
INTRODUCTION
Ascorbate (Vitamin C) is a nutrient essential to the biosynthesis of collagen and L-carnitine and the conversion of dopamine to norepinephrine [1]. Most animals are able to synthesize large quantities of ascorbate, but humans lost this ability due to mutations of the gulonolactone oxidase gene [2]. It is well known that an acute lack of ascorbate leads to scurvy and causes death [3] and ascorbate is an attractive marker of fruit and vegetable intake because these foods are the primary sources of dietary vitamin C [4], which ascorbate functions as a potent reducing agent that efficiently quenches potentially damaging free radicals [5]. It is very interesting that Cameron suggested that high doses of ascorbate could provide a clinical benefit for cancer patients [6, 7]. In vitro studies have shown that pharmacologic ascorbate is effective in a large panel of tumor cell lines [8, 9] and that increasing tumor cells’ generation of hydrogen peroxide (H2O2) might be used to induce ascorbate-specific cytotoxicity [10]. Pharmacologic ascorbate has been found to mediate the mitochondrial release of cytochrome C, which leads to H2O2-mediated activation of the caspase cascade and apoptotic process and thence to a significant decrease in the growth rate of some solid tumors [11–14]. Herst investigated that 5 mM ascorbate in combination with radiation killed more glioblastoma multiforme primary cells by increasing oxidative DNA damage than either treatment alone [15]. However, the effect of high doses of intravenous ascorbate in the treatment of cancer has been controversial although there is growing evidence that intravenous high-dose ascorbate has been found to improve the health-related quality of life of terminal cancer patients [16,17].
On another hand, some normal human cells (lymphocytes, monocytes, fibroblasts and normal breast cells) and some types of human cancer cells (breast cancer cells and glioblastoma multiforme cells) are more resistant to ascorbate than others [8, 9]. Moreover, ascorbate penetrating the tissue at a slightly lower rate than mannitol and travelling via the paracellular route were also found [18]. Compared with human renal carcinoma TC-1 cells, higher intracellular glutathione concentration was detected in glioblastoma multiform tumor T98G cells, which were more resistant to ascorbate than TC-1 cells [9]. Sinnberg also found that ascorbate resistance was associated with the expression of HIF1α and oxygen pressure [19]. Therefore, the use of ascorbate as an anticancer agent and ascorbate-induced resistance warrants further study. The mechanisms of drug resistance in cancer cells have been thoroughly studied at the gene transcription levels by cDNA microarrays, which is a high-throughput system developed to monitor the expression of many genes in a single experiment and to identify genes differentially expressed in multidrug-resistant cancer cells and those of their parental cells [20–22]. In this study, we used transcriptomic profiling, quantitative real-time polymerase chain reaction (qRT-PCR), and antioxidant enzyme activity assays to clarify the molecular mechanism of ascorbate-induced resistance in Burkitt lymphoma cells.
RESULTS
Cytotoxic response of JLPS and JLPR cells to ascorbate or H2O2
Using the MTT assay, we found that JLPR cells were highly resistant to ascorbate and H2O2 at IC50 values of 1250 μM and 32 μM, respectively (Figure 1A, Figure 1B, and Table 1). High-performance liquid chromatography analysis revealed that JLPR cells had a larger amount of ascorbate than JLPS cells did (Figure 1C). JLPS cells incubated with 1 mM docosahexanoic acid (DHA) for 1 h had a similar amount of ascorbate as JLPR cells did (Figure 1C). Because cellular ascorbate cannot induce the death of JLP cells, there was almost no difference in cell viability rates between JLPS cells treated with ascorbate and JLPS cells treated with ascorbate and DHA (Figure 1A). JLPS cells pretreated with CAT acquired some resistance to ascorbate (Figure 1A). After 48 h, JLPR cells that had not been maintained in 1 mM ascorbate lost little H2O2 resistance (Figure 1B). These results suggested that ascorbate efflux has an antioxidation function that protects cells from H2O2-induced cytotoxicity and that ascorbate resistance is associated with progressive accumulation of reactive oxygen species.
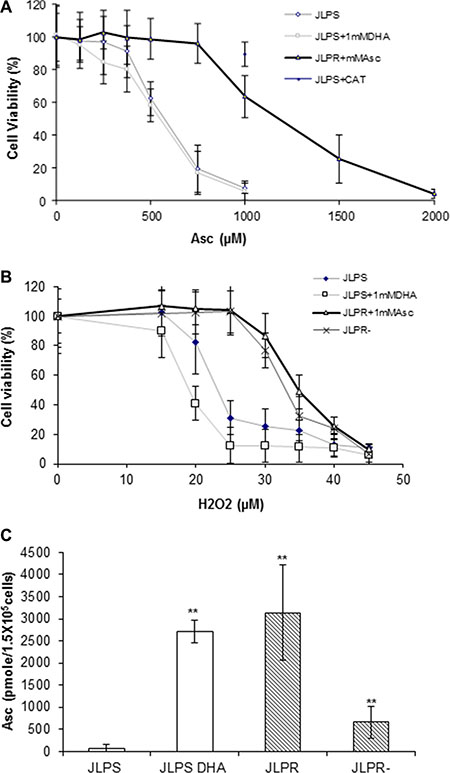
Figure 1: Cytotoxicity analysis of ascorbate and H2O2 with the measurement of intracellular ascorbate in JLPR cells and JLPS cells. (A) Viability rates of JLPS cells, JLPS cells pretreated with DHA (JLPS+DHA), JLPR cells, and JLPS cells pretreated with CAT following treatment with increasing concentrations of ascorbate. (B) Viability rates of JLPS cells, JLPS+DHA, JLPR cells, and JLPR cells that had not been maintained in ascorbate (JLPR−) following treatment with increasing concentrations of H2O2. (C) Posttreatment concentrations of ascorbate in JLPS cells, JLPS+DHA cells, JLPR cells, and JLPR− cells.
Table 1: Cytotoxicity studies of JLP cells by MTT assay (IC50)
Drug |
JLP S |
JLPR |
Resistance indexa |
|---|---|---|---|
IC50 |
IC50 |
||
Ascorbate |
375 μM |
1250 μM |
3.33 |
H2O2 |
16 μM |
32 μM |
2.00 |
Resistance index a: the ratio of the IC50 for JLPR treatment with ascorbate/ H2O2 to the IC50 for JLPS treatment with ascorbate.
Increased glucose uptake in JLPR cells with activation of glucose transport
The amount of 2-DG uptake at different times in JLPR cells was higher than that in JLPS cells (Figure 2A). To determine whether the changes in glucose transport occurred at the level of gene transcription, we analyzed the expressions of Glut1 and Glut3. Real-time PCR revealed that the expression levels of Glut1 and Glut3 in JLPR cells were 1.72- and 1.77-fold higher, respectively, than those in JLPS cells (Figure 2B). We found that following treatment with ascorbate and H2O2, the amount of glucose uptake in JLPS cells was usually significantly lower than that in JLPR cells (P < 0.05). The amount of 2-DG uptake was decreased by 78.57% (from 35 to 7.5 pmoles 2-DG/min/106 cells) in JLPS cells treated with 1000 μM ascorbate or 25 μM H2O2 (Figure 2C and 2D). Our results demonstrated that ascorbate treated with cancer cells by the role of H2O2 in mediating glucose uptake activation in JLPS cells. On the other hand, the resistant JLPR cells maintained high glucose uptake (about 55 pmoles 2-DG/min/106 cells) despite being treated in 1 mM ascorbate (Figure 2C) or 25 μm H2O2 (Figure 2D). These results suggested that ascorbate resistance actively restores glucose uptake against H2O2-induced apoptosis and necrosis.
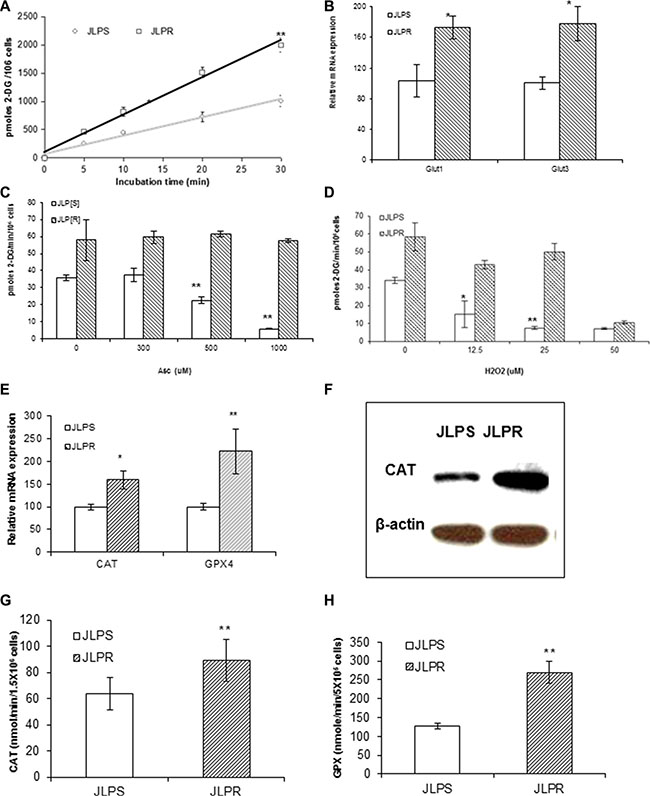
Figure 2: Metabolomic assay and qRT-PCR analysis in JLPR cells and JLPS cells. (A) 2-DG uptake at different times. (B) Real-time PCR analysis of Glut1 and Glut3 mRNA. (C) 2-DG uptake in cells treated with increasing concentrations of ascorbate. (D) 2-DG uptake in cells treated with increasing concentrations of H2O2. Antioxidant enzyme analysis in JLPS and JLPR cells. (E) qRT-PCR analysis of CAT and GPX4 gene expression. (F) Western blot analysis of CAT protein expression levels. (G) CAT activity. (H) GPX4 activity.
Increased levels of antioxidant enzyme (CAT and GPX) mRNA, protein, and activity in JLPR cells
qRT-PCR was used to assess the levels of antioxidant enzyme mRNA expression. The expression levels of CAT and GPX4 mRNA in JLPR cells were higher than those in the JLPS cells (Figure 2E). Immunoblots analysis revealed CAT protein levels to be higher in JLPR cells than JLPS cells (Figure 2F). The CAT and GPX activities of JLPR cells (CAT: 59.6 nmoles/min/106 cells; GPX: 538.6 nmoles/min/106 cells) were 1.5 and 2 times as high as those of JLPS cells (CAT: 42.3 nmoles/min/106 cells; GPX: 253.6 nmoles/min/106 cells), respectively (Figure 2G and Figure 2H). These results were consistent with the findings regarding mRNA and protein levels. These findings indicated that the activities of the CAT and GPX enzymes are involved in protecting cells against oxidative stress, which suggests that overexpression of CAT and GPX confers resistance to ascorbate.
Ascorbate and H2O2 trigger similar models of cell cycling, but JLPR cells do not arrest in the G2/M phase induced by ascorbate or H2O2
Propidium iodide staining and flow cytometry were used to assess the DNA content of untreated and ascorbate- or H2O2-treated JLPS cells and JLPR cells. Following treatment with 1 mM ascorbate or 20 μM H2O2, JLPS cells were arrested predominantly in the G2/M phase; a few cells arrested in the G1 phase (Figure 3A and 3B). These results demonstrated that JLPS cells arrest at the G2/M phase, whereas JLPR cells do not, which implied that JLPR cells are more effective in removing H2O2 from the treatment medium. Interestingly, the cell cycling of JLPS cells treated with ascorbate was basically identical to that of JLPS cells treated with H2O2. These findings indicated that JLPS cells, but not JLPR cells, arrest in the G2/M phase in response to ascorbate or H2O2 and that ascorbate and H2O2 trigger similar models of cell killing in JLPS cells.
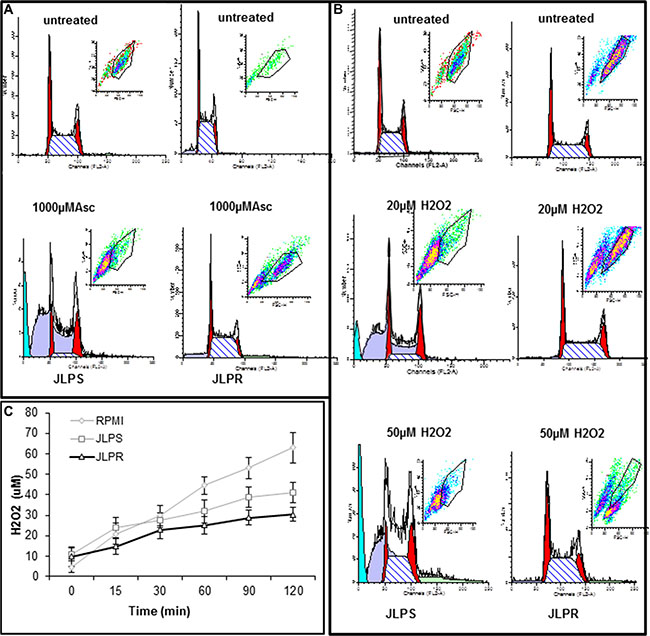
Figure 3: Cellular DNA content in JLPS and JLPR cells following treatment with ascorbate and H2O2. (A) Cells following treatment with 0 μM or 1000 μM ascorbate. (B) Cells following treatment with 0 μM, 20 μM or 50 μM H2O2. (C) H2O2 production in RPMI 1640 medium alone, containing JLPS cells, or containing JLPR cells. Ascorbate (1 mM) was added and medium was incubated at room temperature for up to 120 min. Data are means ± standard deviations of three independent experiments.
Ascorbate generates extracellular H2O2, which induces cancer cells’ cytotoxic pro-oxidant stress
An O2 electrode assay was used to measure the amount of H2O2 in complete culture medium with or without JLPS or JLPR cells after dissolution of ascorbate. The assay revealed that the amount of H2O2 in the presence of cells, especially JLPR cells, was less than that in medium alone (Figure 3C). The amount of H2O2 plateaued in JLPS cells treated with ascorbate for 1 h or more. These findings indicated that an ascorbate oxidation process produces extracellular H2O2. In the presence of JLPS cells in RPMI medium, 1 mM ascorbate generated 38 μM H2O2, arrested JLPS cells in the G2/M phase, and induced 90% cytotoxicity. In the presence of JLPR cells, 1 mM ascorbate generated 30 μM H2O2 but did not arrest cells in the G2/M phase or cause cell death (Figure 3A).
Transcriptomic profiles in JLP cells
To identify possible mechanisms of ascorbate resistance, we performed gene microarray profiling of JLPS and JLPR cells. A genome-wide analysis of the gene transcripts expressed in both cell lines was performed using agilent array. Hybridization of the microarray data revealed that 1.82% of the cDNAs exhibited more than a 1.5-fold change in expression level (P ≤ 0.05). The genes and genetic networks involved in ascorbic resistance are described in Tables 2 and 3, respectively. The microarray experiments also revealed that the following genes and genetic networks play roles in acquired ascorbate resistance: the double-strand break repair genes TOP2B and XRCC5; ferritin genes FTL, FTH1, and FTHL2; antioxidation gene GPX4; major histocompatibility complex (MHC) class I genes human leukocyte antigen HLA-A, HLA-G, and HLA-H; and asparagine synthetase; the downregulation of the histone genes HIST1H2AE, HIST1H2BK, H2BFS, and HIST1H4B; histone deacetylase (HDAC) complex subunit SAP18; heat shock protein (HSP) genes HSPH1 and HSPE1; FLAP; the inhibition of tumor necrosis and apoptosis signaling by inhibiting the expression of high-mobility group protein box 1 (HMGB1) and v-Myc myelocytomatosis viral oncogene homolog (MYC); and the increased expression of activating transcription factor 5 (ATF5) at the gene transcription level (Supplementary Table S1).
Table 2: Genetic networks associated with ascorbate-resistance in JLPR cells
Associated network functions |
Score |
Focus molecules |
Molecules in network |
|---|---|---|---|
Cell-to-cell signaling and interaction, hematological system development and function, immune cell trafficking |
47 |
18 |
Akt, ASNS, ATF5, BCR, c-Myc/N-Myc, C5orf13, CD3, CD79B, Ck2, E2f,ERK1/2, FTL, HIST1H2AB/HIST1H2AE, HIST1H2BJ/ HIST1H2BK, HISTONE, HLA-A, HMGB1, HSPH1, IFN Beta, IGLL1/IGLL5, Immunoglobulin, Insulin, Interferon alpha, KIAA0020, MHC CLASS I (family), MYC, NFkB (complex), PI3K (complex), RPS7, SAP18, SSX1, TCL1A, TKT, TOP2B, Ubiquitin |
Cell death, cell-to-cell signaling and interaction, hematological system development and function |
22 |
10 |
ALOX5AP, ATF5, BATF, beta-estradiol, CD6, DDIT3, FGF2, Gm4617 /Ptma, GRB2, HIF1A, HLA Class I, HPS1, HPS4, IDH2, IFI30, IFNG, KRT6A, LGMN, MAMLD1, mir-23, miR-199a-5p, MLANA, MYL6, Myosin Light Chain Kinase, NCAN, NCR3, POMP, RAC1, SLK, SSX2, SSX4/SSX4B, Tgtp1, TMEM45A, TNF, ZFP57 |
Table 3: Top 5 canonical pathways involving genes differentially expressed in JLPR cells
Signaling pathway |
P value |
Ratio |
Molecules |
|---|---|---|---|
PI3K signaling in B lymphocytes |
1.95 E-02 |
2:147 (0.014) |
CD79B, ATF5 |
Cdc42 signaling |
2.77 E-02 |
2:180 (0.011) |
MLC, MHC |
DNA methylation and transcriptional repression signaling |
3.14 E-02 |
1:23 (0.043) |
SAP18 |
Polyamine regulation in colon cancer |
3.45 E-02 |
1:29 (0.034) |
C-Myc |
ILK signaling |
3.49 E-02 |
2:193 (0.01) |
C-Myc, Myosin |
Functional networks and pathways of ascorbate-induced drug resistance
The genetic networks and cellular pathways deregulated in JLPR cells were identified using the IPA software program. Expression microarray profiling studies revealed that 45 genes were deregulated in JLPR cells (Supplementary Table S1). A comprehensive network and pathway analysis of the deregulated genes revealed that these genes were associated with two network functions and five canonical pathways relevant to the development of ascorbate resistance in cancer cells. In each of the two genetic networks, the differently expressed genes constituted about half of the molecules involved in network-associated cellular functions and included genes related to cell-to-cell signaling and interaction, cell death, hematological system development and function, and immune cell trafficking (Table 2). These genes are expected to be affected during ascorbate resistance development in Burkitt lymphoma cells. The differentially expressed genes belong to five canonical signaling pathways that are frequently deregulated in ascorbate-resistant cells (Table 3).
qRT-PCR and immunobloting analyses validation of differently expressed oxidative stress genes in JLP cells
qRT-PCR revealed that the antioxidant genes CAT and GPX4 (Figure 2E), the DNA repair genes XRCC5 and TOP2B, and asparagine synthetase (ASNS), GNB1, SSX3, ATF5, HLA-A, and FTL were upregulated in JLPR cells (Figure 4A). Moreover, qRT-PCR and western blot analyses revealed that the expressions of HMGB1, c-Myc, HIST1H4, CD74, CD79B, and HSPH1 were suppressed in JLPR cells (Figure 4A and Figure 4B). The functional activity assay showed that the activity of CAT and GPX increased in resistant JLPR cells adapted to up to 1 mM ascorbate (Figure 2G and Figure 2H).
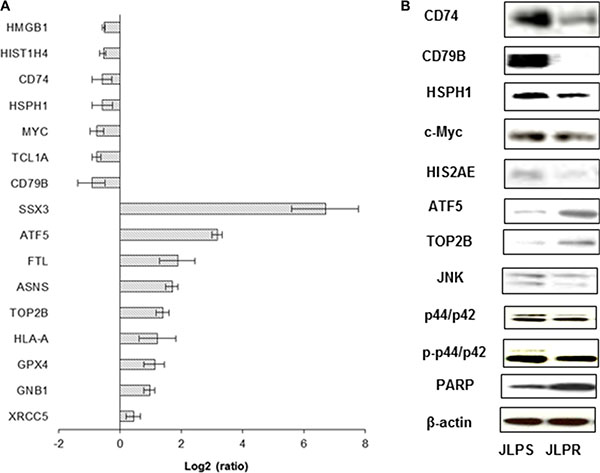
Figure 4: Real-time PCR and western blot analyses of differentially expressed genes and protein expression levels of JLPS and JLPR cells treated with ascorbate. (A) Real-time PCR analysis of HMGB1, HIST1H4, CD74, HSPH1, c-Myc, TCL1A, CD79B, SSX3, ATF5, ASNS, FTL, TOP2B, HLA-A, XRCC5, GNB1, ATP5, and GPX4. Results were normalized using 18S ribosomal RNA. (B) Western blot analysis of CD74, CD79B, ATF5, TOP2B, HSPH1, HIS2AE, JNK, p44/p42, p-p44/p42, PARP, and c-Myc expression in JLPS and JLPR cells. β-actin was used as the loading control.
DISCUSSION
In the present study, we demonstrated that resistance to ascorbate reduced drug accumulation, decreased apoptosis, and increased glucose transport, anti-oxidation activity, and DNA repair. Compared with JLPS cells, JLPR cells generated smaller amounts of ROS than JLPS cells with ascorbate treatment (Figure 3C). Produced intracellularly as a byproduct of oxidative metabolism, under normal conditions H2O2 is reduced to water by CAT, GPXs, and peroxiredoxins [23]. In contrast, JLPR cells had a wide array of enzymatic and non-enzymatic antioxidants, including GPX, CAT, and glutathione. Other studies have revealed that CAT activity increases in H2O2-resistant fibroblasts and that cell death is prevented by cellular or extracellular CAT [24, 25]. Overexpression of the corresponding genes may dramatically increase cells’ resistance to the ascorbate-induced oxidative damage of lipids, proteins, and DNA.
In our study, we found that JLPR cells were resistant to H2O2 and that the effects elicited by ascorbate or H2O2 in these cells were strikingly similar. Moreover, ascorbate and H2O2 exposed to JLPR cells resulted in reduced cell cycle arrest in G2/M phase, but increased glucose uptake compared with that in JLPS cells. We also found that Glut1 and Glut3 were primarily responsible for glucose transport in JLPR cells and that the amounts of these transporters were higher in JLPR than JLPS cells. Smith et al. found that increased glucose transport was associated with fluorouracil resistance in MCF7 cells and that Glut1 was increased in fluorouracil-resistant cells [26]. These results imply that JLPR cells overcame the role of H2O2 in mediating glucose uptake activation by activation of the expression of glucose transporters. We also investigated whether ascorbate treatment of JLPR and JLPS cells resulted in different patterns of gene regulation with oxidative stress pathways induced by reactive oxygen species. After treatment with ascorbate, the expression of c-Myc was downregulated in JLPR cells but overexpressed in JLPS cells (Figure 4D). c-Myc activation can induce sufficient DNA damage to elicit a stress response and induce the accumulation of cells with a 4N DNA content [27], which is consistent with our finding that JLPS cells treated with ascorbate induced sufficient DNA damage to cause cell cycle arrest in the G2/M phase. Following treatment with ascorbate, cleaved PARP was present in JLPS cells and native PARP was overexpressed in JLPR cells. We further analyzed the functional networks and gene pathways involved in ascorbate resistance. Although only five genes (CD79B, ATF5, Myosin, MHC, and SAP18) with similar and overlapping signature patterns were represented in the signaling pathways (phosphoinositide kinase signaling, Cdc42 signaling, DNA methylation and transcriptional repression signaling, polyamine regulation signaling, and integrin-linked kinase signaling), each of their gene products are functionally involved in controlling cell growth and cell death and have been suggested to play roles in ascorbate resistance in JLPR cells. Real-time PCR and immunoblot analysis confirmed the expression of ATF5 in JLPR cells. ATF5, which is part of the ATF/cAMP-response element-binding protein gene family, may play a role in protecting cells from amino acid limitation or arsenite-induced oxidative stress. Previous studies have shown that ATF5 loss-of-function induces glioma cell death in culture and in vivo [28, 29]. Taken together, these results suggested that ATF5 induces ascorbate resistance in lymphoma cells by activating transcription factors.
In conclusion, we found that in parental JLPS cells—but not in JLPR cells—treatment with pharmacologic ascorbate or H2O2 induced cell cycle arrest in the G2/M phase and PARP cleavage, decreased glucose uptake, and activated c-Myc pathway. In addition, the gene expression profiles of JLPS cells and JLPR cells were different. The differentially expressed genes in microarrays, which mediated multiple cytotoxic pro-oxidant stress pathways, were also verified at the levels of mRNA, protein, and functional activity by real-time PCR, western blotting, and enzyme activity analyses, respectively. These target genes from vitamin C–induced resistant cells may serve as novel biomarkers for identifying cancer patients who may benefit from pharmacologic ascorbate treatment.
MATERIALS AND METHODS
JLPS and JLPR Burkitt lymphoma cells
The Burkitt lymphoma cell line JLPS and ascorbate-resistant JLPR cells were maintained in RPMI 1640 medium (Mediatech, Inc., Herndon, VA, USA) with 10% inactivated FBS, 2 mM L-glutamine, and 50 μM β-mercaptoethanol. JLPR cells were maintained with 1 mM ascorbate. To develop cells resistant to ascorbate, we incubated JLPS cells with ascorbate in a stepwise manner, gradually increased from 100 μM to 1 mM over 6 months.
O2 electrode assay of H2O2 concentration
The amount of H2O2 in the culture medium was measured using an O2 electrode (Hansatech Instruments, Ltd., Norfolk, UK) as described previously (24). Tested samples of cell culture medium that had been treated with 1 mM ascorbate or H2O2 for various times were placed in the reaction cells. After equilibration of the samples, 100 μl of catalase (CAT) solution (10,000 U/ml) was microinjected into the cells. Calibration curves were obtained by measuring the amount of O2 released in the medium.
Assessment of glucose transport
After JLPS and JLPR cells (2 × 106) were treated with ascorbate or H2O2, the cells were washed with PBS and incubated with 250 nM 2-deoxy-D-[1,2-3H] glucose (2-DG) (Sigma, St. Louis, MO, USA). The cells were washed with ice-cold PBS containing phloretin (100 μM), and dissolved in 300 μl of 0.1 N NaOH, 1% CHAPS. 2-DG uptake was measured using a scintillation counter.
Measurement of intracellular ascorbate, CAT, and glutathione peroxidase (GPX)
After JLPS and JLPR cells were incubated with ascorbate, the cells were washed with PBS and harvested. The cell pellets were resuspended in 60% methanol and centrifuged at 15,300 rpm for 20 min. A HPLC system was used to assess the amount of intracellular ascorbate. For CAT activity, JLPS and JLPR cells (1.5 x 106 cells) were centrifuged at 1,000 g for 10 min at 4°C. The cell pellet was sonicated in 300 μl of cold buffer comprising 50 mM potassium phosphate and 1 mM ethylenediaminetetraacetic acid and centrifuged at 10,000 g for 15 min. CAT activity in 20 μl of the resulting supernatant was assessed using a CAT assay kit (Calbiochem, San Diego, CA, USA) according to the manufacturer’s instructions. Intracellular GPX4 activity was measured using a cellular GPX assay kit (Calbiochem) according to the manufacturer’s instructions.
Transcriptomic profiles
After quality assessment using the Agilent NanoChip Bioanalyzer assay, total RNA from cell lines was labeled using Agilent’s Low RNA Input Labeling Kit, which involves reverse transcribing the mRNA to produce cDNA and then transcribing in the presence of Cy3-CTP or Cy5-CTP to produce labeled cRNA. Labeled cell line cRNA was paired with the differentially labeled normal colonic epithelial cRNA and, using the Agilent In situ Hybridization Kit, hybridized to the Agilent 44K Whole Genome Oligo Microarray (G4112A) for 17 hours at 60°C according to the manufacturer’s protocol. The arrays washed in 2X SSC, 0.005% Triton × 102 for 10 minutes, 0.006 × SSPE, 0.005% N-lauroylsarcosine and coated with Agilent’s Stabilization and Drying Solution. Arrays were scanned on Agilent DNA Microarray scanner.
Pathway analysis
To determine the potential specific pathways based on changes in gene expression, we used the Ingenuity Pathway Analysis (IPA) software program (Ingenuity, Redwood City, CA) as described previously [21, 30].
CONFLICTS OF INTEREST
None
GRANT SUPPORT
This research was supported by this work was sponsored by the grant (16PJ1408800) from the Shanghai Pujiang Program, Shanghai, and the grant RCJJP21 (J.W.) from Shanghai Public Health Clinical Center, China.
REFERENCES
1. Rebouche CJ. Ascorbic acid and carnitine biosynthesis. The American journal of clinical nutrition. 1991; 54:1147S-52S.
2. Nishikimi M, Fukuyama R, Minoshima S, Shimizu N, Yagi K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J Bio Chem. 1994; 269:13685–13688.
3. Li Y, Schellhorn HE. New developments and novel therapeutic perspectives for vitamin C. The Journal of nutrition. 2007; 137: 2171–2184.
4. Padayatty SJ, Levine M. Fruit and vegetables: think variety, go ahead, eat! Am J Clin Nutr. 2008; 87:5–7.
5. Arrigoni O, De Tullio MC. Ascorbic acid: much more than just an antioxidant. Biochim Biophys Acta. 2002;1569: 1–9.
6. Cameron E, Rotman D. 1Ascorbic Acid Cell Proliferation and Cancer. Lancet. 1972; 1: 542.
7. Cameron E, Campbell A. 1The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem Biol Interact. 1974; 9:285–315.
8. Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, Shacter E, Levine M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci USA. 2005; 102:13604–13609.
9. Makino Y, Sakagami H, Takeda M. Induction of cell death by ascorbic acid derivatives in human renal carcinoma and glioblastoma cell lines. Anticancer Res. 1999; 19:3125–3132.
10. González MJ1, Miranda-Massari JR, Mora EM, Guzmán A, Riordan NH, Riordan HD, Casciari JJ, Jackson JA, Román-Franco A. Orthomolecular oncology review: ascorbic acid and cancer 25 years later. Integrative cancer therapies. 2005; 4:32–44.
11. Park S, Han SS, Park CH, Hahm ER, Lee SJ, Park HK, Lee SH, Kim WS, Jung CW, Park K, Riordan HD, Kimler BF, Kim K, et al. L-Ascorbic acid induces apoptosis in acute myeloid leukemia cells via hydrogen peroxide-mediated mechanisms. Int J Biochem Cell Biol. 2004; 36:2180–2195.
12. Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, Khosh DB, Drisko J, Levine M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci USA. 2008; 105:11105–11109.
13. Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, Taghiyev AF, Du C, Knudson CM, Cullen JJ. Mechanisms of Ascorbate-Induced Cytotoxicity in Pancreatic Cancer. Clin Cancer Res. 2010; 16:509–520.
14. Verrax J, Calderon PB. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radical Bio Med. 2009; 47:32–40.
15. Herst PM, Broadley KW, Harper JL, McConnell MJ. Pharmacological concentrations of ascorbate radiosensitize glioblastoma multiforme primary cells by increasing oxidative DNA damage and inhibiting G2/M arrest. Free Radic Biol Med. 2012; 52: 1486–1493.
16. Mastrangelo D, Massai L, Lo Coco F, Noguera NI, Borgia L, Fioritoni G, Berardi A, Iacone A, Muscettola M, Pelosi E, Castelli G, Testa U, Di Pisa F, et al. Cytotoxic effects of high concentrations of sodium ascorbate on human myeloid cell lines. Ann Hematol. 2015; 94: 1807–1816.
17. Yeom CH, Jung GC, Song KJ. Changes of terminal cancer patients’ health-related quality of life after high dose vitamin C administration. J Korean Med Sci. 2007; 22:7–11.
18. Kuiper C, Vissers MC, Hicks KO. Pharmacokinetic modeling of ascorbate diffusion through normal and tumor tissue. Free Radic Biol Med. 2014; 77: 340–352.
19. Sinnberg T, Noor S, Venturelli S, Berger A, Schuler P, Garbe C, Busch C. The ROS-induced cytotoxicity of ascorbate is attenuated by hypoxia and HIF-1alpha in the NCI60 cancer cell lines. J Cell Mol Med. 2014; 18:530–541.
20. Wang J, Tai LS, Tzang CH, Fong WF, Guan XY, Yang M. 1p31, 7q21 and 18q21 chromosomal aberrations and candidate genes in acquired vinblastine resistance of human cervical carcinoma KB cells. Oncol Rep. 2008; 19:1155–1164.
21. Wang J, Chan JYW, Fong CC, Tzang CH, Fung KP, Yang MS. Transcriptional analysis of doxorubicin-induced cytotoxicity and resistance in human hepatocellular carcinoma cell lines. Liver Int. 2009; 29:1338–1347.
22. Schena M, Shalon D, Davis RW, Brown PO. Quantitative Monitoring of Gene-Expression Patterns with a Complementary-DNA Microarray. Science. 1995; 270:467–470.
23. Rhee SG, Yang KS, Kang SW, Woo HA, Chang TS. Controlled elimination of intracellular H2O2: Regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid Redox Signal. 2005; 7:619–626.
24. Spitz DR, Li GC, Mccormick ML, Sun Y, Oberley LW. Stable H2O2-Resistant Variants of Chinese-Hamster Fibroblasts Demonstrate Increases in Catalase Activity. Radiat Res. 1988; 114:114–124.
25. Sestili P, Brandi G, Brambilla L, Cattabeni F, Cantoni O. Hydrogen peroxide mediates the killing of U937 tumor cells elicited by pharmacologically attainable concentrations of ascorbic acid: Cell death prevention by extracellular catalase or catalase from cocultured erythrocytes or fibroblasts. J Pharmacol Exp Ther. 1996; 277:1719–1725.
26. Smith TAD, Sharma RI, Wang WG, Welch AE, Schweiger LF, Collie-Duguid ESR. Decreased [F-18] fluoro-2-deoxy-D-glucose incorporation and increased glucose transport are associated with resistance to 5FU in MCF7 cells in vitro. Nucl Med Biol. 2007; 34: 955–960.
27. Felsher DW, Zetterberg A, Zhu J, Tlsty T, Bishop JM. Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc Natl Acade Sci USA. 2000; 97: 10544–10548.
28. Watatani Y, Ichikawa K, Nakanishi N, Fujimoto M, Takeda H, Kimura N, Hirose H, Takahashi S, Takahashi Y. Stress-induced translation of ATF5 mRNA is regulated by the 5′-untranslated region. J Biol Chem. 2008; 283: 2543–2553.
29. Angelastro JM, Canoll PD, Kuo J, Weicker M, Costa A, Bruce JN, Greene LA. Selective destruction of glioblastoma cells by interference with the activity or expression of ATF5. Oncogene. 2006; 25: 907–916.
30. Wang J, Sen S. MicroRNA functional network in pancreatic cancer: from biology to biomarkers of disease. Journal of biosciences. 2011; 36: 481–491.