cancer. Oncogene. 1997; 15(5):579-584.
5. Martin ME, Perez MI, Redondo C, Alvarez MI, Salinas M and Fando JL. 4E binding protein 1 expression is inversely correlated to the progression of gastrointestinal cancers. The international journal of biochemistry & cell biology. 2000; 32(6):633-642.
6. Wang S, Lloyd RV, Hutzler MJ, Rosenwald IB, Safran MS, Patwardhan NA and Khan A. Expression of eukaryotic translation initiation factors 4E and 2alpha correlates with the progression of thyroid carcinoma. Thyroid. 2001; 11(12):1101-1107.
7. Berkel HJ, Turbat-Herrera EA, Shi R and de Benedetti A. Expression of the translation initiation factor eIF4E in the polyp- cancer sequence in the colon. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2001; 10(6):663-666.
8. Bauer C, Brass N, Diesinger I, Kayser K, Grasser FA and Meese E. Overexpression of the eukaryotic translation initiation factor 4G (eIF4G-1) in squamous cell lung carcinoma. International journal of cancer Journal international du cancer. 2002; 98(2):181-185.
9. Mayeur GL and Hershey JW. Malignant transformation by the eukaryotic translation initiation factor 3 subunit p48 (eIF3e). FEBS letters. 2002; 514(1):49-54.
10. Yang HS, Jansen AP, Komar AA, Zheng X, Merrick WC, Costes S, Lockett SJ, Sonenberg N and Colburn NH. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol Cell Biol. 2003; 23(1):26-37.
11. Graff JR, Konicek BW, Vincent TM, Lynch RL, Monteith D, Weir SN, Schwier P, Capen A, Goode RL, Dowless MS, Chen Y, Zhang H, Sissons S, Cox K, McNulty AM, Parsons SH, et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. The Journal of clinical investigation. 2007; 117(9):2638-2648.
12. Rousseau D, Gingras AC, Pause A and Sonenberg N. The eIF4E-binding proteins 1 and 2 are negative regulators of cell growth. Oncogene. 1996; 13(11):2415-2420.
13. Avdulov S, Li S, Michalek V, Burrichter D, Peterson M, Perlman DM, Manivel JC, Sonenberg N, Yee D, Bitterman PB and Polunovsky VA. Activation of translation complex eIF4F is essential for the genesis and maintenance of the malignant phenotype in human mammary epithelial cells. Cancer cell. 2004; 5(6):553-563.
14. Das S, Ghosh R and Maitra U. Eukaryotic translation initiation factor 5 functions as a GTPase-activating protein. J Biol Chem. 2001; 276(9):6720-6726.
15. Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE and Hinnebusch AG. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol. 2001; 21(15):5018-5030.
16. Brostrom CO, Chin KV, Wong WL, Cade C and Brostrom MA. Inhibition of translational initiation in eukaryotic cells by calcium ionophore. J Biol Chem. 1989; 264(3):1644-1649.
17. Raveh T, Hovanessian AG, Meurs EF, Sonenberg N and Kimchi A. Double-stranded RNA-dependent protein kinase mediates c-Myc suppression induced by type I interferons. J Biol Chem. 1996; 271(41):25479-25484.
18. Aktas H, Fluckiger R, Acosta JA, Savage JM, Palakurthi SS and Halperin JA. Depletion of intracellular Ca2+ stores, phosphorylation of eIF2alpha, and sustained inhibition of translation initiation mediate the anticancer effects of clotrimazole. Proc Natl Acad Sci U S A. 1998; 95(14):8280-8285.
19. Brewer JW, Hendershot LM, Sherr CJ and Diehl JA. Mammalian unfolded protein response inhibits cyclin D1 translation and cell-cycle progression. Proc Natl Acad Sci U S A. 1999; 96(15):8505-8510.
20. Palakurthi SS, Fluckiger R, Aktas H, Changolkar AK, Shahsafaei A, Harneit S, Kilic E and Halperin JA. Inhibition of translation initiation mediates the anticancer effect of the n-3 polyunsaturated fatty acid eicosapentaenoic acid. Cancer Res. 2000; 60(11):2919-2925.
21. Dever TE. Gene-specific regulation by general translation factors. Cell. 2002; 108(4):545-556.
22. Palakurthi SS, Aktas H, Grubissich LM, Mortensen RM and Halperin JA. Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor gamma and mediated by inhibition of translation initiation. Cancer Res. 2001; 61(16):6213-6218.
23. Kozak M. Structural features in eukaryotic mRNAs that modulate the initiation of translation. J Biol Chem. 1991; 266(30):19867-19870.
24. Kozak M. Circumstances and mechanisms of inhibition of translation by secondary structure in eucaryotic mRNAs. Mol Cell Biol. 1989; 9(11):5134-5142.
25. Lodish HF. Model for the regulation of mRNA translation applied to haemoglobin synthesis. Nature. 1974; 251:385-388.
26. Heinrich R and Rapoport TA. Mathematical modelling of translation of mRNA in eucaryotes; steady state, time-dependent processes and application to reticulocytes. J Theor Biol. 1980; 86(2):279-313.
27. Kozak M. An analysis of vertebrate mRNA sequences: intimations of translational control. J Cell Biol. 1991; 115(4):887-903.
28. Kozak M. Determinants of translational fidelity and efficiency in vertebrate mRNAs. Biochimie. 1994; 76(9):815-821.
29. Pain VM. Initiation of protein synthesis in eukaryotic cells. Eur J Biochem. 1996; 236(3):747-771.
30. Harding HP, Zhang Y, Bertolotti A, Zeng H and Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular cell. 2000; 5(5):897-904.
31. Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD and Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Molecular cell. 2001; 7(6):1153-1163.
32. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M and Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Molecular cell. 2000; 6(5):1099-1108.
33. Benzaquen LR, Brugnara C, Byers HR, Gatton-Celli S and Halperin JA. Clotrimazole inhibits cell proliferation in vitro and in vivo. Nature medicine. 1995; 1(6):534-540.
34. Lytton J, Westlin M and Hanley MR. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J Biol Chem. 1991; 266:17067-17071.
35. Gill DL, Waldron RT, Rys-Sikora KE, Ufret-Vincenty CA, Graber MN, Favre CJ and Alfonso A. Calcium pools, calcium entry, and cell growth. Biosci Rep. 1996; 16(2):139-157.
36. Miyawaki A, Griesbeck O, Heim R and Tsien RY. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci U S A. 1999; 96(5):2135-2140.
37. Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF and Hinnebusch AG. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992; 68(3):585-596.
38. Hinnebusch AG. Gene-specific translational control of the yeast GCN4 gene by phosphorylation of eukaryotic initiation factor 2. Mol Microbiol. 1993; 10(2):215-223.
39. Chen T, Ozel D, Qiao Y, Harbinski F, Chen L, Denoyelle S, He X, Zvereva N, Supko JG, Chorev M, Halperin JA and Aktas BH. Chemical genetics identify eIF2alpha kinase heme-regulated inhibitor as an anticancer target. Nat Chem Biol. 2011; 7(9):610-616.
40. Ziegeler G, Ming J, Koseki JC, Sevinc S, Chen T, Ergun S, Qin X and Aktas BH. Embryonic lethal abnormal vision-like HuR-dependent mRNA stability regulates post-transcriptional expression of cyclin-dependent kinase inhibitor p27Kip1. J Biol Chem. 2010; 285(20):15408-15419.
41. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S and Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Molecular cell. 2001; 7(6):1165-1176.
42. Kozak M. An analysis of 5’-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987; 15(20):8125-8148.
43. Norrish AE, Skeaff CM, Arribas GL, Sharpe SJ and Jackson RT. Prostate cancer risk and consumption of fish oils: a dietary biomarker- based case-control study. Br J Cancer. 1999; 81(7):1238-1242.
44. Rhodes LE, Shahbakhti H, Azurdia RM, Moison RM, Steenwinkel MJ, Homburg MI, Dean MP, McArdle F, Beijersbergen Van Henegouwen GM, Epe B and Vink AA. Effect of eicosapentaenoic acid, an omega-3 polyunsaturated fatty acid, on UVR-related cancer risk in humans. An assessment of early genotoxic markers. Carcinogenesis. 2003; 24(5):919-925.
45. Tapiero H, Ba GN, Couvreur P and Tew KD. Polyunsaturated fatty acids (PUFA) and eicosanoids in human health and pathologies. Biomed Pharmacother. 2002; 56(5):215-222.
46. Demetri GD, Fletcher CD, Mueller E, Sarraf P, Naujoks R, Campbell N, Spiegelman BM and Singer S. Induction of solid tumor differentiation by the peroxisome proliferator- activated receptor-gamma ligand troglitazone in patients with liposarcoma. Proc Natl Acad Sci U S A. 1999; 96(7):3951-3956.
47. Sequeira SJ, Ranganathan AC, Adam AP, Iglesias BV, Farias EF and Aguirre-Ghiso JA. Inhibition of proliferation by PERK regulates mammary acinar morphogenesis and tumor formation. PloS one. 2007; 2(7):e615.
48. Bjornsti MA and Houghton PJ. Lost in translation: dysregulation of cap-dependent translation and cancer. Cancer cell. 2004; 5(6):519-523.
49. Dua K, Williams TM and Beretta L. Translational control of the proteome: relevance to cancer. Proteomics. 2001; 1(10):1191-1199.
50. Fels DR and Koumenis C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer biology & therapy. 2006; 5(7):723-728.
51. Chen L, Aktas BH, Wang Y, He X, Sahoo R, Zhang N, Denoyelle S, Kabha E, Yang H, Freedman RY, Supko JG, Chorev M, Wagner G and Halperin JA. Tumor suppression by small molecule inhibitors of translation initiation. Oncotarget. 2012; 3(8):869-881.
52. Gelev V, Aktas H, Marintchev A, Ito T, Frueh D, Hemond M, Rovnyak D, Debus M, Hyberts S, Usheva A, Halperin J and Wagner G. Mapping of the auto-inhibitory interactions of protein kinase R by nuclear magnetic resonance. J Mol Biol. 2006; 364(3):352-363.
53. Clementi E, Scheer H, Zacchetti D, Fasolato C, Pozzan T and Meldolesi J. Receptor-activated Ca2+ influx. Two independently regulated mechanisms of influx stimulation coexist in neurosecretory PC12 cells. J Biol Chem. 1992; 267(4):2164-2172.
54. Miyawaki A, Llopis J, Heim R, McCaffery JM, Adams JA, Ikura M and Tsien RY. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature. 1997; 388(6645):882-887.
55. Aktas H and Halperin JA. Translational regulation of gene expression by omega-3 fatty acids. J Nutr. 2004; 134(9):2487S-2491S.
56. Aktas H, Cai H and Cooper GM. Ras links growth factor signaling to the cell cycle machinery via regulation of cyclin D1 and the Cdk inhibitor p27KIP1. Mol Cell Biol. 1997; 17(7):3850-3857.