
For a tumor to arise, it has long been common understanding that a critical degree of accumulating factors jointly contribute to clonal evolution. These include but are not limited to (i) a proliferation advantage mediated in part by the activation of proto-oncogenes (e.g. MYC, RAS, and/or PI3K-AKT-mTOR pathway components) and (ii) the disturbance of cellular control mechanisms due to mutations in tumor suppressor genes involved in cell cycle control (TP53, CHEK2), DNA repair (BRCA1, BRCA2), or proliferation-restrictive signaling (PTEN). When present at germline level, these variations constitute a potential shortcut in clonal evolution and predispose affected individuals to tumor development. However, impaired DNA repair mechanisms, aberrant cell cycle control, and genomic instability alone may not be sufficient for malignant transformation as rapid proliferation can only be possible in the presence of sufficient macromolecule supply for building blocks and ATP for energy. Accumulative evidence suggests that, in order for tumors to emerge and particularly to evolve into a more aggressive state, a third component (iii), intertwined with (i) and (ii), may be mandatory - the metabolic reprogramming of tumor cells including their surrounding stromal environment. This metabolic switch has been entitled one of the new “hallmarks of cancer” [1], expanding the original set of hallmarks [2] along with further upgrades such as chronic inflammation, escape from the immune system, and genomic instability. Metabolome analyses on tumor tissue and cancer patients’ biofluids aim for a better understanding of metabolic reprogramming and malignant transformation in tumor cells, and integration of the underlying mechanisms and signaling pathways is expected to help elucidate overall tumor pathogenesis as well as breast cancer pathogenesis in particular, the latter constituting the main focus of the present review.
Metabolic tumor models and tumor biology
Originally described by Otto Warburg [3], metabolic reprogramming in cancer cells was thought to involve a shift in energy metabolism away from an oxidative towards a glycolytic one - even under aerobic conditions - subsequently termed the “Warburg effect” or “aerobic glycolysis”. Dysfunctional mitochondrial oxidative phosphorylation (OXPHOS) was proposed to play a major causative role in this process. However, in recent years this concept of mitochondrial respiratory impairment has been challenged and a growing body of evidence now suggests that, indeed, high mitochondrial activity and even a dependence on mitochondrial metabolism - as well as glycolysis - is essential for rapid tumor cell proliferation. This principle (reviewed in [4]) is supported by the observation that depletion of mitochondrial DNA (mtDNA) halts cancer cell proliferation and reverses tumorigenicity. The original Warburg effect can instead be found in cells belonging to the surrounding stromal tumor microenvironment, e.g. cancer-associated fibroblasts (CAFs), which are believed to stand in a quasi-symbiotic relationship with cancer cells (Figure 1). In an autophagy-associated paracrine manner, CAFs supply adjacent cancer cells with anapleurotic substrates such as lactate, pyruvate, and ketone bodies derived from their own excessive glycolytic activity. Hereby, they induce OXPHOS in adjacent cancer cells, enabling them to produce ample amounts of ATP as well as supplying necessary macronutrients for proliferation. This model of two-compartment tumor metabolism and the dynamic interplay or “near-parasitic” metabolic symbiosis between cancer cells and CAFs has been redefined as the “reverse Warburg effect” [5] or “metabolic coupling”. Apart from providing a foundation for better interpretation of tumor-associated metabolome analyses, this concept may have broad implications for (i) the elucidation of fundamental aspects of tumor etiology and pathogenesis, (ii) the enhancement of diagnostic accuracy and classification, and (iii), above all, the development of preventive and therapeutic approaches, focusing in parallel on (a) basic metabolic root tumor mechanisms as well as (b) precise personalized treatment according to individual metabolic tumor-specific aberrations.
Mechanistically, metabolic reprogramming (Figure 1) of fibroblasts into a CAF phenotype is mediated by cancer cells through the generation and secretion of high amounts of hydrogen peroxide (H2O2), which creates a pseudohypoxic state in adjacent stroma cells, mimicking nutrient and oxygen depletion [6]. Cancer cells are protected from excessive damage by reactive oxygen species (ROS) through the simultaneous upregulation of innate protective antioxidant pathways via mechanisms such as nuclear factor, erythroid 2 like 2 (NFE2L2) stabilization (also known as NRF2) and generation of reduced glutathione (GSH) [7, 8]. Physiologically, a state of energy starvation - in particular a shortage of glucose or oxygen - leads to the following signaling scenario (Figure 2A). The lack of fuel for OXPHOS results in lower ATP output and increased AMP/ATP ratios, subsequently activating AMP-activated protein kinase (AMPK) and resulting in (i) the activation of glycolysis through (a) phosphorylation of phosphofructokinase 2 (PFK2), and (b) elevated glucose uptake by means of glucose transporter type 4 (GLUT4) expression, (ii) the activation of fatty acid oxidation through inhibition of acetyl-CoA-carboxylase-beta (ACCB), (iii) the induction of anti-proliferative signaling through an enhancement of AMPK’s inhibitory effect on mechanistic target of rapamycin (mTOR), and (iv) the onset of an autophagic, self-digestive condition through direct phosphorylation of unc-51-like kinase 1 (ULK1) in order to recycle any nonessential cell components for nutrients and for the highest possible energy exploitation [9] (http://www.kegg.jp/kegg-bin/highlight_pathway?scale=1.0&map=map04152&keyword=sirt1; 02/05/2016). Conceivably, all of these signaling reactions are of major advantage to a cell or an entire organism under conditions of starvation. In a tumor setting (Figure 2B), however, cancer cells divert this mechanism towards their own advantage by mimicking a state of energy depletion in surrounding cells even though nutrient supply is ample and constantly triggers the growth-promoting, pro-proliferative PI3K-AKT-mTOR pathway via growth factors such as insulin and insulin-like growth factor 1 (IGF1). Whereas in a state of authentic nutrient and oxygen depletion (Figure 2A) mTOR signaling is physiologically downregulated by AMPK, the ROS-mediated pseudohypoxic state cancer cells provoke in surrounding fibroblasts (Figure 2B) results in simultaneous and persistent activation of autophagy-promoting AMPK signaling, on the one hand, and elevated mTOR signaling, on the other hand - an unphysiological condition, leading to macromolecule abundance and catabolism.
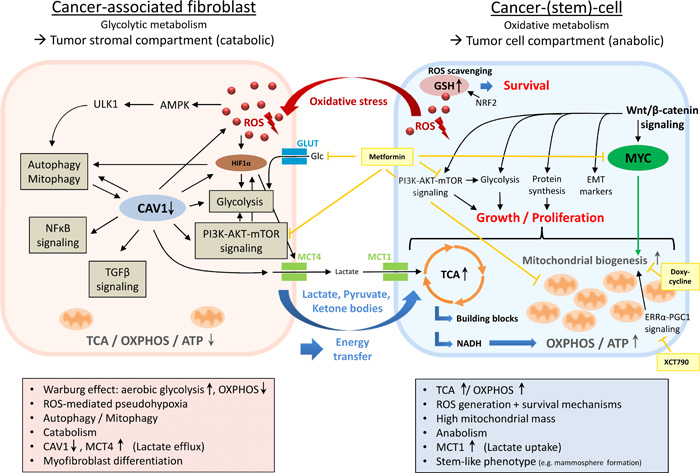
Figure 1: Metabolic coupling in two-compartment tumor metabolism. In this model of metabolic symbiosis, the dynamic interplay between distinct tumor compartments (i.e. the tumor stromal compartment and the tumor cell compartment) enables cancer cells to acquire anapleurotic substrates for energy production and proliferation through paracrine supply of glycolytic end products derived from cancer-associated fibroblasts (CAFs). Cancer cells mediate metabolic reprogramming of fibroblasts into CAFs via the secretion of ROS, creating a pseudohypoxic state in adjacent stroma cells. Due to the simultaneous upregulation of innate antioxidant pathways (e.g. NRF2 signaling, GSH replenishment), cancer cells protect themselves from excessive oxidative damage. In the tumor stromal compartment, ROS accumulation initiates fibroblast-to-myofibroblast differentiation and transformation into a pro-inflammatory, catabolic phenotype producing high amounts of macromolecules (e.g. lactate, pyruvate, ketone bodies) due to a major bioenergetic emphasis on aerobic glycolysis (Warburg effect). The ROS-triggered loss of CAV1 plays a vital role in this process by promoting the upregulation of HIF1α, NFκB, TGFβ, glycolysis, autophagy/mitophagy, and oxidative stress in a feed-forward cycle manner. Loss of CAV1 is associated with high MCT4 expression - a marker of lactate efflux and aerobic glycolysis. Anapleurotic substrates derived from CAFs’ excessive glycolytic activity are subsequently transferred into adjacent cancer cells where they induce TCA/OXPHOS, enabling the generation of ample amounts of ATP as well as macronutrients for proliferation. High mitochondrial mass, a new feature of the stem-like phenotype associated with upregulated Wnt/β-catenin-, MYC-, and ERRα-PGC1-signaling, reflects cancer cells’ reliance on oxidative metabolism. AMPK, AMP-activated protein kinase; CAV1, caveolin 1; EMT, epithelial-mesenchymal transition; ERRα, estrogen-related receptor alpha; Glc, glucose; GLUT, glucose transporter; GSH, glutathione (reduced form); HIF1α, hypoxia-inducible factor 1-alpha; MCT1, monocarboxylate transporter 1; MCT4, monocarboxylate transporter 4; NRF2, nuclear factor E2-related factor 2; OXPHOS, oxidative phosphorylation; PGC1, peroxisome proliferator-activated receptor γ coactivator 1; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; ULK1, unc-51-like kinase 1; Light yellow boxes, medical interventions.
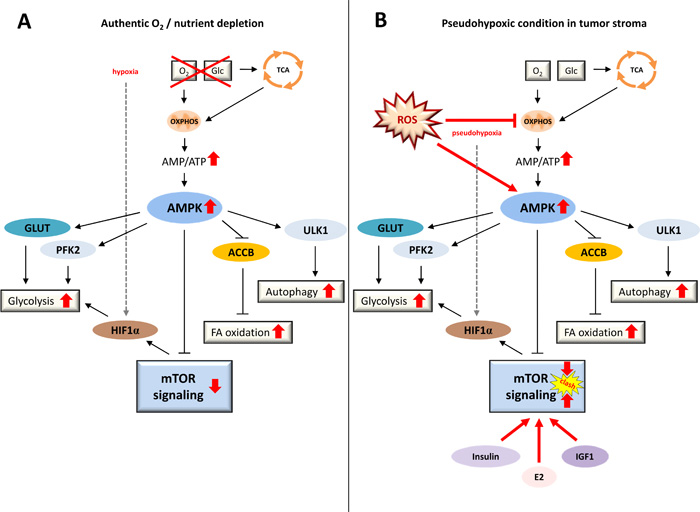
Figure 2: Conditions of authentic energy starvation versus tumor-associated pseudohypoxia. A. Under physiological conditions, a state of energy starvation leads to a lack of fuel for OXPHOS resulting in lower ATP output, increased AMP/ATP ratios, and activated AMPK, ultimately promoting glycolysis, fatty acid oxidation, autophagy and anti-proliferative signaling via mTORC1 inhibition. These signaling reactions are of major advantage to a cell or an organism under conditions of starvation. B. In a tumor setting, in contrast, cancer cells mimic a state of energy depletion in the tumor stromal compartment, even though nutrient supply is ample and growth factors constantly trigger pro-proliferative PI3K-AKT-mTOR signaling. Thus, in a state of authentic nutrient and oxygen depletion (A) mTOR signaling is physiologically downregulated by AMPK, whereas in a state of pseudohypoxia provoked in fibroblasts by surrounding cancer cells (B) autophagy-promoting AMPK signaling clashes with elevated PI3K-AKT-mTOR signaling - a condition that leads to a major excess of macromolecules. ACCB, acetyl-CoA-carboxylase-beta; AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; E2, estradiol; FA oxidation, fatty acid oxidation; Glc, glucose; GLUT, glucose transporter; HIF1α, hypoxia-inducible factor 1-alpha; IGF1, insulin-like growth factor 1; mTOR, mechanistic target of rapamycin; OXPHOS, oxidative phosphorylation; PFK2, phosphofructokinase 2; ROS, reactive oxygen species; TCA, tricarboxylic acid cycle; ULK1, unc-51-like kinase 1.
The more steps towards this process are made, the easier it may be for tumor-initiating cells (i.e. cancer stem cells (CSCs)) to succeed in creating a catabolic stroma environment for their own purpose (Figure 3). This means that any type of non-physiologically elevated prolonged ROS generation, overactive PI3K-AKT-mTOR signaling, and/or favoring of glycolytic flux over OXPHOS may set the conditions for a metabolic switch towards tumorigenesis. As will be explained later in detail, this may include germline BRCA1 deficiency, TP53 mutations, and PTEN loss. Moreover, activation of proto-oncogenes (e.g. RAS, NFKB1, TGFB1) as well as loss of tumor-suppressor genes (e.g. BRCA1) in cancer cells have been shown to be sufficient to induce metabolic reprogramming of the fibroblast compartment via ROS generation, and this transformation could be rescued by antioxidants such as N-acetylcysteine [10-12]. Furthermore, well-established environmental modulators of cancer predisposition such as cigarette smoke [13] and ethanol exposure [14] have been demonstrated to specifically induce the CAF phenotype. Since the common basis of all of these contributing factors appears to be endured oxidative stress and chronic inflammation, further environmental or intrinsic determinants causing continuous ROS generation may play a vital role in the emergence of a pro-inflammatory, pro-tumorigenic stromal environment. These include chronic microbial or viral infections, radiation, toxic chemicals, obesity and/or hypercaloric diets, autoimmune disease, and chronic allergen exposure (reviewed in [15]). A majority of the above factors has been associated with disease-related epigenetic changes as well [16], and, interestingly, many of the CAF features are also major contributors to the aging process [17] - age being a clear risk factor for tumor development.
While findings and hypotheses regarding tumor biology have been inconsistent and manifold, it is clearly understood that tumor tissue exhibits overwhelming morphological and physiological intertumoral and intratumoral heterogeneity in its cellular origin, cell surface markers, gene expression profiling, metabolic features, and proliferative as well as metastatic potential [18]. This is likely a reflection of the extremely adaptive nature that allows cancer cells to adjust to individual microenvironmental conditions including alterable nutrient and oxygen supplies. This adaptive quality may also be the cause for pharmacological resistances many tumors appear to acquire during treatment. The cellular phenotype presenting the highest degree of adaptability in a tumor is the CSC-phenotype, which is increasingly made responsible for minimal residual disease, relapse, and metastasis [19-21]. Resistances to redox stress and to chemotherapeutics are considered characteristics of “stemness” [22, 23]. In fact, chemotherapy (CTx) and radiotherapy (RTx) were found to promote a CSC-like phenotype by increasing genotoxic stress and ROS levels [24-26]. CSCs have particular cell surface markers and are capable of sphere formation (e.g. mammosphere growth in breast cancer stem cells), which is one of the hallmark characteristics of stem cells. The expression of genes governing mitochondrial function, autophagy, and lysosome activity has been shown to be upregulated, and a strong reliance on mitochondrial OXPHOS is evident in cells featuring a CSC-like phenotype [27]. Importantly, high mitochondrial mass has recently been introduced as a new feature of the stem-like anabolic phenotype, in which upregulated Wnt/β-catenin signaling promotes the induction of mitochondrial proteins, glycolytic enzymes, the protein synthesis machinery, epithelial-mesenchymal transition (EMT) markers, and increased mitochondrial mass and activity as well as mammosphere formation [28] (Figure 1). Mitochondrial biogenesis is governed by the ERRα-PGC1 signaling pathway, which has been found to enhance mammosphere formation and can be blocked by a specific inhibitor, XCT790, through the suppression of signaling pathways such as sonic hedgehog, TGFβ-SMAD, STAT3, and Wnt-signaling [29]. Induction of a stem-like phenotype in bulk cancer cells has recently been observed by Ning et al. in pancreatic cancer cell lines cultured in stem cell medium [30].
Likewise, the induction of a CAF phenotype (Figure 1, Figure 3) in stromal fibroblasts through cancer cells has been described to be mediated by several mechanisms, including miRNAs, TGFβ secretion, and ROS generation promoting HIF1α accumulation [31-33]. The CAF phenotype can, however, also be introduced by CTx [34], comprising proteomic changes in a number of factors including proteins involved in autophagy, inducers of inflammation, metabolic enzymes, antioxidants, and myofibroblast differentiation markers. This generates a catabolic tumor stroma, which strikingly correlates with relapse, metastasis and reduced overall survival. During transformation, fibroblasts undergo myofibroblast differentiation - usually a process initiated upon tissue damage and resulting in fibroblast proliferation and wound healing - and begin to express, among others, α-smooth muscle actin (α-SMA) as a specific myofibroblast marker. The loss of Caveolin 1 (CAV1), which is triggered upon initiation of oxidative stress from adjacent cancer cells and implemented by means of autophagolysosomal degradation, appears to play a major role in the onset of the myofibroblast phenotype [35]. CAV1 loss mediates the upregulation of NFκB and HIF1α activity and ultimately promotes TGFβ signaling, glycolysis, autophagy, and oxidative stress in a feed-forward cycle manner. In CAFs, loss of CAV1 is typically inversely associated with high monocarboxylate transporter 4 (MCT4) expression - a marker of lactate efflux and aerobic glycolysis [36, 37], which is upregulated in a HIF1α-dependent manner and shows strict correlation with poor overall survival, particularly so in triple-negative breast cancer (TNBC) patients [38].
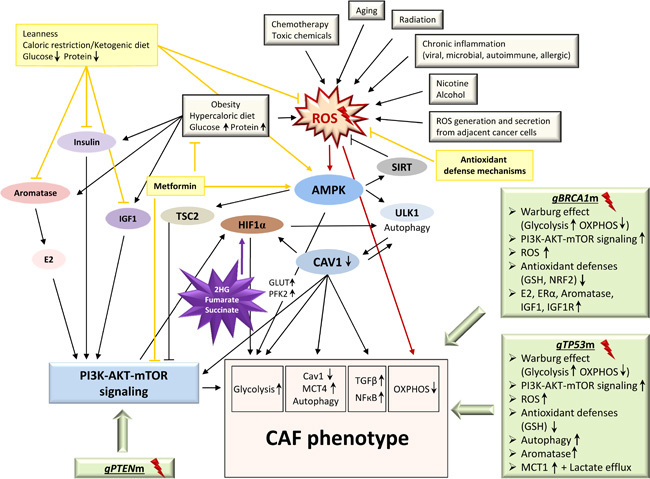
Figure 3: The CAF phenotype: onset and triggers. The onset of a pro-inflammatory, pro-tumorigenic stromal environment by metabolic reprogramming of fibroblasts into a CAF phenotype can be triggered by a variety of factors including genetic (light green boxes) and environmental (light grey boxes) aspects. The growing burden of accumulating factors likely assists cancer-(stem)-cells in creating a catabolic, glycolytic, and autophagic stromal environment to their needs. Consequently, any type of prolonged ROS accumulation, overactive PI3K-AKT-mTOR signaling, or favoring of glycolytic flux over OXPHOS may help set the conditions for a metabolic switch. This likely includes germline aberrations in tumor suppressor genes known to play a role in hereditary breast cancer, e.g. BRCA1, TP53 and PTEN (light green boxes), as cells lacking the respective functional proteins harbor Warburg-like metabolic features consistent with the CAF phenotype. Well-established environmental cancer predisposition factors (light grey boxes) such as nicotine and ethanol exposure but also chemotherapeutic agents specifically induce the CAF phenotype. Further ROS-generating determinants, e.g. chronic microbial or viral infections, radiation, toxic chemicals, obesity, autoimmune disease, chronic allergen exposure, as well as the aging process itself may set the stage for the evolution of a CAF phenotype, predisposing the organism to tumor development. 2HG, 2-hydroxyglutarate; AMPK, AMP-activated protein kinase; CAF, cancer-associated fibroblast; CAV1, caveolin 1; E2, estradiol; ERα, estrogen receptor alpha; gBRCA1m, germline BRCA1 mutation; GLUT, glucose transporter; gPTENm, germline PTEN mutation; GSH, glutathione (reduced form); gTP53m, germline TP53 mutation; HIF1α, hypoxia-inducible factor 1-alpha; IGF1, insulin-like growth factor 1; IGF1R, insulin-like growth factor 1 (IGF-1) receptor; MCT1, monocarboxylate transporter 1; MCT4, monocarboxylate transporter 4; NRF2, nuclear factor E2-related factor 2; OXPHOS, oxidative phosphorylation; PFK2, phosphofructokinase 2; ROS, reactive oxygen species; SIRT, sirtuins; TSC2, tuberous sclerosis complex 2; ULK1, unc-51-like kinase 1; Light yellow boxes, medical/lifestyle interventions; Purple star, oncometabolites.
The individual cell adaptability and overall inter-tumoral and intra-tumoral heterogeneity makes it extremely difficult for scientists and clinicians to properly treat cancer patients, as each may need highly specific individual handling, and tendencies for relapse and metastases depend on the primary success or failure in eliminating every single tumor cell. Yet, in light of accumulating evidence for what may possibly depict specific overall parallels in tumor metabolism - central pathway alterations and metabolic switches that can consistently be found in the vast majority of tumors - it may be reasonable to consider common, fundamental metabolic root mechanisms of tumorigenesis and, thus, treatment. The aforementioned model of two-compartment tumor metabolism between cancer cells and tumor stroma may be a significant contribution to such attempts and may pose new opportunities for cancer treatment. Current studies are indicative of both compartments - that is the catabolic/glycolytic (including CAFs) versus the anabolic/oxidative (including CSCs) compartment - contributing to tumor initiation and progression, and also of both compartments needing mandatory parallel therapeutic targeting in order to prevent relapse and metastatic disease.
Thus far, it is not entirely clear if (i) the reverse Warburg effect may, to a greater or lesser extent, play a role in each tumor, if (ii) strict intertumoral categorization into different metabolic tumor phenotypes, including Warburg-like and reverse-Warburg-like, could be applied, if (iii) these two metabolic phenotypes demonstrate plasticity and adaptability in response to outside triggers and within the process of clonal evolution, or if (iv) even the great mass of bulk cancer cells may comprise completely opposing metabolisms, differing from one cell to the next - some glycolytic-catabolic in the manner of CAFs, others oxidative-anabolic in the manner of CSCs. Therapeutically, however, this may not make a difference as long as each cell could be assigned to either one of the two metabolic compartments and as long as both these metabolic extremes were to be targeted simultaneously.
Breast tumor metabolome
To elucidate cancer cell metabolism, a number of tumor metabolome analyses utilizing differing methods have been conducted. However, given the intratumoral heterogeneity in virtually any tumor (including the immediate co-existence of cancer cells, CAFs, normal fibroblasts, macrophages and other immune cells, vascular endothelial cells, adipocytes, neurons and so forth), interpretation of metabolome analyses on a slice of tumor tissue may need to be approached with care. The likelihood of receiving data containing a mixture of all of the above cells’ metabolites (some showing “normal” metabolism, some exhibiting Warburg-like and some oxidative metabolism) - which may overall be equational to single metabolite peaks and therefore tamper results - appears to be very high. In order to overcome this burden and to receive unbiased, clearly referable data, tumor tissue may have to be prepared and dissected into separate cell types via fluorescence-activated cell sorting (FACS) methods prior to metabolome analyses. Since it is also quite likely that bulk cancer cells immediately adjacent to each other may comprise completely variable metabolic features, single-cell metabolomics analyses could alternatively be applied to CAFs, CSCs, and separate bulk cancer cells. As such analyses are yet waiting to be conducted, we will review current knowledge from tumor metabolome analyses performed thus far.
Despite the metabolic heterogeneity and partly discrepant results, certain metabolic patterns tend to be distinguishable in tumor breast tissue in comparison to normal breast tissue. Whereas the molecular subtypes described from gene expression profiling do not appear to clearly overlap with metabolic profiles, reproducible metabolic signatures do allow the distinction of estrogen receptor-positive (ER+) from estrogen receptor-negative (ER-) tumors [39-41]. Overall, increased glucose consumption and strong lactate accumulation, even more so in ER- than in ER+ breast cancer, is the most typical trait observed in breast cancer and in cancer tissue overall, standing in line with the increased glycolytic activity of both CAFs and cancer cells. At the same time, tumors show an increase in fatty acid synthesis including the immediate consumption of free fatty acids for membranes. An activation of nucleotide and protein biosynthesis for DNA replication and cellular proliferation can be observed. Next to the increase in glycolysis, one can often find additional characteristic catabolic changes, such as drastically increased glutamine consumption in combination with glutamate accumulation, commonly referred to as the “glutamine addiction” phenotype [42]. The glutamate/glutamine ratio (GGR) correlates with estrogen receptor (ER) status in breast cancer (“glutamate enrichment” being found in 56 % of ER+ and 88 % of ER- breast cancer, opposing 2.2 % of normal breast tissue), with tumor grading (GGR as a MYC-associated marker of aggressive disease), and with tyrosine metabolism [43].
In terms of more specific markers, a wide range of molecules, enzymes, and metabolite ratios has been suggested as oncometabolites or diagnostic and prognostic biomarkers, many of which can only be touched upon in this review and often still lack functional explanation. For instance, breast tumors generally tend to display high concentrations of taurine, choline, and glycine, the latter of which has been suggested as an erb-b2 receptor tyrosine kinase 2 (ERBB2)-associated marker of aggressiveness; also, serine auxotrophy, particularly in aggressive ER- or TNBC, has been observed [40, 44-46]. ER- breast cancer metabolomes generally tend to demonstrate more drastic and characteristic features compared to ER+ breast cancer: elevated β-alanine (suggested as the strongest differentiating marker between ER- and ER+ breast cancer), an accumulation of 2-hydroxyglutarate (2HG), GSH, branched-chain amino acids, carnitines, and strongly elevated cholines (in particular phosphocholine and phospholipids) combined with elevated glycerol-3-phosphate acyltransferase (GPAM) activity has been detected [39-41, 43, 47]. The uptake of amino acids is usually elevated in breast tumors, particularly that of N-acetyl-aspartate [41]; however, a number of amino acids has been shown to be reduced in several breast cancer cell lines in comparison to a reference breast cell line [48], indicating heightened consumption. Furthermore, an accumulation of kynurenine and a shift within the kynurenine/tryptophan ratio can often be observed, particularly in basal-like breast cancer [40]. Kynurenine is an immunomodulatory metabolic intermediate generated during tryptophan degradation and NAD+/nicotinic acid biosynthesis. Lactate, physiologically produced excessively under hypoxic conditions, acts immunomodulatory as well, fuels angiogenesis and metastasis, and constitutes a major contribution to establishing the relationship between tumor cells and tumor microenvironment [6, 49]. Prostaglandins, which are derived from aberrantly increased cyclooxygenase-2 (COX2) in breast tumors and which function as oncogenic lipid messengers [50-52], link breast cancer to inflammatory processes. A high ratio of glycerophosphocholine to phosphocholine (GPC/PCho) has been proposed as a marker of basal-like and luminal B breast tumors but has yielded discrepant results (reviewed in [53]). In turn, the cytidine-5-monophosphate/pentadecanoic acid ratio has been demonstrated to be a fairly reliable discriminator between cancerous and normal tissue, detecting cancer with a sensitivity of 94.8 % and a specificity of 93.9 % [54].
The interplay between 2HG, MYC, and glutamine catabolism
Metabolic diversity between individuals originates from exogenic as well as endogenic influencing factors, including germline or somatic mutations, and the resulting metabolic state may or may not have a decisive impact on disease predisposition in humans. More precisely, the question as to whether metabolic alterations merely represent byproducts from oncogenesis, whether they function as “reactive” mediators antagonizing the oncogenic process, or whether they could indeed be etiologically seminal for tumorigenesis - quasi acting as oncometabolites - will need to be further addressed in detail. However, in the context of rare hereditary metabolic diseases, termed “inborn errors of metabolism” (IEMs), metabolic changes have been shown to be capable of initiating malignant transformation [55]. In this regard, the focus has primarily been set on three oncometabolites - namely fumarate, succinate, and MYC-associated 2HG - the latter of which will be the focus of the below paragraph (Figure 4, Key factor 1). These oncometabolites accumulate in the presence of defective mitochondrial tricarboxyl acid (TCA) cycle enzymes fumarate hydratase (FH), succinate-dehydrogenase (SDH), and isocitrate-dehydrogenase 2 (IDH2) as well as the cytosolic isocitrate-dehydrogenase 1 (IDH1). Their accumulation results in competitive inhibition of alpha-ketoglutarate (αKG)-dependent dioxygenases and, among other consequences, leads to extensive epigenetic alteration as well as HIF1α stabilization, which raises the expression levels of glycolytic enzymes. Additionally, fumarate modifies the function of thiol-containing compounds such as glutathione.
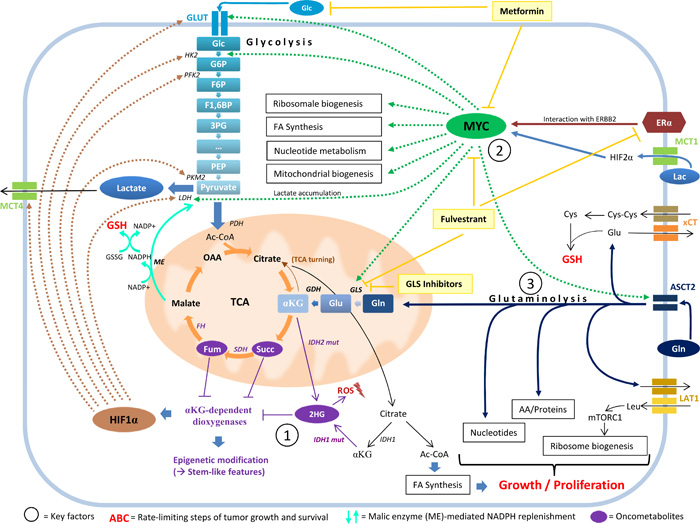
Figure 4: The interplay between 2HG, MYC, and glutamine catabolism. MYC pathway activation, glutaminolysis, and the oncometabolite 2HG stand in a functional relationship to each other and are associated with stem-like features in cancer cells. 2HG accumulation (Key factor 1) inhibits αKG-dependent dioxygenases, which results in stabilization of HIF1α and in extensive epigenetic alteration. These methylation-specific changes cause a block of cell differentiation and an increase in the expression of stem and progenitor cell markers. 2HG accumulation is often associated with MYC pathway activation (Key factor 2) in breast cancer cells. MYC maintains stem cell pluripotency and simultaneously influences major metabolic pathways implicated in overactive tumor cell metabolism, including mitochondrial biogenesis. MYC expression is affected by the interaction of ERα with ERBB2 and strongly influences glutamine metabolism by regulating glutaminase expression. A metabolic switch towards activated glutaminolysis is mediated by extracellular lactate uptake via MCT1, stabilizing HIF2α and transactivating MYC, which subsequently triggers the expression of glutamine transporter ASCT2 and of glutaminase (GLS). Glutaminolysis (Key factor 3) contributes to cancer cell metabolism by fueling OXPHOS through the replenishment of TCA cycle intermediates, supplying building blocks for proliferation, replenishing GSH pools for ROS-scavenging (which involves the xCT cystine/glutamate antiporter for cysteine acquisition and the generation of NADPH by malic enzyme to reduce GSSG back to GSH), coupling glutamine export with leucine-import (which mobilizes mTORC1 and stimulates ribosome biosynthesis), and promoting 2HG accumulation via its precursors glutamate and αKG. 2HG, 2-hydroxyglutarate; 3PG, 3-phosphoglycerate; AA, amino acids; Ac-CoA, acetyl coenzyme A; ASCT2, ASC amino-acid transporter 2; αKG, alpha-ketoglutarate; Cys, cysteine; Cys-Cys, cystine; ERα, estrogen receptor alpha; ERBB2, erb-b2 receptor tyrosine kinase 2; F1,6BP, fructose 1,6-bisphosphate; F6P, fructose 6-phosphate; FA synthesis, fatty acid synthesis; FH, fumarate hydratase; Fum, fumarate; G6P, glucose 6-phosphate; GDH, glutamate dehydrogenase; Glc, glucose; Gln, glutamine; GLS, glutaminase; Glu, glutamate; GLUT, glucose transporter; GSH, glutathione (reduced form); GSSG, glutathione disulfide (oxidized form); HIF1α, hypoxia-inducible factor 1-alpha; HIF2α, hypoxia-inducible factor 2-alpha; HK2, hexokinase 2; IDH1/2, isocitrate-dehydrogenase 1/2; IDH1/2 mut, isocitrate-dehydrogenase 1/2 mutant; Lac, lactose; LAT1, L-type amino acid transporter 1; LDH, lactate dehydrogenase; Leu, leucine; ME, malic enzyme; MCT1, monocarboxylate transporter 1; MCT4, monocarboxylate transporter 4; mTORC1, mechanistic target of rapamycin complex 1; NADP+/NADPH, nicotinamide adenine dinucleotide phosphate (oxidized and reduced form); OAA, oxaloacetate; PDH, pyruvate dehydrogenase; PEP, phosphoenolpyruvate; PFK2, phosphofructokinase 2; PKM2, pyruvate kinase isozyme M2; ROS, reactive oxygen species; SDH, succinate dehydrogenase; Succ, succinate; TCA, tricarboxylic acid cycle; xCT/SLC7A11, solute carrier family 7 (xc- system), member 11, cystine/glutamate transporter; Light yellow boxes, medical interventions.
Notably, 2HG has emerged as a main focus of interest, particularly in the context of glioma and leukemia, which tend to accumulate 2HG by means of IDH1 or IDH2 mutations [56, 57]. It is important to note the enantiomer-specific effect of D-2-hydroxyglutarate (D-2HG) versus L-2-hydroxyglutarate (L-2HG) - both having been implicated in tumorigenesis - whereas ‘2HG’ usually refers to the sum of the two. As not all data distinguishes between D-2HG and L-2HG, we will refer to the cited data in the original manner of the study. Breast tumors, in comparison to normal breast tissue, have been shown to comprise high levels of 2HG - more than 200-fold in ER- breast cancer (basal-like/mesenchymal tumors showing the highest concentrations) and to a smaller (around 20-fold) extent in ER+ breast cancer [40]. In the context of breast tumors, however, mutations in IDH enzymes have not been identified, which is why alternative mechanisms of 2HG accumulation are being discussed (e.g. alcohol dehydrogenase, iron containing 1 (ADHFE1) overexpression or D-2-hydroxyglutarate dehydrogenase (D2HGDH) downregulation) [41]. Intracerebral 2HG injection in rats (both D-2HG and L-2HG) has resulted in increased oxidative damage on lipids, proteins, and DNA, paralleled by the suppression of antioxidative defense mechanisms [58, 59]. As mentioned above, both D-2HG and L-2HG have been shown to inhibit αKG-dependent dioxygenases, including prolyl hydroxylase domain (PHD) enzymes (which usually help degrade HIF1α), histone demethylases, and the ten-eleven translocation (TET) enzyme family [60]. The latter hydroxylases 5-methyl-cytosine (5mC) to 5-hydroxy-methyl-cytosine (5hmC), causing a dramatic shift in the 5hmC/5mC ratio and a genome-wide DNA hypermethylation phenotype [57, 60, 61]. Notably, these changes in histone and DNA methylation have been shown to cause a block of cell differentiation and an increase in the expression of stem and progenitor cell markers, indicating a switch to a stem-like phenotype in cancer cells (Figure 4, Key factor 1). Consistent with this, a striking overlap of 2HG accumulation and MYC pathway activation (Figure 4, Key factor 2) can be observed [41]. As MYC is known to be essential in maintaining stem cell pluripotency [62] and as it antagonizes re-differentiation of neoplastic cells into typical tissue-specific cells [63], these findings appear to be of utmost relevance for tumorigenesis. In comparison to other oncogenes, MYC is unique in the way that it simultaneously influences all cellular pathways that cumulatively contribute to overactive tumor cell metabolism: ribosome biogenesis, glycolysis, glutaminolysis, fatty acid synthesis, nucleotide metabolism, lactate accumulation, and - possibly most suggestive of a stem-like phenotype - mitochondrial biogenesis [64, 65]. This wide range of functions is reflected in a strong metabolic signature in tumors that harbor MYC pathway activation [41]. The MYC pathway even provides means for bypassing PI3K signaling, which is why resistance to PI3K pathway inhibitors often goes alongside MYC amplification [66, 67]. About 40-45 % of all breast tumors exhibit MYC overexpression [68], which is associated with unfavorable prognostic markers. MYC is a target of WNT/β-catenin signaling, which induces epigenetic repression of BRCA1 expression by means of snail family zinc finger 2 (SNAI2) and is associated with basal-like breast cancer and EMT, including metastasis [69]. Possibly offering the most important contribution to a major metabolic switch in tumor tissue, MYC strongly influences glutamine metabolism (Figure 4, Key factor 3) by regulating glutaminase (GLS) expression through miRNAs [70, 71]. A subgroup of the most 2HG-rich breast tumors showed a specific methylation pattern characterized by hypomethylation of the IDH2 locus and IDH2 overexpression; this subtype was more prevalent in patients of Afro-American descent with ER-, stem cell-like breast cancer, GLS overexpression, and reduced overall survival [41]. As MYC overexpression increases, and MYC knockdown decreases intracellular 2HG levels, but 2HG levels also decrease following GLS inhibition [41], a functional relationship between MYC, glutamine catabolism and 2HG accumulation is likely. Recent findings have highlighted a possible mechanism which may enable cancer cells to induce a metabolic switch towards activated glutaminolysis [72] (Figure 4, upper right corner): extracellular lactate uptake by means of monocarboxylate transporter 1 (MCT1) first stabilizes HIF2α, which then transactivates MYC in a pseudohypoxic response. This subsequently triggers the expression of glutamine transporter ASCT2 and of GLS, promoting glutamine uptake and catabolism. Glutamine catabolism, in turn, contributes to cancer cell metabolism in multiple ways, possibly provoking “glutamine addiction” in cancer cells (Figure 4, Key factor 3): (i) Glutamine fuels OXPHOS [73] and, thus, the very metabolic feature cancer cells - in contrast to CAFs - appear to depend on. Inhibition or knockdown of GLS has resulted in reduced OXPHOS, leading to an arrest in cell proliferation and activated mitochondrial apoptosis [73]. The mechanism by which glutaminolysis exerts its effect on OXPHOS is thought to mainly involve replenishment of TCA cycle intermediates by converting glutamine to glutamate and further to αKG; this way glutamine serves as the major carbon source for the TCA cycle. (ii) Glutamine supplies building blocks for cell proliferation by serving as a nitrogen source for nucleotides and amino acids and by supporting fatty acid synthesis through a process termed TCA turning (a partly reverse running TCA cycle) [74, 75]. (iii) Glutamine promotes the replenishment of reduced glutathione pools for ROS scavenging and subsequent escape from ROS-induced cell death, helping tumor cells to survive. This mechanism involves (a) the xCT (SLC7A11) antiporter for cysteine/glutamate exchange (Figure 4, right rim), which is expressed on one third of TNBC in vivo [76] and accounts for cysteine acquisition and subsequent use in GSH generation (reviewed in [77]). GSH consists of only three amino acids, these being glycine, cysteine, and glutamate, the last of which is directly synthesized from glutamine. Moreover (b), glutaminolysis is a means for NADPH generation via the conversion of glutamine to pyruvate by malic enzyme, which is necessary to reduce the oxidized form of glutathione - glutathione disulfide (GSSG) - back to its reduced form - GSH - , and thus replenish GSH pools [78] (Figure 4, turquoise arrows). (iv) Intracellular glutamine levels are crucial for quick proliferation in the way that glutamine export is coupled with an import of leucine, which mobilizes mechanistic target of rapamycin complex 1 (mTORC1) and stimulates ribosome biosynthesis [64]. (v) Glutamine catabolism is also involved in the aberrant accumulation of 2HG since αKG, glutamate, and glutamine serve as precursors for this oncometabolite. The aforementioned link between 2HG and a de-differentiation into a stem-like phenotype thus appears to complement a feed-forward cycle promoting CSC maintenance.
Glutaminase inhibitors have already been used successfully in preclinical studies inhibiting the proliferation of TNBC but not of ER+ breast cancer cell lines [79, 80]. By means of “glutamine deprivation”, MYC expression can be reduced in cell cultures and apoptosis can selectively be triggered in MYC-transformed cells [81]. As MYC expression can also be significantly influenced by the interaction of estrogen receptor alpha (ERα) with ERBB2, and as MYC-associated glutamine dependence has been linked to aromatase inhibitor (AI) resistance in ER+ breast cancer, fulvestrant - an ERα downregulator that also inhibits MYC expression and glutaminase - has been suggested as a therapeutic agent in AI-resistant ER+ breast tumors [82] and may experience future repurposing for further indications (Figure 4, upper right corner).
Metabolic signatures introduced by mutational landscape
Studies focusing on metabolic and pathway analyses in breast cancer cell lines either harboring (knock-in) or lacking (knock-out) a specific functional wild-type tumor suppressor gene can help elucidate metabolic changes associated with mutations in single genes or pathways. Differential studies on the metabolome of non-tumorous breast tissue derived from patients carrying a specific mutation in a breast cancer predisposition gene versus non-mutation carriers can also help elucidate metabolic alterations in those cells. Genes such as BRCA1, BRCA2, and TP53, but also PTEN, PIK3CA, CHEK2, RAD51C, and ATM are of major interest in this regard. Strikingly, results indicate a close relationship between tumor suppressor genes typically mutated in breast cancer and Warburg-like metabolism in those cells lacking the functional protein. In a germline setting, it is conceivable that a reduced dosage of functional protein or a dominant-negative effect of the mutated over the wild-type protein may not only drive tumor-initiating cells in acquiring a malignant phenotype, but may also result in a Warburg-like metabolic switch in the stromal environment. This, in turn, may predispose the organism to tumor development by setting the stage for the evolution of a CAF phenotype (Figure 3).
For instance, mutations in genetic components of the PI3K-AKT-mTOR-PTEN pathway usually result in strong basal activation of mTOR signaling, which is one of the most characteristic features of the CAF phenotype. Up to 44 % of all breast cancer subtypes bear PI3K pathway aberrations such as PIK3CA, PIK3R1, AKT1, and PTEN mutations, in particular luminal A (53.4 %) and ERBB2-enriched (47 %) subtypes, although much less common in the basal-like (10 %) subtype [83]. Research continuously focuses on this signaling pathway, as its activation appears to be an overall prerequisite for tumor formation. The pathway acts in an essential pro-proliferative and anti-apoptotic manner, stimulating protein and fatty acid synthesis, activating glycolysis, and promoting lactate efflux [84, 85]. Stimulation of glycolytic flux is accomplished by the upregulation of hexokinase 2 (HK2) and HIF1A expression mediated by AKT1 and mTORC1, respectively. Loss of PTEN, which physiologically counteracts PI3K function by dephosphorylating phosphatidylinositol (PI) 3,4,5-triphosphate to PI-4,5-bisphosphate, additionally impairs mitochondrial function via the interplay between PTEN and TP53 [86].
TP53 mutations, in turn, which are amongst the most frequent mutational events in cancers and can be found in about 37 % of all breast tumors and in up to 80 % of basal-like and ERBB2-enriched tumors [83], have also been demonstrated to cause the metabolic switch towards glycolysis in a Warburg-like manner (Figure 3). This mechanism involves the downstream TP53 target genes TP53-induced glycolysis and apoptosis regulator (TIGAR) and synthesis of cytochrome c oxidase-2 (SCO2). Whereas SCO2 regulates the cytochrome c oxidase complex, which is critical in OXPHOS [87], TIGAR operates as a fructose-2,6-bisphosphatase, resulting in the inhibition of glycolysis and the promotion of the pentose phosphate pathway (PPP) [88, 89]. Elevated NADPH levels due to activated PPP promote GSH synthesis and, thus, help to scavenge intracellular ROS, inhibiting autophagy [90, 91]. Loss of TIGAR, in turn, raises intracellular ROS levels and induces autophagy, in line with the autophagic CAF phenotype. In addition, wild-type TP53 is known to inhibit mTORC1 and its downstream targets, ultimately impairing protein synthesis and proliferation. In Li-Fraumeni syndrome (MIM 151623), which is a disorder caused by heterozygous TP53 germline mutations predisposing the subject to multiple malignancies including breast cancer, it is conceivable that the aforementioned major impact on energy metabolism, pro-proliferative signaling, and regulation of cellular survival mechanisms may result in stromal tissue that assists tumor initiating cells by presenting an environment according to their needs. Notably, one particular finding may further substantiate an impact of TP53 loss on metabolic coupling in tumors. TP53 is known to repress monocarboxylate transporter 1 (MCT1) expression, while TP53 deficiency results in elevated MCT1 expression [92]; however, the consequences of altered MCT1 expression levels appear to depend on the metabolic setting. Under hypoxic conditions, as mimicked in CAFs, elevated MCT1 expression due to loss of TP53 promotes lactate export, whereas under conditions of additional glucose deprivation - as may be simulated in cancer cells due to heightened energy demands - MCT1 expression promotes lactate import. Through this differentiated mechanism, cancer cells could profit greatly from the lactate efflux originating from adjacent CAFs.
Similar phenomena in terms of creating a “tumor-friendly” environment in stroma tissue have recently been observed concerning the tumor suppressor gene BRCA1 (Figure 3). BRCA1-associated breast cancer is often triple-negative or basal-like, which commonly confers poor prognosis [93]. Although mainly known for its role in homologous recombination DNA repair, BRCA1 has increasingly been attracting attention for various further functions, including its extensive role in cellular energy metabolism and in the regulation of oxidative stress [94, 95]. A BRCA1-mutated breast cancer cell line transfected with wild-type BRCA1 has displayed the reversal of Warburg-like metabolic features, including an activation of OXPHOS and an impairment of glycolytic flux via the inhibition of the expression of genes that all play major roles in glycolysis (e.g. SLC2A1, HK1, HK2, PFKFB3, and LDHA) [94]. BRCA1 also interacts with HIF1α, AKT1, MYC, and TP53, which have well-established roles in the regulation of glycolysis. Changes in lipid metabolism point to increased beta-oxidation and decreased fatty acid synthesis by means of acetyl-CoA-carboxylase-alpha (ACCA) inhibition through BRCA1 via stabilization of the phosphorylated and inactive form of ACCA, pACCA; free fatty acids and ketone bodies are consumed, and the generated acetyl-CoA may subsequently be fed into the TCA cycle [40, 94, 96]. CoA, acetyl-CoA, and several acylcarnitines are positively associated with BRCA1 mRNA levels, while various lipids (long chained fatty acids and membrane components) and amino acids are inversely correlated with BRCA1 transcription levels [40]. All of the above associations are consistent with a strong antagonizing impact of BRCA1 on Warburg-like metabolism.
Another interesting relationship exists between BRCA1 and nicotinamide-adenine-dinucleotide (NAD+): BRCA1 knockdown results in elevated NAD+ levels, whereas BRCA1 mRNA levels conversely increase with available NAD+ [97]. NAD+ is a weak competitive inhibitor of complex I of OXPHOS (NADH dehydrogenase) [98]. In the stroma of BRCA1 mutation carriers, low levels of BRCA1 resulting in elevated NAD+ supplies may thus exert an inhibitory effect on OXPHOS, contributing to the CAF phenotype. As Poly(ADP-ribose)-Polymerase (PARP) enzymes also use NAD+ as a substrate for their functioning in base excision repair, and as PARP inhibitors - suggested specifically for the treatment of BRCA1/2-mutated cancers - raise the pool of available NAD+, this association may deserve particular attention.
Moreover, BRCA1 greatly influences endocrine parameters. Aromatase expression is inhibited by BRCA1 in a manner similar to TP53 (Li-Fraumeni patients exhibit raised aromatase expression [99]), therefore limiting estradiol (E2) production [100]. BRCA1 impairs the transcriptional activity of ERα, and overexpression of BRCA1 subsequently inhibits the expression of 90 % of estrogen-inducible genes [101]. Furthermore, BRCA1 reduces expression levels of insulin-like growth factor 1 receptor (IGF1R) and its ligand IGF1 [102, 103]. Accordingly, BRCA1 germline mutation carriers are exposed to higher serum E2 levels (particularly in luteal phase) [104, 105], exhibit a higher activity of ERα and elevated IGF1R expression levels in estrogen-stimulated cells [106], and show intratumoral accumulation of IGF1 [107] combined with a decrease in IGF1 serum concentrations (likely due to increased binding to IGF1R) [100]. While an excess supply of E2 and also IGF1/2 upregulates BRCA1 expression in a compensative manner in BRCA1 wild-type cells [108, 109], this mechanism is impaired in mutation carriers. Consequently, the PI3K-AKT-mTOR pathway is stressed in multiple ways in BRCA1-deficient cells: E2 directly activates PI3K and AKT1, IGF1 activates the pathway by binding to IGF1R, and ERα provokes the phosphorylation of AKT1, while the inhibition of the latter by BRCA1 is hampered [110-112]. Phosphorylated and activated AKT1, in turn, stimulates the expression of ERα in a feed-forward mechanism [113]. If, in addition to this, insulin levels happened to be raised (e.g. in a pre-diabetic metabolic state), this would constitute further direct activation of the pathway as well as an increase in E2 levels [114].
Furthermore, antioxidative signaling pathways, including glutathione metabolism, are extensively upregulated by BRCA1. Direct interaction of BRCA1 with NRF2 - the “master regulator of antioxidant responses”, which is activated and stabilized by this interaction - induces the expression of various antioxidant enzymes that protect the cell from ROS and therefore prevent oxidative DNA damage [115]. NRF2 represents a Janus face in tumor biology [116]: while, on the one hand, helping to prevent malignant transformation in the first place (Nrf2 deficiency results in tumor initiation in mice [117], and loss of NRF2 activity due to BRCA1 deficiency has been suggested to contribute to (early-onset) carcinogenesis [95]), excessive NRF2 activation helps tumor cells, on the other hand, to escape apoptosis by upregulating ROS-scavenging antioxidant signaling such as glutathione replenishment. Indeed, NRF2 is known to be strongly upregulated in many tumors and to be associated with aggressive disease and bad prognosis [7]. Gorrini et al. [112] addressed this issue and created an interesting hypothesis concerning the E2-associated tissue specificity of BRCA1-deficient tumors, as follows. Estrogen abundance induces NRF2 accumulation in a PI3K-AKT-mTOR pathway-dependent manner (PI3K-pathway inhibition prevents NRF2 activation). In a physiological setting, this represents a reasonable mechanism, since the generation of estrogen metabolites (e.g. 4-OHE1/E2 or reactive estrogen quinones resulting from oxidation) can induce oxidative DNA damage by forming mutagenic depurinizing adenine/guanine-estrogen-DNA-adducts [118]. Under normal circumstances, an NRF2 accumulation would subsequently be followed by an increase in BRCA1 expression [119], initiating the repair of DNA damage parallel to ROS scavenging and cell survival. This is obviously not possible in BRCA1-deficient cells (and may additionally be impaired in wild-type BRCA1 cells by mechanisms such as epigenetic repression or NAD+ depletion). Hence, PI3K hyperactivation (either by means of constant stimulation, or by means of PTEN deficiency, which can often be found in BRCA1-mutated tumors [120]) coupled with E2 stimulation (which is tissue-specific and generally elevated in BRCA1 mutation carriers) may lead to NRF2 accumulation in BRCA1-/- pre-tumor cells (germline first hit and somatic second hit), helping the cell to escape ROS-induced cell death. Combined with the lack of BRCA1 DNA repair efficiency and resultant genomic instability, this may ultimately induce malignant transformation. Tissue types that are not stimulated by estrogens for the lack of appropriate receptor abundance would not be affected by this mechanism and, thus, were to induce ROS-associated cell death in the case of somatic second hit and randomly generated BRCA1-/- cells.
Finally, BRCA1 has been identified as an important player in autophagy, in the sense that BRCA1 deficiency promotes Beclin 1-dependent autophagic pathways in response to metabolic stresses (e.g. serum starvation or endoplasmatic reticulum stress) [121]. In BRCA1-deficient cancer cells, this stress-mitigating mechanism helps to prevent cell death, supporting cancer cell survival. In stromal cells of BRCA1 mutation carriers, in turn, these findings point to an innate enhancement of autophagic pathways, which is in agreement with the CAF phenotype.
In conclusion, BRCA1 impairment can be expected to result in Warburg-like metabolism, activation of PI3K signaling, elevated ROS signaling, impaired antioxidant response, and enhanced autophagy - the very features of the CAF phenotype. As silencing of BRCA1 expression by means of promoter hypermethylation has also been suggested as an important event in the pathogenesis of sporadic breast cancer disease [122-124], particularly so in TNBC, these findings may have broad implications beyond hereditary breast cancer pathogenesis.
Blood-based metabolome analyses and biomarkers
Besides focusing on tumor tissue and adjacent non-tumorous tissue, it also appears promising to metabolically analyze peripheral white blood cells as well as correlated serum or plasma samples of (i) hereditary breast cancer patients, classified by the affected gene, versus (ii) corresponding mutation carriers who have not (yet) developed cancer, and (iii) a healthy control collective without history of familial breast cancer. Similar to the influence single nucleotide polymorphisms (SNPs) are known to exert on blood metabolite levels [125, 126], systemic metabolic changes resulting from specific germline mutations, possibly setting the stage for tumor formation, are conceivable and may be reflected in these particular analyses. A comparison between diseased versus non-diseased individuals affected by the same germline mutation would be of particular value, preferably focusing on individuals from the same family lineage in order to reduce metabolic noise stemming from overall genetic variability. Likewise, systemic metabolic changes that may predispose an individual to breast cancer development in a sporadic setting and which may be reflected by changes in biofluids or peripheral blood cells are of high interest and have partly been addressed already. At this point, however, it is difficult to securely attribute the metabolic variation detected in serum or plasma of cancer patients either to changes having predisposed the individual to tumor formation in the first place, or to changes being the result of tumor formation. Longitudinal epidemiologic studies will help elucidate this matter. For environmental factors known to contribute to sporadic breast cancer risk, e.g. smoking, alcohol consumption, or BMI, a strong correlation between an exposure to these factors and blood metabolites has already been confirmed [127-131].
As for finding adequate biomarkers to predict disease occurrence and disease status, no single marker but rather specific sets of markers, and multiple versions thereof, have been suggested from serum and plasma metabolome analyses on breast cancer patients independent of mutational status. While these marker sets vary substantially between each other, they do consistently include modifications within glycolysis parameters, phospholipid metabolism, fatty acid metabolism, and amino acid metabolism (reviewed in [53]). One study by Asiago et al. [132] presented a set of 11 markers (namely formate, histidine, prolin, choline, tyrosine, 3-HB, lactate, glutamic acid, N-acetyl-glycine, 3-hydroxy-2-methyl-butanoic acid, and nonanedioic acid) being able to predict recurrent breast cancer disease with a sensitivity of 86 % and a specificity of 84 %. In about half of these cases, this prediction could be made more than a year prior to clinical diagnosis for relapse. Similar advances were achieved in a study by Tenori et al. [133], in which lower levels of histidine and higher levels of glucose, lactate, tyrosine, and lipids were able to predict metastatic disease in 83.7 % of cases, which were predominantly ER-. Likewise, Jobard et al. [134] identified a set of nine metabolites (histidine, acetoacetate, glycerol, pyruvate, N-acetyl glycoproteins (NAC 1 and 2), mannose, glutamate, and phenylalanine) associated with advanced metastatic disease. Three fatty acids (C22:0, C24:0, and C18:2n6 - namely behenic acid, lignoceric acid, and linoleic acid, respectively) were identified to be significantly reduced in plasma of premenopausal and postmenopausal breast cancer patients [135]. Yet another study suggested mainly two markers, lysophosphatidylethanolamine and ceramide, to play an important role in invasive ductal carcinoma [136]. Notably, low circulating levels of the amino acid aspartate have recently been correlated with breast cancer occurrence, and this association was specific to breast tumors as opposed to other malignancies such as gastric or colorectal cancers [137]. Since breast tumors tend to accumulate high intracellular levels of aspartate, increased tumor uptake has been suggested to attribute for low circulating levels of this amino acid. In conjunction with this, it is important to note that, in addition to heightened uptake, aspartate biosynthesis has been identified to be intrinsically elevated in cancer cells and is discussed as one of the chief requirements underlying excessive proliferation as well as redox balance for survival [138, 139].
To the best of our knowledge, untargeted metabolome analyses have not yet been specifically conducted on serum/plasma samples of BRCA1 or BRCA2 mutation carriers. Serum proteomic analyses on BRCA1 mutation carriers have, however, revealed great differences between the protein expression profile of (i) individuals who developed a tumor within the next 3 years, possibly harboring a tumor already at baseline, (ii) those who stayed tumor-free within 7 years of follow-up (until trial closure), and (iii) a cohort of sporadic breast cancer patients; in contrast, accurate discrimination was not possible between healthy controls versus tumor-free BRCA1 mutation carriers [140]. Another more recent proteomic study [141] on BRCA1 mutation carriers of a specific founder mutation suggested a BRCA1-specific proteomic profiling signature and the downregulation of the protein gelsolin as a promising biomarker differentiating between diseased and non-diseased individuals.
Without question, it would be desirable to identify diagnostic and prognostic biomarkers of even superior sensitivity and specificity to the above metabolite ratios in serum/plasma of breast cancer patients (mutation carriers as well as non-mutation carriers). Importantly, these need to be suitable for clinical use and will have to prove reliable in the long run, reducing false-positive and false-negative results to a minimum, which is of particular relevance for such a widespread disease of high overall prevalence.
Preventive and therapeutic measures
For preventive action regarding mutation carriers in particular but also the general public, deep knowledge of aberrant metabolic signaling and enzyme/substrate modification will give important clues about etiologically significant factors in need for targeting on a long-term basis. Ideally, altered signaling resulting from reduced expression of tumor suppressor genes could be antagonized on a metabolic level downstream of genomic and transcriptomic events, balancing the aberration’s consequences. By identifying appropriate, targeted means of intervening in misbalanced biochemical signaling patterns (e.g. via drugs such as small molecular PI3K-AKT-mTOR inhibitors, glutaminase inhibitors, fulvestrant, metformin, but possibly also by means of glutamine and glucose restriction, specific substrate substitution, antioxidant modification, and general lifestyle changes), one may eventually get down to decreasing tumor rates in genetically predisposed individuals. This includes BRCA1/2 mutation carriers, whose sole option for actively reducing tumor burden currently involves risk-reducing surgery (mastectomy, adnexectomy).
At the same time it is indispensable to find suitable therapeutic means once tumors have arisen. In particular, this concerns TNBC patients for whom, as of date, no standard established treatment line exists due to the absence of specific target receptors. TNBC patients account for about 15 % of all breast cancer cases [93], and about 10-15 % of these harbor germline BRCA1 mutations [142]. According to genomic profiling, TNBC can further be classified into two major subgroups, BRCA1-like and non-BRCA1-like. A study by Severson et al. [143] recently revealed that about 55 % of the analyzed TNBC samples could be classified as BRCA1-like, including around 15 % exhibiting BRCA1 promoter hypermethylation and 10 % harboring a BRCA1 germline mutation. These BRCA1-like tumors were found to frequently harbor somatic TP53 mutations, whereas non-BRCA1-like tumors frequently harbored mutations in PI3K pathway components such as PIK3CA. This implies that separate subgroups within TNBC may need entirely different treatment (e.g. PARP inhibitors for BRCA1-like tumors and PI3K pathway inhibitors for non-BRCA1-like tumors). Moreover, a few other studies concerning TNBC have recently identified a number of mutated genes, particularly DNA repair genes and cell cycle regulators, in BRCA1/2 mutation-negative TNBC [144-146]. Thus, further splitting of the two major subgroups into additional sub-categories appears likely and may reveal more distinct metabolic information and therapeutic options.
Within the context of personalized medicine, approaching each tumor, each patient, and each mutation carrier on an individual basis according to metabolomic and genomic profiling will eventually enable customized preventive and therapeutic measures. Whereas gene-therapeutic means - still rather utopic for the time being - would certainly depict the most causal treatment for mutation carriers, metabolic modification occurring downstream of the genomic level should attempt to tackle biochemical imbalances as far up the root as possible. The further downstream the intervention takes place, the more likely it will be that only part of the matter is solved and that the tumor cell finds a way to bypass the intervention by means of metabolic adaptation, thus, acquiring resistance. However, in order to fully evaluate, assess, and lastly benefit from each tumor’s individuality, it is indispensable to thoroughly comprehend root metabolic mechanisms of tumor cell proliferation, cell growth, and cell survival, as well as rate-limiting steps thereof (Figure 4, bright red text). This will allow creating a more complete overall picture of tumor biology, in which individual aberrations can subsequently be assessed in a differentiated manner.
These root metabolic mechanisms and their rate-limiting steps require in-depth discussion and will likely be subject of ongoing future debate and research. In terms of (i) tumor cell survival, it appears reasonable to suggest the ROS-scavenging potential of tumor cells being one of the most essential rate-limiting steps. Some tumors, particularly ER- ones, may achieve this goal by upregulating MYC - either through MYC amplification or by means of MYC overexpression (e.g. due to great amounts of lactate import from adjacent stroma cells) -, which should result in activated glutaminolysis and subsequent GSH production, conferring a “glutamine addiction” phenotype. Other tumors, mainly ER+ ones, may achieve the same goal by the simple upregulation of estrogen receptors in an estrogen-rich setting, leading to (a) NRF2 accumulation in the presence of an activated PI3K pathway and, thus, elevated antioxidant response, and to (b) MYC expression [147] through a process involving an upstream enhancer and the activator protein 1 (AP-1) transcription factor, leading to similar consequences as described above. The effect is the same. However, these mechanisms may be adaptable and even overlap to some extent, which would make therapeutic targeting even more challenging. For instance, once ER+ tumor cells find themselves in an estrogen-deprived environment due to antiestrogen therapy such as tamoxifen, some of these cells - especially CSCs - might theoretically be capable of adapting to this challenge by switching to an alternative way of accumulating GSH (e.g. glutamine consumption). On the other hand, once glutamine-dependent tumors were to be targeted by glutaminase inhibition, they might adapt to alternative ROS scavenging mechanisms, such as NRF2 upregulation. Hence, simultaneous targeting of multiple possible adaptation methods tackling the same root process may represent the most promising approach in order to prevent adaptation in the first place - very similar, in fact, to finding the right combination of antibiotics for secure and fast eradication of multi-resistant bacteria while preventing further resistances.
The rate-limiting step to (ii) tumor cell proliferation and growth, in turn, likely includes mitochondrial biogenesis and, thus, ATP generation through mitochondrial OXPHOS in the cancer cell compartment, supported by macronutrient supply from the adjacent glycolytic stromal compartment. Yet again, MYC activation appears to be an important player, as mitochondrial biogenesis is triggered by MYC [148]. In light of the two-compartment model of cancer, it appears absolutely indispensable to target both compartments simultaneously and stringently. By conventional chemotherapeutic or radiotherapeutic approaches, this major goal is not achieved: whereas bulk cancer cells may be drastically reduced, CSCs are left to survive and the CAF phenotype is reinforced by great amounts of ROS originating from CTx, which has been suggested to pave the way for relapse and metastasis and to correlate with poor prognosis [34]. Notably, both compartments appear to rely heavily on activated PI3K-AKT-mTOR signaling, which is a major advantage for therapeutic targeting. Therefore, current strategies are focused on small molecular PI3K-pathway inhibitors such as everolimus, which is in use in clinical trials and has already been approved for specific indications in breast cancer patients.
Metformin, another strong mTOR inhibitor and a widely used oral antidiabetic drug, has lately been receiving considerable attention for its cancer-preventive and cancer-therapeutic effects [149-151] and is on its way to being repurposed for preventive and therapeutic use in cancer patients - an example of “drug repositioning” [152]. Metformin is mainly known to impair gluconeogenesis in liver tissue. However, another mechanism of action can be attributed to the inhibition of complex 1 of the respiratory chain [153, 154], which promotes AMPK activation and, ultimately, glycolysis (in fact, one very rare but feared side effect of metformin is lactic acidosis evolving from activated glycolysis combined with impaired gluconeogenesis from lactate). This appears contradictory on first glance. If a tumor was to thrive on Warburg metabolism alone, as has been suggested previously, this mechanism would not be expected to antagonize but instead to drive tumor progression. In the context of two-compartment reverse Warburg tumor metabolism, however, the potential mechanism of action appears obvious: while antagonizing both compartments by strongly and directly inhibiting mTOR, metformin additionally inhibits OXPHOS in cancer cells relying on mitochondria for energy production. In fact, CSCs have been shown to be particularly sensitive towards metformin treatment [30]. Consistent with this, metformin has also been found to downregulate MYC mRNA by inducing the expression of ribonuclease DICER1 and upregulating miRNA-33a in an AMPK-dependent manner [155]. There is evidence, however, that metformin’s action in promoting cancer cell death is significantly enhanced when glucose availability is drastically restricted [156]. Metformin itself lowers plasma glucose levels only by about 3 mM (55 mg/dl) in diabetics [157] and does not have any significant glucose-lowering effect in non-diabetics [158]. As patient compliance for strict glucose restriction may not be very high (e.g. adherence to ketogenic or calorie-restricted diets - though probably most natural for humans over the course of millions of years of evolution), an alternative option might be the combined administration of metformin with 2-deoxyglucose, which is taken up in place of glucose but cannot subsequently be used for glycolytic catabolism [159-162]. The enhanced cytotoxicity of this drug combination to cancer cells has been shown for a broad spectrum of cancer models - in vitro and in vivo [163] - but has, to the best of our knowledge, only been shown experimentally to this date. However, since both drugs have been widely used in clinical diagnostic or therapeutic settings for decades, clinical application of the drug combination should be able to follow promptly. Metformin has further been shown to greatly improve the anti-cancer efficacy of chemotherapeutic drugs commonly used in breast cancer, often allowing reduced dosage regimens [164, 165]. Moreover, combinations with PI3K pathway inhibitors, e.g. rapamycin and everolimus, and with tamoxifen, trastuzumab, and erlotinib have revealed synergistic effects in breast cancer treatment [166-171]. Most likely being of great advantage for the majority of cancers, metformin appears to be particularly well suited for the needs of BRCA1 germline mutation carriers [149] for the following reasons. (i) BRCA1 is imitated in its role in ACCA inhibition (as metformin activates AMPK, which in turn inhibits ACCA), thereby impairing fatty acid synthesis. (ii) Metformin acts selectively toxic on TP53-deficient cells (as is almost always the case in BRCA1-associated tumors). (iii) Metformin antagonizes PI3K-AKT-mTOR signaling (e.g. due to additional PTEN deficiency) through mTOR inhibition. (iv) Metformin antagonizes IGF1R overexpression via disruption of androgen signaling, aromatase (and thus E2 generation), and ER expression levels [151, 172, 173], substantially attenuating the double burden on PI3K signaling in BRCA1 germline mutation carriers.
Another potential treatment option to specifically target mitochondrial biogenesis in CSCs has recently been described by Lamb et al. for a long-known class of drugs [174]. Antibiotics known for targeting bacterial protein biosynthesis, including erythromycins and tetracyclines, inhibit mitochondrial biogenesis as a known side effect. Intriguingly, the authors have shown that these antibiotic drugs can be used to specifically eradicate CSCs in 12 different cancer cell lines across eight different tumor types including breast cancer, effectively treating cancer as a “single disease of stemness”. To trace down chemo-resistant CSCs after applied CTx and to target them in their individual vulnerable hotspots, the same group has suggested an innovative method for personalized cancer treatment [175]. First, mitochondrial fluorescent staining is applied to primary tumor or metastatic tissue in order to estimate mitochondrial mass (cells with high mitochondrial mass are also specifically enriched with CSC markers). Then, cells are sorted by flow cytometry in order to isolate mito-high cells (CSCs). Finally, these cells can be tested for sensitivity towards a broad range of chemotherapeutics, mitochondria-targeting antibiotics, metformin or other biguanides, and further drugs. This exciting new treatment concept will likely be a major focus of future research.
Conclusions and outlook
There have been several attempts to correlate molecular breast cancer subtypes with major metabolic features. For instance, Lloyd et al. [176] proposed to classify ER+ versus ER- tumors into glucose-dependent versus glutamine-dependent, respectively. However, results from a recent study by Willmann et al. [48] largely contradict this proposal. In this study, the analyzed ER+ breast cancer cell line BT-474 showed the lowest glucose consumption and the lowest lactate levels in comparison to all other investigated cell lines, which were ER-. Timmerman et al. [76] also showed that molecular subtypes of breast cancer cell lines do not neatly separate into glucose or glutamine high and low consumers. It is possible that metabolic studies on cell culture models of breast cancer may be heavily biased due to unphysiological conditions such as altered composition and concentration of the cell medium or lack of inclusion of in vivo tumor microenvironment, which excludes an important player in tumor metabolism. Thus, cells that only show moderate glucose consumption in an in vitro setting may be ones that - in an in vivo setting - substantially rely on macronutrients obtained from adjacent CAFs, rather than on their own glycolytic flux, and may therefore be more adapted to alternative fuel sources besides glucose. On the other hand, cells that show high glucose consumption in vitro may either be more autonomous in an in vivo setting or may profit from adjacent CAFs’ metabolism in addition to their own and subsequently exhibit a more aggressive phenotype in vivo. Martinez-Outschoorn et al. [10] have attempted to diminish this problem by mimicking the breast tumor microenvironment utilizing an in vitro co-culture model of human breast cancer cells and immortalized human fibroblasts. Regarding the potential metabolic heterogeneity between cancer cells existing in immediate adjacency to each other in an in vitro as well as an in vivo setting, cell sorting via FACS prior to fractionized metabolome analyses on separate cell types, or even single-cell metabolome analyses, may be mandatory. Great metabolic heterogeneity, as a possible reflection of the individual adaptability of cancer cells, may further demonstrate the necessity to focus in parallel on (i) joint, central pathways that appear rate-limiting to each tumor independent of individual adaptations (e.g. mitochondrial OXPHOS in cancer cells, glycolysis particularly in CAFs, and PI3K signaling in both), and (ii) individual customized treatment by means of specific enzyme and substrate substitution following individual metabolome analyses or through tumor cell sorting with subsequent drug sensitivity screening in a manner similar to antibiogram screening.
As ROS signaling appears to play a major initiating role in the onset of both CAF and CSC phenotypes, one is prone to wonder why patients of distinct mitochondrial disease, which often leads to high amounts of ROS, do not appear to exhibit excessive tumor incidence rates at a young age already. In fact, taking into account the above findings, innate dysfunctional mitochondria could even be protective to the organism in regard to tumor formation, since highly efficient OXPHOS - possibly a prerequisite for rapid proliferation - is essentially impaired. Mitophagy defects, acquired or innate, may indeed confer a dual protective mechanism as (i) CAFs may be hampered in exporting nutrients resulting from autophagic degradation, and (ii) cancer cells may be hindered in acquiring highly productive, powerful mitochondrial mass. Interestingly, for Parkinson’s disease, in which defective mitophagy has been found to play a major pathogenic role, a negative correlation between disease prevalence and the incidence of most cancers has indeed been described (reviewed in [177]).
Alas, deeper understanding of the metabolic mechanisms involved in tumorigenesis - perhaps above all the conditioning of stroma cells towards an oxidation-rich, glycolytic, pro-inflammatory, and pro-tumorigenic state - may serve as a basis not only for further elucidation of oncogenic processes, but potentially also for the illumination of a large number of chronic “non-communicable” diseases, interlinking metabolic syndrome and cardiovascular disease, neurodegenerative and neuro-psychiatric disorders, autoimmune and chronic inflammatory disease [178]. All of these conditions have been implicated in similar aging-related metabolic changes, notably aberrant ROS accumulation, inflammation, mitochondrial dysfunction, autophagy-related changes, and an imbalance between pro-proliferative and pro-apoptotic signaling. Alterations within the corresponding underlying cellular pathways, which are physiologically meant to assist regulatory processes (e.g. wound healing, germ-fighting, tissue regeneration, cell survival strategies, and simple growth), present a major focus of ongoing trans-sectoral research and point towards hereditary factors and environmental modifiers being equally contributive operating forces in driving metabolic variance. Further research focusing on disease-associated metabolomic aberrations in a major effort to finding preventive and therapeutic measures against the diseases of our time - in particular common but devastating malignancies such as breast cancer - is warranted.
Abbreviations
2HG-2-hydroxyglutarate, 3PG-3-phosphoglycerate, AA-amino acids, Ac-CoA-acetyl coenzyme A, ACCB-acetyl-CoA-carboxylase-beta, AMPK-AMP-activated protein kinase, ASCT2-ASC amino-acid transporter 2, AMP-adenosine monophosphate, ATP-adenosine triphosphate, αKG-alpha-ketoglutarate, CAF-cancer-associated fibroblast, CAV1-caveolin 1, Cys-cysteine, Cys-Cys-cystine, E2 -estradiol, EMT-epithelial-mesenchymal transition, ERα-estrogen receptor alpha, ERBB2-erb-b2 receptor tyrosine kinase 2, ERRα-estrogen-related receptor alpha, F1,6BP-fructose 1,6-bisphosphate, F6P-fructose 6-phosphate, FA oxidation-fatty acid oxidation, FA synthesis-fatty acid synthesis, FH-fumarate hydratase, Fum-fumarate, G6P-glucose 6-phosphate, gBRCA1m-germline BRCA1 mutation, GDH-glutamate dehydrogenase, Glc-glucose, Gln-glutamine, GLS-glutaminase, Glu-glutamate, GLUT-glucose transporter, gPTENm-germline PTEN mutation, GSH-glutathione (reduced form), GSSG-glutathione disulfide (oxidized form), gTP53m-germline TP53 mutation, HIF1α-hypoxia-inducible factor 1-alpha, HIF2α-hypoxia-inducible factor 2-alpha, HK2-hexokinase 2, IDH1/2-isocitrate-dehydrogenase 1/2, IDH1/2 mut-isocitrate-dehydrogenase 1/2 mutant, IGF1-insulin-like growth factor 1, IGF1R-insulin-like growth factor 1 (IGF-1) receptor, Lac-lactose, LAT1-L-type amino acid transporter 1, LDH-lactate dehydrogenase, Leu-leucine, ME-malic enzyme, MCT1-monocarboxylate transporter 1, MCT4-monocarboxylate transporter 4, mTOR-mechanistic target of rapamycin, NADP+/NADPH-nicotinamide adenine dinucleotide, phosphate-(oxidized and reduced form), NFĸB-nuclear factor-ĸB, NRF2-nuclear factor E2-related factor 2, OAA-oxaloacetate, OXPHOS-oxidative phosphorylation, PDH-pyruvate dehydrogenase, PEP-phosphoenolpyruvate, PFK2-phosphofructokinase 2, PGC1-peroxisome proliferator-activated receptor γ, coactivator 1, PKM2-pyruvate kinase isozyme M2, ROS-reactive oxygen species, SDH-succinate dehydrogenase, SIRT-sirtuins, Succ-succinate, TGFβ-transforming growth factor beta, TCA-tricarboxylic acid cycle, TSC2-tuberous sclerosis complex 2, ULK1-unc-51-like kinase 1, xCT/SLC7A11-solute carrier family 7 (xc- system), member 11, cystine/glutamate transporter.
Acknowledgments
The authors gratefully acknowledge Daniel Penkert, Stephanie Schubert, Lisa Pahl, and Claudia Davenport for critically reading the manuscript.
Conflicts of interest
The authors have no conflicts of interest to disclose.
References
1. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
2. Hanahan D and Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57-70.
3. Warburg O. On the origin of cancer cells. Science. 1956; 123:309-314.
4. Wallace DC. Mitochondria and cancer. Nature reviews Cancer. 2012; 12:685-698.
5. Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F and Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009; 8:3984-4001.
6. Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F and Lisanti MP. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011; 10:2504-2520.
7. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011; 475:106-109.
8. Shanware NP, Mullen AR, DeBerardinis RJ and Abraham RT. Glutamine: pleiotropic roles in tumor growth and stress resistance. J Mol Med (Berl). 2011; 89:229-236.
9. Kim J, Kundu M, Viollet B and Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature cell biology. 2011; 13:132-141.
10. Martinez-Outschoorn UE, Balliet R, Lin Z, Whitaker-Menezes D, Birbe RC, Bombonati A, Pavlides S, Lamb R, Sneddon S, Howell A, Sotgia F and Lisanti MP. BRCA1 mutations drive oxidative stress and glycolysis in the tumor microenvironment: implications for breast cancer prevention with antioxidant therapies. Cell Cycle. 2012; 11:4402-4413.
11. Martinez-Outschoorn UE, Curry JM, Ko YH, Lin Z, Tuluc M, Cognetti D, Birbe RC, Pribitkin E, Bombonati A, Pestell RG, Howell A, Sotgia F and Lisanti MP. Oncogenes and inflammation rewire host energy metabolism in the tumor microenvironment: RAS and NFkappaB target stromal MCT4. Cell Cycle. 2013; 12:2580-2597.
12. Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, Howell A, Aquila S, Ando S, Martinez-Outschoorn U, Sotgia F and Lisanti MP. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: connecting TGF-beta signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle. 2012; 11:3019-3035.
13. Salem AF, Al-Zoubi MS, Whitaker-Menezes D, Martinez-Outschoorn UE, Lamb R, Hulit J, Howell A, Gandara R, Sartini M, Galbiati F, Bevilacqua G, Sotgia F and Lisanti MP. Cigarette smoke metabolically promotes cancer, via autophagy and premature aging in the host stromal microenvironment. Cell Cycle. 2013; 12:818-825.
14. Sanchez-Alvarez R, Martinez-Outschoorn UE, Lin Z, Lamb R, Hulit J, Howell A, Sotgia F, Rubin E and Lisanti MP. Ethanol exposure induces the cancer-associated fibroblast phenotype and lethal tumor metabolism: implications for breast cancer prevention. Cell Cycle. 2013; 12:289-301.
15. Reuter S, Gupta SC, Chaturvedi MM and Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free radical biology & medicine. 2010; 49:1603-1616.
16. Ho SM, Johnson A, Tarapore P, Janakiram V, Zhang X and Leung YK. Environmental epigenetics and its implication on disease risk and health outcomes. ILAR journal / National Research Council, Institute of Laboratory Animal Resources. 2012; 53:289-305.
17. Sampson N, Berger P and Zenzmaier C. Therapeutic targeting of redox signaling in myofibroblast differentiation and age-related fibrotic disease. Oxidative medicine and cellular longevity. 2012; 2012:458276.
18. Marusyk A and Polyak K. Tumor heterogeneity: causes and consequences. Biochimica et biophysica acta. 2010; 1805:105-117.
19. Meads MB, Gatenby RA and Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nature reviews Cancer. 2009; 9:665-674.
20. Ghiaur G, Gerber J and Jones RJ. Concise review: Cancer stem cells and minimal residual disease. Stem Cells. 2012; 30:89-93.
21. Yoshida GJ and Saya H. Therapeutic strategies targeting cancer stem cells. Cancer science. 2016; 107:5-11.
22. Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, Oshima M, Ikeda T, Asaba R, Yagi H, Masuko T, Shimizu T, Ishikawa T, Kai K, Takahashi E, Imamura Y, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer cell. 2011; 19:387-400.
23. Yae T, Tsuchihashi K, Ishimoto T, Motohara T, Yoshikawa M, Yoshida GJ, Wada T, Masuko T, Mogushi K, Tanaka H, Osawa T, Kanki Y, Minami T, Aburatani H, Ohmura M, Kubo A, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nature communications. 2012; 3:883.
24. Cahu J, Bustany S and Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell death & disease. 2012; 3:e446.
25. Ogasawara MA and Zhang H. Redox regulation and its emerging roles in stem cells and stem-like cancer cells. Antioxidants & redox signaling. 2009; 11:1107-1122.
26. Yoshida GJ and Saya H. Inversed relationship between CD44 variant and c-Myc due to oxidative stress-induced canonical Wnt activation. Biochemical and biophysical research communications. 2014; 443:622-627.
27. Viale A, Pettazzoni P, Lyssiotis CA, Ying H, Sanchez N, Marchesini M, Carugo A, Green T, Seth S, Giuliani V, Kost-Alimova M, Muller F, Colla S, Nezi L, Genovese G, Deem AK, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014; 514:628-632.
28. Lamb R, Bonuccelli G, Ozsvari B, Peiris-Pages M, Fiorillo M, Smith DL, Bevilacqua G, Mazzanti CM, McDonnell LA, Naccarato AG, Chiu M, Wynne L, Martinez-Outschoorn UE, Sotgia F and Lisanti MP. Mitochondrial mass, a new metabolic biomarker for stem-like cancer cells: Understanding WNT/FGF-driven anabolic signaling. Oncotarget. 2015; 6:30453-30471. doi: 10.18632/oncotarget.5852.
29. De Luca A, Fiorillo M, Peiris-Pages M, Ozsvari B, Smith DL, Sanchez-Alvarez R, Martinez-Outschoorn UE, Cappello AR, Pezzi V, Lisanti MP and Sotgia F. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget. 2015; 6:14777-14795. doi: 10.18632/oncotarget.4401.
30. Ning X, Du Y, Ben Q, Huang L, He X, Gong Y, Gao J, Wu H, Man X, Jin J, Xu M and Li Z. Bulk pancreatic cancer cells can convert into cancer stem cells(CSCs) in vitro and two compounds can target these CSCs. Cell Cycle. 2016; 15.403-412.
31. Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME and Lengyel E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer discovery. 2012; 2:1100-1108.
32. Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, Richardson M, Rigaill G, Parrini MC, Lucchesi C, Bellanger D, Stern MH, Dubois T, Sastre-Garau X, Delattre O, Vincent-Salomon A, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO molecular medicine. 2010; 2:211-230.
33. Wen S, Niu Y, Yeh S and Chang C. BM-MSCs promote prostate cancer progression via the conversion of normal fibroblasts to cancer-associated fibroblasts. International journal of oncology. 2015; 47:719-727.
34. Peiris-Pages M, Smith DL, Gyorffy B, Sotgia F and Lisanti MP. Proteomic identification of prognostic tumour biomarkers, using chemotherapy-induced cancer-associated fibroblasts. Aging (Albany NY). 2015; 7:816-38. doi: 10.18632/aging.100808.
35. Martinez-Outschoorn UE, Sotgia F and Lisanti MP. Caveolae and signalling in cancer. Nature reviews Cancer. 2015; 15:225-237.
36. Dimmer KS, Friedrich B, Lang F, Deitmer JW and Broer S. The low-affinity monocarboxylate transporter MCT4 is adapted to the export of lactate in highly glycolytic cells. The Biochemical journal. 2000; 350 Pt 1:219-227.
37. Ullah MS, Davies AJ and Halestrap AP. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. The Journal of biological chemistry. 2006; 281:9030-9037.
38. Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, Sneddon S, Martinez-Outschoorn UE, Sotgia F and Lisanti MP. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012; 11:1108-1117.
39. Budczies J, Brockmoller SF, Muller BM, Barupal DK, Richter-Ehrenstein C, Kleine-Tebbe A, Griffin JL, Oresic M, Dietel M, Denkert C and Fiehn O. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: alterations in glutamine and beta-alanine metabolism. Journal of proteomics. 2013; 94:279-288.
40. Tang X, Lin CC, Spasojevic I, Iversen ES, Chi JT and Marks JR. A joint analysis of metabolomics and genetics of breast cancer. Breast cancer research. 2014; 16:415.
41. Terunuma A, Putluri N, Mishra P, Mathe EA, Dorsey TH, Yi M, Wallace TA, Issaq HJ, Zhou M, Killian JK, Stevenson HS, Karoly ED, Chan K, Samanta S, Prieto D, Hsu TY, et al. MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. The Journal of clinical investigation. 2014; 124:398-412.
42. Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB and Thompson CB. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:18782-18787.
43. Budczies J, Pfitzner BM, Gyorffy B, Winzer KJ, Radke C, Dietel M, Fiehn O and Denkert C. Glutamate enrichment as new diagnostic opportunity in breast cancer. International journal of cancer. 2015; 136:1619-1628.
44. Bezabeh T, Ijare OB, Nikulin AE, Somorjai RL and Smith IC. MRS-based Metabolomics in Cancer Research. Magnetic resonance insights. 2014; 7:1-14.
45. Cao MD, Lamichhane S, Lundgren S, Bofin A, Fjosne H, Giskeodegard GF and Bathen TF. Metabolic characterization of triple negative breast cancer. BMC cancer. 2014; 14:941.
46. Li M, Song Y, Cho N, Chang JM, Koo HR, Yi A, Kim H, Park S and Moon WK. An HR-MAS MR metabolomics study on breast tissues obtained with core needle biopsy. PloS one. 2011; 6:e25563.
47. Brockmoller SF, Bucher E, Muller BM, Budczies J, Hilvo M, Griffin JL, Oresic M, Kallioniemi O, Iljin K, Loibl S, Darb-Esfahani S, Sinn BV, Klauschen F, Prinzler J, Bangemann N, Ismaeel F, et al. Integration of metabolomics and expression of glycerol-3-phosphate acyltransferase (GPAM) in breast cancer-link to patient survival, hormone receptor status, and metabolic profiling. Journal of proteome research. 2012; 11:850-860.
48. Willmann L, Schlimpert M, Halbach S, Erbes T, Stickeler E and Kammerer B. Metabolic profiling of breast cancer: Differences in central metabolism between subtypes of breast cancer cell lines. Journal of chromatography B. 2015; 1000:95-104.
49. Kennedy KM, Scarbrough PM, Ribeiro A, Richardson R, Yuan H, Sonveaux P, Landon CD, Chi JT, Pizzo S, Schroeder T and Dewhirst MW. Catabolism of exogenous lactate reveals it as a legitimate metabolic substrate in breast cancer. PloS one. 2013; 8:e75154.
50. Glynn SA, Prueitt RL, Ridnour LA, Boersma BJ, Dorsey TM, Wink DA, Goodman JE, Yfantis HG, Lee DH and Ambs S. COX-2 activation is associated with Akt phosphorylation and poor survival in ER-negative, HER2-positive breast cancer. BMC cancer. 2010; 10:626.
51. Lanza-Jacoby S, Miller S, Flynn J, Gallatig K, Daskalakis C, Masferrer JL, Zweifel BS, Sembhi H and Russo IH. The cyclooxygenase-2 inhibitor, celecoxib, prevents the development of mammary tumors in Her-2/neu mice. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2003; 12:1486-1491.
52. Ristimaki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, Joensuu H and Isola J. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer research. 2002; 62:632-635.
53. Gunther UL. Metabolomics Biomarkers for Breast Cancer. Pathobiology. 2015; 82:153-165.
54. Budczies J, Denkert C, Muller BM, Brockmoller SF, Klauschen F, Gyorffy B, Dietel M, Richter-Ehrenstein C, Marten U, Salek RM, Griffin JL, Hilvo M, Oresic M, Wohlgemuth G and Fiehn O. Remodeling of central metabolism in invasive breast cancer compared to normal breast tissue - a GC-TOFMS based metabolomics study. BMC genomics. 2012; 13:334.
55. Erez A and DeBerardinis RJ. Metabolic dysregulation in monogenic disorders and cancer - finding method in madness. Nature reviews Cancer. 2015; 15:440-448.
56. Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau LM, Rabinowitz JD, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009; 462:739-744.
57. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer cell. 2010; 18:553-567.
58. da Rosa MS, Joao Ribeiro CA, Seminotti B, Teixeira Ribeiro R, Amaral AU, Coelho Dde M, de Oliveira FH, Leipnitz G and Wajner M. In vivo intracerebral administration of L-2-hydroxyglutaric acid provokes oxidative stress and histopathological alterations in striatum and cerebellum of adolescent rats. Free radical biology & medicine. 2015; 83:201-213.
59. da Rosa MS, Seminotti B, Amaral AU, Parmeggiani B, de Oliveira FH, Leipnitz G and Wajner M. Disruption of redox homeostasis and histopathological alterations caused by in vivo intrastriatal administration of D-2-hydroxyglutaric acid to young rats. Neuroscience. 2014; 277:281-293.
60. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell. 2011; 19:17-30.
61. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012; 483:474-478.
62. Varlakhanova NV, Cotterman RF, deVries WN, Morgan J, Donahue LR, Murray S, Knowles BB and Knoepfler PS. myc maintains embryonic stem cell pluripotency and self-renewal. Differentiation. 2010; 80:9-19.
63. Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH and Felsher DW. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004; 431:1112-1117.
64. Dang CV. Links between metabolism and cancer. Genes & development. 2012; 26:877-890.
65. Li F, Wang Y, Zeller KI, Potter JJ, Wonsey DR, O’Donnell KA, Kim JW, Yustein JT, Lee LA and Dang CV. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Molecular and cellular biology. 2005; 25:6225-6234.
66. Ilic N, Utermark T, Widlund HR and Roberts TM. PI3K-targeted therapy can be evaded by gene amplification along the MYC-eukaryotic translation initiation factor 4E (eIF4E) axis. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:E699-708.
67. Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, Yang J, Semaan DJ, Chen C, Fox EA, Gray NS, Monahan J, Schlegel R, Beroukhim R, Mills GB and Zhao JJ. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nature medicine. 2011; 17:1116-1120.
68. Chen Y and Olopade OI. MYC in breast tumor progression. Expert review of anticancer therapy. 2008; 8:1689-1698.
69. Wu ZQ, Li XY, Hu CY, Ford M, Kleer CG and Weiss SJ. Canonical Wnt signaling regulates Slug activity and links epithelial-mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109:16654-16659.
70. Dang CV. Rethinking the Warburg effect with Myc micromanaging glutamine metabolism. Cancer research. 2010; 70:859-862.
71. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and Dang CV. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009; 458:762-765.
72. Perez-Escuredo J, Dadhich RK, Dhup S, Cacace A, Van Hee VF, De Saedeleer CJ, Sboarina M, Rodriguez F, Fontenille MJ, Brisson L, Porporato PE and Sonveaux P. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle. 2016; 15:72-83.
73. Jacque N, Ronchetti AM, Larrue C, Meunier G, Birsen R, Willems L, Saland E, Decroocq J, Thiago TT, Lambert M, Poulain L, Hospital MA, Sujobert P, Joseph L, Chapuis N, Lacombe C, et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. 2015; 126:1346-1356.
74. Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, Lopez-Larrubia P, Stephanopoulos G and Iliopoulos O. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell metabolism. 2013; 17:372-385.
75. Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC and Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108:19611-19616.
76. Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, Hu M, Chan DA, Ethier SP, van ‘t Veer LJ, Polyak K, McCormick F and Gray JW. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer cell. 2013; 24:450-465.
77. Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. Journal of experimental & clinical cancer research. 2015; 34:111.
78. DeBerardinis RJ and Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010; 29:313-324.
79. Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, Mackinnon AL, Parlati F, Rodriguez ML, Shwonek PJ, Sjogren EB, Stanton TF, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Molecular cancer therapeutics. 2014; 13:890-901.
80. Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV and Cerione RA. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer cell. 2010; 18:207-219.
81. Yuneva M, Zamboni N, Oefner P, Sachidanandam R and Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. The Journal of cell biology. 2007; 178:93-105.
82. Chen Z, Wang Y, Warden C and Chen S. Cross-talk between ER and HER2 regulates c-MYC-mediated glutamine metabolism in aromatase inhibitor resistant breast cancer cells. The Journal of steroid biochemistry and molecular biology. 2015; 149:118-127.
83. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61-70.
84. Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM and Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer research. 2004; 64:3892-3899.
85. Manning BD and Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007; 129:1261-1274.
86. Napoli E, Ross-Inta C, Wong S, Hung C, Fujisawa Y, Sakaguchi D, Angelastro J, Omanska-Klusek A, Schoenfeld R and Giulivi C. Mitochondrial dysfunction in Pten haplo-insufficient mice with social deficits and repetitive behavior: interplay between Pten and p53. PloS one. 2012; 7:e42504.
87. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F and Hwang PM. p53 regulates mitochondrial respiration. Science. 2006; 312:1650-1653.
88. Bensaad K, Cheung EC and Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. The EMBO journal. 2009; 28:3015-3026.
89. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E and Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006; 126:107-120.
90. Filomeni G, De Zio D and Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell death and differentiation. 2015; 22:377-388.
91. Desideri E, Filomeni G and Ciriolo MR. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy. 2012; 8:1769-1781.
92. Boidot R, Vegran F, Meulle A, Le Breton A, Dessy C, Sonveaux P, Lizard-Nacol S and Feron O. Regulation of monocarboxylate transporter MCT1 expression by p53 mediates inward and outward lactate fluxes in tumors. Cancer research. 2012; 72:939-948.
93. Foulkes WD, Smith IE and Reis-Filho JS. Triple-negative breast cancer. The New England journal of medicine. 2010; 363:1938-1948.
94. Privat M, Radosevic-Robin N, Aubel C, Cayre A, Penault-Llorca F, Marceau G, Sapin V, Bignon YJ and Morvan D. BRCA1 induces major energetic metabolism reprogramming in breast cancer cells. PloS one. 2014; 9:e102438.
95. Yi YW, Kang HJ and Bae I. BRCA1 and Oxidative Stress. Cancers. 2014; 6:771-795.
96. Moreau K, Dizin E, Ray H, Luquain C, Lefai E, Foufelle F, Billaud M, Lenoir GM and Venezia ND. BRCA1 affects lipid synthesis through its interaction with acetyl-CoA carboxylase. The Journal of biological chemistry. 2006; 281:3172-3181.
97. Li D, Chen NN, Cao JM, Sun WP, Zhou YM, Li CY and Wang XX. BRCA1 as a nicotinamide adenine dinucleotide (NAD)-dependent metabolic switch in ovarian cancer. Cell Cycle. 2014; 13:2564-2571.
98. Vinogradov AD. NADH/NAD+ interaction with NADH: ubiquinone oxidoreductase (complex I). Biochimica et biophysica acta. 2008; 1777:729-734.
99. Wang X, Docanto MM, Sasano H, Lo C, Simpson ER and Brown KA. Prostaglandin E2 inhibits p53 in human breast adipose stromal cells: a novel mechanism for the regulation of aromatase in obesity and breast cancer. Cancer research. 2015; 75:645-655.
100. Kim J, Johnson L, Skrzynia C, Buchanan A, Gracia C and Mersereau JE. Prospective multicenter cohort study of estrogen and insulin-like growth factor system in BRCA mutation carriers. Cancer causes & control. 2015; 26:1087-1092.
101. Xu J, Fan S and Rosen EM. Regulation of the estrogen-inducible gene expression profile by the breast cancer susceptibility gene BRCA1. Endocrinology. 2005; 146:2031-2047.
102. Maor SB, Abramovitch S, Erdos MR, Brody LC and Werner H. BRCA1 suppresses insulin-like growth factor-I receptor promoter activity: potential interaction between BRCA1 and Sp1. Molecular genetics and metabolism. 2000; 69:130-136.
103. Kang HJ, Yi YW, Kim HJ, Hong YB, Seong YS and Bae I. BRCA1 negatively regulates IGF-1 expression through an estrogen-responsive element-like site. Cell death & disease. 2012; 3:e336.
104. Hong H, Yen HY, Brockmeyer A, Liu Y, Chodankar R, Pike MC, Stanczyk FZ, Maxson R and Dubeau L. Changes in the mouse estrus cycle in response to BRCA1 inactivation suggest a potential link between risk factors for familial and sporadic ovarian cancer. Cancer research. 2010; 70:221-228.
105. Widschwendter M, Rosenthal AN, Philpott S, Rizzuto I, Fraser L, Hayward J, Intermaggio MP, Edlund CK, Ramus SJ, Gayther SA, Dubeau L, Fourkala EO, Zaikin A, Menon U and Jacobs IJ. The sex hormone system in carriers of BRCA1/2 mutations: a case-control study. The Lancet Oncology. 2013; 14:1226-1232.
106. Maor S, Yosepovich A, Papa MZ, Yarden RI, Mayer D, Friedman E and Werner H. Elevated insulin-like growth factor-I receptor (IGF-IR) levels in primary breast tumors associated with BRCA1 mutations. Cancer letters. 2007; 257:236-243.
107. Hudelist G, Wagner T, Rosner M, Fink-Retter A, Gschwantler-Kaulich D, Czerwenka K, Kroiss R, Tea M, Pischinger K, Kostler WJ, Attems J, Mueller R, Blaukopf C, Kubista E, Hengstschlager M and Singer CF. Intratumoral IGF-I protein expression is selectively upregulated in breast cancer patients with BRCA1/2 mutations. Endocrine-related cancer. 2007; 14:1053-1062.
108. Maor S, Papa MZ, Yarden RI, Friedman E, Lerenthal Y, Lee SW, Mayer D and Werner H. Insulin-like growth factor-I controls BRCA1 gene expression through activation of transcription factor Sp1. Hormone and metabolic research. 2007; 39:179-185.
109. Werner H and Bruchim I. IGF-1 and BRCA1 signalling pathways in familial cancer. The Lancet Oncology. 2012; 13:e537-544.
110. Yu Z, Gao W, Jiang E, Lu F, Zhang L, Shi Z, Wang X, Chen L and Lv T. Interaction between IGF-IR and ER induced by E2 and IGF-I. PloS one. 2013; 8:e62642.
111. Xiang T, Ohashi A, Huang Y, Pandita TK, Ludwig T, Powell SN and Yang Q. Negative Regulation of AKT Activation by BRCA1. Cancer research. 2008; 68:10040-10044.
112. Gorrini C, Gang BP, Bassi C, Wakeham A, Baniasadi SP, Hao Z, Li WY, Cescon DW, Li YT, Molyneux S, Penrod N, Lupien M, Schmidt EE, Stambolic V, Gauthier ML and Mak TW. Estrogen controls the survival of BRCA1-deficient cells via a PI3K-NRF2-regulated pathway. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:4472-4477.
113. Martin MB, Franke TF, Stoica GE, Chambon P, Katzenellenbogen BS, Stoica BA, McLemore MS, Olivo SE and Stoica A. A role for Akt in mediating the estrogenic functions of epidermal growth factor and insulin-like growth factor I. Endocrinology. 2000; 141:4503-4511.
114. Bruchim I, Sarfstein R and Werner H. The IGF Hormonal Network in Endometrial Cancer: Functions, Regulation, and Targeting Approaches. Frontiers in endocrinology. 2014; 5:76.
115. Gorrini C, Baniasadi PS, Harris IS, Silvester J, Inoue S, Snow B, Joshi PA, Wakeham A, Molyneux SD, Martin B, Bouwman P, Cescon DW, Elia AJ, Winterton-Perks Z, Cruickshank J, Brenner D, et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. The Journal of experimental medicine. 2013; 210:1529-1544.
116. Rotblat B, Melino G and Knight RA. NRF2 and p53: Januses in cancer? Oncotarget. 2012; 3:1272-1283. doi: 10.18632/oncotarget.754.
117. Satoh H, Moriguchi T, Takai J, Ebina M and Yamamoto M. Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer research. 2013; 73:4158-4168.
118. Gaikwad NW, Yang L, Pruthi S, Ingle JN, Sandhu N, Rogan EG and Cavalieri EL. Urine biomarkers of risk in the molecular etiology of breast cancer. Breast cancer. 2009; 3:1-8.
119. Wang Q, Li J, Yang X, Sun H, Gao S, Zhu H, Wu J and Jin W. Nrf2 is associated with the regulation of basal transcription activity of the BRCA1 gene. Acta biochimica et biophysica Sinica. 2013; 45:179-187.
120. Saal LH, Gruvberger-Saal SK, Persson C, Lovgren K, Jumppanen M, Staaf J, Jonsson G, Pires MM, Maurer M, Holm K, Koujak S, Subramaniyam S, Vallon-Christersson J, Olsson H, Su T, Memeo L, et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nature genetics. 2008; 40:102-107.
121. Tang MK, Kwong A, Tam KF, Cheung AN, Ngan HY, Xia W and Wong AS. BRCA1 deficiency induces protective autophagy to mitigate stress and provides a mechanism for BRCA1 haploinsufficiency in tumorigenesis. Cancer letters. 2014; 346:139-147.
122. Zhang L and Long X. Association of BRCA1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Scientific reports. 2015; 5:17869.
123. Yamashita N, Tokunaga E, Kitao H, Hitchins M, Inoue Y, Tanaka K, Hisamatsu Y, Taketani K, Akiyoshi S, Okada S, Oda Y, Saeki H, Oki E and Maehara Y. Epigenetic Inactivation of BRCA1 Through Promoter Hypermethylation and Its Clinical Importance in Triple-Negative Breast Cancer. Clinical breast cancer. 2015; 15:498-504.
124. Al-Moghrabi N, Nofel A, Al-Yousef N, Madkhali S, Bin Amer SM, Alaiya A, Shinwari Z, Al-Tweigeri T, Karakas B, Tulbah A and Aboussekhra A. The molecular significance of methylated BRCA1 promoter in white blood cells of cancer-free females. BMC cancer. 2014; 14:830.
125. Shin SY, Fauman EB, Petersen AK, Krumsiek J, Santos R, Huang J, Arnold M, Erte I, Forgetta V, Yang TP, Walter K, Menni C, Chen L, Vasquez L, Valdes AM, Hyde CL, et al. An atlas of genetic influences on human blood metabolites. Nature genetics. 2014; 46:543-550.
126. Draisma HH, Pool R, Kobl M, Jansen R, Petersen AK, Vaarhorst AA, Yet I, Haller T, Demirkan A, Esko T, Zhu G, Bohringer S, Beekman M, van Klinken JB, Romisch-Margl W, Prehn C, et al. Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels. Nature communications. 2015; 6:7208.
127. Xu T, Holzapfel C, Dong X, Bader E, Yu Z, Prehn C, Perstorfer K, Jaremek M, Roemisch-Margl W, Rathmann W, Li Y, Wichmann HE, Wallaschofski H, Ladwig KH, Theis F, Suhre K, et al. Effects of smoking and smoking cessation on human serum metabolite profile: results from the KORA cohort study. BMC medicine. 2013; 11:60.
128. Gu F, Derkach A, Freedman ND, Landi MT, Albanes D, Weinstein SJ, Mondul AM, Matthews CE, Guertin KA, Xiao Q, Zheng W, Shu XO, Sampson JN, Moore SC and Caporaso NE. Cigarette smoking behaviour and blood metabolomics. International journal of epidemiology. 2015.
129. Jaremek M, Yu Z, Mangino M, Mittelstrass K, Prehn C, Singmann P, Xu T, Dahmen N, Weinberger KM, Suhre K, Peters A, Doring A, Hauner H, Adamski J, Illig T, Spector TD, et al. Alcohol-induced metabolomic differences in humans. Translational psychiatry. 2013; 3:e276.
130. Floegel A, Wientzek A, Bachlechner U, Jacobs S, Drogan D, Prehn C, Adamski J, Krumsiek J, Schulze MB, Pischon T and Boeing H. Linking diet, physical activity, cardiorespiratory fitness and obesity to serum metabolite networks: findings from a population-based study. International journal of obesity. 2014; 38:1388-1396.
131. Moore SC, Matthews CE, Sampson JN, Stolzenberg-Solomon RZ, Zheng W, Cai Q, Tan YT, Chow WH, Ji BT, Liu DK, Xiao Q, Boca SM, Leitzmann MF, Yang G, Xiang YB, Sinha R, et al. Human metabolic correlates of body mass index. Metabolomics. 2014; 10:259-269.
132. Asiago VM, Alvarado LZ, Shanaiah N, Gowda GA, Owusu-Sarfo K, Ballas RA and Raftery D. Early detection of recurrent breast cancer using metabolite profiling. Cancer research. 2010; 70:8309-8318.
133. Tenori L, Oakman C, Morris PG, Gralka E, Turner N, Cappadona S, Fornier M, Hudis C, Norton L, Luchinat C and Di Leo A. Serum metabolomic profiles evaluated after surgery may identify patients with oestrogen receptor negative early breast cancer at increased risk of disease recurrence. Results from a retrospective study. Molecular oncology. 2015; 9:128-139.
134. Jobard E, Pontoizeau C, Blaise BJ, Bachelot T, Elena-Herrmann B and Tredan O. A serum nuclear magnetic resonance-based metabolomic signature of advanced metastatic human breast cancer. Cancer letters. 2014; 343:33-41.
135. Chiu HH, Tsai SJ, Tseng YJ, Wu MS, Liao WC, Huang CS and Kuo CH. An efficient and robust fatty acid profiling method for plasma metabolomic studies by gas chromatography-mass spectrometry. Clinica chimica acta. 2015; 451:183-190.
136. Cui M, Wang Q and Chen G. Serum metabolomics analysis reveals changes in signaling lipids in breast cancer patients. Biomedical chromatography. 2016; 30:42-47.
137. Xie G, Zhou B, Zhao A, Qiu Y, Zhao X, Garmire L, Shvetsov YB, Yu H, Yen Y and Jia W. Lowered circulating aspartate is a metabolic feature of human breast cancer. Oncotarget. 2015; 6:33369-33381. doi: 10.18632/oncotarget.5409.
138. Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E and Vander Heiden MG. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell. 2015; 162:552-563.
139. Yang Y. Enhancing doxorubicin efficacy through inhibition of aspartate transaminase in triple-negative breast cancer cells. Biochemical and biophysical research communications. 2016; 473:1295-1300.
140. Becker S, Cazares LH, Watson P, Lynch H, Semmes OJ, Drake RR and Laronga C. Surfaced-enhanced laser desorption/ionization time-of-flight (SELDI-TOF) differentiation of serum protein profiles of BRCA-1 and sporadic breast cancer. Annals of surgical oncology. 2004; 11:907-914.
141. Scumaci D, Tamme L, Fiumara CV, Pappaianni G, Concolino A, Leone E, Faniello MC, Quaresima B, Ricevuto E, Costanzo FS and Cuda G. Plasma Proteomic Profiling in Hereditary Breast Cancer Reveals a BRCA1-Specific Signature: Diagnostic and Functional Implications. PloS one. 2015; 10:e0129762.
142. Foulkes WD, Stefansson IM, Chappuis PO, Begin LR, Goffin JR, Wong N, Trudel M and Akslen LA. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. Journal of the National Cancer Institute. 2003; 95:1482-1485.
143. Severson TM, Peeters J, Majewski I, Michaut M, Bosma A, Schouten PC, Chin SF, Pereira B, Goldgraben MA, Bismeijer T, Kluin RJ, Muris JJ, Jirstrom K, Kerkhoven RM, Wessels L, Caldas C, et al. BRCA1-like signature in triple negative breast cancer: Molecular and clinical characterization reveals subgroups with therapeutic potential. Molecular oncology. 2015; 9:1528-1538.
144. Ollier M, Radosevic-Robin N, Kwiatkowski F, Ponelle F, Viala S, Privat M, Uhrhammer N, Bernard-Gallon D, Penault-Llorca F, Bignon YJ and Bidet Y. DNA repair genes implicated in triple negative familial non-BRCA1/2 breast cancer predisposition. American journal of cancer research. 2015; 5:2113-2126.
145. Kriegsmann M, Endris V, Wolf T, Pfarr N, Stenzinger A, Loibl S, Denkert C, Schneeweiss A, Budczies J, Sinn P and Weichert W. Mutational profiles in triple-negative breast cancer defined by ultradeep multigene sequencing show high rates of PI3K pathway alterations and clinically relevant entity subgroup specific differences. Oncotarget. 2014; 5:9952-9965. doi: 10.18632/oncotarget.2481.
146. Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, Powers J, Rathbun K, Stopfer JE, Zhu J, Bradbury AR, Simon MS, DeMichele A, Domchek SM and Nathanson KL. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genetics in medicine. 2015; 17:630-638.
147. Wang C, Mayer JA, Mazumdar A, Fertuck K, Kim H, Brown M and Brown PH. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol Endocrinol. 2011; 25:1527-1538.
148. Morrish F and Hockenbery D. MYC and mitochondrial biogenesis. Cold Spring Harbor perspectives in medicine. 2014; 4(5). pii: a014225.
149. Kumar NB, Vadaparampil ST, Mahajan N, Lilienfeld HS, Lee JH, Laronga C, Hakam A, Hein JJ, Egan KM, Arun B and Pal T. Metformin- A Promising Agent for Chemoprevention in Carriers. Hereditary genetics. 2012; 1(2). pii: 104.
150. Lord SR, Patel N, Liu D, Fenwick J, Gleeson F, Buffa F and Harris AL. Neoadjuvant Window Studies of Metformin and Biomarker Development for Drugs Targeting Cancer Metabolism. Journal of the National Cancer Institute Monographs. 2015; 2015:81-86.
151. Rice S, Pellat L, Ahmetaga A, Bano G, Mason HD and Whitehead SA. Dual effect of metformin on growth inhibition and oestradiol production in breast cancer cells. International journal of molecular medicine. 2015; 35:1088-1094.
152. Ashburn TT and Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nature reviews Drug discovery. 2004; 3:673-683.
153. Owen MR, Doran E and Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. The Biochemical journal. 2000; 348 Pt 3:607-614.
154. Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS and Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014; 3:e02242.
155. Blandino G, Valerio M, Cioce M, Mori F, Casadei L, Pulito C, Sacconi A, Biagioni F, Cortese G, Galanti S, Manetti C, Citro G, Muti P and Strano S. Metformin elicits anticancer effects through the sequential modulation of DICER and c-MYC. Nature communications. 2012; 3:865.
156. Zhuang Y, Chan DK, Haugrud AB and Miskimins WK. Mechanisms by which low glucose enhances the cytotoxicity of metformin to cancer cells both in vitro and in vivo. PloS one. 2014; 9:e108444.
157. DeFronzo RA and Goodman AM. Efficacy of metformin in patients with non-insulin-dependent diabetes mellitus. The Multicenter Metformin Study Group. The New England journal of medicine. 1995; 333:541-549.
158. Rizkalla SW, Elgrably F, Tchobroutsky G and Slama G. Effects of metformin treatment on erythrocyte insulin binding in normal weight subjects, in obese non diabetic subjects, in type 1 and type 2 diabetic patients. Diabete & metabolisme. 1986; 12:219-224.
159. Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, Le Marchand-Brustel Y, Giorgetti-Peraldi S, Cormont M, Bertolotto C, Deckert M, Auberger P, Tanti JF and Bost F. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer research. 2010; 70:2465-2475.
160. Lea MA, Chacko J, Bolikal S, Hong JY, Chung R, Ortega A and desbordes C. Addition of 2-deoxyglucose enhances growth inhibition but reverses acidification in colon cancer cells treated with phenformin. Anticancer research. 2011; 31:421-426.
161. Matsuo J, Tsukumo Y, Saito S, Tsukahara S, Sakurai J, Sato S, Kondo H, Ushijima M, Matsuura M, Watanabe T and Tomida A. Hyperactivation of 4E-binding protein 1 as a mediator of biguanide-induced cytotoxicity during glucose deprivation. Molecular cancer therapeutics. 2012; 11:1082-1091.
162. Sandulache VC, Ow TJ, Pickering CR, Frederick MJ, Zhou G, Fokt I, Davis-Malesevich M, Priebe W and Myers JN. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer. 2011; 117:2926-2938.
163. Cheong JH, Park ES, Liang J, Dennison JB, Tsavachidou D, Nguyen-Charles C, Wa Cheng K, Hall H, Zhang D, Lu Y, Ravoori M, Kundra V, Ajani J, Lee JS, Ki Hong W and Mills GB. Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models. Molecular cancer therapeutics. 2011; 10:2350-2362.
164. Iliopoulos D, Hirsch HA and Struhl K. Metformin decreases the dose of chemotherapy for prolonging tumor remission in mouse xenografts involving multiple cancer cell types. Cancer research. 2011; 71:3196-3201.
165. Soo JS, Ng CH, Tan SH, Malik RA, Teh YC, Tan BS, Ho GF, See MH, Taib NA, Yip CH, Chung FF, Hii LW, Teo SH and Leong CO. Metformin synergizes 5-fluorouracil, epirubicin, and cyclophosphamide (FEC) combination therapy through impairing intracellular ATP production and DNA repair in breast cancer stem cells. Apoptosis. 2015; 20:1373-1387.
166. Liu H, Scholz C, Zang C, Schefe JH, Habbel P, Regierer AC, Schulz CO, Possinger K and Eucker J. Metformin and the mTOR inhibitor everolimus (RAD001) sensitize breast cancer cells to the cytotoxic effect of chemotherapeutic drugs in vitro. Anticancer research. 2012; 32:1627-1637.
167. Wang Y, Wei J, Li L, Fan C and Sun Y. Combined Use of Metformin and Everolimus Is Synergistic in the Treatment of Breast Cancer Cells. Oncology research. 2014; 22:193-201.
168. Zakikhani M, Blouin MJ, Piura E and Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast cancer research and treatment. 2010; 123:271-279.
169. Ma J, Guo Y, Chen S, Zhong C, Xue Y, Zhang Y, Lai X, Wei Y, Yu S, Zhang J and Liu W. Metformin enhances tamoxifen-mediated tumor growth inhibition in ER-positive breast carcinoma. BMC cancer. 2014; 14:172.
170. Vazquez-Martin A, Oliveras-Ferraros C, Del Barco S, Martin-Castillo B and Menendez JA. The anti-diabetic drug metformin suppresses self-renewal and proliferation of trastuzumab-resistant tumor-initiating breast cancer stem cells. Breast cancer research and treatment. 2011; 126:355-364.
171. Lau YK, Du X, Rayannavar V, Hopkins B, Shaw J, Bessler E, Thomas T, Pires MM, Keniry M, Parsons RE, Cremers S, Szabolcs M and Maurer MA. Metformin and erlotinib synergize to inhibit basal breast cancer. Oncotarget. 2014; 5:10503-10517. doi: 10.18632/oncotarget.2391.
172. Malaguarnera R, Sacco A, Morcavallo A, Squatrito S, Migliaccio A, Morrione A, Maggiolini M and Belfiore A. Metformin inhibits androgen-induced IGF-IR up-regulation in prostate cancer cells by disrupting membrane-initiated androgen signaling. Endocrinology. 2014; 155:1207-1221.
173. Schuler KM, Rambally BS, DiFurio MJ, Sampey BP, Gehrig PA, Makowski L and Bae-Jump VL. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer medicine. 2015; 4:161-173.
174. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F and Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015; 6:4569-4584. doi: 10.18632/oncotarget.3174.
175. Farnie G, Sotgia F and Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget. 2015; 6:30472-30486. doi: 10.18632/oncotarget.5401.
176. Lloyd SM, Arnold J and Sreekumar A. Metabolomic profiling of hormone-dependent cancers: a bird’s eye view. Trends in endocrinology and metabolism. 2015; 26:477-485.
177. Devine MJ, Plun-Favreau H and Wood NW. Parkinson’s disease and cancer: two wars, one front. Nature reviews Cancer. 2011; 11:812-823.
178. Camps J. (2014). Oxidative stress and inflammation in non-communicable diseases - molecular mechanisms and perspectives in therapeutics. (New York, NY: Springer Berlin Heidelberg).