
INTRODUCTION
The extremely poor outcomes associated with pancreatic ductal adenocarcinoma (PDAC) are related to its high frequency of genetic mutations, existence of interdependent signaling pathways, and the presence of a dense stromal compartment encapsulating the tumor [1, 2] [3]. Elevated tumor interstitial fluid pressure and decreased vascularity is partly responsible for the poor penetration and delivery of therapeutic agents in PDAC [4]. Emerging evidence indicates that the extensive stroma of PDAC not only impedes drug delivery, but also plays a role in promoting tumor growth, facilitating invasion and metastasis, and mediating therapeutic resistance of PDAC [5, 6]. Recent evidence indicates that crosstalk between the tumor and stromal elements of the tumor microenvironment (TME) mediates cancer progression and metastasis [7]. The pancreatic tumor-associated stroma consists of several cell types, including immune cells, endothelial cells and stellate cells. These stromal cell types display a dynamic interaction with each other, as well as with PDAC cells [5, 6]. Precursor pancreatic intraepithelial neoplasia (PanIN) lesions are known to progress and develop into invasive PDAC. A reactive stroma accompanies this transition from PanIN, resulting in a fibrotic desmoplasia with a hallmark of immune cell infiltration [8]. However, the mechanism through which tumor-associated stromal cells promote the growth and progression of PDAC from PanINs is not well understood.
The myofibroblast-like pancreatic stellate cells (PSCs) reside in a quiescent state in the normal pancreas, but transition to an activated state under pathological conditions such as inflammation or cancer [5, 6, 9, 10]. Numerous studies have shown that PDAC is often associated with the infiltration of immune cells that creates an inflamed microenvironment enriched with growth factors, cytokines, endotoxins, a hypoxic TME and an increased oxidant stress level, all of which contribute to the activation of PSCs [11, 12]. Yet, how PSCs activate intercellular signaling in PDAC cells remains to be elucidated.
Cytokines represent therapeutically targetable mediators of PDAC progression. Specifically, IL-6 is a multifunctional cytokine that has been shown to be elevated in the serum of patients with PDAC [13] and confirmed to promote PDAC development in a mouse model [14]. STAT3, a downstream signaling mediator of IL-6, is activated in stromal and myeloid cells and is a critical element in the formation of a pre-metastatic niche, leading to increased metastasis [15–17]. We have previously shown that the activated STAT3 correlates with poor prognosis in patients with PDAC [18–20]. Previous studies have shown that IL-6 from macrophages and myeloid cells in the TME activate STAT3 signaling in PDAC [14] [21]. We have demonstrated a mechanistic rationale for activated STAT3 as a biomarker of therapeutic resistance in PDAC [1, 2]. The level of STAT3 expression in PDAC cell lines correlates with resistance to treatment, and the level of phosphorylated STAT3 expression in human pancreatic tissues correlates positively with PDAC progression and negatively with overall survival [20]. These data confirm the functional role of activated STAT3 signaling in the development of PDAC.
The purpose of this study was to determine the influence of cytokine-induced signaling between PSCs and neoplastic cells at the earliest stages of pancreatic cancer development. This study demonstrates for the first time that PSCs actively secrete IL-6, which in turn activates STAT3 signaling in PanIN cells, and promotes the invasive and tumorigenic capacity of these pancreatic precursor lesions as well as already established PDAC cells.
RESULTS
Phosphorylation of STAT3 is increased with the progression of PDAC
The baseline expression of total and phospho-STAT3 (pSTAT3) (Figure 1, left panel) was examined in a human pancreatic stellate cell line (hPSC), immortalized pancreatic ductal epithelial cell line HPDE6-E6E7 (H6c7), and in PDAC cell lines (BxPC3 and PANC1) (Figure 1A). Mouse pancreatic stellate cells (mPSC) and mouse PanIN cells from KC (LSL-KrasG12D/+; Pdx1Cre/+) mice, primary PDAC (PDA) and liver metastasis (LMP) cell lines derived from the KPC (LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+) mice [22] were also examined for baseline total and pSTAT3 expression (Figure 1A, right panel). As shown in Figure 1A, pSTAT3 levels were low to undetectable in normal and benign pancreatic epithelial cells and PanIN cells, but increased with the progression of pancreatic neoplasia from PanIN cells to primary PDAC to liver metastasis in both human and mouse cell lines.
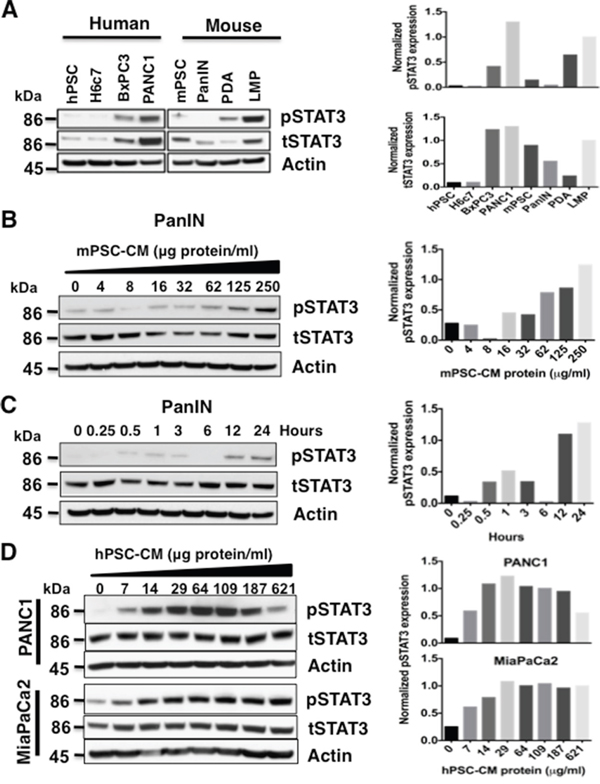
Figure 1: Pancreatic stellate cell (PSC) conditioned media (CM) activates STAT3. A. Baseline STAT3 level was obtained by immunoblotting total cell lysates from human (left panel) PSC (hPSCs), pancreatic ductal cells (H6c7), and PDAC cells lines (BxPC3, PANC1), as well as mouse (right panel) PSC (mPSCs), PanIN, PDA and liver metastasis (LMP) cell lines. Only the tumor cell lines expressed high levels of pSTAT3 at baseline. Densitometry analyses for the immunoblots from pSTAT3 (top panel) and tSTAT3 (bottom panel) after normalization to actin protein is shown to the right. B and C. Serial exposure to concentrated mPSC-CM results in activation of STAT3 in mouse PanIN cells in a dose- (B) and time- (C) dependent manner. Densitometry analyses of (B) and (C) pSTAT3 immunoblots normalized to tSTAT3 are shown in the graphs to the right. D. PANC1 and MiaPaCa2 cells also demonstrate a dose- dependent increase in STAT3 activation when exposed to hPSC-CM (left panel). Densitometry analyses of pSTAT3 immunoblots normalized to tSTAT3 for PANC1 (top panel) and MiaPaCa2 (bottom panel) cells are displayed in graphs to the right.
Pancreatic stellate cell - conditioned medium (PSC-CM) protein activates STAT3 signaling in PanIN and PDAC cells
PSCs are known to produce numerous cytokines, which in turn can regulate oncogenic signaling pathways. To study the role of PSCs in activating STAT3 signaling, mouse PanIN cells were exposed to mouse PSC-CM (mPSC-CM) concentrates in a dose- (0-250 μg protein/ml) and time- (0-24 hours, 50 μg protein/ml) dependent manner (Figure 1B and 1C). Phosphorylation of STAT3 in these cell lysates was determined by western blot. Serial exposure to concentrated mPSC-CM in PanIN cells and human PSC-CM (hPSC-CM, 0-621 μg/ml) in human PDAC PANC1 and MiaPaCa2 cells increased pSTAT3 levels in both a concentration (Figure 1B and 1D) and time-dependent (Figure 1C and Supplementary Figure S1) manner. Downregulation of pSTAT3 expression in PANC1 cells treated with 621 μg protein/ml hPSC-CM protein occurred in the last lane, suggesting an effective cell type specific concentration window.
PSC secreted IL-6 and serum IL-6 is increased in Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) and LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+ (KPC) mice
PSCs secrete factors that can support the growth and survival of tumor cells [23]. Cytokine array analysis of supernatants from PSCs showed significantly higher concentrations (1000-fold) of IL-6 compared with other cytokines (data not shown). IL-6 activates STAT3 signaling [24, 25]. We therefore studied the effects of PSC secreted IL-6 on STAT3 signaling in PanIN and PDAC cells. IL-6 levels were measured by ELISA in serially diluted samples of conditioned media (CM) collected from human and mouse PSCs (Figure 2). The concentration of PSC secreted IL-6 increased in concordance with the concentration of total protein content in the conditioned media. Overall, mPSC-CM (Figure 2A) demonstrated lower concentration ranges of IL-6 than hPSC-CM (Figure 2B) relative to total protein level. Exposure to higher concentrations of PSC-CM protein and therefore higher IL-6 concentrations, resulted in increased expression of pSTAT3 in PDAC cells (Figure 1D), except in PANC1 cells when pSTAT3 expression peaked at a relative IL-6 concentration of 64 μg/ml compared with 621 μg/ml in MiaPaCa2 cells.
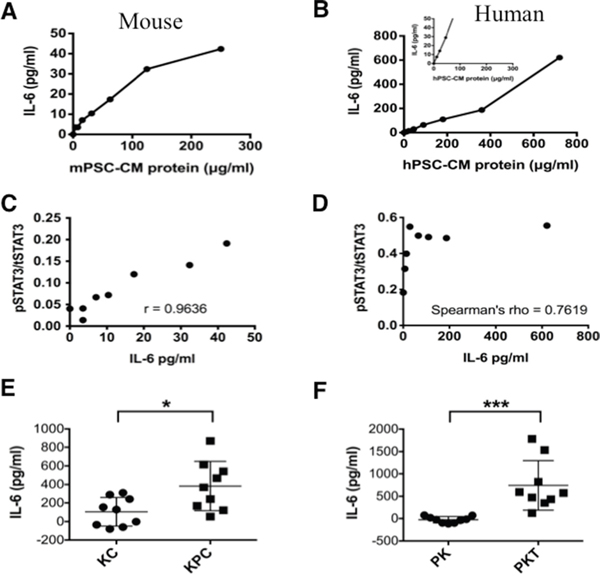
Figure 2: IL-6 concentration in mouse and human PSC-CM. Levels of IL-6 were measured by ELISA relative to total protein levels in serially diluted CM secreted from mPSCs A. and hPSCs B. Inset for hPSCs (B) shows the same concentration range as provided for the mPSCs (A). The Relationship between recombinant IL-6 concentration and STAT3 activation; C. The ratio of pSTAT3 to tSTAT3 levels in mouse PanIN cells was determined by quantifying pSTAT3 and tSTAT3 luminescence on the immunoblots from Figure 1B (ImageJ) and performing a Pearson’s correlation (p < 0.001). D. The relationship between IL-6 concentration and ratio of pSTAT3 to tSTAT3 levels in human MiaPaCa2 cells was determined by quantifying pSTAT3 and tSTAT3 luminescence on the immunoblots from Figure 1C (ImageJ) and performing a Spearman’s correlation (p < 0.001). E. and F. In vivo analysis of IL-6 from the serum collected from LSL-KrasG12D/+; Pdx1Cre/+ (KC) and LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+ (KPC) mice (E) or Ptf1aCre/+; LSL-KrasG12D/+ (PK) and Ptf1aCre/+; LSL-KrasG12D/+; Tgfbr2flox/flox (PKT) mice (F). Serum from 3 mice was analyzed in triplicates (n=9). * – p<0.05; *** – P<0.001.
Exposure of mouse PanIN cells to IL-6 resulted in a significant concentration-dependent positive linear association between the pSTAT3/tSTAT3 ratio and IL-6 concentration (Pearson’s Correlation; r = 0.9636, p < 0.001, Figure 2C). MiaPaCa2 cells, which have a high baseline expression of pSTAT3 [20], also exhibited a significant, but nonlinear, dose response relationship between IL-6 exposure and pSTAT3/tSTAT3 ratio (Spearman’s rho = 0.7619, p = 0.028, Figure 2D).
To further determine the systemic effects of IL-6 in the progression of pancreatic neoplasia, we compared the level of serum IL-6 in KC and PK mice (without PDAC) with those of KPC and PKT mice (with PDAC) respectively. Serum IL-6 levels were significantly higher in KPC (Figure 2E) and PKT (Figure 2F) mice when compared with their respective KC and PK control mice. In Figure 1A (right panel) we show that PDA and LMP lines derived from KPC mice have increased pSTAT3 expression compared with PanIN cells derived from KC mice, further corroborating the roles of IL-6 and activated STAT3 signaling in the progression of PDAC from PanINs.
IL-6 secreted from PSCs activates STAT3 signaling in PDAC cells
To gain further insight into the ability of PSC secreted IL-6 to act as a critical mediator driving STAT3 activation in PDAC, PANC1 and BxPC3 cells were exposed to hPSC-CM with and without an IL-6 neutralizing antibody or the Jak/STAT3 inhibitor AZD1480. Pre-treatment of human PDAC cells with AZD1480 inhibited hPSC-CM (100μg protein/ml) mediated phosphorylation of STAT3 (Figure 3A). Treatment of hPSC-CM with an IL-6 neutralizing antibody effectively reduced the IL-6 concentration in the PSC-CM to IL-6 concentrations seen in serum-free control medium (Supplementary Figure S2). Exposure of IL-6 antibody-depleted hPSC-CM to PDAC cells also substantially reduced hPSC-CM mediated phosphorylation of STAT3 (Figure 3B). These results indicate PSC secreted IL-6 activates STAT3 signaling in PDAC cells.
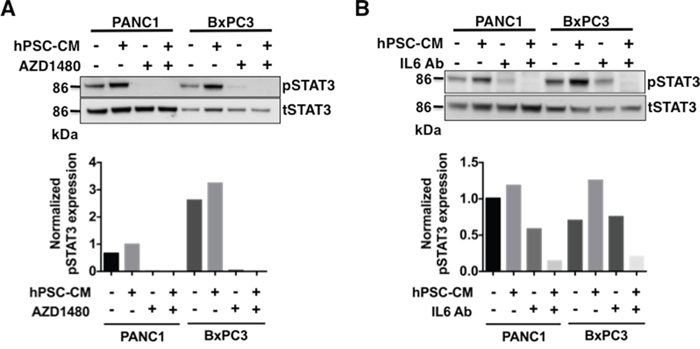
Figure 3: Pharmacological inhibition of JAK/STAT3 signaling or blocking IL-6 inhibits phosphorylation of STAT3 in hPSC-CM protein PDAC treated cells. PANC1 and BxPC3 cells were treated with hPSC-CM with or without JAK/STAT3 inhibitor AZD1480 (100 nmol/L) A. or IL-6 neutralizing antibody B. At the end of the study, cell lysates were analyzed for total STAT3 and phospho-STAT3 levels by immunoblot analysis. Densitometry analyses of pSTAT3 normalized to tSTAT3 was shown in the bottom panels of A and B. AZD1480 or IL-6 Ab treatment inhibited hPSC-CM induced activation of STAT3.
Neutralization of IL-6 abrogates PSC-CM induced cell invasion and anchorage independent growth
STAT3 activation enhances the invasive ability of tumor cells [14, 26]. To determine if IL-6-mediated activation of STAT3 was able to enhance invasive ability of PDAC cells, PANC1 and BxPC3 cells were seeded in the upper chamber of a matrigel coated transwell filter with the lower chambers filled with hPSC-CM (100μg protein/ml) or control medium in the presence or absence of IL-6 neutralizing antibody (1:400 dilution). Exposure to hPSC-CM significantly enhanced invasiveness of PANC1 (Figure 4A) and BxPC3 (Supplementary Figure S3) cells (p<0.05), while antibody mediated neutralization of IL-6 inhibited this effect to baseline levels (p<0.05). These data support a role for PSC secreted IL-6 as a critical factor associated with enhanced cellular invasion of PDAC cells.
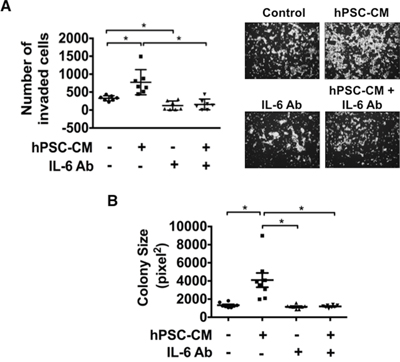
Figure 4: Blocking IL-6 attenuates hPSC-CM induced cell invasion and colony size. PANC1 cells were treated with hPSC-CM (100μg total protein/ml) with or without IL-6 neutralizing antibody (IL-6 Ab). Cell invasion A. and colony size B. were analyzed as detailed in Materials and Methods. Cell invasion images demonstrate representative pictures from each treatment group (A, right panel). Cell invasion results represent the average number of cells in seven fields in triplicate inserts. Colony size results are represented by eight photographs analyzed from triplicate wells. * – P < 0.05.
In order to assess whether PSC-CM stimulates anchorage-independent PDAC growth, soft agar colony formation assays were conducted using PANC1 cells treated with serum free control medium or hPSC-CM (100μg protein/ml) with or without the addition of IL-6 neutralizing antibody (Figure 4B). Exposure to hPSC-CM significantly increased the size of colonies formed while blockade of IL-6 with the neutralizing antibody significantly reduced hPSC-CM induced colony size to baseline levels (p<0.05), suggesting that PSC secreted IL-6 plays a role in supporting tumor growth.
PSC secreted IL-6 stimulates cell invasion of precursor mouse PanIN cells in a STAT3 dependent manner
PanINs are precursor lesions of PDAC. Having shown that PSC secreted IL-6 enhances the tumorigenicity of PDAC cells via STAT3 signaling, we next sought to determine if IL-6 exposure can stimulate the invasive capabilities of non-invasive PanIN cells. Exposure of PanIN cells to mPSC-CM significantly enhanced cell invasion (Figure 5A). Interestingly, the enhanced and sustained phosphorylation of STAT3 seen with exposure to mPSC-CM (Figure 1B) correlated with the increased cell invasiveness induced by exposure to mPSC-CM. The invasive effect was significantly inhibited with an IL-6 neutralizing antibody (Figure 5B). To further determine if mPSC-CM mediated cell invasion was dependent on STAT3 activity, PanIN cells were treated with the JAK/STAT3 inhibitor AZD1480. Treatment with AZD1480 resulted in significantly attenuated mPSC-CM induced invasion of PanIN cells to control levels (Figure 5B). These results confirm that PSC secreted IL-6, enhances the invasive capacity of pre-cancerous PanIN cells in a STAT3 dependent manner. Our results further emphasize the tumor-stromal cross-talk that exists between PSCs and tumor cells, which not only enhances tumor progression of PDAC cells, but also stimulates the invasive capabilities of non-malignant PanIN lesions.
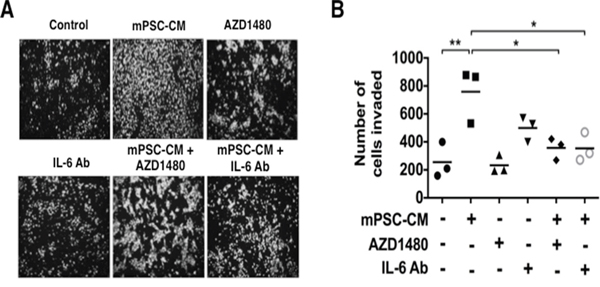
Figure 5: Mouse PSC-CM mediates activation of PanIN cell invasion through IL6-JAK-STAT3 signaling. Mouse PanIN cells were treated with mPSC-CM (100μg total protein/ml) with or without AZD1480 (100 nmol/L) or IL-6 neutralizing Ab and analyzed for cell invasion. A. Images demonstrate representative pictures from each treatment group. B. PanIN cell invasion analysis results showed that either IL-6 neutralizing Ab or AZD1480 significantly blocked mPSC-CM induced activation of cell invasion. * – P < 0.05; ** – P < 0.01.
DISCUSSION
We have previously shown that activated STAT3 is a biomarker and a key mediator of therapeutic resistance in PDAC [1, 2] [26]. In this study, we demonstrate a positive correlation expression of phosphorylated STAT3 in the progression of pancreatic neoplasia from normal ductal cells to precancerous PanIN cells to PDAC (Figure 1A); supporting the significant role that STAT3 plays in the progression of PDAC. Importantly, tumor associated PSCs have clearly been demonstrated to play a key role in PDAC-associated fibrogenesis by secreting components of the extracellular matrix (ECM) and tumor associated stroma [27, 28]. We have previously shown that STAT3 inhibition results in remodeling of the tumor stroma that is associated with increased drug delivery and cytotoxic therapeutic response of PDAC tumors in an aggressive mouse model of PDAC [20], further validating STAT3 as a novel and viable therapeutic PDAC target [3, 6, 9, 29–32].
Our current study shows that serum from KPC and PKT mice have higher levels of serum IL-6 then their control KC and PK counterparts respectively. Furthermore, IL-6 secreted from PSCs stimulates STAT3 signaling in both the mouse pancreatic precursor, noninvasive PanIN cells as well as in primary human PDAC cell lines. Secretion of IL-6 by PSCs directly enhances the tumorigenic potential of PanIN cells and stimulates progression of pre-neoplastic lesions to an invasive phenotype in a STAT3 dependent manner. This important finding defines a novel mechanism of PSC secreted IL-6 mediated activation of STAT3 signaling, as a critical regulator of tumor initiation and progression of PDAC.
This study further highlights the importance of cross-talk signaling processes and functional cell-ECM communication between the neoplastic cells and their microenvironment. This tug-of-war allows the dense stroma surrounding a growing tumor to recruit helper PSCs as well as other protective cell types of cellular immunity providing an environmentally supported transition into a more aggressive and metastatic phenotype. As we demonstrate, IL-6 plays a fundamental role in this process (Figures 2, 3, 4 and 5). IL-6 secreted from either human PSCs or mouse PSCs (Figure 2), appears to be a key factor that mediates the cross-talk between PSCs and PDAC cells. Neutralization of IL-6 with a ligand-specific antibody effectively reduces the overall free/active IL-6 levels in PSC-CM, blocks PSC-CM protein induced activation of STAT3 (Figure 3B), and abrogates PSC-CM enhanced growth and invasion of PDAC cells (Figures 4, 5). These findings reinforce the direct role of IL-6/STAT3 mediated PSC-PDAC interaction in promoting invasion and progression of pancreatic cancer. Additional IL-6 targeted therapeutics are under-going clinical trials for the treatment of renal cell cancer, prostate cancer, lymphoma and multiple myeloma [33]. Our data provides important pre-clinical information and rationale for targeting IL-6/STAT3 signaling in pancreatic cancer.
Accumulating evidence supports the notion that the tumor-associated stroma not only causes inefficient drug delivery, but also creates a cytokine and growth factor enriched microenvironment, which often results in enhanced proliferation and invasion of PDAC [6, 9, 20]. PSCs have been shown to induce PDAC cell resistance to chemotherapy [34, 35], which may be partly mediated by PSC secreted factors. Our data confirms the role of PSC secreted IL-6 in supporting the initiation and progression of PDAC. Therefore, therapeutic targeting of PSCs and/or IL-6 may provide a novel approach for anti-fibrotic and anti-neoplastic therapy, which could suppress IL-6 induced STAT3 activation and contribute to reduced invasiveness and tumorigenicity of PDAC. Supporting our findings, clinical studies have shown that elevated IL-6 serum levels correlate with worse patient outcomes [13, 36], and targeting IL-6 signaling with a neutralizing antibody has shown promising results in a clinical trial for the treatment of ovarian cancer [33].
Collectively, our results demonstrate for the first time that the IL-6/STAT3 signaling axis is a key element of tumor-stromal interaction between PSCs and tumor cells, playing a critical role in the progression from pre-neoplastic PanINs to PDAC. These findings demonstrate a novel role of PSCs in supporting an inflammatory TME and extend the evidence of crosstalk between the tumor stroma and diseased epithelium. Novel therapies focusing on both of these cell types and the TME should achieve better therapeutic responses.
MATERIALS AND METHODS
Cell lines, cell culture and reagents
Mouse PanIN cell line derived from the LSL-KrasG12D/+; Pdx1Cre/+ (KC) GEMM. Primary PDAC (PDA) and liver metastasis (LMP) cell lines derived from the LSL-KrasG12D/+; Trp53R172H/+; Pdx1Cre/+ (KPC) mouse models of PDAC [22] (kindly provided by Dr. Andrew Lowy, UC San Diego, CA) and maintained as previously described [37]. Ptf1aCre/+;LSL-KrasG12D/+ (PK) and Ptf1aCre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice were maintained as previously described [20]. The immortalized human pancreatic ductal cell line HPDE6-E6E7 (H6c7) was kindly provided by Dr. M.S. Tsao [38] and was maintained in in keratinocyte growth media (Invitrogen, Carlsbad, CA) supplemented with human epidermal growth factor and bovine pituitary extract. Human PDAC cell lines MiaPaCa2, PANC1 and BxPC3 were obtained from the American Type Culture Collection (ATCC). Tumor cells were maintained according to ATCC guidelines. Immortalized mouse PSCs and human PSCs were a gift from Dr. Anna Means (Vanderbilt University, Nashville, TN) and Dr. Ralf Jesnowski (German Cancer Research Center, Germany), respectively [18, 19]. AZD1480 was provided by AstraZeneca (Wilmington, DE). For in vitro experiments, AZD1480 was dissolved in 100% DMSO to prepare a 10 mM stock and stored at -20°C. Total STAT3 (124H6) and phospho-STAT3 (Y705) antibodies were purchased from Cell Signaling Technology (Denver, MA).
Enzyme-linked immunosorbent assay (ELISA)
Serum levels of IL-6 was measured using human and mouse IL-6 Quantikine ELISA Kit (R&D Systems Inc, Minneapolis, MN) according to the manufacturer’s instructions.
Collection of human and mouse PSCs-conditioned medium (CM)
Human PSCs (hPSCs) and mouse PSCs (mPSCs) were grown on culture flasks until 70% to 80% confluent and then cultured in serum-free medium (SFM) for 24 hours at 37°C. Conditioned medium (CM) was collected separately from hPSCs and mPSCs and stored at -80°C in multiple rounds. Human and mouse collections were separately pooled. PSCs were maintained in culture with fresh media added twice weekly. CM and culture medium was concentrated using Millipore centriprep centrifugal filters (EMD Millipore, Billerica, MA) with 10k cut off and centrifuged at 2000 rpm, 4°C to concentrate. The concentrated CM and culture medium was quantified for the protein concentration and the aliquots were stored at -80°C. In the following experiments, PSC-CM and culture medium protein was diluted with serum-free medium (SFM) as indicated to evaluate the effect of PSCs secretions on the mouse PanIN and human PDAC cells.
Western blot analysis
Western blot analysis was performed using standard methods previously described [39]. In brief, after treatment, cells were washed and scraped off the culture dishes. Cell pellets were collected by centrifuge and lysed in RIPA buffer (0.1% SDS, 50 mMTris HCl, 150 mMNaCl, 1% NP-40, and 0.5% Na deoxycholate) with protease inhibitor cocktail (Sigma, St. Louis, MO) and PhosSTOP phosphatase inhibitor (Roche, Indianapolis, IN, USA). Cell lysates were sonicated and centrifuged to collect supernatant. The collected supernatant was quantified for protein concentration measurements by Bio-Rad protein assay kit (Bio-Rad, Herculus, CA). The same quantities of protein were loaded from all the samples onto a SDS-PAGE gel. After running the gel, the proteins were transferred to a PVDF membrane and probed for the proteins of interest. Immunoblots were quantified using ImageJ and analysis was performed with Prism software (Graphpad Software Inc., La Jolla, CA).
IL-6 neutralization assay
The IL-6 level in PSC-CM protein was determined by using a commercial ELISA according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN). The neutralization was performed at 1:400 ratios of IL-6 antibody (Abcam, Cambridge, MA) and PSC-CM protein (50 μg/ml) with the mixture incubation at 4°C overnight on a rocker.
Cell invasion assay
Cell inserts (8.0 μm pore size membrane; Corning) were first coated with Matrigel (BD Biosciences, San Jose, CA) at a dilution of 3.3 ng/ml in SFM. Subsequently, 3 × 104 cells, previously deprived of serum (0.1%) for 24 hours, were seeded on the Matrigel in the upper chamber in SFM, while the lower chamber contained media with PSC-CM protein with/without IL-6 neutralizing antibody and/or AZD1480. Standard culture medium was collected as detailed above and was used as control medium. After 48 hours, non-motile cells at the top of the filter were removed and the cells in the bottom chamber were fixed with methanol and stained with DAPI and counted using immunofluorescence microscopy. Results represent the average number of cells in 2 or 3 fields per membrane in triplicate inserts.
Soft agar assays
5x104 cells were suspended in media containing 0.33% Select Agar (Invitrogen, Carlsbad, CA) and plated on a bottom layer of media containing 0.5% Select Agar. Plates were incubated at 37°C for 2-3 weeks prior to imaging. IL-6 neutralizing antibody and/or hPSC-CM, and/or mPSC-CM protein was added directly into the top agar layer during plating. Colonies were photographed and quantified using ImageJ and analysis was performed with Prism software.
Statistical and correlation analysis
Descriptive statistics including mean values and s.d. were calculated. Each treatment group was compared using one-way ANOVA and Tukey’s multiple comparison test using Prism software (Graphpad Software Inc., La Jolla, CA). Results are shown as values of mean ± s.d. unless otherwise indicated. Pearson’s and Spearman’s correlation analysis were conducted in Stata 14 (StataCorp LP, College Station, TX).
Abbreviations
Pancreatic ductal adenocarcinoma (PDAC); pancreatic stellate cells (PSCs); human pancreatic stellate cells (hPSCs); mouse pancreatic stellate cells (mPSCs); signal transducer and activator of transcription 3 (STAT3); myeloid derived suppressor cells (MDSCs); tumor microenvironment (TME); pancreatic intraepithelial neoplasia cell line (PanIN); liver metastases line (LMP); human pancreatic ductal epithelial cell line (H6c7).
ACKNOWLEDGMENTS
The Authors thank Dr. Vikas Dudeja for serum from KPC mice.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
GRANT SUPPORT
This work was supported by the NIH Grants CA161976, P50 95103 GI Special Program of Research Excellence Grant (SPORE), 5P30DK058404-08 Vanderbilt Digestive Disease Research Center (DDRC) Translational Award and NCI Cancer Center Support Grant #P30 CA068485-1 to N. B. Merchant, and CA209536 to N.S. Nagathihalli. Core services were performed through the NIH DDRC P30DK058404 and Sylvester Comprehensive Cancer Center (SCCC) support grants (N.B. Merchant and N.S. Nagathihalli).
REFERENCES
1. Nagaraj NS, Smith JJ, Revetta F, Washington MK, Merchant NB. Targeted inhibition of SRC kinase signaling attenuates pancreatic tumorigenesis. Mol Cancer Ther. 2010; 9:2322-2332.
2. Nagaraj NS, Washington MK, Merchant NB. Combined Blockade of Src Kinase and Epidermal Growth Factor Receptor with Gemcitabine Overcomes STAT3-Mediated Resistance of Inhibition of Pancreatic Tumor Growth. Clin Cancer Res. 2011; 17:483-493.
3. Korc M. Pancreatic cancer-associated stroma production. Am J Surg. 2007; 194:S84-86.
4. Welter M, Rieger H. Interstitial fluid flow and drug delivery in vascularized tumors: a computational model. PLoS One. 2013; 8:e70395.
5. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007; 117:50-59.
6. Erkan M. Understanding the stroma of pancreatic cancer: co-evolution of the microenvironment with epithelial carcinogenesis. J Pathol. 2013; 231:4-7.
7. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. 2007; 7:41-51.
8. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007; 67:9518-9527.
9. Tang D, Wang D, Yuan Z, Xue X, Zhang Y, An Y, Chen J, Tu M, Lu Z, Wei J, Jiang K, Miao Y. Persistent activation of pancreatic stellate cells creates a microenvironment favorable for the malignant behavior of pancreatic ductal adenocarcinoma. Int J Cancer. 2013; 132:993-1003.
10. Apte MV, Pirola RC, Wilson JS. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Frontiers in physiology. 2012; 3:344.
11. Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andren-Sandberg A, Domellof L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993; 328:1433-1437.
12. Marzoq AJ, Giese N, Hoheisel JD, Alhamdani MS. Proteome variations in pancreatic stellate cells upon stimulation with proinflammatory factors. J Biol Chem. 2013; 288:32517-32527.
13. Okada S, Okusaka T, Ishii H, Kyogoku A, Yoshimori M, Kajimura N, Yamaguchi K, Kakizoe T. Elevated serum interleukin-6 levels in patients with pancreatic cancer. Jpn J Clin Oncol. 1998; 28:12-15.
14. Lesina M, Kurkowski MU, Ludes K, Rose-John S, Treiber M, Kloppel G, Yoshimura A, Reindl W, Sipos B, Akira S, Schmid RM, Algul H. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011; 19:456-469.
15. Deng J, Liu Y, Lee H, Herrmann A, Zhang W, Zhang C, Shen S, Priceman SJ, Kujawski M, Pal SK, Raubitschek A, Hoon DS, Forman S, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012; 21:642-654.
16. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014; 14:736-746.
17. Corcoran RB, Contino G, Deshpande V, Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA, Bardeesy N. STAT3 plays a critical role in KRAS-induced pancreatic tumorigenesis. Cancer Research. 2011; 71:5020-5029.
18. Jesnowski R, Furst D, Ringel J, Chen Y, Schrodel A, Kleeff J, Kolb A, Schareck WD, Lohr M. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: deactivation is induced by matrigel and N-acetylcysteine. Laboratory investigation. 2005; 85:1276-1291.
19. Blaine SA, Ray KC, Branch KM, Robinson PS, Whitehead RH, Means AL. Epidermal growth factor receptor regulates pancreatic fibrosis. American journal of physiology Gastrointestinal and liver physiology. 2009; 297:G434-441.
20. Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, Chen X, Walsh AJ, Skala MC, Moses HL, Merchant NB. Signal Transducer and Activator of Transcription 3, Mediated Remodeling of the Tumor Microenvironment Results in Enhanced Tumor Drug Delivery in a Mouse Model of Pancreatic Cancer. Gastroenterology. 2015; 149:1932-1943.
21. Niemand C, Nimmesgern A, Haan S, Fischer P, Schaper F, Rossaint R, Heinrich PC, Muller-Newen G. Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J Immunol. 2003; 170:3263-3272.
22. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005; 7:469-483.
23. Erkan M, Reiser-Erkan C, Michalski CW, Kong B, Esposito I, Friess H, Kleeff J. The impact of the activated stroma on pancreatic ductal adenocarcinoma biology and therapy resistance. Current molecular medicine. 2012; 12:288-303.
24. Shi C, Washington MK, Chaturvedi R, Drosos Y, Revetta FL, Weaver CJ, Buzhardt E, Yull FE, Blackwell TS, Sosa-Pineda B, Whitehead RH, Beauchamp RD, Wilson KT, Means AL. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab Invest. 2014; 94:409-421.
25. Nagathihalli NS, Beesetty Y, Reyzer M, Shi C, Caprioli R, Merchant NB. STAT3 Mediates Regulation of the Tumor Microenvironment in Pancreatic Cancer. Pancreas. 2011; 40:1342-1342.
26. Devarajan E, Huang S. STAT3 as a central regulator of tumor metastases. Current molecular medicine. 2009; 9:626-633.
27. Apte MV, Park S, Phillips PA, Santucci N, Goldstein D, Kumar RK, Ramm GA, Buchler M, Friess H, McCarroll JA, Keogh G, Merrett N, Pirola R, Wilson JS. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004; 29:179-187.
28. Bachem MG, Schunemann M, Ramadani M, Siech M, Beger H, Buck A, Zhou S, Schmid-Kotsas A, Adler G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005; 128:907-921.
29. Kim JH, Ho SB, Montgomery CK, Kim YS. Cell lineage markers in human pancreatic cancer. Cancer. 1990; 66:2134-2143.
30. Fujita H, Ohuchida K, Mizumoto K, Egami T, Miyoshi K, Moriyama T, Cui L, Yu J, Zhao M, Manabe T, Tanaka M. Tumor-stromal interactions with direct cell contacts enhance proliferation of human pancreatic carcinoma cells. Cancer Sci. 2009; 100:2309-2317.
31. Ebrahimi B, Tucker SL, Li D, Abbruzzese JL, Kurzrock R. Cytokines in pancreatic carcinoma: correlation with phenotypic characteristics and prognosis. Cancer. 2004; 101:2727-2736.
32. Lu J, Zhou S, Siech M, Habisch H, Seufferlein T, Bachem MG. Pancreatic stellate cells promote hapto-migration of cancer cells through collagen I-mediated signalling pathway. Br J Cancer. 2014; 110:409-420.
33. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev. 2012; 38:904-910.
34. Muerkoster S, Wegehenkel K, Arlt A, Witt M, Sipos B, Kruse ML, Sebens T, Kloppel G, Kalthoff H, Folsch UR, Schafer H. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004; 64:1331-1337.
35. Miyamoto H, Murakami T, Tsuchida K, Sugino H, Miyake H, Tashiro S. Tumor-stroma interaction of human pancreatic cancer: acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas. 2004; 28:38-44.
36. Schultz NA, Christensen IJ, Werner J, Giese N, Jensen BV, Larsen O, Bjerregaard JK, Pfeiffer P, Calatayud D, Nielsen SE, Yilmaz MK, Hollander NH, Wojdemann M, Bojesen SE, Nielsen KR, Johansen JS. Diagnostic and Prognostic Impact of Circulating YKL-40, IL-6, and CA 19.9 in Patients with Pancreatic Cancer. PLoS One. 2013; 8:e67059.
37. Thomas RM, Toney K, Fenoglio-Preiser C, Revelo-Penafiel MP, Hingorani SR, Tuveson DA, Waltz SE, Lowy AM. The RON receptor tyrosine kinase mediates oncogenic phenotypes in pancreatic cancer cells and is increasingly expressed during pancreatic cancer progression. Cancer Research. 2007; 67:6075-6082.
38. Liu N, Furukawa T, Kobari M, Tsao MS. Comparative phenotypic studies of duct epithelial cell lines derived from normal human pancreas and pancreatic carcinoma. Am J Pathol. 1998; 153:263-269.
39. Nagathihalli NS, Beesetty Y, Lee W, Washington MK, Chen X, Lockhart AC, Merchant NB. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-alpha: impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res. 2014; 74:2062-2072.