
INTRODUCTION
Squamous cell carcinoma of urinary bladder (SCCB), though accounting less than 10% of primary bladder carcinomas, has showed frequent relapse (>50%) and metastasis compared to urothelial carcinoma (UC) [1, 2], highlighting the complex and yet poorly understood mechanisms of SCCB.
Recently, an increasing number of studies have revealed the presence of tumor heterogeneity, which holds a challenge to cancer diagnosis and therapy. It described as modifiability within tumors, and diverse outcomes and therapeutic responses are correlated to different tumor stages, genetic lesions and expression programs [3–5]. Alternatively, cells generated from one tumor commonly comprise distinct mutations, or display diverse phenotypic, or epigenetic status [6–8]. Previous studies identified that UC mostly exhibit mixed histologies within a single tumor, with squamous components being the most common ones [9–12]. However, the definition of SCCB should be reserved exclusively for those with and only with squamous components [13]. And yet for all that, it remains unknown whether intratumoral heterogeneity takes responsibility for the treatment failure and metastasis of SCCBs [14].
Single-cell RNA-seq (scRNA-seq) analysis has been demonstrated to be an efficient method to reveal gene-expression heterogeneity and uncover characteristics of each subpopulation within a tumor at single cell level [15, 16]. In this study, we performed the single-cell tagged reverse transcription (STRT) on a SCCB case and has demonstrated the complexity of the genetic patterns within the tumor, providing a better comprehending of heterogeneous expression profile in SCCB.
RESULTS
Clinicopathology of the case
The subject was initially diagnosed with bladder tumor by computed tomography (CT) and further confirmed as SCCB by cystoscope biopsy (Supplementary Figure S1). No other organ metastasis was identified before operation by systematic examination. No neoadjuvant or nor adjuvant chemotherapy was administered pre-operation. Macroscopically, the tumor was 3.0 cm in diameter and located in posterior wall of urinary bladder. After radical cystectomy, histological examination demonstrated that the specimen infiltrated to the outside of the bladder wall and the adjacent intestinal canal was also infiltrated, with no lymph node metastasis found. The patient died six months after the operation because of intestinal metastasis.
Data description and quality control (QC)
To investigate the intratumoral heterogeneity systematically, we isolated individual cells by Fluorescence Activated Cell Sorting (FACS) from a piece of fresh radically resected and dissociated human SCCB as well as normal urothelial tissues, and performed scRNA-seq based on the STRT method (75 tumor cells, 18 normal cells and 3 negative control) [17]. The mean sequencing depth of all samples is 0.38M reads/sample (ranged from 0.01M~2.42M), which is sufficient for STRT method to detect gene expression profile. The average mapping rate of all samples is 64.87% (ranged from 28.4%~91.9%). We attached these details in Supplementary Table S1. In addition, we filtered the cells with detected genes less than 3000, retaining 67 tumor cells and 7 normal cells, respectively (Supplementary Figure S2). Principal Component Analysis (PCA) indicated no normal cell pollution in tumor cells (Supplementary Figure S3). These cells were chosen for further analysis.
Analyses of differentially expressed genes of cancer
As bulk approaches commonly fail to uncover the subtle but potentially biologically significant differences between seemingly identical cells (Figure 1a), we compared the differences between cancer and normal at single cells level by differentially expressed gene (DEG) analysis using NOISeq (Supplementary Table S2 and Supplementary Figure S4, probability > 0.999) [20]. We noticed many cell cycle correlated genes, such as CDK1 (↑), CDC26 (↑), CCND1 (↑) and SMAD3 (↓). Cancer is demonstrated as the pathological manifestation of unbounded cell division. And CCND1 is a crucial regulator of the G1 progression, regarded as a dominant positive regulator of the G1 restriction point [21]. Upregulation of CCND1 was uncovered in various cancers, indicating its potential effects on tumorigenesis process, providing a therapeutic target of this patient. In addition, to further find the pivotal gene sets that function in the tumorigenesis of SCCB, we paid attention to the significant pathways (Figure 1b) that were enriched by DEGs. Compared to the normal cells, we identified that pathways in cancer, p53 signaling, cell cycle and bladder cancer pathways were significantly enriched (Supplementary Figure S5 and S6, Supplementary Table S3). Enormous studies have proved that tumor suppressor gene TP53 and signaling pathway were imputed to the tumorigenesis and therapy resistance process and, therefore, has been regarded as a vital cellular drug target [22, 23].
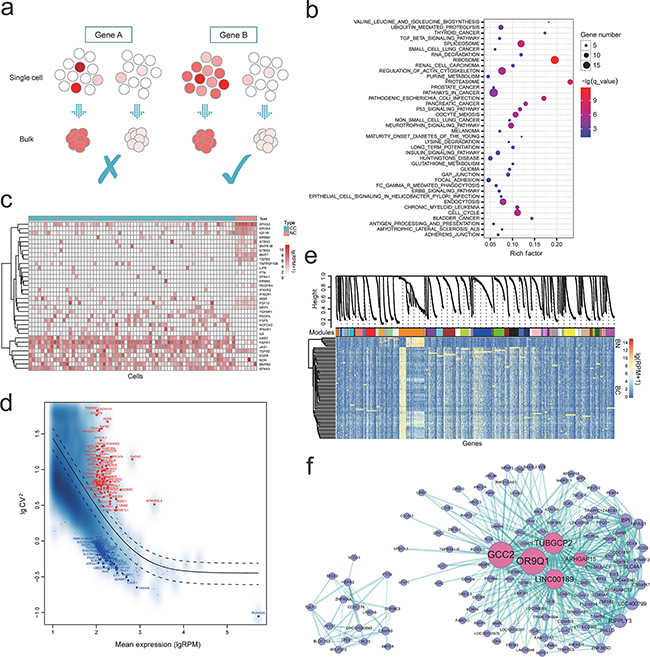
Figure 1: a. Bulk approaches were conducted on a “population level” by using the average transcriptomes of millions of cells, frequently fail to uncover the subtle but potentially biologically significant differences between seemingly identical cells, while single-cell transcriptomics will uncover the gene expression at single cell level; b. Pathway enrichment of these differentially expressed genes; c. Variation in expression of RTKs pathway in tumor and normal single cells population; d. Coefficient of variation analyses of these genes highly expressed in cancer cells; according to the variability of these highly expressed genes (RPM > 100), the most 50 variably (red) and most 50 stably (blue) expressed genes were marked, individually; e. Gene co-expression modules derived from 74 single cells based on expression level (modules are distinguished by colors); f. Hub-gene network of the “darkorange” module in e, and the size of the dots represents hubness. Pink highlights the genes being discussed in the text.
Intra-tumor heterogeneity
We first examined gene expression profile of pathways that have been widely reported in bladder cancer or squamous-cell carcinoma, such as receptor tyrosine kinases (RTKs) (Figure 1c) and epigenetic pathways [24, 25], which are important therapeutic targets for bladder cancer (Supplementary Figure S7) [26, 27], as well as the MAPK, JAK-STAT, Notch, PI3K and VEGF pathways (Supplementary Figure S8-S12). We identified obvious mosaic expression pattern of genes in these pathways, such as EGFR, TGFB2, IL6ST, BMPR2, PDGFA, FGFR1, FGF12, NOTCH2, JAG1, HSPA9, etc. These findings indicated that the expression level heterogeneity was existed across individual tumor cells of SCCB, inconsistent with the features of SCCB definition (purity component), which may compromise therapies targeting specific signaling.
Correlations between individual tumor cells from the same tumor showed a broad spread (correlation coefficient r ~ 0.15 to 0.89) (Supplementary Figure S13), consistent with intratumoral heterogeneity. However, no obvious subpopulation was identified within tumor cells. We calculated the coefficient of variation (CV) of each gene to uncover their contribution to intra-tumor heterogeneity (mean RPM > 10, Figure 1d). According to the variability of these highly expressed genes (RPM > 100), we extracted the most 100 variably and most 100 stably expressed gene sets, individually for further analyses. Recent studies showed that cell cycle is a confounding factor of expression heterogeneity [28, 29]. As expected, the variably expressed gene group contains many cell cycle related genes. In addition, six genes were significantly enriched in MAPK signaling pathway (RPS6KA1, RAC2, CACNG4, CACNA1E, CACNA1H and MAPKAPK5, p = 1.93×10-5, FDR = 3.59×10-3), which was identified as a pivotal pathway for human cancer cell survival, dissemination and resistance to drug therapy [30], suggesting the potential role of this pathway on the intratumor heterogeneity formation of SCCB. In contrast, majority of these stably expressed genes were housekeeping genes and enriched in ubiquitin mediated proteolysis, proteasome pathways, results consistent with our expectations.
Gene co-expression network analysis
To understand the co-expression profile between genes at a system level, we performed Weighted Gene Co-expression Network Analysis (WGCNA) using the expression profile of all single cells [31]. We selected 5530 genes with high variability (mean RPM > 10, SD > 100) for co-expression analysis, and detected 48 different co-expressed modules (Figure 1e). We focused on the top 5 largest modules (“darkorange”, “blue”, “brown”, “yellow” and “green”) for further analysis. Several widely reported cancer related pathways were uncovered within the five modules, such as pathways in cancer, VEGF and MAPK signaling pathways. The largest module (“darkorange”) containing highly differentially expressed genes between cancer and normal single cells, was enriched in both cancer and cell cycle pathways (p = 6.9×10-6, FDR = 5.95×10-5, Supplementary Figure S14, Supplementary Table S4) while the second largest module (“blue” in Figure 1e) were dominated by the neurotrophic signaling pathway (p = 7.27×10-9, FDR = 1.35×10-6), spliceosom (p = 1.02×10-7, FDR = 9.45×10-6) and pathways in cancer (p = 4.76, FDR = 3.49×10-6, Supplementary Figure S15, Supplementary Table S4). In addition, the pathway enrichment of the other three modules were presented in Supplementary Figure S16-S18 and Supplementary Table S4.
Hub-gene-network analysis
Hub-gene-network analysis of these modules demonstrated hierarchical organizations of highly connected genes in individual modules, through which key controlling (hub) genes in the modular network can be uncovered. Several significant hub genes of the “darkorange” module were uncovered, such as GCC2 (↓), OR9Q1 (↓), TUBGCP2 (↓), LINC00189 (↓) and ARHGAP15 (↓) (Figure 1f). With these genes rarely reported in previous cancer studies, it remains an intriguing question for future research. We recognized LINC00189, is one of the major regulators of “darkorange” module network, potentially participating in the tumorigenesis of SCCB. In addition, ArhGAP15 is a potential regulator of Rac1, a member of the Ras superfamily of GTPases involved in signaling pathways controlling cell proliferation and apoptosis [32]. Previous study identified that ArhGAP15 gene knocking out influences the apoptosis induced by ethanol in bovine fibroblast cells [33].
On the other hand, “hub genes” of four other modules contain POU2F3 (↑), CENPH (↑), PCSK6 (↑) and HERC2 (↑), BCAR3 (↑), DOHH (↑), NLK (↑), SCN2A (↑), CACNA2D3 (↓) and CTNND2 (↓), which were commonly reported related to cancer (Supplementary Figure S19). Especially, POU2F3 is a keratinocyte-specific POU transcription factor expressed in stratified squamous epithelia [34, 35], and its expression is tied to squamous epithelial stratification [36]. POU2F3 activates genes encoding cytokeratin 10 and SPRR2A, POU2F3, promoting keratinocyte proliferation and enhancing keratinocyte differentiation, and subsequently contribute to epidermal stratification [34, 37, 38]. Besides, down-regulation of POU2F3 was reported correlated to the process of both cervical intraepithelial neoplasia (CIN) and cervical cancer (CC) [39].
DISCUSSION
In the present work, we conducted scRNA-seq of individual cells generated from one tumor-normal paired SCCB case. We observed obvious differences in expression patterns of genes and pathways between cancer and normal single cells, which may contribute to SCCB processing. We revealed the intratumor heterogeneity within SCCB single cells in expression, and considered that genes encompassed in MAPK signaling pathway involved in cancer evolution and heterogeneity formation. With the combination of co-expression network modules of SCCB single cells and “hub gene” analysis, several “hub genes” were identified, such as POU2F3 (↑), NKD1 (↑) and ARHGAP15 (↓), and the expression networks could collapse when their expression was interfered.
As bulk approaches, which conducted on a “population level” by using the average transcriptomes of millions of cells, frequently fail to uncover the subtle but potentially biologically significant differences between seemingly identical cells. Single-cell transcriptomics will contribute to the reconstitution of temporal transcription networks during developmental processes [40] or when cells are exposed to external stimuli [29], either of which can be masked at the population level. Here we demonstrated the significant DEGs between cancer and normal single cells population, and identified several interesting pathways, including pathways in cancer, cell cycle and P53 signaling pathways. Cancer is usually described as a pathological manifestation of uncontrolled cell division [41]. We noticed several cell cycle related genes such as CDK1 (↑), CCND1 (↑) and SMAD3 (↓), that have been widely investigated in various cancers. Therefore, it has been anticipated for long that our understanding of the basic principles of cell cycle control would lead to effective cancer therapies.
WGCNA is used to uncover the gene co-expression modules and therapeutic targets in SCCB. Several computational methods have been proposed for combining biological pathway information and gene sets into transcriptome data analysis, such as gene set enrichment analysis (GSEA) [42]. WGCNA shares the philosophy of GSEA to concentrate on gene sets as against to individual gene, and gene sets of the modules are tracked back to the RNA-seq data by applying unsupervised clustering. The expression profiles of intramodular hub genes within an interesting module are highly correlated to dozens of targets (in present data, r2 > 0.999). Although these targets are statistically equivalent, they may differ biologically. In our data, we revealed a batch of interesting “hub genes” of each co-expression module, including GCC2 (↓), OR9Q1 (↓), LINC00189 (↓), ARHGAP15 (↓), POU2F3 (↑) and NKD1 (↑) [39, 43–45]. Some of them were commonly investigated in cancer, while others were rarely reported, such as GCC2 and LINC00189, which may hold significant role in regulating specific module networks. Of these hub genes, we noticed POU2F3 is reported as a pivotal factor in the complex regulatory network of differentiation, especially tied to squamous epithelial stratification [36]. In addition, POU2F3 was also reported might be a cancer-related tumor suppressor in both intraepithelial neoplasia and cervical cancer [39], highlighted the importance of POU2F3 in bladder squamous cell carcinoma. However, further functional validations are necessary for these hub genes, such as RNAi tests, biomarkers availability tests and druggability tests. Comprehending how broadly cancer-related modules interact with specific molecular lesions in an individual cancer patient is critical to identify new molecular targets.
Single-cell transcriptomics offers us unprecedented chance to master the transcriptional stochasticity and cellular heterogeneity, which are important for maintaining cell functions and for promoting disease progression or treatment response, while these details are commonly masked in bulk-cell studies. STRT is a PCR-based multiplexed scRNA-seq method performed on the Illumina platform, regarding as a good method to detect single cell expression profile in a high throughput and low cost way offered by the early barcoding strategy. However, because STRT quantifies transcripts through reads mapping to 5’ ends of mRNA [46], the data could not be used to reveal genetic variations, such as somatic point mutations and structural variations (gene fusion, alternative splicing, etc.). Several other limitations should also be illustrated as follow: 1) the veracity. Single cell RNA-seq comprises sequential steps of target cell isolation, RNA extraction, fragmentation, and reverse transcription. Each step introduces biases and artifacts that may influence the coverage, accuracy, and timeliness of transcript expression and therefore disrupt both the internal characterization and quantification of transcripts. It is therefore important to control the quality of the data prior to including the datasets in a meaningful global study; 2) owing to the limitation of STRT method (basing on the STRT method, we barcoded mRNA of each cell and reversed them into cDNA respectively, and then perform cDNA amplification after pool them together), we cannot validate specific genes identified by the analyses from the amplification products by qPCR. Currently, the STRT method was modified by introducing “unique molecular identifiers (UMI)” and implying on a microfluidic platform (Fluidigm C1 AutoPrep), which substantially decreased the bias of PCR and increased the stability of RNA amplification [47]. Recently, Simone Picelli et al. [48] optimized the method of Smart-seq named Smart-seq2 in terms of improved sensitivity, accuracy and full-length coverage across transcripts and decreased cost. Furthermore, Tamar Hashimshony et al. [49] made significant improvements that makes CEL-Seq2 uniquely suited to scRNA-Seq analysis in terms of economics, resolution, and ease of use. In addition to external molecule controls, improved single-cell chemistry and physics [50, 51], incorporation of molecular barcoding system [18] have significantly decreased the noise level within each study.
Taken together, we have leveraged single cell RNA-Seq to characterize heterogeneous gene expression profiles within a SCCB case and interrelated their transcriptional, functional, and genetic diversity. In addition, “hub gene” networks are uncovered by WGCNA, a powerful method in providing useful information associated with cell-type specificity. The discovery of these modules should lead to a better comprehension of the molecular features of different cell types along the differentiation and evaluation of SCCB in the future. These findings are to incur fundamental recalibration on tumor biology and therapeutic strategies.
MATERIALS AND METHODS
Human tumor specimens
Surgically resected squamous cell carcinoma of urinary bladder (SCCB) specimens were collected at Shenzhen Second People’s Hospital with approval by the Institutional Review Board. The fresh tissues (cancer and normal control specimens from one patient) were minced (5-10mm in maximum dimension) during surgery, and kept in cryopreservation medium (10% DMSO+90% DMEM medium with 30% FBS) under -80°C.
Cell isolation and mRNA sequencing
5 mL of STRT buffer (20 mM Tris-HCl at pH 8.0, 75 mM KCl, 6 mM MgCl2, 0.02% Tween-20) with 400 nM STRT-V3-T30 and 1:50,000 Life Technologies ERCC Spike-In Mix 1 were added into each well of the 96-well plate. The frozen tissues were thawed and digested into single cell suspension using 0.1% collagenase I (1mg/ml, 200U/ml) and 0.05% collagenase IV (0.1mg/ml, 20U/ml) for 2 hours under 37°C. Both single tumor cells and normal cells were sorted into the 96-well plate by Fluorescence-activated cell sorting (FACS) (75 tumor cells and 18 normal cells, 3 negative control). All of the following STRT steps including cDNA amplification and library construction were according to the protocols set by Saiful Islam et al. [17] The final cDNA library was sequenced on an Illumina HiSeq2000 using the customized sequencing primer STRT-SEQ-V3. Single-end reads of 50bp were generated along with 8-bp index reads corresponding to the cell-specific barcodes. The sequencing data from this study have been submitted to the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra) under accession no. SRP078083.
Data processing
Data analysis pipeline was basically followed the previous methods [18]. Generally, raw reads were separated into different FASTQ files by barcode, and trimmed the low-quanlity bases and polluted adapter sequence, and reads that less than 25 bp or contain more than 6 sequential “A” at 3′ site. The “valid STRT reads” were mapped to the human genome (GRCh37, http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/) using Tophat 2.0.12 with default parameters expect “--bowtie1”. Unmapped reads were discarded. Then, a homemade Perl script was used to count the reads that align to exons and in the sense orientation toward the expression value for each annotated feature in the NCBI 37.1 assembly. Finally, the reads count of each gene were normalized to Reads Per Million (RPM), which is different from the commonly used RPKM measure that we do not divided reads count by the gene length, because each mRNA gives rise to a single sequenceable fragment near its 5’ end. To exclude poor quality cells, single cells with expressed genes more than 3000 were retained for downstream analysis.
Differential expression analysis
The DEG analysis was performed between tumor cells and normal cells using NOISeq with recommended parameters (“noiseqbio” function), which can take the RPM matrix as input [19]. Genes with probability > 0.999 were extracted as confident differentially expressed genes. The “MD plot” were generated using the “DE.plot” function of NOISeq with parameters “q = 0.999, graphic = “MD”)”.
Heterogeneity analysis of single cells
Cell-to-cell similarity was quantified as Pearson’s Correlation Coefficient, and the variability of gene A expression among single cells were estimated as the coefficient of variation (CV), which was generated as the standard deviation of gene A expression divided by the mean expression of gene A among single cells.
Co-expression network analysis
To minimize the influence of the stochastic differences during the experimental and sequencing procedures, genes with a mean of RPM less than 10 or a variance of RPM less than 100 were filtered out. The co-expression networks were constructed from the single cells using the weighted co-expression network analysis (WGCNA) R package. Soft power parameter was estimated and used to derive a pairwise distance matrix for selected genes using the topological overlap measure, and the dynamic hybrid cut method with minimum module size of 30 genes was used to detect clusters. The node centrality, defined as the sum of within-cluster connectivity measures, was used to rank genes for “hubness” within each cluster. Hard threshold of edge distances were set separately to limit the numbers of edges and nodes, and the constructed network of each module were denoted using Cystoscape 3.2.1.
ACKNOWLEDGMENTS
We thank Prof. F.Q. (Fengjie Qi) for their help in confirming the pathologic figures in the present work.
CONFLICTS OF INTEREST
The authors disclose no potential conflicts of interest.
GRANT SUPPORT
The authors gratefully acknowledge the financial supports by the National Natural Science Foundation of China (Grant No. 81301740 and 81502207), 973 program (No. 2015CB755500, No. 2014CB745200, and Innovation Program of Shenzhen (Grant No. JCYJ20130401114715714; No. CXZZ20140826163906370; No. JCYJ201304011149 28183; No. JSGG20130411091246833; No. CXZZ2013051 653248144; No. 20120612131638712).
REFERENCES
1. Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994; 30: 774-8.
2. Czarnecki D, Staples M, Mar A, Giles G, Meehan C. Metastases from squamous cell carcinoma of the skin in southern Australia. Dermatology. 1994; 189: 52-4.
3. Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, Kamiyama M, Hruban RH, Eshleman JR, Nowak MA, Velculescu VE, Kinzler KW, Vogelstein B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010; 467: 1114-7. doi: 10.1038/nature09515.
4. Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, Metzeler KH, Poeppl A, Ling V, Beyene J, Canty AJ, Danska JS, Bohlander SK, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011; 17: 1086-93. doi: 10.1038/nm.2415.
5. Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008; 321: 1807-12. doi: 10.1126/science.1164382.
6. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011; 472: 90-4. doi: 10.1038/nature09807.
7. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366: 883-92. doi: 10.1056/NEJMoa1113205.
8. Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012; 488: 527-30. doi: 10.1038/nature11344.
9. Wasco MJ, Daignault S, Zhang Y, Kunju LP, Kinnaman M, Braun T, Lee CT, Shah RB. Urothelial carcinoma with divergent histologic differentiation (mixed histologic features) predicts the presence of locally advanced bladder cancer when detected at transurethral resection. Urology. 2007; 70: 69-74. doi: 10.1016/j.urology.2007.03.033.
10. Erdemir F, Tunc M, Ozcan F, Parlaktas BS, Uluocak N, Kilicaslan I, Gokce O. The effect of squamous and/or glandular differentiation on recurrence, progression and survival in urothelial carcinoma of bladder. Int Urol Nephrol. 2007; 39: 803-7. doi: 10.1007/s11255-006-9151-0.
11. Jozwicki W, Domaniewski J, Skok Z, Wolski Z, Domanowska E, Jozwicka G. Usefulness of histologic homogeneity estimation of muscle-invasive urinary bladder cancer in an individual prognosis: a mapping study. Urology. 2005; 66: 1122-6. doi: 10.1016/j.urology.2005.06.134.
12. Shah RB, Montgomery JS, Montie JE, Kunju LP. Variant (divergent) histologic differentiation in urothelial carcinoma is under-recognized in community practice: impact of mandatory central pathology review at a large referral hospital. Urol Oncol. 2013; 31: 1650-5. doi: 10.1016/j.urolonc.2012.04.009.
13. EbleJL, SauterG, EpsteinJI, SesterhennIA. Pathology and genetics of tumours of the urinary system and male genital organs. World Health Organization Classification of Tumours. 2004.
14. Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013; 501: 355-64. doi: 10.1038/nature12627.
15. Tang F, Barbacioru C, Nordman E, Bao S, Lee C, Wang X, Tuch BB, Heard E, Lao K, Surani MA. Deterministic and stochastic allele specific gene expression in single mouse blastomeres. PLoS One. 2011; 6: e21208. doi: 10.1371/journal.pone.0021208.
16. Kurimoto K, Yabuta Y, Ohinata Y, Ono Y, Uno KD, Yamada RG, Ueda HR, Saitou M. An improved single-cell cDNA amplification method for efficient high-density oligonucleotide microarray analysis. Nucleic Acids Res. 2006; 34: e42. doi: 10.1093/nar/gkl050.
17. Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P, Linnarsson S. Highly multiplexed and strand-specific single-cell RNA 5' end sequencing. Nat Protoc. 2012; 7: 813-28. doi: 10.1038/nprot.2012.022.
18. Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lonnerberg P, Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014; 11: 163-6. doi: 10.1038/nmeth.2772.
19. Tarazona S, Garcíaalcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Research. 2011; 21: 2213-23.
20. Tarazona S, Furio-Tari P, Turra D, Pietro AD, Nueda MJ, Ferrer A, Conesa A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015; 43: e140. doi: 10.1093/nar/gkv711.
21. Strauss M, Lukas J, Bartek J. Unrestricted cell cycling and cancer. Nat Med. 1995; 1: 1245-6.
22. Bellmunt J, Guix M. New agents for bladder cancer. Ann Oncol. 2010; 21 Suppl 7: vii56-8. doi: 10.1093/annonc/mdq367.
23. Liu HC, Ma F, Shen Y, Hu YQ, Pan S. Overexpression of SMAR1 Enhances Radiosensitivity in Human Breast Cancer Cell Line MCF7 via Activation of p53 Signaling Pathway. Oncol Res. 2015; 22: 293-300. doi: 10.3727/096504015X14424348426035.
24. Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, Fakhry C, Xie TX, Zhang J, Wang J, Zhang N, El-Naggar AK, Jasser SA, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011; 333: 1154-7. doi: 10.1126/science.1206923.
25. Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, Dean M, Huang Y, Jia W, Zhou Q, Tang A, Yang Z, Li X, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet. 2013; 45: 1459-63. doi: 10.1038/ng.2798.
26. Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol. 2014; 9: 1-25. doi: 10.1146/annurev-pathol-011110-130324.
27. Sebova K, Fridrichova I. Epigenetic tools in potential anticancer therapy. Anticancer Drugs. 2010; 21: 565-77. doi: 10.1097/CAD.0b013e32833a4352.
28. Wu L, Zhang X, Zhao Z, Wang L, Li B, Li G, Dean M, Yu Q, Wang Y, Lin X, Rao W, Mei Z, Li Y, et al. Full-length single-cell RNA-seq applied to a viral human cancer: applications to HPV expression and splicing analysis in HeLa S3 cells. Gigascience. 2015; 4: 51. doi: 10.1186/s13742-015-0091-4.
29. Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, Trombetta JJ, Gennert D, Gnirke A, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013; 498: 236-40. doi: 10.1038/nature12172.
30. De Luca A, Maiello MR, D'Alessio A, Pergameno M, Normanno N. The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opin Ther Targets. 2012; 16 Suppl 2: S17-27. doi: 10.1517/14728222.2011.639361.
31. Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005; 4: Article17. doi: 10.2202/1544-6115.1128.
32. Embade N, Valeron PF, Aznar S, Lopez-Collazo E, Lacal JC. Apoptosis induced by Rac GTPase correlates with induction of FasL and ceramides production. Mol Biol Cell. 2000; 11: 4347-58.
33. Ullah I, Lee HY, Kim MJ, Shah SA, Badshah H, Kim TH, Chung HJ, Yang BC, Kim MO. Rho GTPase activating protein 15 (arhGAP15) siRNA effect apoptosis-induced by ethanol in bovine fibroblast cells. Pak J Pharm Sci. 2013; 26: 605-10.
34. Andersen B, Schonemann MD, Flynn SE, Pearse RV, 2nd, Singh H, Rosenfeld MG. Skn-1a and Skn-1i: two functionally distinct Oct-2-related factors expressed in epidermis. Science. 1993; 260: 78-82.
35. Goldsborough AS, Healy LE, Copeland NG, Gilbert DJ, Jenkins NA, Willison KR, Ashworth A. Cloning, chromosomal localization and expression pattern of the POU domain gene Oct-11. Nucleic Acids Res. 1993; 21: 127-34.
36. Andersen B, Schonemann MD, Flynn SE, Pearse RV, Singh H, Rosenfeld MG. Skn-1a and Skn-1i: two functionally distinct Oct-2-related factors expressed in epidermis. Science. 1993; 262: 78-82.
37. Fischer DF, Gibbs S, van De Putte P, Backendorf C. Interdependent transcription control elements regulate the expression of the SPRR2A gene during keratinocyte terminal differentiation. Mol Cell Biol. 1996; 16: 5365-74.
38. Hildesheim J, Kuhn U, Yee CL, Foster RA, Yancey KB, Vogel JC. The hSkn-1a POU transcription factor enhances epidermal stratification by promoting keratinocyte proliferation. J Cell Sci. 2001; 114: 1913-23.
39. Zhang Z, Huettner PC, Nguyen L, Bidder M, Funk MC, Li J, Rader JS. Aberrant promoter methylation and silencing of the POU2F3 gene in cervical cancer. Oncogene. 2006; 25: 5436-45. doi: 10.1038/sj.onc.1209530.
40. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014; 32: 381-6. doi: 10.1038/nbt.2859.
41. Wayne AS, Giralt S, Kroger N, Bishop MR. Proceedings from the National Cancer Institute's Second International Workshop on the Biology, Prevention, and Treatment of Relapse after Hematopoietic Stem Cell Transplantation: introduction. Biol Blood Marrow Transplant. 2013; 19: 1534-6. doi: 10.1016/j.bbmt.2013.08.016.
42. A S, P T, VK M, S M, BL E, MA G, A P, SL P, TR G, ES L, JP M. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences. 2012; 102: 15545-50.
43. Wei X, Zhang D, Dou X, Niu N, Huang W, Bai J, Zhang G. Elevated 14,15- epoxyeicosatrienoic acid by increasing of cytochrome P450 2C8, 2C9 and 2J2 and decreasing of soluble epoxide hydrolase associated with aggressiveness of human breast cancer. BMC Cancer. 2014; 14: 841. doi: 10.1186/1471-2407-14-841.
44. Lv ZD, Zhang L, Liu XP, Jin LY, Dong Q, Li FN, Wang HB, Kong B. NKD1 down-regulation is associated with poor prognosis in breast invasive ductal carcinoma. Int J Clin Exp Pathol. 2015; 8: 4015-21.
45. Zhang S, Wang Y, Dai SD, Wang EH. Down-regulation of NKD1 increases the invasive potential of non-small-cell lung cancer and correlates with a poor prognosis. BMC Cancer. 2011; 11: 186. doi: 10.1186/1471-2407-11-186.
46. Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011; 21: 1160-7. doi: 10.1101/gr.110882.110.
47. S I, A Z, S J, G LM, P Z, M K, P L, S L. Quantitative single-cell RNA-seq with unique molecular identifiers. Nature Methods. 2014; 11: 163-6.
48. Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014; 9: 171-81. doi: 10.1038/nprot.2014.006.
49. Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O, Dor Y, Regev A, Yanai I. CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 2016; 17: 77. doi: 10.1186/s13059-016-0938-8.
50. Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Reports. 2012; 2: 666-73.
51. Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nature Reviews Genetics. 2011; 12: 87-98.