
INTRODUCTION
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer, accounting for 25% of all pediatric tumors [1]. Despite continued refinement of childhood ALL subtype classification and improved risk-based treatment strategies, survival rates remain significantly lower among high-risk patients [2]. Pediatric T-cell ALL (T-ALL) represents 10–15% of ALL cases [3] with a quarter of the patients experiencing relapse, and lower post-relapse survival compared to the more common B-lineage ALL [1]. Interestingly, a threefold higher incidence is observed among males [4], however the biological implications underlying this gender bias remain poorly understood. Despite the introduction of intensified chemotherapy protocols, very few inroads into new therapeutic approaches for these high-risk patients have been made. Recent studies [5–8] have shown that further classification of T-ALL could reveal new diagnostic markers and provide alternative targeted treatment options.
Immunophenotypic and gene expression signature analyses revealed a limited number of T-ALL subtypes based largely on differential expression of surface antigen markers and oncogene expression signatures related to stage-specific T-cell developmental arrest [3, 9, 10]. Gene fusions involving the juxtaposition of transcription factor proto-oncogenes under the control of T-cell specific enhancers located in the TCRB (7q34) or TCRA-TCRD (14q11) have been shown to be essential driver events in T-ALL and further define molecular subtypes [9]. Additional recurrent, as well as cryptic chromosomal rearrangement events that lead to T-cell specific proto-oncogene activation have also been described and some have shown prognostic significance. For instance, CALM-AF10 resulting from the t(10;11)(p13;q14-21) translocation is one of the most frequent fusion events found in 10% of childhood T-ALL cases and has been associated with poor prognosis, particularly among immature T-ALL patients [11, 12].
Recent studies have used comprehensive genomic approaches to gain further insight into the mutational landscape of T-ALL and have led to the identification of novel disease mechanisms [6, 8] and recurrent somatic alterations with pathogenic relevance. The most prevalent are constitutive activation of NOTCH1 signaling, observed in up to 60% of T-ALL patients [13], and loss of the CDKN2A/p16INK4a (chromosome 9p21) locus [14], occurring in up to 70% of cases. Loss of function mutations in FBXW7 are also frequent in T-ALL (about 15% of cases) and contribute to sustained NOTCH1 activation by preventing its proteasomal degradation in the nucleus [15]. Other frequently altered gene/pathway categories in T-ALL include signal transduction (PTEN, JAK1, JAK3, NF1, NRAS, IL7R and FLT3), transcription factors (WT1, LEF1, ETV6, GATA3 and BCL11B) as well as chromatin remodeling (EZH2, SUZ12, EED and KDM6A/UTX) [15]. Such studies have also identified a distinct, very aggressive T-ALL subtype defined by very early arrest in T-cell development [16, 17, 5]. These early T-cell precursor ALLs (ETP-ALLs) were genetically characterized by activating mutations in genes regulating cytokine and RAS signaling, inactivating mutations in hematopoietic development genes and histone-modifying genes [5].
These observations suggest that large, integrative efforts will continue to yield further insight into childhood T-ALL, particularly given that the pathogenesis of this disease and of its various subtypes cannot be entirely explained by the data currently available. In this study, we used a combination of exome and transcriptome sequencing, as well as high-density genotyping to characterize 30 childhood T-ALLs. We identified common recurrent mutations in known T-ALL genes (e.g. NOTCH1, PHF6, FBXW7 and JAK3) as well as novel somatic mutations in genes involved in RNA splicing (U2AF1), chromatin remodeling (KMT2C/MLL3) and of particular interest given the observed male gender bias, in X-linked genes MED12 and USP9X. Additional functional studies provide evidence of the potential implication of these newly identified mutations in childhood T-ALL pathogenesis. Overall, our findings indicate a need for further large-scale genomic investigations to refine patient stratification and optimize treatment strategies in childhood T-ALL.
RESULTS
The genomic landscape of childhood T-ALL
Partially overlapping data from cytogenetic analysis, whole exome and genome sequencing, ultra-deep targeted re-sequencing, and RNA sequencing were available at diagnosis for 30 childhood T-ALL patients (matched normal-tumor) and at relapse for two of these patients (Figure 1 and Table 1). Immunophenotyping and gene expression data, when available, were used to classify patients according to T-cell maturation status (Table 1, Supplementary Figure S1, Supplementary Table S1, S2 Table and Supplementary Information). Seven patients (324, 432, 706, 716, 748, 791 and 879) clustered as early immature T-ALLs, among whom 2 patients (791 and 879) showed immunophenotype and expression markers indicative of an early T-cell precursor ALL (ETP-ALL) phenotype [16]. Thirteen patients (340, 341, 437, 544, 547, 636, 647, 693, 727, 743, 744, 759 and 849) were classified as mature T-ALLs and 10 patients could not be classified due to insufficient data (Table 1).
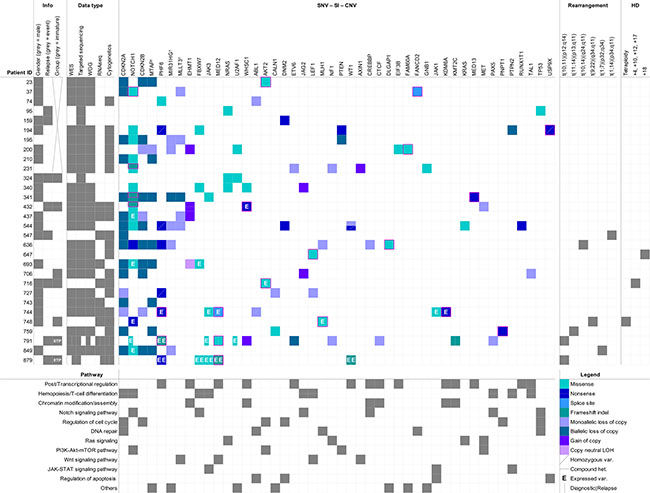
Figure 1: Overview of the SNVs, small indels and structural variations identified among 30 childhood T-ALL patients. Genes (top) are ordered according to the number of events identified in the cohort (descending order from left to right). Pathways (bottom) are ordered according to the number of genes identified as mutated (descending order from top to bottom). The ‘’Info’’ and ‘’Data type’’ sections respectively inform about the clinical data (gender, relapse status and differentiation group classification) and the type of informative data (WES, targeted sequencing, WGG, RNA-seq, cytogenetics) available for each patient. ETP-ALL cases are indicated directly in the corresponding square of the ‘’Group’’ column in the ‘’Info’’ section. SNVs and Indels unreferenced in COSMIC v72 are indicated by a pink frame and were considered as novel. The black cross stands for missing data. †: genes included in the deleted 9p region with tumor suppressor genes CDKN2A/B. WES: Whole Exome Sequencing; WGG: Whole Genome Genotyping; SNV: Single Nucleotide Variant; SI: Small Indel; CNV: Copy Number Variant; HD: Hyperdiploidy.
Table 1: Childhood T-ALL patient clinical information
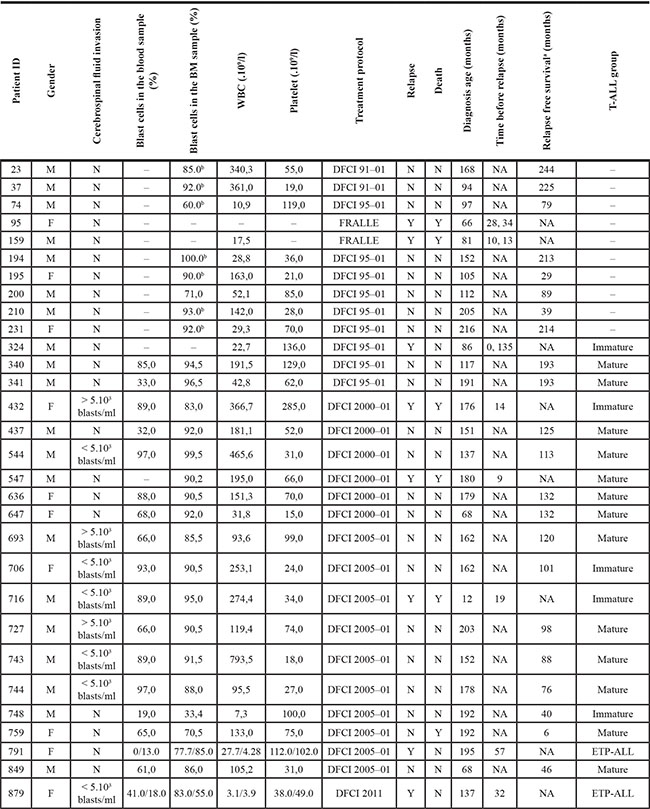
aPeriod during which the patient was followed after diagnosis and presented no relapse event. bBlast counts estimated from genotyping data analysis (Methods). BM: bone marrow; WBC: white blood cell; NA: not applicable; N: no; Y: yes; ETP: early T-cell precursor ALL; (–): missing data.
We identified several structural chromosomal abnormalities within our cohort (Figure 1 and Supplemental Information). Mature T-ALL patients 547, 759 and 636 were shown to carry translocations t(1;14)(p34;q11), t(11;14)(p13;q11) and t(10;14)(q24;q11) respectively, leading to the juxtaposition of the TAL1, LMO2 and TLX1/HOX11 oncogenes to the T-cell receptor alpha/delta (TCRA/D) at locus 14q11 [18–20]. We also identified a rarer translocation t(1;7)(p32;q34)/TRB-TAL1 in the mature T-ALL patient 849 [9] as well as the well-known t(10;11)(p12;q14) CALM-AF10 translocation in both ETP-ALL cases 791 and 879 and a t(9;22)(q34;q11.2)/BCR-ABL in the immature T-ALL patient 748 which is very rare in T-ALL (~1%) [21, 3]. Above 80% of the patients with available expression data showed activation of at least one (proto-)oncogene such as TAL1, TLX3, FLT3, LMO2, LYL1 and PIM1 with no evidence of a related fusion event (Supplementary Figure S1A and Supplementary Table S2). For example, LMO2 was shown to be upregulated in the mature T-ALL patient 547 and four early immature T-ALL patients including both ETP-ALLs (432, 748, 791 and 879), while none of these patients were identified as carriers of a LMO2 activating translocation. LYL1 was upregulated in all but one early immature T-ALL patient (716) without an associated translocation.
On average, we identified 29 somatic SNVs/indels and 37 somatic CNVs per tumor (Figure 1 and Supplementary Table S3). Based on strict filtering criteria (Methods), we identified a total of 68 candidate driver SNVs/indels (55 distinct mutations) across 28 genes among the 30 pediatric T-ALL patients and all patients harboured at least one candidate driver mutation (Figure 1 and Supplementary Table S3). RNA-seq data, when available, confirmed expression of 84% of the mutated alleles (21/25) (Figure 1, Supplementary Figure S2). ‘’Hemopoiesis/T-cell differentiation’’ was the most frequently altered pathway among the cohort with 80% of patients carrying mutations in 9 genes affecting this pathway. ‘’Post/Transcriptional regulation’’ (14 genes), ‘’Chromatin modification/assembly’’ (6 genes), ‘’Notch signaling” (6 genes) and ‘’Regulation of cell cycle’’ (6 genes) were also found to be frequently altered. 34 of the reported candidate driver mutations were previously reported (COSMIC 72) among which 29 in hematopoietic malignancies (COSMIC v72) including 26 in known T-ALL driver genes such as FBXW7, JAK1, JAK3, PHF6, KDM6A and NOTCH1. These variations were mostly clonal (mean variant allele frequency-VAF = 0.48, standard deviation-SD = 0.10), confirming their presence in the majority of tumor cells at diagnosis and their initiating role in T-ALL (Supplementary Figure S3 and Supplemental Information). Ras pathway mutations had significantly lower frequencies compared to these common drivers with a mean VAF = 0.33 (SD = 0.11) (p = 0.006, Mann-Whitney-U test) (Supplementary Figure S3 and Supplementary Information). Subclonality of these mutations corroborates previous reports [22–24] that describe a secondary role for Ras mutations in T-ALL occurring later in tumor progression. Fifteen patients (50%) harbored NOTCH1 mutations and two thirds (12/18) of identified NOTCH1 mutations were located in exons 26 and 27, coding for the extracellular heterodimerization domain and were mainly missense mutations (11/12). The remaining 6 NOTCH1 mutations were located in exon 34 coding for the C-terminal PEST (4/6) and transactivation (2/6) domains and consisted mainly of truncating mutations (5/6), as previously reported [25]. Recurrent events also involved the tumor suppressor locus 9p21 with CDKN2A/2B (p16INK4a and p15INK4b) deletions occurring in 17 (57%) of our childhood T-ALL cases (Figure 1 and Supplementary Table S3), of which 14 were biallelic. For 5/17 patients, the 9p21 deletion event included the hematopoiesis regulator MLLT3 [26], 6/17 included the long non-coding RNA MIR31HG recently shown to regulate CDKN2A expression [27], and 9/17 included the methylthioadenosine phosphorylase MTAP. Although the associations require validations in larger cohorts, patients mutated for CDKN2A had significantly less chance of relapse (p = 0.0121, Fisher’s Exact test). Interestingly, immature T-ALL patients (early immature T-ALL and ETP-ALL together) had significantly less CDKN2A alterations (p = 0.0044, Fisher’s Exact test) and a higher risk of relapse (p = 0.0072, Fisher’s Exact test).
Twenty-one variations had not previously been reported and thus were considered as novel T-ALL driver mutations, including 3 in NOTCH1 (p.N1603K, p.P2475fs and p.L2326fs) as well as novel predicted deleterious mutations in the well-known X-linked PHF6 (p.E221* and p.G226fs) and KDM6A/UTX (p.Q692*) [8, 28] (Figure 1 and Supplementary Table S3). Among these novel putative childhood T-ALL drivers, we identified a number of mutations in chromatin remodeling genes EHMT1 (n = 4), WHSC1 (n = 3) and KMT2C/MLL3 (n = 1), a recurrent missense mutation (p.R35L) in the first zinc finger of splicing factor U2AF1 (n = 3), as well as loss of ABL1 (n = 2) and in MLH1 (n = 2), both involved in DNA repair. Two patients had gain of copy of JAG2 which functions in the Notch signaling pathway, and two novel missense mutations (p.Q79K and p.R208K) were identified in the oncogenic serine/threonine-protein kinase AKT2 involved in cell cycle regulation. We also identified two novel candidate driver genes on the X chromosome: a novel nonsense mutation in the deubiquitinating protease USP9X (p.Q117*) in male patient 194; and 3 novel mutations in MED12, a member of the Mediator complex involved in regulating RNA polymerase II-dependent transcription, were found in male patient 744 (splice site mutation g.chrX:70339329T>C) and female ETP-ALL patients 791 (missense mutation p.R1989H) and 879 (frameshift insertion p.V167fs). Mutations in USP9X and MED12 presented VAFs (mean = 0.97, SD = 0.04) that were similar to the known driver mutations in PHF6 and KDM6A/UTX (mean = 0.97, SD = 0.05) (p = 1.0000) (Supplementary Figure S3 and Supplementary Information).
The novel U2AF1 p.R35L mutation alters pre-mRNA splicing in human T-cells
The novel recurrent p.R35L missense mutation was located in a zinc finger (ZnF) domain of the U2AF1 (U2 small nuclear RNA auxiliary factor 1) gene, coding for a member of the spliceosome machinery involved in pre-mRNA processing (Figure 2A and Supplementary Table S3). Three patients (200, 324 and 791) carried the predicted damaging mutation; patient 200 was unclassified and the other two were classified as early immature T-ALLs (324 and the ETP-ALL 791) and experienced relapse. An additional cohort consisting of 8 adult relapsed T-ALL patients was used for further screening of newly identified somatic driver candidates and revealed a clonal p.R35L mutation (VAF = 0.53) in the relapse genome of a 32 years old female case (patient P6, Methods and Supplementary Table S4).
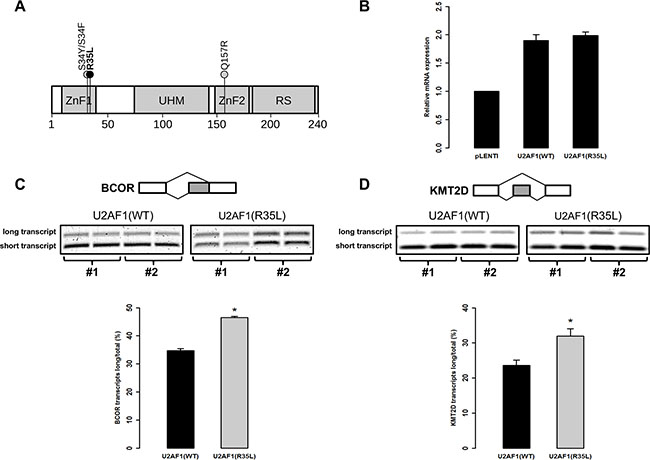
Figure 2: Mutation p.R35L in U2AF1 alters pre-mRNA splicing in human T-cells. (A) Schematic representation of U2AF1 predicted protein including zinc fingers 1 and 2 (ZnF1, ZnF2), the U2AF homology motif (UHM) and the arginine-serine rich domain (RS). The black circle indicates the location of the new mutation p.R35L identified here in 3 T-ALL cases. The grey circles indicate previously identified recurrent mutations in U2AF1. The amino acid scale is indicated at the bottom. (B) The expression of WT or R35L U2AF1 transgenes in Jurkat cells was measured by qPCR and reported to the expression of the endogeneous gene measured in cells infected with the empty vector (pLENTI). The alternative splice site utilization of BCOR (C) and KMT2D/MLL2 (D) mRNAs were measured in infected cells as previously described [32]. RT-PCRs were performed in duplicate on cDNAs obtained from two different infections (#1 and #2) of WT or R35L U2AF1 transgenes in Jurkat cells and PCR products were electrophoresed on agarose gel stained with SYBR Safe (top left). Quantification of each isoform was done by densitometry using Image J software (version 1.49) and the mean ratio of the long isoforms of BCOR and KMT2D/MLL2 reported to the total of both long and short isoforms (right). Statistical significance was determined by a two-tailed Mann-Whitney U test; P-values < 0.05 are represented by one asterisk.
The previously identified U2AF1 p.S34F mutation (Figure 2A), located in the same ZnF in myelodysplastic syndrome (MDS) patients, was shown to disrupt splicing of a number of cancer-relevant genes leading to overall dysregulation of several downstream pathways including epigenetic regulation and DNA damage response [29, 30–32]. To assess the putative impact of the novel p.R35L mutant on alternative splice site utilization, we tested for the presence of quantifiable isoforms among known U2AF1 targets (BCOR, KMT2D/MLL2, KDM6A/UTX and PICALM) [32] in human T lymphocyte (Jurkat) cells. BCOR and KMT2D/MLL2 were the only target genes with two identifiable isoforms (data not shown). Using site-directed mutagenesis, we created mutant T-cell lines overexpressing either WT or p.R35L mutated U2AF1 (Figure 2B) and showed alternative splice site usage at both BCOR and KMT2D/MLL2 loci that were specific to p.R35L (p = 0.0286 for both genes, Mann-Whitney U test) (Figure 2C). These data suggest a comparable effect for the p.R35L mutation leading to disrupted splicing and further support a role for functional U2AF1 mutations in abnormal hematopoiesis, including childhood T-ALL.
Novel X-linked drivers of T-ALL
In 8 T-ALL patients (74, 194, 544, 636, 727, 744, 791 and 879), we identified 4 known somatic mutations (p.I314T, p.R116*, p.R225*, p.Y303*) and 2 novel mutations (p.E221*, p.G226fs) in the well-known X-linked driver gene PHF6 (Figure 3A). One mature T-ALL (744) also carried a novel nonsense mutation in KDM6A/UTX (p.Q692*) which was recently characterized as a T-ALL X-linked driver gene [8, 28] (Figure 3B). We observed no gender bias in the distribution of PHF6 mutations in our cohort (p = 1.0000) with 25% (5/20) of male patients harboring mutations compared to 30% (3/10) of females. This is in line with recent observations [25, 33] that PHF6 alone is unlikely to account for the higher incidence of T-ALL among males.
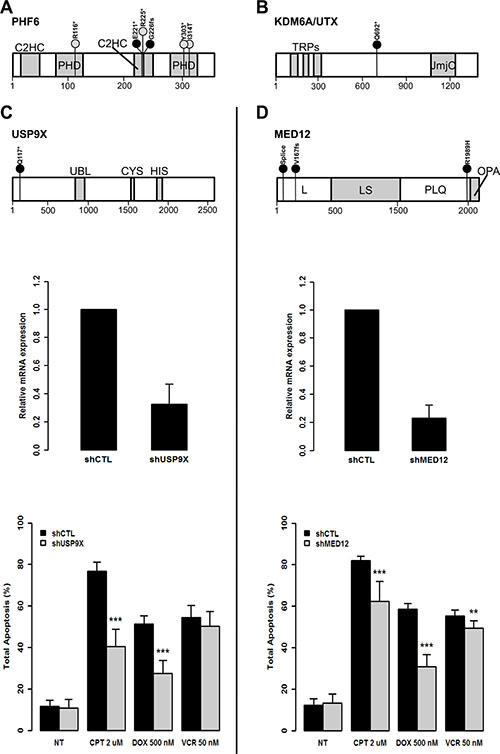
Figure 3: The loss of function of USP9X and MED12 protects from induced apoptosis in leukemic T-cells. (A) Schematic representation of PHF6 predicted protein including the two PHD type zinc finger domains (PHD) and the two CysCysHisCys type zinc finger domains (C2HC). Black circles indicate the location of new mutations. Grey circles indicate previously referenced mutations identified in this childhood T-ALL cohort. The size of the bars are representative of mutation frequencies in the cohort (PHF6 p.R225* was identified in three cases while all others were identified in one case). The amino acid scale is indicated at the bottom. (B) Schematic representation of KDM6A/UTX predicted protein including three tetratricopeptide repeat domains (TRPs) and the Jumanji C domain (JmjC). (C, top) Schematic representation of USP9X protein including the ubiquitin-like module (Ubl) and the USP-definitive cysteine and histidine box catalytic motifs (CYS and HIS). (D, top) Schematic representation of MED12 including the leucine-rich domain (L), the leucine- and serine-rich domain (LS), the proline-, glutamine-, leucine-rich domain (PQL) and the opposite paired domain (OPA). ShRNAs were used to knockdown expression of: USP9X (C, middle) and MED12 (D, middle) in Jurkat cells. Residual mRNA levels after shRNA transduction were measured by RT-qPCR. GAPDH was used as calibrator and non-mammalian shRNAs (shCTL) were used as control for normalization. In vitro apoptosis assays show overall reduced levels of apoptosis associated with knockdown of USP9X (C, bottom) and MED12 (D, bottom). DNA damage-induced apoptosis was provoked with 2 μM of Camptothecin (CPT) for 17 hours or 500 nM of Doxorubicine (DOX) for 18 hours or 50 nM of Vincristine (VCR) for 24 hours. Non-treated (NT) transduced cells were used as controls. Overall apoptosis levels were measured after 30 minutes of annexin V (AnV) propidium iodide (PI) double staining (AnV+/PI–, AnV+/PI+, and AnV–/PI+). Statistical significance was determined by a two-tailed Mann-Whitney U test; **P-values < 0.01; ***P-values < 0.001.
Our genomic investigation revealed 2 new candidate X-linked drivers of T-ALL: USP9X and MED12. For USP9X, we identified a novel truncating mutation (p.Q117*) in exon 5 carried by the unclassified male patient 194 suggesting a tumor suppressor role (Figure 3C).
As for MED12, we identified 3 novel somatic mutations (Figure 3D). One was located at the donor splice site of exon 2 (g.chrX:70339329T>C) in mature male patient 744 (Figure 4A). RNA-seq performed at diagnosis, as well as RT-PCR assays, confirmed that the mutated donor site was skipped leading to an aberrant form of the mature MED12 mRNA where exon 2 was lost (Figure 4B and 4C). This is the first MED12 mutation shown to lead to exon skipping in cancer. Interestingly, we also identified a missense mutation (p.R1989H) in exon 41 and a frameshift mutation (p.V167fs) in exon 14 of MED12 in the two female ETP-ALL cases 791 and 879, respectively. While, MED12 was previously shown to be subject to X-inactivation, it has also been shown that inactivation can be tissue-specific [34]. Through allelic expression analysis, we showed biallelic expression of a germline synonymous SNP (rs5030619) in the female case 879 (Supplementary Figure S4), suggesting that MED12 could indeed escape X-inactivation in T-ALL.
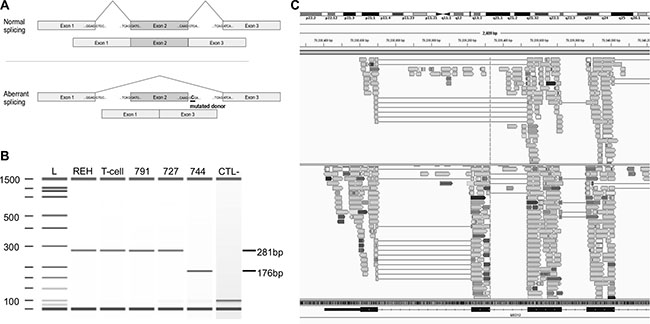
Figure 4: Mutation g.chrX:70339329T>C in MED12 causes the splicing of exon 2. (A) Schematic representation of the normal (top) and aberrant splicing (bottom) of MED12 transcripts from the normal and the identified mutant gene. In the presence of the mutation, the donor splice site of exon 2 (underlined) is skipped and exon 1 is directly spliced to exon 3, leading to an aberrant mature form of MED12 mRNA lacking exon 2. (B) Synthetic electrophoresis gel of MED12 PCR products (Methods). cDNAs were synthesized from mature RNA extracted from patient 744 mutated at g.chrX:70339329T>C, REH cells, mature T-cells (CD19-CD3+) isolated from cord blood samples and two T-ALL patients, WT for this position (791 and 727). Amplified fragments of 281 bp (WT samples) and 176 bp (mutated sample) were analyzed using Agilent 2100 Bioanalyzer. The 105 bp difference corresponds to the skipped exon 2. L: ladder; CTL-: negative control. (C) Screenshot of the Integrative Genomics Viewer (IGV) window presenting aligned RNA-seq data obtained from cases 744 (top) and a third T-ALL WT patient (693, bottom). The dotted line is centered on the donor splice site of exon 2 of MED12 (g.chrX:70339329). Case 744 shows aberrant splicing of exon 2, while 693 was WT for this position and shows a mature transcript that includes the exon 2.
To further test the impact of the loss of function of MED12 and USP9X in driving T-ALL development or maintenance, we performed in vitro small hairpin RNA (shRNA) assays in human T lymphocyte (Jurkat) cells (Figure 3C and 3D) and assessed aberrant proliferation and apoptosis resistance. While no significant changes in proliferation were observed (Supplementary Figure S5), knockdown of both genes led to a reproducible and significant reduction of apoptosis following Camptothecin (CPT) and at least one relevant chemotherapy drug treatment, Doxorubicine (DOX) or Vincristine (VCR), compared to control (Figure 3C and 3D and Supplementary Figure S6) (p = 3.375E-06 and 4.114E-05 after CPT, p = 4.095E-04 and 4.114e-05 after DOX, p = 3.767E-01 and 5.636E-03 after VCR for shUSP9X and shMED12 cells respectively, two-tailed Mann-Whitney U test). Although the observed effect might be context-dependent or cell-type specific, these results provide evidence that reduced activity of these new candidate X-linked driver genes could perturb normal T-cell development, confer a treatment resistance and point to a potential contribution to the observed gender bias in T-ALL among males [4].
DISCUSSION
In this study we performed comprehensive genomic characterization of 30 pediatric T-cell ALL patients, including 7 early immature T-ALLs (of which 2 were ETP-ALLs), 13 mature T-ALLs, and 10 patients for whom the maturation status of T-ALL cells could not be determined. We identified mutations (novel and known) and analyzed expression profiles of common driver genes, further highlighting the heterogeneity and distinct characteristics of T-ALL. We identified recurrent mutations in novel childhood T-ALL genes and functionally characterized 3 new candidate driver genes (U2AF1, MED12 and USP9X). Overall, mutation rates in this childhood T-ALL cohort were similar to those previously reported [14, 33, 35–39]. For example, 15 (50%) and 16 (53%) patients harboured NOTCH1 mutations and CDKN2A/2B deletions, respectively. Immature T-ALL patients harboured significantly less CDKN2A deletions and experienced more relapse events compared to mature T-ALL cases, as previously published [16]. Our data also suggested a correlation between CDKN2A deletions and positive outcome with only one CDKN2A-deleted patient suffering a relapse event against 15 who did not, suggesting potential clinical utility of CDKN2A for risk-based stratification of T-ALL patients.
Another recurrently mutated T-ALL gene in our cohort was PHF6. Seven patients harbored PHF6 mutations with a higher mutational rate compared to those previously reported for pediatric T-ALL (26.7% vs. 16–17.1%) [40, 33]. Interestingly, while previous analysis of the gender distribution of X-linked PHF6 mutations showed higher prevalence among males (32.0% vs. 2.5% of females) [40], we found here a slightly higher proportion of PHF6 mutated females (30.0% vs. 25% of males). The E3-ubiquitin ligase FBXW7 had a mutation frequency of 13.3% (n = 4) in our childhood T-ALL cohort, similar to previous reports (8.6–19.1%) [33, 41, 42, 35–39]. We also observed recurrent mutations in the JAK3 oncogene (13.3% vs. 5–15.3%), which is the most frequently targeted gene of the JAK-STAT pathway in T-ALL [5, 33]. Additional known T-ALL driver genes also found to be mutated in our cohort include KDM6A/UTX (3.4% vs. 4.5–14%) [33, 28], JAK1 (3.4% vs. 4.5%) [33], PTPN2 (3.4% vs. 3.6%) [33] and LEF1 (6.9% vs. 7.2–17.0%) [33, 43]. T-ALL genes that were mutated less frequently in our cohort, compared to previous studies, include WT1 (6.7% vs. 13.2–16.2%) [44, 33], PTEN (3.4% vs. 10–11.7%) [45, 46, 5, 33] and DNM2 (6.9% vs. 10.8– 18.0%) [5, 33]. And despite previous reports of childhood T-ALL [5, 8, 47] we found no mutation in the polycomb repressive complex 2 (PRC2), CNOT3, RPL10 and IL7R in our cohort.
T-ALL is characteristically more common among males. Recent studies have characterized T-ALL drivers in non-autosomal regions of chromosome X such as PHF6, RPL10 and KDM6A/UTX [40, 8, 28]. However, PHF6 and RPL10 are subject to X inactivation in females [40, 8], and KDM6A/UTX has a relatively low frequency (4.5 to 14%) in T-ALL [33, 28], therefore these genes alone cannot explain the observed skewed male:female ratio [48]. We identified additional novel X-linked candidate driver mutations in USP9X and MED12 that were shown to co-occur with PHF6 mutations in our childhood T-ALL cohort. Both genes have previously been implicated in diverse cancer types but have never been associated with T-ALL. USP9X belongs to the Ub-specific protease family and targets multiple proteins including SMURF1 [49], MCL1 [50] and Smad4 [51] and has been shown to escape X-inactivation [52]. USP9X has been demonstrated to positively regulate T-cell receptor signaling and to be required for T-cell function [53]. The oncogenic driving potential of USP9X was confirmed in several tumor types: loss of function of the gene in chronic myelogenous leukemia [54], hepatocellular [55] or colorectal carcinoma [56], bladder cancer [57] and B-cell ALL [58] leads to increased sensitivity to chemotherapy and to apoptosis. On the other hand, tumor suppressor functions have also been described in pancreatic adenocarcinoma [59, 60] and gingivo-buccal oral squamous cell carcinoma [61], highlighting the context-dependent role of USP9X in oncogenesis. The p.Q117* truncating mutation identified here in a single male patient (194) suggests a tumor suppressor role for USP9X in T-ALL, and the strong decrease in apoptosis observed following knockdown of the gene and chemotherapy drug treatment further corroborates the anti-oncogenic role of USP9X in childhood T-ALL and suggests a possible involvement on treatment resistance. As for MED12, it was altered in 10% of our T-ALL patients and was as frequently mutated as the known T-ALL gene NRAS. The loss-of-function mutations in MED12 (p.V167fs and splice site g.chrX:70339329T>C) were both shown to be expressed, supporting a functional role of these mutations in T-ALL. Somatic mutations in MED12 exon 2 splice sites have previously been identified in breast cancer and were shown to cause intron retention [62]. MED12, along with MED13, Cyclin C, and CDK8 or CDK19, is a member of the kinase module of Mediator, a multisubunit complex required for regulation of RNA polymerase II-dependent transcription [63]. In line with our observations, MED12 was recently characterized as a tumor suppressor in uterine leiomyomas, prostate cancer, chronic lymphocytic leukemia and breast fibroadenoma [64–68]. Mutations in exon 2 showed a particularly high rate of somatic alterations (70% of uterine leiomyomas) [64] and have been shown to disrupt the direct interaction of MED12 with the cyclin C-CDK8 leading to reduced Mediator activity [66]. Here, MED12 appears to escape X-inactivation. In addition we showed that loss of MED12 in human T-cells leads to decreased apoptosis levels provoked by chemotherapy drugs, further corroborating its role as tumor suppressor in childhood T-ALL and highlighting its putative relapse driving potential in leukemogenesis. This is in line with recent results showing that MED12 repression induces resistance to multiple cancer drugs through TGF-βR signaling regulation [69].
USP9X and MED12 mutations were detected in the predominant clone at diagnosis and the two patients for whom we had relapse sequencing data available showed the presence of mutations in the major clone at relapse. These results indicate an early clonal selection supporting a possible functional role in early phases of T-ALL development and a possible implication in relapse. While further investigations are required to fully decipher the mechanisms underlying the observed USP9X and MED12 induced anti-apoptotic effects, the identification of two novel X-linked tumor suppressor genes in pediatric T-ALL and their co-occurrence with known X-linked driver gene mutations (PHF6) suggests cooperating effects of X-linked mutations in T-ALL onset. The presence of several T-ALL tumor suppressor genes on the X chromosome, most of which escape X-inactivation, substantially increases the odds for males to develop the disease and could therefore explain the higher incidence of T-ALL among male children, provided that mutated forms of these proteins do not create dominant effects. However the underlying disease mechanisms associated with these alleles remains to be determined.
We also identified a novel recurrent T-ALL mutation (p.R35L) in U2AF1 in 3 patients, including 2 immature cases. And through screening of additional adult relapsed T-ALL patients we confirmed recurrence of this novel mutation even in adults. U2AF1 is a common mutational target in several cancer types, such as MDS where 11% of cases are mutated [70, 71]. The importance of the spliceosome machinery in leukemogenesis has been demonstrated [70, 72] and mutations in U2AF1 were recently shown to predict poor prognosis in patients with de novo acute myeloid leukemia [73]. The identified p.R35L mutation was previously reported in 2 cases of myeloid neoplasms [74, 29], but to the best of our knowledge, U2AF1 has never been associated with T-ALL prior to this study. U2AF1 has a U2AF homology motif allowing the heterodimerization with U2AF2 [75], an arginine-serine (RS) domain required for RNA high-affinity binding [76], and two ZnF domains. The p.R35L mutation identified here, as well as recurrent MDS mutations at residues S34 and Q157, fall within the ZnF domains. Although the role of these domains remains elusive, our functional studies support a function in the splicing mechanism with the p.R35L mutation leading to alternative splice site usage in known target genes such as the transcriptional corepressor BCOR, associated with poor prognosis in MDS when altered [77], or the H3K4 methyltransferase KMT2D/MLL2 that have been show to play a role in hematopoiesis [78]. KMT2C/MLL3 and KMT2D/MLL2 methyltransferases along with the histone demethylase KDM6A/UTX are members of the activating MLL2-KDM6A/UTX complex [79], their deregulation due to cooperating mutations or aberrant splicing events, could provide an alternative mechanism to recurrent alterations of PRC2 members EZH2, SUZ12 or EED in T-ALL. Mutations in U2AF1 typically occur early in the founding clone and are present in the major clone at diagnosis [80–82], however p.R35L was subclonal in our 3 patients. This U2AF1 mutation would appear therefore to be a late, secondary event in the developmental history of these T-ALL cases. The fact that this mutation is subclonal could explain its absence in other T-ALL studies, particularly in the event of low tumor purity or extensive intra-tumor heterogeneity. It should be noted that in ETP-ALL patient 791, p.R35L was present in a very minor subclone (VAF = 0.09) and was lost at relapse, which in this case, could render its functional role questinable. However, in addition to U2AF1 p.R35L, patient 791 also harbored a frameshift insertion p.Y816fs in the methyltransferase KMT2C/MLL3, that was carried by another subclone at diagnosis (VAF = 0.28) and presented a positive shift of frequency at relapse (VAF = 0.39). KMT2C/MLL3 and KMT2D/MLL2 methyltransferases, along with the histone demethylase KDM6A/UTX, are members of the activating MLL2-KDM6A/UTX complex involved in promoting chromatin remodeling [79]; cooperating mutations and aberrant splicing events leading to altered histone modification could contribute to disease pathogenesis.
Additional epigenetic regulators that were mutated in this childhood T-ALL cohort include: EHMT1, WHSC1, KTM2C/MLL3, CTCF, CREBBP and KDM6A/UTX. EHMT1 codes for a H3K9 methyltransferase and is a member of the E2F6 repressor complex. To the best of our knowledge this gene has never been associated with T-ALL, although previous reports demonstrated overexpression of EHMT1 associated with poor prognosis in esophageal cancer and treatment resistance in chronic myeloid leukemia [83, 84]. We did not identify loss-of-function mutations in known T-ALL genes EZH2, SUZ12 or EED, but we identified activating somatic events in the H3K27 methyltransferase WHSC1 (MMSET, NSD2). WHSC1 is a well-known and recurrent target for mutation in pediatric B-ALL as well as adult T-ALL [85, 86], however somatic disruption of WHSC1 in pediatric T-ALL is less frequent [86]. WHSC1 was mutated at higher frequency in our cohort, compared to previous reports in adult T-ALL (10.3% vs 4.9%) [86]. The gain of copy identified in the ETP-ALL case 791 as well as the recurrent p.E1099K mutation found in the mature case 340 lead to enhanced activation of WHSC1 which correlates with increased H3K36 and decreased H3K27 methylation and an open chromatin state across the genome. Activating WHSC1 mutations mimic the described PCR2 loss-of-function mutations in ALL and could alter normal lymphoid differentiation and cell survival and support an oncogenic role for WHSC1 in childhood T-ALL [87]. However the p.S231* loss of function mutation identified in the early immature patient 432 is difficult to interpret. Haploinsufficiency of WHSC1 accounts for the core phenotypes of Wolf-Hirschhorn syndrome including facial appearance, mental retardation, growth delay and seizures [88]. This stop codon mutation could simply be a passenger event in 432 or provide a specific advantage given this patient’s genetic background. The KMT2C/MLL3 frameshift mutation (p.Y816fs) in patient 791 had never been identified in hematological malignancies before. KMT2C/MLL3 haploinsufficiency was shown to impair differentiation of hematopoietic stem and progenitor cells and to provoke resistance to conventional chemotherapy [89]. This resistance might explain the emergence of subclonal KMT2C/MLL3 p.Y816fs positive cells in patient 791 at relapse. Finally, as recently reported in T-ALL [33], we identified the loss of a complete copy of the transcriptional repressor CTCF in one ETP-ALL patient, as well as the histone and non-histone acetyltransferase CREBBP in one mature case (636). CTCF was recently demonstrated to be a tumor suppressor [90] and its haploinsufficiency to lead to an increased variability in CpG methylation genome-wide. Tumors with hemizygous loss of CTCF showed increased aggressiveness, as observed here for ETP-ALL patient 791. The tumor suppressor gene CREBBP is a frequent mutational target in hematological malignancies and mutations in this gene are associated with increased risk of relapse in ALL [91], though patient 636 did not suffer relapse. Overall, these results support an important role for chromatin modification in T-ALL [92, 93, 5].
In conclusion, through integrated whole-exome, transcriptome, as well as targeted re-sequencing and genotyping investigation of 30 childhood T-ALL patients, we showed that each patient carried a unique combination of known and novel somatic alterations, including SNVs, indels, CNVs, and chromosomal rearrangements. We observed a number of uncommon and novel mutations in the early immature cases of our cohort, particularly in the two ETP-ALL patients who harbored 80% of newly-identified candidate driver mutations. We characterized a recurrent mutation in the spliceosome member U2AF1 and demonstrated its impact on alternative splicing of cancer-relevant genes, further suggesting the importance of aberrant splicing in leukemogenesis. We also identified MED12 and USP9X as putative new X-linked drivers and provided evidence of the functional impact of their loss in T-cells supporting a potential role for these genes in the male-biased sex ratio observed in T-ALL. These results further highlight the underlying complexity of the genomic landscape of T-ALL, and the pressing need of larger integrative studies in well-defined cohorts to increase understanding of the biological mechanisms that contribute to T-ALL and its various subtypes.
MATERIALS AND METHODS
Study subjects
All study subjects were French-Canadians of European descent. Incident cases were diagnosed in the Division of Hematology-Oncology at the Sainte-Justine Hospital (Montreal, Canada) as part of the Quebec childhood ALL cohort (QcALL) [94]. This childhood T-ALL cohort (n = 30) consisted of 20 males and 10 females, with a mean age at diagnosis of 11.8 years. All were classified as high-risk patients and treated accordingly under FRALLE and DFCI protocols depending on year of diagnosis (Table 1). Eight patients experienced relapse after a median time of 21 months post-induction, of which five patients did not survive post-relapse, and one case (759) was refractory to induction chemotherapy and died of cerebral hemorrhage at 6 months after diagnosis.
Whole-exome sequencing and variant identification
Whole exome sequencing (WES) was performed on 24 matched normal-tumor T-ALL patients (Figure 1). DNA was extracted from bone marrow samples (at diagnosis) and peripheral blood samples (after remission) (Table 1) using standard protocols [95]. Whole exomes were captured in solution with Agilent’s SureSelect Human All Exon (38 Mb or 50 Mb), Nextera Rapid Capture Exome Enrichment kit (case 791) or SureSelectXT Clinical Research Exome (case 879) kits according to the manufacturer’s protocol and sequenced on the Life Technologies SOLiD 4/5500 System (paired-end: 50 × 35 bp, mean coverage on targeted region = 35X) and for cases 791 and 879 (paired-end: 100 × 100 bp, mean coverage on targeted region = 120X). Reads obtained from SOLiD 4/5500 and HiSeq 2500 systems were aligned to the Hg19 reference genome using LifeScope Genomic Analysis Software and Bowtie2 (version 2.2.3) [96] respectively. PCR duplicates were removed using Picard [97]. Genotype quality score recalibration was performed using the Genome Analysis ToolKit (GATK) [98]. Sequencing metrics were obtained using the DepthOfCoverage option in GATK. After filtering out low quality reads, pileup files were created using SAMtools [99]. Somatic single nucleotide variants (SNVs) and small indels were called from pileup files using SNooPer, a highly versatile machine learning approach that uses Random Forest classification models and integrates matched normal-tumor data to accurately call somatic variants in low-depth sequencing data (Spinella et al. in revision, software available upon request).
Targeted sequencing
Ultra-deep targeted re-sequencing was performed on all candidate somatic driver mutations using the Illumina TruSeq Custom Amplicon assay as per the manufacturer’s instructions (Figure 1). Illumina DesignStudio was used to design custom oligos targeting the select mutated regions (available upon request). Genomic DNA from bone marrow at diagnosis and from blood at remission was used to validate somatic hits identified from WES (24 cases) or to screen identified variant positions in 3 additional cases with insufficient DNA for WES. PCR purification was performed with Ampure Beads for 150bp amplicon size selection. Double stranded amplicons were pooled, quantified by qPCR and sequenced on the Illumina HiSeq2500 device (paired-end: 2 × 100 bp) to reach a mean coverage of 2,500X. Reads were aligned to the Hg19 reference genome using Bowtie2 (version 2.2.3) [96]. Cleaned BAM files were used to create pileup files using SAMtools [99]. A modified version of SNooPer was used to screen mutations at the targeted positions directly in the pileup file, and to compare normal and tumoral information to confirm the somatic origin of the validated mutations (details available upon request).
Because NOTCH1 mutations were difficult to identify in the exome sequencing data due to insufficient local coverage, Sanger sequencing was performed targeting recurrently mutated regions of NOTCH1 in T-ALL [13]. These included the N terminal region of the heterodimerization (HD) domain on exon 26; the C terminal region of the HD domain on exon 27; the proline, glutamic acid, serine, threonine-rich (PEST) domain; and the C terminal region of the transcriptional activation domain (TAD) on exon 34. Primers used are listed in Supplementary Table S5. Chromatograms were analysed using the Sequencher software (Gene Codes) (Supplementary Figure S7).
An additional cohort consisting of 8 adult relapsed T-ALL patients from the Princess Margaret Cancer Centre, University Health Network (Toronto, Canada) (Supplementary Table S4) was used for further screening of newly identified somatic driver candidates in USP9X, MED12 and U2AF1. Ultra-deep targeted sequencing was performed on the 5 somatic mutations identified in these 3 genes on Illumina MiSeq system (McGill University and Génome Québec Innovation Centre); primers used are available upon request. The analysis of sequencing data was performed as described above.
RNA-sequencing and variant identification
RNA-sequencing (RNA-seq) was performed on 11 T-ALL patients (Figure 1) with suitable RNA quantity and quality. Total RNA was extracted from bone marrow samples at diagnosis for patients 432, 437, 547, 693, 716, 743, 744, 748, 791 and 849 using the mirVana Isolation kit (Ambion) according to the manufacturer’s protocol. The Allprep DNA/RNA Mini kit (Qiagen) was used for relapse samples in patients 791 and 879. For patient 744, mature RNA was also purified using the Ambion’s MicroPoly(A)Purist kit (Small Scale mRNA Purification Kit P/N AM1919). Following a DNAse I treatment, total or mature RNA samples were quantified by NanoDrop ND1000 (Thermo-Fisher Scientific) and RNA quality was assessed using the Agilent 2100 Bioanalyzer (Agilent). Ribosomal ribonucleic acid (rRNA) were depleted using the Invitrogen RiboMinus Eukaryote kit (Life Technologies). cDNA libraries were prepared using the SOLiD Total RNA-seq kit (diagnosis samples) and the Illumina TruSeq Stranded Total RNA kit (relapse samples) based on manufacturer’s protocol and sequenced on the Life Technologies SOLiD 4/5500 System (paired-end: 50 × 35 bp) or the Illumina HiSeq 2500 System (paired-end: 100 × 100 bp). Reads obtained from SOLiD 4/5500 and HiSeq 2500 systems were aligned to the Hg19 reference genome using LifeScope Genomic Analysis Software (Whole Transcriptome Analysis pipeline, default parameters) and STAR aligner (version 2.5) [100] respectively. Remaining ribosomal sequences were filtered out. Recalibration of the genotype quality scores was performed using the Genome Analysis ToolKit (GATK) [98]. Cleaned BAM files were used to create pileup files using SAMtools [99]. SNVs and small indels identified from WES were screened in RNA-seq pileup files using a modified version of SNooPer. Reads Per Kilobase per Million mapped reads (RPKM) were calculated for 22,292 genes using the R bioconductor package edgeR [101]. Heatmaps were constructed using the heatmap.2 library of the gplots R package using RPKM values of genes of interest. Breakdancer (version 1.1.2) [102] with a minimum mapping quality (q) set to 30 was used to confirm rearrangements identified by cytogenetics or to call new events from RNA-seq data.
Whole genome high-density SNV genotyping and copy number variant identification
Whole genome genotyping (WGG) data were available for 23 T-ALL patients (Figure 1). Normal and tumor samples from these patients were genotyped using Illumina’s HumanOmni 2.5-Quad or HumanOmni2.5-Octo SNP bead arrays (McGill University and Genome Quebec Innovation Centre, Montreal, Quebec). Extracted genomic DNA was processed according to the Illumina Infinium HD Assay Ultra protocol. BeadChips were imaged on Illumina’s iScan System with iScan Control Software (v3.2.45). The Genotyping Module (Version 1.9.4) of the Illumina GenomeStudio software (V2011.1) was used for raw data normalization, genotype clustering and calling, with default. ASCAT version 2.2 [103] was used to evaluate the sample purity, to evaluate tumor ploidy and to identify tumor-specific copy number variants (CNVs) or copy-neutral loss of heterozygosity (LOH).
CDKN2A gene allelic status was further evaluated in all T-ALL patients at diagnosis using PCR. Each 25 μl reaction contained 50 ng of template DNA, 1X KOD Buffer, 1.5 mM MgSO4, 200 μM dNTPs, 0.3 μM of each primer (listed in Supplementary Table S5) and 0.5U of KOD Hot Start DNA Polymerase (Millipore). Cycling parameters used were: 95°C 2 min; 40 cycles (95°C 20 sec, 58°C 10 sec, 70°C 10 sec). Amplified fragments of 368 bp were visualized using standard gel electrophoresis. Electrophoresis gels are available upon request.
Variant annotation and prioritization of cancer driver gene mutations
ANNOVAR (version 2015Jun17) [104] and Oncotator (version 1.8) [105] were used to annotate somatic splice site variants, non-synonymous SNVs and frameshift small indels. Variants were queried against publically available datasets such as 1000 Genomes [106], NHLBI GO Exome Sequencing Project (ESP) [107] and Exome Aggregation Consortium (ExAC) [108] to filter out common polymorphisms (minor allele frequency > 0.01). The Catalogue of Somatic Mutations in Cancer (COSMIC, version 72) [109] was used to evaluate prior implications in cancer. We classified each mutated gene as either tumor suppressor genes (TSGs) or oncogenes based on Vogelstein’s 20/20 rule [110] on COSMIC v72 data (Supplementary Table S3). The predicted functional impact of non-synonymous variants and small indels was assessed using Sift (version 1.03) [111], Polyphen2 (version 2.2.2) [112], MutationTaster 2 [113] and the Cancer-specific High-throughput Annotation of Somatic Mutations tool (CHASM version 3.0) [114] (Supplementary Table S3). Variants presenting a score ≤ 0.05 were considered as damaging by Sift (D). Mutations with a Polyphen2 score between 0.447 and 0.909 were predicted as possibly damaging (P) while a score > 0.909 was considered as damaging. Mutations with a MutationTaster score > 0.9 were also considered as damaging. Finally, for CHASM classification we used the Blood-Lymphocyte training set and a Benjamini and Hochberg’s adjusted false discovery rate (FDR) ≤ 0.20, to prioritize mutations based on their predicted driver potential. To identify candidate driver mutations we filtered events and kept only somatic alterations that were: i) missense mutations predicted to be driver by CHASM [114]; or to be damaging by at least two of the other three prediction algorithms; or identified as recurrent in COSMIC v72 [109]; ii) splice site and nonsense mutations that were predicted to be damaging by MutationTaster and Sift, respectively; or located in a tumor suppressor gene; or identified as recurrent in COSMIC; iii) frameshift indels and deletions located in a TSG and gain of copies located in an oncogene; iv) genes with more than one mutation (SNV, indel or CNV) in our cohort but previously never associated with pediatric T-ALL were considered as new candidate driver genes. Based on these filtering criteria, we identified somatic gene mutations with putative functional effects in driving T-ALL development, some of which we further investigated (USP9X, MED12 and U2AF1).
Cell lines
Human acute T-cell leukemia-derived Jurkat Tet-On cells (630915, Clontech) and REH human leukemia cells (#CRL-8286, A.T.C.C.) were grown in RPMI-1640 medium (Wisent) supplemented with 10% fetal bovine serum, 100 IU/ml penicillin and 100 μg/ml streptomycin (Wisent). The highly transfectable HEK293T cells were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (Wisent). Cells were routinely maintained at 37°C in a humidified atmosphere composed of 95% air and 5% CO2 and provided with fresh medium every 2 to 3 days.
RT-PCR validation of alternative splicing in MED12
Total RNA was extracted (as above) from the patient’s bone marrow at diagnosis, as well as from REH (human B leukemia) cells, mature T cells (CD3+/CD19-) isolated from cord blood samples, and from two g.chrX:70339329TT wild type (WT) patients (727 and 791), used as controls. RNA was reverse transcribed into cDNA using the Ovation® qPCR System (NuGEN Technologies). PCR were performed using KOD Polymerase as described above. Amplified fragments were analyzed on the Agilent 2100 Bioanalyzer Instrument and by Sanger sequencing (McGill University and Genome Quebec Innovation Centre).
ShRNA-mediated gene knockdown
Lentivirus-mediated gene-specific small hairpin RNAs (shRNAs) were used to knockdown expression of 2 candidate driver genes: USP9X, and MED12, in Jurkat (human T leukemia) cells. The complete list of MISSION® shRNAs (Sigma-Aldrich) used to silence target gene expression are listed in Supplementary Table S6 (at least 3 for each gene). shRNA target sequences were subcloned into either the lentiviral vector pLKO.1-puro (TRC1 version) or pLKO.5-puro (TRC2 version). Briefly, using Polyethylenimine (PEI; Polysciences), plasmids and packaging vectors (6 μg of pRSV-Rev, 7.8 μg of plasmid pMD2.VSVG, 15 μg of pMDL, and 9 μg of shRNA in pLKO-puro plasmid) were co-transfected into HEK293T cells to generate respective lentivirus. Supernatants containing lentiviruses were harvested 48 h post-transfection. 1x10E6 Jurkat cells were infected with 1 ml of supernatant in the presence of 5 μg/mL of polybrene (Sigma-Aldrich). 72 hours post-infection, cells were screened with 2 μg/μl puromycin (Sigma-Aldrich) for two weeks to select for shRNA-knockdown cells. Three biologically independent replicates were carried out for each target gene. The expression of target genes was measured by quantitative PCR (qPCR).
Total RNA was extracted from infected cells using RNeasy mini kit (Qiagen). 500 ng of total RNA were reverse transcribed using the M-MLV reverse transcriptase (Thermo Fisher Scientific) and qPCR amplifications (triplicates) were performed on the ABI PRISM 7000 Sequence Detection System (Thermo Fisher Scientific) in a total volume of 25 μl as follows: 5 μl of cDNA (diluted 1:5), 0.2 μM of each primers (listed in Supplementary Table S5), and 1X SYBR Green PCR Master Mix (Thermo Fisher Scientific). The cycling parameters were: 95°C 10 min; 40 cycles [95°C 15 sec, 62°C 1 min], followed by a denaturation curve at 60°C. GAPDH was used as reference gene. Expression values were calculated by the 2−(ΔΔCt) formula previously described [115].
Apoptosis assay
Apoptosis was measured using the Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, cells were seeded at 5x10E5 cells/mL in their culture medium and treated with 2 μM of Camptothecin for 17 hours or 500 nM of Doxorubicine for 18 hours or 50 nM of Vincristine for 24 hours, to promote DNA damage-induced apoptosis. Following incubation with Annexin V Alexa Fluor® and propidium iodide (PI) at room temperature for 30 min, stained cells were immediately analyzed by flow cytometry. The percentage of apoptotic cells was measured on the FACS Fortessa using the BD FACSDiva software (BD Biosciences) according to manufacturer’s guidelines. At least three independent experiments were carried out for each biological replicate.
U2AF1 p.R35L splicing assay
The pOTB7 plasmid containing U2AF1 cDNA was purchased from Harvard PlasmID Repository (clone # HsCD00321863) and subcloned into the Gateway compatible vector pDONR221. The R35L mutation was introduced into the cDNA sequence of U2AF1 using the QuickChange II XL Site Directed Mutagenesis kit (Agilent) with primers listed in Supplementary Table S5. Sanger sequencing (McGill University and Genome Quebec Innovation Centre) was performed to confirm the presence of the mutation. WT or R35L U2AF1 coding sequences were then subcloned into the Gateway lentiviral vector pLenti CMV Puro DEST (w118–1) using LR clonase Enzyme mix (Thermo Fisher Scientific). Lentiviruses were generated and Jurkat cells infected (two biologically independent replicates) as described above. Total RNA was extracted from infected cells using RNeasy Mini Kit and treated with RNase-Free DNase Set (Qiagen), and cDNAs were generated using M-MLV reverse transcriptase (Thermo Fisher Scientific). Overexpression of WT or R35L U2AF1 in Jurkat cells was measured by qPCR amplification as described above. To validate alternative splice site utilization at the BCOR and KMT2D/MLL2 target genes, as described elsewhere [32], RT-PCR was performed on cDNAs in duplicates using KOD Hot Start Polymerase as described previously with primers listed in Supplementary Table S5. PCR products were electrophoresed in agarose gel stained with SYBR Safe (Thermo Fisher Scientific), and quantified by densitometry using Image J software (version 1.49).
Statistical tests
Significance of observations was assessed with R using two-tailed Fisher’s exact test or Mann-Whitney-U test when appropriate.
ACKNOWLEDGMENTS
The authors are indebted to the patients and their parents for participating in this study. We thank Dr. Stéphane Gobeil (Centre Hospitalier de l’Université Laval, Quebec, QC, Canada) and Dr. Christian Beausejour (CHU Sainte-Justine, Montreal, QC, Canada) who respectively provided some of the shRNAs and the HEK293T cells used for this study. Patient tissue samples were provided by the CHU Sainte-Justine Pediatric Cancer biobank. WES and RNA-seq were performed at the Integrated Clinical Genomics Centre In Pediatrics, CHU Sainte-Justine; WGG, Sanger sequencing and part of the ultra-deep targeted sequencing were performed at the McGill University and Genome Quebec Innovation Centre; Computations were made on the supercomputer Briarée from Université de Montréal, managed by Calcul Québec and Compute Canada.
CONFLICTS OF INTEREST
The authors (JFS, PC, CR, VS, MO, SL, PSO, TS, JH, MM and DS) declare no conflicts of interest.
GRANT SUPPORT
This study was supported by research funds provided by the Terry Fox Research Institute and the Canadian Institutes for Health Research. JFS is the recipient of a Réseau de médecine génétique appliquée (RMGA) Fellowship. DS holds the François-Karl-Viau Research Chair in Pediatric Oncogenomics. The operation of the Calcul Québec supercomputer is funded by the Canada Foundation for Innovation (CFI), NanoQuébec, RMGA and the Fonds de recherche du Québec - Nature et technologies (FRQ-NT).
Authors’ contributions
DS is the principal investigator and takes primary responsibility for the paper. JFS, JH, and DS contributed to the conception and design of the study. TS performed DNA and RNA extractions for patient samples. PC, CR, SL and MO were involved in sample and library preparation for whole-exome, RNA-seq and targeted sequencing. VS and PSO contributed to data processing. JFS performed bioinformatics analysis, data integration and analysis. PC carried out functional assays. MM provided the samples of the additional cohort of adult T-ALL patients. JFS drafted the paper and JFS, JH, PC and DS contributed to interpretation of the data and were involved in critical revision of the manuscript. All authors approved the final version.
REFERENCES
1. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008; 371:1030–43.
2. Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008; 322:1377–80.
3. Ferrando AA1, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, Lander ES, Golub TR, Look AT. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002; 1:75–87.
4. Goldberg JM, Silverman LB, Levy DE, Dalton VK, Gelber RD, Lehmann L, Cohen HJ, Sallan SE, Asselin BL. Childhood T-cell acute lymphoblastic leukemia: the Dana-Farber Cancer Institute acute lymphoblastic leukemia consortium experience. J Clin Oncol. 2003; 21:3616–22.
5. Zhang J1, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012; 481:157–63.
6. Lindqvist CM, Nordlund J, Ekman D, Johansson A, Moghadam BT, Raine A, Övernäs E, Dahlberg J, Wahlberg P, Henriksson N, Abrahamsson J, Frost BM, Grandér D, et al. The mutational landscape in pediatric acute lymphoblastic leukemia deciphered by whole genome sequencing. Hum Mutat. 2015; 36:118–28.
7. Ntziachristos P, Tsirigos A, Welstead GG, Trimarchi T, Bakogianni S, Xu L, Loizou E, Holmfeldt L, Strikoudis A, King B, Mullenders J, Becksfort J, Nedjic J, et al. Contrasting roles of histone 3 lysine 27 demethylases in acute lymphoblastic leukaemia. Nature. 2014; 514:513–7.
8. De Keersmaecker K, Atak ZK, Li N, Vicente C, Patchett S, Girardi T, Gianfelici V, Geerdens E, Clappier E, Porcu M, Lahortiga I, Lucà R, Yan J, et al. Exome sequencing identifies mutation in CNOT3 and ribosomal genes RPL5 and RPL10 in T-cell acute lymphoblastic leukemia. Nat Genet. 2013; 45:186–90.
9. Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia. 2006; 20:1496–510.
10. Rothenberg EV, Moore JE, Yui MA. Launching the T-cell-lineage developmental programme. Nat Rev Immunol. 2008; 8:9–21.
11. Dreyling MH, Schrader K, Fonatsch C, Schlegelberger B, Haase D, Schoch C, Ludwig W, Löffler H, Büchner T, Wörmann B, Hiddemann W, Bohlander SK. MLL and CALM are fused to AF10 in morphologically distinct subsets of acute leukemia with translocation t(10;11): both rearrangements are associated with a poor prognosis. Blood. 1998; 91:4662–7.
12. Asnafi V, Radford-Weiss I, Dastugue N, Bayle C, Leboeuf D, Charrin C, Garand R, Lafage-Pochitaloff M, Delabesse E, Buzyn A, Troussard X, Macintyre E. CALM-AF10 is a common fusion transcript in T-ALL and is specific to the TCRgammadelta lineage. Blood. 2003; 102:1000–6.
13. Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004; 306:269–71.
14. Hebert J, Cayuela JM, Berkeley J, Sigaux F. Candidate tumor-suppressor genes MTS1 (p16INK4A) and MTS2 (p15INK4B) display frequent homozygous deletions in primary cells from T- but not from B-cell lineage acute lymphoblastic leukemias. Blood. 1994; 84:4038–44.
15. Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest. 2012; 122:3398–406.
16. Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, Cheng C, Su X, Rubnitz JE, Basso G, Biondi A, Pui CH, Downing JR, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009; 10:147–56.
17. Neumann M, Heesch S, Gökbuget N, Schwartz S, Schlee C, Benlasfer O, Farhadi-Sartangi N, Thibaut J, Burmeister T, Hoelzer D, Hofmann WK, Thiel E, Baldus CD. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012; 2:e55.
18. Xia Y, Brown L, Tsan JT, Yang CY, Siciliano MJ, Crist WM, Carroll AJ, Baer R. The Translocation (I ; I4)(p34;q I I) in Human T-cell Leukemia: Chromosome Breakage 25 Kilobase Pairs Downstream of the TALI Protooncogene. Genes Chromosomes Cancer. 1992; 4:211–6.
19. Cheng JT1, Yang CY, Hernandez J, Embrey J, Baer R. The chromosome translocation (11;14)(p13;q11) associated with T cell acute leukemia. Asymmetric diversification of the translocational junctions. J Exp Med. 1990; 171:489–501.
20. Zutter M, Hockett RD, Roberts CW, McGuire EA, Bloomstone J, Morton CC, Deaven LL, Crist WM, Carroll AJ, Korsmeyer SJ. The t(10;14)(q24;q11) of T-cell acute lymphoblastic leukemia juxtaposes the delta T-cell receptor with TCL3, a conserved and activated locus at 10q24. Proc Natl Acad Sci U S A. 1990; 87:3161–5.
21. Pui CH, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004; 350:1535–48.
22. Tartaglia M, Martinelli S, Cazzaniga G, Cordeddu V, Iavarone I, Spinelli M, Palmi C, Carta C, Pession A, Aricò M, Masera G, Basso G, Sorcini M, et al. Genetic evidence for lineage-related and differentiation stage-related contribution of somatic PTPN11 mutations to leukemogenesis in childhood acute leukemia. Blood. 2004; 104:307–13.
23. Case M, Matheson E, Minto L, Hassan R, Harrison CJ, Bown N, Bailey S, Vormoor J, Hall AG, Irving JA. Mutation of genes affecting the RAS pathway is common in childhood acute lymphoblastic leukemia. Cancer Res. 2008; 68:6803–9.
24. Irving J, Matheson E, Minto L, Blair H, Case M, Halsey C, Swidenbank I, Ponthan F, Kirschner-Schwabe R, Groeneveld-Krentz S, Hof J, Allan J, Harrison C, et al. RAS pathway mutations are highly prevalent in relapsed childhood acute lymphoblastic leukaemia, are frequently relapse-drivers and confer sensitivity to MEK Inhibition. Blood. 2013; 122:823.
25. Wang NJ, Sanborn Z, Arnett KL, Bayston LJ, Liao W, Proby CM, Leigh IM, Collisson EA, Gordon PB, Jakkula L, Pennypacker S, Zou Y, Sharma M, et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci U S A. 2011; 108:17761–6.
26. Pina C, May G, Soneji S, Hong D, Enver T. MLLT3 regulates early human erythroid and megakaryocytic cell fate. Cell Stem Cell. 2008; 2:264–73.
27. Montes M, Nielsen MM, Maglieri G, Jacobsen A, Højfeldt J, Agrawal-Singh S, Hansen K, Helin K, van de Werken HJ, Pedersen JS, Lund AH. The lncRNA MIR31HG regulates p16(INK4A) expression to modulate senescence. Nat Commun. 2015; 6:6967.
28. Van der Meulen J, Sanghvi V, Mavrakis K, Durinck K, Fang F, Matthijssens F, Rondou P, Rosen M, Pieters T, Vandenberghe P, Delabesse E, Lammens T, De Moerloose B, et al. The H3K27me3 demethylase UTX is a gender-specific tumor suppressor in T-cell acute lymphoblastic leukemia. Blood. 2015; 125:13–21.
29. Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013; 122:999–1006.
30. Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015; 25:14–26.
31. Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, Kielkopf CL, Ley TJ, Walter MJ, et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia. 2015; 29:909–17.
32. Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, Liu T, Griffith M, McGrath S, et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell. 2015; 27:631–43.
33. Vicente C, Schwab C, Broux M, Geerdens E, Degryse S, Demeyer S, Lahortiga I, Elliott A, Chilton L, La Starza R, Mecucci C, Vandenberghe P, Goulden N, et al. Targeted sequencing identifies association between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica. 2015; 100:1301–10.
34. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005; 434:400–4.
35. Clappier E, Collette S, Grardel N, Girard S, Suarez L, Brunie G, Kaltenbach S, Yakouben K, Mazingue F, Robert A, Boutard P, Plantaz D, Rohrlich P, et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia. 2010; 24:2023–31.
36. Kox C, Zimmermann M, Stanulla M, Leible S, Schrappe M, Ludwig WD, Koehler R, Tolle G, Bandapalli OR, Breit S, Muckenthaler MU, Kulozik AE. The favorable effect of activating NOTCH1 receptor mutations on long-term outcome in T-ALL patients treated on the ALL-BFM 2000 protocol can be separated from FBXW7 loss of function. Leukemia. 2010; 24:2005–13.
37. Zuurbier L, Homminga I, Calvert V, te Winkel ML, Buijs-Gladdines JG, Kooi C, Smits WK, Sonneveld E, Veerman AJ, Kamps WA, Horstmann M, Petricoin EF 3rd, Pieters R, et al. NOTCH1 and/or FBXW7 mutations predict for initial good prednisone response but not for improved outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on DCOG or COALL protocols. Leukemia. 2010; 24:2014–22.
38. Mansur MB, Hassan R, Barbosa TC, Splendore A, Jotta PY, Yunes JA, Wiemels JL, Pombo-de-Oliveira MS. Impact of complex NOTCH1 mutations on survival in paediatric T-cell leukaemia. BMC Cancer. 2012; 12:9.
39. Jenkinson S, Koo K, Mansour MR, Goulden N, Vora A, Mitchell C, Wade R, Richards S, Hancock J, Moorman AV, Linch DC, Gale RE. Impact of NOTCH1/FBXW7 mutations on outcome in pediatric T-cell acute lymphoblastic leukemia patients treated on the MRC UKALL 2003 trial. Leukemia. 2013; 27:41–7.
40. Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, De Moerloose B, Philippé J, González-García S, Toribio ML, Taghon T, Zuurbier L, Cauwelier B, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010; 42:338–42.
41. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007; 204:1813–24.
42. Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007; 204:1825–35.
43. Gutierrez A, Sanda T, Ma W, Zhang J, Grebliunaite R, Dahlberg S, Neuberg D, Protopopov A, Winter SS, Larson RS, Borowitz MJ, Silverman LB, Chin L, et al. Inactivation of LEF1 in T-cell acute lymphoblastic leukemia. Blood. 2010; 115:2845–51.
44. Tosello V, Mansour MR, Barnes K, Paganin M, Sulis ML, Jenkinson S, Allen CG, Gale RE, Linch DC, Palomero T, Real P, Murty V, Yao X, et al. WT1 mutations in T-ALL. Blood. 2009; 114:1038–45.
45. Palomero T, Sulis ML, Cortina M, Real PJ, Barnes K, Ciofani M, Caparros E, Buteau J, Brown K, Perkins SL, Bhagat G, Agarwal AM, Basso G, et al. Mutational loss of PTEN induces resistance to NOTCH1 inhibition in T-cell leukemia. Nat Med. 2007; 13:1203–10.
46. Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, Dahlberg S, Neuberg D, Moreau LA, Winter SS, Larson R, Zhang J, Protopopov A, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. 2009; 114:647–50.
47. Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, Asp P, Hadler M, Rigo I, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012; 18:298–301.
48. Aries, Gutierrez. An X-linked tumor suppressor in T-ALL. Blood. 2015; 125:3–4.
49. Xie Y, Avello M, Schirle M, McWhinnie E, Feng Y, Bric-Furlong E, Wilson C, Nathans R, Zhang J, Kirschner MW, Huang SM, Cong F. Deubiquitinase FAM/USP9X interacts with the E3 ubiquitin ligase SMURF1 protein and protects it from ligase activity-dependent self-degradation. J Biol Chem. 2013; 288:2976–85.
50. Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, Maecker H, O’Rourke K, Bazan F, Eastham-Anderson J, Yue P, Dornan D, Huang DC, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010; 463:103–7.
51. Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, Martello G, Stinchfield MJ, Soligo S, Morsut L, Inui M, Moro S, Modena N, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009; 136:123–35.
52. Zhang Y, Castillo-Morales A, Jiang M, Zhu Y, Hu L, Urrutia AO, Kong X, Hurst LD. Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol Biol Evol. 2013; 30:2588–601.
53. Park Y, Jin HS, Liu YC. Regulation of T cell function by the ubiquitin-specific protease USP9X via modulating the Carma1-Bcl10-Malt1 complex. Proc Natl Acad Sci U S A. 2013; 110:9433–8.
54. Sun H, Kapuria V, Peterson LF, Fang D, Bornmann WG, Bartholomeusz G, Talpaz M, Donato NJ. Bcr-Abl ubiquitination and Usp9x inhibition block kinase signaling and promote CML cell apoptosis. Blood. 2011; 117:3151–62.
55. Hu H, Tang C, Jiang Q, Luo W, Liu J, Wei X, Liu R, Wu Z. Reduced ubiquitin-specific protease 9X expression induced by RNA interference inhibits the bioactivity of hepatocellular carcinoma cells. Oncol Lett. 2015; 10:268–72.
56. Harris DR, Mims A, Bunz F. Genetic disruption of USP9X sensitizes colorectal cancer cells to 5-fluorouracil. Cancer Biol Ther. 2012; 13:1319–24.
57. Cui J, Sun W, Hao X, Wei M, Su X, Zhang Y, Su L, Liu X. EHMT2 inhibitor BIX-01294 induces apoptosis through PMAIP1-USP9X-MCL1 axis in human bladder cancer cells. Cancer Cell Int. 2015; 15:4.
58. Zhou M, Wang T, Lai H, Zhao X, Yu Q, Zhou J, Yang Y. Targeting of the deubiquitinase USP9X attenuates B-cell acute lymphoblastic leukemia cell survival and overcomes glucocorticoid resistance. Biochem Biophys Res Commun. 2015; 459:333–9.
59. Pérez-Mancera PA, Rust AG, van der Weyden L, Kristiansen G, Li A, Sarver AL, Silverstein KA, Grützmann R, Aust D, Rümmele P, Knösel T, Herd C, Stemple DL, et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature. 2012; 486:266–70.
60. Mann KM, Ward JM, Yew CC, Kovochich A, Dawson DW, Black MA, Brett BT, Sheetz TE, Dupuy AJ; Australian Pancreatic Cancer Genome Initiative, Chang DK, Biankin AV, Waddell N, et al. Sleeping Beauty mutagenesis reveals cooperating mutations and pathways in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2012; 109:5934–41.
61. India Project Team of the International Cancer Genome Consortium. Mutational landscape of gingivo-buccal oral squamous cell carcinoma reveals new recurrently-mutated genes and molecular subgroups. Nat Commun. 2013; 4:2873.
62. Lim WK, Ong CK, Tan J, Thike AA, Ng CC, Rajasegaran V, Myint SS, Nagarajan S, Nasir ND, McPherson JR, Cutcutache I, Poore G, Tay ST, et al. Exome sequencing identifies highly recurrent MED12 somatic mutations in breast fibroadenoma. Nat Genet. 2014; 46:877–80.
63. Malik S, Roeder RG. The metazoan Mediator co-activator complex as an integrative hub for transcriptional regulation. Nat Rev Genet. 2010; 11:761–72.
64. Mäkinen N, Mehine M, Tolvanen J, Kaasinen E, Li Y, Lehtonen HJ, Gentile M, Yan J, Enge M, Taipale M, Aavikko M, Katainen R, Virolainen E, et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science. 2011; 334:252–5.
65. Je EM, Kim MR, Min KO, Yoo NJ, Lee SH. Mutational analysis of MED12 exon 2 in uterine leiomyoma and other common tumors. Int J Cancer. 2012; 131:E1044–7.
66. Turunen M, Spaeth JM, Keskitalo S, Park MJ, Kivioja T, Clark AD, Mäkinen N, Gao F, Palin K, Nurkkala H, Vähärautio A, Aavikko M, Kämpjärvi K, et al. Uterine leiomyoma-linked MED12 mutations disrupt mediator-associated CDK activity. Cell Rep. 2014; 7:654–60.
67. Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, Nickerson E, Chae SS, Boysen G, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012; 44:685–9.
68. Kämpjärvi K, Park MJ, Mehine M, Kim NH, Clark AD, Bützow R, Böhling T, Böhm J, Mecklin JP, Järvinen H, Tomlinson IP, van der Spuy ZM, Sjöberg J, et al. Mutations in Exon 1 highlight the role of MED12 in uterine leiomyomas. Hum Mutat. 2014; 35:1136–41.
69. Huang S, Hölzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U, Garnett M, Grernrum W, Sun C, Prahallad A, Groenendijk FH, Mittempergher L, Nijkamp W, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell. 2012; 151:937–50.
70. Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, McLellan MD, Dooling DJ, Abbott RM, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2011; 44:53–7.
71. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478:64–9.
72. Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, Ramsay AJ, Beà S, Pinyol M, Martínez-Trillos A, López-Guerra M, Colomer D, Navarro A, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2011; 44:47–52.
73. Hou HA, Liu CY, Kuo YY, Chou WC, Tsai CH, Lin CC, Lin LI, Tseng MH, Chiang YC, Liu MC, Liu CW, Tang JL, Yao M, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget. 2016; 7:9084–101. doi: 10.18632/oncotarget.7000.
74. Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, Sekeres MA, Padgett RA, Tiu RV, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012; 119:3203–10.
75. Zamore PD, Green MR. Identification, purification, and biochemical characterization of U2 small nuclear ribonucleoprotein auxiliary factor. Proc Natl Acad Sci. 1989; 86: 9243–7.
76. Rudner DZ, Breger KS, Kanaar R, Adams MD, Rio DC. RNA binding activity of heterodimeric splicing factor U2AF: at least one RS domain is required for high-affinity binding. Mol Cell Biol. 1998; 18:4004–11.
77. Damm F, Chesnais V, Nagata Y, Yoshida K, Scourzic L, Okuno Y, Itzykson R, Sanada M, Shiraishi Y, Gelsi-Boyer V, Renneville A, Miyano S, Mori H, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013; 122: 3169–77.
78. Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015; 15:334–46.
79. Van der Meulen J, Speleman F, Van Vlierberghe P. The H3K27me3 demethylase UTX in normal development and disease. Epigenetics. 2014; 9: 658–68.
80. Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, Yoshida K, Roller A, Nadarajah N, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014; 28:241–7.
81. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, Shlien A, Groves MJ, Forbes SA, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013; 122:3616–27.
82. Walter MJ, Shen D, Shao J, Ding L, White BS, Kandoth C, Miller CA, Niu B, McLellan MD, Dees ND, Fulton R, Elliot K, Heath S, et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia. 2013; 27:1275–82.
83. Guan X, Zhong X, Men W, Gong S, Zhang L, Han Y. Analysis of EHMT1 expression and its correlations with clinical significance in esophageal squamous cell cancer. Mol Clin Oncol. 2014; 2:76–80.
84. Loh SW, Ng WL, Yeo KS, Lim YY, Ea CK. Inhibition of euchromatic histone methyltransferase 1 and 2 sensitizes chronic myeloid leukemia cells to interferon treatment. PLoS One. 2014; 9:e103915.
85. Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang HE, Taylor JE, Hu M, Englund NP, Yan F, Wang Z, Robert McDonald E 3rd, et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 2013; 45:1386–91.
86. Neumann M, Vosberg S, Schlee C, Heesch S, Schwartz S, Gökbuget N, Hoelzer D, Graf A, Krebs S, Bartram I, Blum H, Brüggemann M, Hecht J, et al. Mutational spectrum of adult T-ALL. Oncotarget. 2015; 6:2754–66. doi: 10.18632/oncotarget.2218.
87. Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, Allegretta M, Okot-Kotber CI, Patel JP, Melnick A, Levine RL, Ferrando A, Mackerell AD Jr, et al. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia. 2014; 28:198–201.
88. Andersen EF, Carey JC, Earl DL, Corzo D, Suttie M, Hammond P, South ST. Deletions involving genes WHSC1 and LETM1 may be necessary, but are not sufficient to cause Wolf-Hirschhorn Syndrome. Eur J Hum Genet. 2014; 22:464–70.
89. Chen C, Liu Y, Rappaport AR, Kitzing T, Schultz N, Zhao Z, Shroff AS, Dickins RA, Vakoc CR, Bradner JE, Stock W, LeBeau MM, Shannon KM, et al. MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell. 2014; 25:652–65.
90. Kemp CJ, Moore JM, Moser R, Bernard B, Teater M, Smith LE, Rabaia NA, Gurley KE, Guinney J, Busch SE, Shaknovich R, Lobanenkov VV, Liggitt D, et al. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 2014; 7:1020–9.
91. Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J, Buetow KH, Pui CH, Baker SD, et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011; 471:235–9.
92. Andersson AK, Ma J, Wang J, Chen X, Gedman AL, Dang J, Nakitandwe J, Holmfeldt L, Parker M, Easton J, Huether R, Kriwacki R, Rusch M, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015; 47:330–7.
93. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Böttcher S, Carter SL, Cibulskis K, Mertens D, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015; 526:525–30.
94. Healy J, Bélanger H, Beaulieu P, Larivière M, Labuda D, Sinnett D. Promoter SNPs in G1/S checkpoint regulators and their impact on the susceptibility to childhood leukemia. Blood. 2007; 109:683–92.
95. Baccichet A, Qualman SK, Sinnett D. Allelic loss in childhood acute lymphoblastic leukemia. Leuk Res. 1997; 21:817–23.
96. Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012; 9:357–9.
97. Picard. Broadinstitute. 2016. [cited 5 Jan 2016]. Available: http://broadinstitute.github.io/picard/.
98. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–303.
99. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009; 25:2078–9.
100. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21.
101. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–40.
102. Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, Shi X, Fulton RS, Ley TJ, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009; 6:677–81.
103. Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W, Weigman VJ, Marynen P, Zetterberg A, Naume B, Perou CM, Børresen-Dale AL, Kristensen VN. Allele-specific copy number analysis of tumors. Proc Natl Acad Sci U S A. 2010; 107:16910–5.
104. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010; 38:e164.
105. Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, Getz G. Oncotator: cancer variant annotation tool. Hum Mutat. 2015; 36:E2423–9.
106. 1000 Genomes Project Consortium, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010; 467:1061–73.
107. Exome Variant Server. NHLBI Exome Sequencing Project (ESP). 2016. [cited 5 Jan 2016]. Available: http://evs.gs.washington.edu/EVS/.
108. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–91. doi: 10.1038/nature19057.
109. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet. 2008; Chapter 10:Unit 10.11.
110. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz Jr. LA, Kinzler KW. Cancer Genome Landscapes. Science. 2013; 339:1546–58.
111. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4:1073–81.
112. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7:248–9.
113. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014; 11:361–2.
114. Carter H, Chen S, Isik L, Tyekucheva S, Velculescu VE, Kinzler KW, Vogelstein B, Karchin R. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009; 69:6660–7.
115. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402–8.