
INTRODUCTION
Aneuploidy is common in solid tumors, with nearly 90% of such tumors being aneuploid [1]. Despite the prevalence of aneuploidy in these cancers, it has not been firmly established if aneuploidy is an early step that drives malignant transformation, results as a consequence of transformation, or what role, if any, oncogenes play in this process. Although many solid tumors are characterized by a specific oncogenic mutation, very few reports examined the role that the resulting oncoprotein plays in driving the initial stages of tumor development, possibly through the acquisition of aneuploidy and in overcoming aneuploidy-dependent growth arrest. In many of these reports, oncogene-induced aneuploidy results from the overexpression of a protein that is central to the chromosome segregation machinery or regulates the process of chromosome segregation [2-6]. Although one report demonstrates that an oncogenic transcription factor, TLX1, is sufficient to induce aneuploidy in T-cell progenitors, the process involves a limited mechanism in which specific genes important for maintaining mitotic checkpoint control are affected [7].
Upon the acquisition of aneuploidy, a cell’s natural response is to attenuate proliferation. Previous work demonstrated that the induction of aneuploidy in diploid or near diploid cell lines resulted in significant proliferative defects [8, 9], raising an apparent contradiction for tumor progression: rapidly proliferating tumor cells are often aneuploid, a state that under normal circumstances is not conducive to proliferation [10]. Therefore, tumor cells must develop a mechanism to overcome aneuploid-dependent proliferative defects and develop an enhanced proliferation rate. However, the mechanism by which an oncogene promotes aneuploidy in pre-cancerous cells, how these cells overcome the adverse proliferative effects associated with this state, and whether post-translational modifications can regulate this process are important, yet unanswered, questions in the development of solid tumors.
Like other solid tumors, aneuploidy is common in Rhabdomyosarcoma (RMS) [11], which accounts for nearly half of childhood soft tissue sarcomas. RMS is comprised of two main subtypes: embryonal (ERMS) and alveolar (ARMS), each defined by its unique histology, clinical presentation, therapy, and prognosis [12]. ARMS, the more aggressive subtype, is primarily defined by the t(2;13)(q35;q14) translocation, which creates the oncogenic fusion protein PAX3-FOXO1 [13, 14]. In addition to the defining cytogenetic abnormality, ARMS tumor cells contain cytogenetic evidence of polyploidy, having cells ranging in chromosome number from hypodiploid to hypertetraploid, with a wide and heterogeneous range of chromosome number in cells within a single patient sample [11, 15-18]. There were also instances of chromosome amplification as double minutes and heterogeneously staining regions, gains and losses through insertions and deletions, derivative chromosomes and additional translocations, and breakpoints around the centromere [11, 15-18]. Despite extensive work understanding the altered molecular characteristics of PAX3-FOXO1 relative to PAX3 [19-25] and the knowledge that phosphorylation of the fusion protein contributes to ARMS tumor phenotypes [26], it is not known what role, if any, the fusion protein plays in the promotion of aneuploidy and chromosomal structural abnormalities, overcoming aneuploidy-dependent proliferative defects, and whether phosphorylation of the fusion protein contributes to this process.
In this study we examine how the expression of PAX3-FOXO1 affects global mRNA and miRNA expression. We are the first to show that the presence of PAX3-FOXO1 is sufficient to alter mRNA and miRNA expression, either through direct regulation of genes or indirectly through downstream effects, subsequently altering protein levels important for multiple aspects of chromosome number and structure, thereby promoting the aneuploid state. Further, we are the first to demonstrate that the presence of an oncogene is sufficient to alter, either directly or indirectly, the expression of multiple growth related mRNA and miRNA to overcome aneuploidy-induced proliferative defects. In addition, inhibition of phosphorylation at Ser201 or Ser205 on PAX3-FOXO1 reversed oncogene dependent mRNA and miRNA changes, and inhibited the proliferation of cells containing chromosomal abnormalities. Our results allow us to propose a model by which the expression and phosphorylation of an oncogene, PAX3-FOXO1, serves as the driving molecular event to initiate the development of a solid tumor by reprograming cells to induce aneuploidy and override aneuploidy-induced growth arrest.
RESULTS
PAX3-FOXO1 is sufficient to alter the expression of mRNA and miRNA to promote aneuploidy
To understand how PAX3-FOXO1 affects global mRNA and microRNA (miRNA) expression, we stably transduced passage-matched mouse primary myoblasts with the MSCV-IRES-puromycin retroviral vector (negative control), or the same retroviral vector expressing FLAG-epitope tagged PAX3 (FLAG-PAX3), or FLAG-PAX3-FOXO1 (Figure 1A), a tag previously shown to not affect Pax3 or Pax3-FOXO1 function [24, 26]. The transduced cells were selected with puromycin; selected cells were harvested from three independent transductions and pooled, resulting in a single mixed population for each individual construct. By utilizing a population of transduced cells, we remove the potential for variability that may occur from clonal effects. The level of PAX3-FOXO1 expression we observed is equivalent to the level of expression of the fusion protein in ARMS tumor cell lines (Supplementary Figure 1 and [27, 28]) and is therefore directly relevant to the role of the oncogenic fusion protein in ARMS. This model allows us to use a physiologically relevant cell system to determine how the sole expression of the oncogenic PAX3-FOXO1 can initiate a cascade of events, both direct and downstream, over a series of proliferation events, to contribute to the initial stages of tumor development with respect to aneuploidy and overcoming aneuploidy-dependent proliferative defects. This model is in direct contrast to tumor cell lines or primary tumor samples in which it is difficult to determine whether these processes result as a “byproduct” of the final oncogenic state.
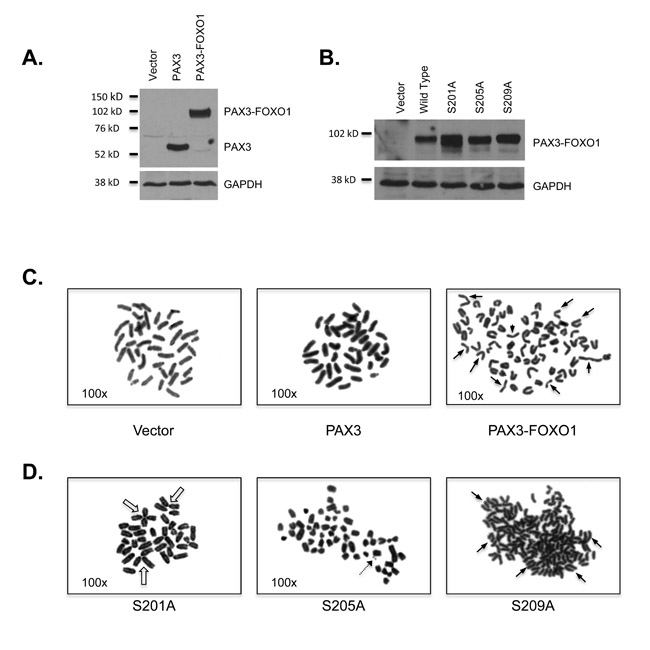
Figure 1: The expression of PAX3-FOXO1 promotes aneuploidy and chromosomal structural abnormalities. Expression of A. PAX3, PAX3-FOXO1 or B. PAX3-FOXO1 phosphomutants. Total extracts were made from stably transduced cells and protein was determined using an antibody specific for PAX3, as described in the Materials and Methods. Representative picture of a metaphase chromosome analysis for C. cells stably transduced with empty vector, PAX3 or PAX3-FOXO1 or D. individual PAX3-FOXO1 phosphomutants. The closed arrows indicate representative sister chromatid dissociation, the open arrow indicate representative telomere association, and the dotted arrow indicate a representative double minute.
We performed mRNA and miRNA deep sequencing on total RNA isolated from three independent growths of stably transduced primary myoblasts and utilized the resulting sequencing data in large-scale comparative transcriptomic analyses, using the on-line Galaxy program or miRNAKey, respectively (see Materials and Methods). Since all stable lines were generated simultaneously and with identical methods, any artifactual effects of the process on mRNA and miRNA expression would be expected to be present in all samples and would therefore by corrected for by our differential analysis. The data used for subsequent studies were limited to 1) mRNA or miRNA displaying statistically significant differences (p < 0.05), 2) mRNA or miRNA present in both data sets being analyzed to rule out artifactual differences resulting from depth of read, and 3) mRNA or miRNA that exhibited at least 2-fold difference in expression either up- or downregulated. Finally, the biological functions of the differentially expressed mRNA or target genes of miRNA were categorized using the Onto-Express gene ontology software (http://vortex.cs.wayne.edu/projects.htm).
We found that in total PAX3-FOXO1 altered the expression of 846 genes, with 562 being upregulated and 284 being downregulated, relative to cells expressing wild-type PAX3 (data not shown). Fifty-nine of these differentially expressed genes have biological functions directly involved with multiple processes important for maintaining chromosome number and structure (Table 1) with 48/59 of these genes being downregulated. The affected genes include kinases that regulate chromosome separation and mitotic progression, cohesins, centromeric proteins, regulators of cytokinesis, components or regulators of the condensin complex, proteins involved with chromosomal segregation, and components of the mitotic spindle. Finally, six of the 59 genes that specifically contribute to promoting aneuploidy contain PAX3-FOXO1 binding sites in their proximal promoters, as previously described [29] (Table 1, carat) and six of these genes have altered gene expression levels consistent with changes seen in human tumor samples [30-33] (Table 1, pound sign).
Table 1a: Differentially expressed mRNA important for promoting aneuploidy by altering genes important for chromosome segregation/cytokinesis, chromosome cohesion/condensing, and mitotic progression.
PROMOTION OF ANEUPLOIDY |
||||
Gene |
Function |
V vs PF* |
P3 vs PF |
V vs P3 |
Chromosome Segregation/Cytokinesis |
||||
ANLN |
Required for cytokinesis |
-2.33 |
-4.21 |
+1.89 |
AURKA |
Regulate chromosome segregation |
-2.29 |
-2.76 |
|
AURKB |
Regulate chromosome segregation |
-5.13 |
||
#CENPF |
Centrosome segregation |
-2.71 |
||
CDC25B |
Required for abscission in cytokinesis |
+2.20 |
||
CORO1C |
Involved in cytokinesis |
-2.27 |
-2.01 |
|
ECT2 |
Nucleotide exchange factor - cytokinesis |
-2.88 |
||
ESPL1 |
Protease in chromosome segregation |
-4.01 |
||
^KLHL13 |
Chromosome segregation |
+2.25 |
||
MYH10 |
Involved in cytokinesis |
-2.09 |
-4.58 |
|
Pard6g |
Cell division and cell polarization |
-3.06 |
||
PRC1 |
Regulator of cytokinesis |
-2.09 |
||
PSRC1 |
Required for normal chromosome segregation |
-2.28 |
-4.15 |
|
Chromosome Cohesion/Condensing |
||||
CDC20B |
Destruction of condensins |
-2.71 |
||
^CEP250 |
Centriole cohesion - chromosome segregation |
-2.57 |
||
NCAPD2 |
Regulatory subunit of condensin complex |
-2.09 |
||
NCAPG2 |
Subunit of condensin complex |
-2.72 |
||
NCAPH |
Subunit of condensin complex |
-3.56 |
||
PDS5B |
Regulator of cohesin |
-2.18 |
||
#RCC1 |
Regulator of chromosome condensation |
-2.27 |
||
^SGOL1 |
Involved with chromosome cohesion |
-5.88 |
+2.76 |
|
SGOL2 |
Prevents premature release of cohesin |
-3.41 |
||
SMC2 |
Critical for mitotic chromosome condensation |
-3.10 |
||
Mitotic Progression |
||||
BUB1 |
Mitotic checkpoint kinase |
-4.71 |
||
BUB1B |
Spindle checkpoint kinase |
-4.04 |
||
CCNA2 |
Cyclin – regulate G2/M progression |
-2.44 |
||
CCNG1 |
Cyclin – regulate G2/M checkpoint |
-2.19 |
-2.66 |
|
CDK1 |
Mitotic cyclin dependent kinase |
-2.46 |
-2.70 |
|
CDK14 |
G2/M checkpoint cyclin dependent kinase |
-4.56 |
||
FoxM1 |
Chromosome maintenance/mitotic division |
-2.45 |
+1.88 |
|
HELLS |
Helicase in mitosis |
-5.25 |
||
^HMGA2 |
Transcriptional regulator in mitosis |
-2.16 |
-4.35 |
|
INCENP |
Centromere protein in mitotic progression |
-2.07 |
||
NEK6 |
Kinase in mitotic progression |
-4.05 |
||
#SIRT2 |
Deacetylase required for mitotic exit |
+2.44 |
||
Gene name, function and fold change for each gene are listed; downregulated genes indicated by negative numbers, upregulated genes indicated by positive numbers. *Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3), ^indicates genes demonstrated to have PAX3-FOXO1 binding sites present in their proximal promoter [29], and #indicates genes that demonstrate similar trends in gene expression changes in human tumor samples [30-33].
Table 1b: Differentially expressed mRNA important for promoting aneuploidy by altering genes important for the segregation machinery.
PROMOTION OF ANEUPLOIDY |
||||
Gene |
Function |
V vs PF* |
P3 vs PF |
V vs P3 |
Segregation Machinery |
||||
BRCA1 |
Centrosomal microtubule nucleation |
-5.76 |
||
CEP41 |
Centrosomal protein |
+2.37 |
+16.62 |
|
CEP128 |
Centrosomal protein |
+7.62 |
||
CEP170B |
Microtubule organization |
-2.19 |
||
CKAP |
Stabilizes microtubules |
-2.43 |
||
KIF11 |
Motor protein in centrosome separation |
-4.67 |
||
KIF20A |
Motor protein in centrosome separation |
-3.45 |
||
KIF23 |
Kinesin – chromosome movement in division |
-3.76 |
+2.11 |
|
^#KIF2A |
Motor protein – normal mitotic progression |
-2.40 |
-2.41 |
|
^KIF3A |
Kinesin – chromosome movement in division |
+3.37 |
||
KNSTRN |
Mitotic spindle component |
-2.39 |
||
NDE1 |
Microtubule organization and mitosis |
-2.22 |
||
NIN |
Important for centrosomal function |
-2.94 |
||
SDCCAG8 |
Centrosome associated protein |
-3.41 |
-4.37 |
|
SFI1 |
Mitotic spindle assembly |
-2.23 |
||
SPAG5 |
Associated with mitotic spindle |
-2.61 |
||
SUV39H1 |
Loss causes chromosome instability |
-2.11 |
-3.05 |
|
TACC3 |
Stabilizes mitotic spindles |
-2.78 |
+2.16 |
|
TOP2A |
Topoisomerase – chromatid separation |
-2.67 |
-3.30 |
|
TOPBP1 |
Topoisomerase binding protein |
-2.01 |
-2.44 |
|
TPX2 |
Mitotic spindle assembly factor |
-2.70 |
-3.43 |
|
#TUBB |
Major component of microtubules |
-2.25 |
||
TUBB2B |
Major component of microtubules |
+3.26 |
||
#TUBB4B |
Major component of microtubules |
-2.07 |
||
Gene name, function and fold change for each gene are listed; downregulated genes indicated by negative numbers, upregulated genes indicated by positive numbers. *Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3), ^indicates genes demonstrated to have PAX3-FOXO1 binding sites present in their proximal promoter [29], and #indicates genes that demonstrate similar trends in gene expression changes in human tumor samples [30-33].
Table 1c: Differentially expressed mRNA important for regulating proliferative control by altering genes for proliferative transcription factors and cell cycle regulatory proteins.
PROLIFERATIVE CONTROL |
||||
Gene |
Function |
V vs PF* |
P3 vs PF |
V vs P3 |
Transcription Factors |
||||
bcl6 |
Suppresses cellular proliferation |
|
+5.94 |
|
#CREB3 |
Promotes cellular proliferation |
+2.25 |
+2.48 |
|
FOXM1 |
Promotes entry into S- and M-phase |
|
-2.45 |
+1.88 |
^#FOXO1 |
Promotes cellular proliferation |
+3.16 |
+2.19 |
|
FOXO4 |
Promotes cellular proliferation |
+3.16 |
+3.84 |
|
^#GADD45A |
Inhibits entry into S-phase |
+2.22 |
|
|
HOXB9 |
Promotes cellular proliferation |
+42.81 |
+9.25 |
|
KLF5 |
Promotes cellular proliferation |
+2.83 |
+2.06 |
|
#MYC |
Promotes cellular proliferation |
+23.59 |
-27.09 |
|
NAB2 |
Negative cofactor – inhibits proliferation |
+2.60 |
|
|
NDN |
Prevents excessive proliferation |
-2.03 |
|
|
#Nupr1 |
Inhibits cellular proliferation |
|
+3.10 |
|
PML |
Promotes cellular proliferation |
-4.86 |
|
|
Suv39h1 |
Promotes cellular proliferation |
-2.11 |
-3.05 |
|
#TFDP1 |
Promotes E2F activity/cellular proliferation |
-2.06 |
|
|
UHRF1 |
Regulates G1/S transition |
-4.00 |
|
|
WRD6 |
G1 arrest – inhibits proliferation |
-2.57 |
||
Cell Cycle Regulatory Proteins |
||||
ARID3A |
Promotes E2F transcription and growth |
+4.53 |
+4.38 |
|
#BTG2 |
Anti-proliferative – inhibits Cyclin D1 |
-2.07 |
|
|
CCNA2 |
Cyclin A2 – G1/S and G2?M progression |
-2.45 |
|
|
#CCND1 |
Cyclin D1 – G1 progression |
-5.46 |
-4.32 |
|
#CCND2 |
Cyclin D2 – G1 progression |
-5.54 |
-4.38 |
|
#CCND3 |
Cyclin D3 – G1 progression |
+4.03 |
|
|
CCNG1 |
Cyclin G1 – negative regulator of cell cycle |
-2.19 |
-2.66 |
|
CCPG1 |
Positive regulator of proliferation |
+2.93 |
||
CCR |
Increase in G1/S – promotes proliferation |
-2.57 |
+2.03 |
|
CDC20 |
Required for completion of mitosis |
-2.27 |
-2.71 |
|
CDC25B |
Required for G2/M progression |
+2.20 |
|
|
CDK1 |
Promotes G2/M progression |
-3.01 |
-2.71 |
|
^#CDK6 |
Promotes G1 progression |
-2.81 |
|
|
CDK14 |
Promotes G1 progression |
-4.56 |
|
|
CDK2AP |
Interacts with CDK2 – promotes proliferation |
+2.48 |
||
cdkn1a (p21) |
Inhibitor of G1 progression |
+2.19 |
|
|
cdkn1c (p57) |
Inhibitor of G1 progression |
+21.71 |
+134.36 |
-5.31 |
cdkn2d (p19) |
Inhibitor of G1 progression |
+7.78 |
+2.46 |
|
DBF4 |
Required for S-phase progression |
-2.57 |
|
|
^#GAS1 |
Block entry into S-phase |
|
+4.26 |
-3.63 |
Rb1 |
Tumor suppressor – inhibits proliferation |
|
+2.10 |
-2.53 |
Mdm2 |
Facilitates G1/S phase transition |
-2.11 |
|
|
NEK6 |
Required for M-phase progression |
|
-4.06 |
|
Gene name, function and fold change for each gene are listed; downregulated genes indicated by negative numbers, upregulated genes indicated by positive numbers. *Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3), ^indicates genes demonstrated to have PAX3-FOXO1 binding sites present in their proximal promoter [29], and #indicates genes that demonstrate similar trends in gene expression changes in human tumor samples [30-33].
Table 1d: Differentially expressed mRNA important for regulating proliferative control by altering genes for growth factors and proliferative receptors.
PROLIFERATIVE CONTROL |
||||
Gene |
Function |
V vs PF* |
P3 vs PF |
V vs P3 |
Growth Factors/Development |
||||
CGREF1 |
Ca+2 binding inhibitor of cellular proliferation |
|
-3.10 |
+2.44 |
CSF1 |
Promotes cellular proliferation |
-5.94 |
-3.29 |
|
#CYR61 |
Promotes cellular proliferation |
-3.29 |
|
|
hdgfrp3 |
May promote cellular proliferation |
|
+6.15 |
|
#IFITM1 |
Suppresses proliferation |
-2.68 |
-2.89 |
|
#Igf2 |
Growth promoting hormone |
|
+19.97 |
-3.72 |
#IGFBP2 |
Inhibits IGF-dependent proliferation |
-8.63 |
||
Igfbp3 |
Interacts with and stabilizes IGF |
|
+4.35 |
-1.74 |
#Igfbp5 |
Interacts with and stabilizes IGF |
+8.28 |
+50.91 |
-5.70 |
RACGAP1 |
Promotes cellular proliferation |
-2.68 |
|
|
^SHB |
Promotes IGF-dependent cellular proliferation |
+2.93 |
+3.23 |
|
#SMAD3 |
Promotes cellular proliferation |
-5.50 |
|
|
SMO |
Promotes cellular proliferation |
|
-2.38 |
|
#TgfB3 |
Anti-proliferative effect |
|
+2.77 |
-2.75 |
Receptor/Signal Transduction |
||||
^#ABI1 |
Negative regulator of proliferation |
-2.41 |
|
|
ADRA1B |
Adranergic receptor – inhibits proliferation |
-4.89 |
+3.34 |
|
#AKAP12 |
Suppresses proliferation |
-7.94 |
-3.86 |
|
AXL |
Promotes cellular proliferation |
-6.06 |
-4.29 |
|
EGFR |
Epidermal growth factor receptor |
-3.32 |
+4.82 |
|
EPS8 |
Involved in promoting EGF pathway |
-9.32 |
+3.58 |
|
Erbb3 |
HER3 –growth factor receptor |
+3.10 |
+6.23 |
-4.06 |
^#FGFR4 |
Receptor for FGF19 – promotes proliferation |
+6.11 |
||
GAREM |
Promotes EGF-receptor proliferation |
+15.78 |
+4.96 |
|
GHR |
Promotes cellular growth |
-4.66 |
+2.28 |
|
#Grb10 |
Negative regulator of proliferation |
+7.11 |
-11.47 |
|
^IGF1R |
IGF1 growth factor receptor |
+2.69 |
-2.13 |
|
IL6ST |
Promotes cellular proliferation |
+2.77 |
+3.05 |
|
^IRS1 |
Involved in insulin/IGF signaling |
-2.14 |
|
|
^#MET |
Hepatocyte growth factor receptor |
+2.91 |
-3.73 |
|
NOTCH2 |
Promotes myoblast proliferation |
+2.38 |
|
|
SPHK2 |
Sphingosine kinase – promotes proliferation |
+2.16 |
|
|
Tgfbr1 |
Anti-proliferative effects |
|
+2.25 |
|
Gene name, function and fold change for each gene are listed; downregulated genes indicated by negative numbers, upregulated genes indicated by positive numbers. *Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3), ^indicates genes demonstrated to have PAX3-FOXO1 binding sites present in their proximal promoter [29], and #indicates genes that demonstrate similar trends in gene expression changes in human tumor samples [30-33].
Table 1e: Differentially expressed mRNA important for regulating proliferative control by altering genes for proliferative enzymes and miscellaneous proliferative proteins.
PROLIFERATIVE CONTROL |
||||
Gene |
Function |
V vs PF* |
P3 vs PF |
V vs P3 |
Enzymatic Activity |
||||
#ADAMTS1 |
Metalloproteinase – promotes proliferation |
+22.01 |
+3.63 |
+5.66 |
BRCA1 |
Tumor suppressor – inhibits proliferation |
-5.78 |
|
|
#DUSP1 |
Phosphatase – inhibits proliferation |
-3.61 |
-2.31 |
|
DUSP4 |
Phosphatase – inhibits proliferation |
-3.07 |
-2.43 |
|
DUSP10 |
Phosphatase – inhibits proliferation |
-2.20 |
||
^#DYRK |
Kinase – inhibits cellular proliferation |
+2.75 |
-3.34 |
|
PDIA4 |
Protein disulfide isomerase – IGFR recycler |
+2.73 |
||
PPP6C |
Phosphatase – restricts G1/S progression |
|
+2.57 |
|
^PRUNE |
Phosphodiesterase – promotes proliferation |
|
+3.32 |
-2.66 |
PTPRK |
Negative regulator of EGFR |
-7.89 |
|
|
TENC1 |
Negative regulator of Akt |
|
+2.45 |
|
#TIMP2 |
Inhibits cellular proliferation |
-2.68 |
|
|
Other |
||||
#CAV1 |
Antiproliferative – downregulates Cyclin Da |
|
+5.31 |
-2.95 |
CRLF3 |
Negative regulator of cell cycle progression |
|
-3.10 |
|
EPB41L3 |
Suppresses proliferation |
-6.32 |
-3.20 |
|
FOSL1 |
Promotes cellular proliferation |
-5.21 |
-4.69 |
|
Gpnmb |
Inhibits cellular proliferation |
-14.52 |
+3.32 |
|
#NPM1 |
Promotes cellular proliferation |
-2.31 |
|
|
PHF10 |
Chromatin remodeler – promotes proliferation |
+2.13 |
+2.99 |
|
SDC1 |
Promotes proliferation |
-2.43 |
-2.33 |
|
UBN1 |
Chromatin remodeler – promotes senescence |
|
+2.10 |
|
Gene name, function and fold change for each gene are listed; downregulated genes indicated by negative numbers, upregulated genes indicated by positive numbers. *Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3), ^indicates genes demonstrated to have PAX3-FOXO1 binding sites present in their proximal promoter [29], and #indicates genes that demonstrate similar trends in gene expression changes in human tumor samples [30-33].
In our miRNA deep sequencing analysis we found a total of 104 miRNAs whose expression changed in a PAX3-FOXO1-dependent manner, 61 of which are downregulated and 43 of which are upregulated (data not shown). Using miRTarBase [34] we found that out of these 104 total miRNA, 19 have validated target genes important for maintaining chromosome number and integrity (10 downregulated, 9 upregulated, Table 2), with validation on miRTarBase by at least two independent experimental methods. In addition to these 19 validated targets, we found using the TargetScan database that 22 additional miRNAs (17 downregulated, 5 upregulated, Table 2) are highly predicted to target genes important for chromosome number and integrity, based on their predicted efficacy of targeting (context score ≥ 85%) [35, 36] or probability of conserved targeting (PCT) [37] [PCT ≥ 0.8], as previously described [38], with several of the genes identified by TargetScan being validated targets in MirTarBase. Combined, the differentially expressed miRNAs target 56 additional genes that promote aneuploidy with many of these genes being targeted by multiple differentially expressed miRNA.
To determine if the PAX3-FOXO1-dependent mRNA and miRNA changes translate into differences in chromosome number and structure, we performed a cytogenetic analysis of proliferating primary myoblasts (Table 3A and Figure 1C) and found that the majority (48/62 cells - 77.4%) of cells transduced with empty vector contain the normal complement of 40 (2N) or 80 (4N) chromosomes, which is consistent with previous reports for the presence of diploidy and tetraploidy in proliferating myoblasts [39]. In a similar manner, a majority of cells (46/50 cells - 92%) ectopically expressing PAX3 contained the normal complement of chromosomes, demonstrating that the process of transduction and ectopic expression of protein does not affect chromosome number or structure. In contrast, nearly all of the cells stably expressing PAX3-FOXO1 (93/103 cells - 90.3%) had hypodiploid (<2N) or hyperdiploid/hypotetraploid (>2N to <4N) chromosome numbers. The numbers of chromosomes seen in individual cells, along with the variation of chromosome numbers between cells correlates will with results seen in ARMS primary tumor samples [11, 15-18].
Table 2a: Differentially expressed miRNA that target genes important for promoting aneuploidy – miRNA downregulated by PAX3-FOXO1 relative to empty vector.
PROMOTION OF ANEUPLOIDY |
|||||
miR |
Target |
Gene Function |
V vs. PF |
P3 vs. PF |
V vs. P3 |
*10a-5p |
HDAC4 |
Regulates chromosome segregation |
-333.33 |
-200.00 |
|
*1a-3p |
Calm1 Calm2 HDAC4 Cdc42 |
Regulates progression of cytokinesis Regulates progression of cytokinesis Regulates chromosome segregation Spindle microtubule attachment |
-18.50 |
-11.11 |
|
*376a-3p |
TTK |
Chromosome alignment at centromere |
-11.08 |
-12.67 |
|
*433-3p |
CEP135 |
Centrosomal protein |
-5.52 |
-3.97 |
|
543-3p |
SIRT1 |
Involved in chromosome maintenance |
-4.50 |
-3.31 |
|
*133a-3p |
Cdc42 HDAC4 |
Spindle microtubule attachment Regulates chromosome segregation |
-3.72 |
-3.62 |
|
148b-3p |
CDC25B |
Induces mitotic progression |
-3.44 |
-3.70 |
|
*19a-3p |
CEP350 MAPRE2 |
Anchors microtubules to centrosome Anchors microtubules to centrosome |
-2.87 |
-2.84 |
|
*351-5p |
NINL SUV39H1 |
Mitotic spindle assembly Methylase important for segregation |
-2.43 |
-2.12 |
|
3099-3p |
KIF3B |
Tethers chromosomes to spindle pole |
-2.36 |
-4.20 |
|
133b-5p |
Myh9 |
Important for cytokinesis |
-2.26 |
-2.89 |
|
504-5p |
CEP170 |
Centrosomal protein |
-2.09 |
-3.87 |
|
*335-5p |
CHFR d4 |
Regulates entry into mitosis Regulates chromosome stability |
-12.50 |
-37.04 |
+2.95 |
486-3p |
PTEN |
Chromosome stability |
-6.70 |
-13.20 |
+1.97 |
*128-3p |
NEK6 PARD6B PDS5B |
Required for chromosome segregation Involved in asymmetrical cell division Important for sister chromatid cohesion |
-2.17 |
-4.85 |
+2.32 |
*339-5p |
MAPRE1 |
Anchors microtubules at centrosome |
-1.93 |
-4.24 |
+2.19 |
*148a-3p |
CamK2 CDC25B CCNF PTEN RCC2 |
Spindle depolarization Induces mitotic progression Inhibitor of centrosome reduplication Chromosome stability Regulates chromosome condensation |
-4.41 |
-2.98 |
|
*29a-3p |
HDAC4 MCL1 Tubb2B KIF3B CEP68 PTEN CDC42BPA MAPRE2 |
Regulates chromosome segregation Inhibits BCL2 and apoptosis Component of microtubules Tethers chromosomes to spindle pole Centrosomal protein Chromosome stability Regulates CDC42 Anchors microtubules to centrosome |
-2.65 |
||
3968 |
ESCO2 |
Establishment of sister chromatid cohesion |
-2.53 |
||
*322-5p |
Arl2 CDC27 |
Regulates centrosome integrity Regulates mitotic progression |
-2.42 |
||
*9-5p |
SIRT1 CCNG1 CDC14B CEP350 |
Involved in chromosome maintenance Associated with G2/M arrest Controls exit of mitosis Centrosomal protein |
-5.75 |
+9.73 |
|
133b-3p |
Pitx3 |
Important for mitotic activity |
-4.83 |
+5.74 |
|
*486-5p |
PTEN |
Chromosome stability |
-2.80 |
+2.09 |
|
*206-5p |
HDAC4 |
Regulates chromosome stability |
-2.63 |
+2.31 |
|
The microRNA, target gene name, function, and fold change for each miRNA are listed; downregulated microRNA indicated by negative numbers, upregulated miRNA indicated by positive numbers. Genes and functions listed in normal font indicate validated targets; genes and functions listed in italics indicate genes highly predicted to be targets of the indicated microRNA. Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3) and the *indicates miRNA that target genes involved with the promotion of aneuploidy and in proliferative control.
Table 2b: Differentially expressed miRNA that target genes important for promoting aneuploidy – miRNA upregulated by PAX3-FOXO1 relative to empty vector.
PROMOTION OF ANEUPLOIDY |
|||||
miR |
Target |
Gene Function |
V vs. PF |
P3 vs. PF |
V vs. P3 |
615-3p |
MAPT |
Determines polarity of the centrosome |
+30.45 |
+6.08 |
+5.01 |
*31-3p |
RHOA |
Signaling protein important for cytokinesis |
+4.19 |
+2.04 |
+2.05 |
*196a-5p |
Hmga2 RCC2 |
Chromosome condensation – G2/M phase Regulator of chromosome condensation |
+24.39 |
+19.33 |
|
92a-1-5p |
TACC2 |
Organizes centrosomal microtubules |
+3.35 |
+3.55 |
|
*301a-3p |
MDM4 CENPO |
Regulates chromosome stability Necessary for chromosome segregation |
+3.07 |
+2.33 |
|
*16-5p |
Arl2 MDM4 G2E3 |
Regulates centrosome integrity Regulates chromosome stability Important for mitotic progression |
+2.07 |
+2.56 |
|
*222-3p |
PTEN |
Chromosome stability |
+2.78 |
+7.02 |
-2.52 |
*221-3p |
PTEN |
Chromosome stability |
+2.10 |
+7.02 |
-2.52 |
*30c-2-5p |
RCC2 CEP350 |
Regulator of chromosome condensation Anchors microtubules at centrosome |
+3.55 |
-2.32 |
|
20a-5p |
Mapk4 PTEN |
Important for cytokinesis Chromosome stability |
+2.01 |
||
*130b-3p |
MAP4 |
Important for chromosome segregation |
+3.39 |
+2.20 |
|
421-3p |
ARHGEF9 |
Regulates spindle microtubule attachment |
+3.22 |
+3.27 |
|
*206-3p |
Hdac4 Tppp CORO1C |
Regulates chromosome segregation Mitotic spindle assembly Potential role in cytokinesis |
+2.42 |
+4.49 |
|
*15b-5p |
Arl2 CDC25A |
Regulates centrosome integrity Induces mitotic progression |
+2.86 |
||
183-5p |
KIF2A |
Microtubule associated protein – mitosis |
+2.05 |
||
*Let-7g-5p |
HMGA2 |
Transcriptional regulator in mitosis |
+2.04 |
-2.15 |
|
The microRNA, target gene name, function, and fold change for each miRNA are listed; downregulated microRNA indicated by negative numbers, upregulated miRNA indicated by positive numbers. Genes and functions listed in normal font indicate validated targets; genes and functions listed in italics indicate genes highly predicted to be targets of the indicated microRNA. Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3) and the *indicates miRNA that target genes involved with the promotion of aneuploidy and in proliferative control.
Table 2c: Differentially expressed miRNA that target genes important for proliferative control – miRNA downregulated by PAX3-FOXO1 relative to empty vector.
PROLIFERATIVE CONTROL |
|||||
miR |
Target |
Gene Function |
V vs. PF |
P3 vs PF |
V vs P3 |
*10a-5p |
HDAC4 |
Histone deacetylase 4 – pro-proliferative |
-333.33 |
-200.00 |
|
143-3p |
Kras Ptn |
G-protein coupled receptor Secreted growth factor – mitogenic |
-22.22 |
-17.85 |
|
*1a-3p |
HDAC4 IGF1 Igf1R PDGFA TIMP3 |
Pro-proliferative Insulin like growth factor - proliferative Insulin like growth factor receptor Growth factor – promotes proliferation Inhibits cellular proliferation |
-18.50 |
-11.11 |
|
133a-5p |
INSR FGF1 FGFR1 |
Insulin receptor – proliferative Growth factor ligand – proliferative Growth factor ligand – proliferative |
-16.76 |
-12.67 |
|
*376a-3p |
CDK2 IGF1R |
Cyclin dependent kinase – G1/S Insulin like growth factor 1 receptor |
-11.08 |
-13.56 |
|
*433-3p |
GRB2 |
EGF-dependent proliferation |
-5.52 |
-3.97 |
|
*133a-3p |
Spry1 CCND2 Igf1R EGFR |
Antagonist of the FGF pathway Cyclin D2 – cell cycle regulation Insulin like growth factor 1 receptor Epidermal growth factor receptor |
-3.72 |
-3.62 |
|
362-3p |
CDKN1A Rb1 HBEGF |
Cyclin dependent kinase inhibitor (p57) Cell cycle regulatory protein Growth factor with EGFR and ERRB2 |
-3.67 |
-3.21 |
|
*19a-3p |
CDKN1A CCND1 MDM4 MAPK1 GRB10 IGFBP3 CCND2 |
Cyclin dependent kinase inhibitor (p57) Cyclin D1 – G1/S progression Promotes cellular proliferation Kinase – promotes proliferation Histone deacetylase 4 – proliferative Negative regulator of proliferation Interacts with and stabilizes IGF Cyclin D2 – promotes cell cycle |
-2.87 |
-2.84 |
|
*34b-5p |
E2F3 |
Promotes cell cycle progression |
-2.60 |
-2.43 |
|
*351-5p |
E2F2 |
Promotes cell cycle progression |
-2.43 |
-2.12 |
|
*335-5p |
Rb1 |
Tumor suppressor – cell cycle regulator |
-12.50 |
-37.04 |
+2.95 |
145a-5p |
Hoxa9 IRS1 Kras CCND2 |
Homeobox transcription factor Insulin signaling pathway Protooncogene – proliferative Cylcin D2 – cell cycle regulation |
-7.59 |
-14.83 |
+1.95 |
335-3p |
IGF1R |
IGF1 receptor – pro-proliferative |
-5.85 |
-15.87 |
+2.72 |
*128-3p |
Trim71 FoxP1 c-Met IRS1 SOS1 |
E3-ubiquitin ligase –G1/S transition Inhibits proliferation Hepatocyte growth factor receptor Involved with insulin signaling Promotes cellular proliferation |
-2.17 |
-4.85 |
+2.32 |
*339-5p |
Kdm6b |
Histone demethylase – pro-proliferative |
-1.93 |
-4.24 |
+2.19 |
*148a-3p |
Kdm6b Dnmt1 ERBB3 CDC25B CDKN1B E2F7 |
Histone demethylase – pro-proliferative DNA methyltransferase – pro-proliferative Human epidermal growth factor receptor Required for G2/M progression Inhibitor of G1 progression Promotes cell cycle progression |
-4.41 |
-2.98 |
|
*181c-5p |
KRAS E2F7 |
Promotes cell cycle progression Proto-oncogene – proliferative |
-2.34 |
-3.95 |
|
149-3p |
E2F1 MYBL2 |
Cell cycle regulatory transcription factor Proliferative transcription factor |
-2.34 |
+2.99 |
-7.04 |
*29a-3p |
CDK6 PDGFRB PDGFA FOXO3 PDGFB |
Promotes G1 progression Promotes proliferation Promotes proliferation Promotes proliferation Promotes proliferation |
-2.65 |
||
*322-5p |
CDC27 |
Promotes M-phase progression |
-2.42 |
||
340-5p |
MET |
Hepatocyte growth factor receptor |
-2.06 |
||
450a-5p |
DUSP10 |
Negative regulator of proliferation |
-2.03 |
||
*9-5p |
CDKN1A Hes1 FGF5 BRAF CDC25A IGFBP3 FOXO1 FOXO3 HDAC5 |
Cyclin dependent kinase inhibitor (p21) Promotes proliferation Growth factor – promotes proliferation Pro-proliferative kinase Cell Cycle regulatory protein Interacts with and stabilizes IGF Promotes cellular proliferation Promotes cellular proliferation Promotes cellular proliferation |
-5.75 |
+9.73 |
|
*486-5p |
FOXO1 |
Promotes cellular proliferation |
-2.80 |
+2.09 |
|
*206-5p |
HDAC4 |
Histone deacetylase 4 – pro-proliferative |
-2.63 |
+2.31 |
|
345-5p |
CDKN1A IGFBP5 |
Cyclin dependent kinase inhibitor (p21) Interacts with and stabilizes IGF |
-2.39 |
+3.09 |
|
23b-3p |
Hes1 MET |
Promotes proliferation Hepatocyte growth factor receptor |
-2.10 |
+2.19 |
|
The microRNA, target gene name, function, and fold change for each miRNA are listed; downregulated microRNA indicated by negative numbers, upregulated miRNA indicated by positive numbers. Genes and functions listed in normal font indicate validated targets; genes and functions listed in italics indicate genes highly predicted to be targets of the indicated microRNA. Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3) and the *indicates miRNA that target genes involved with the promotion of aneuploidy and in proliferative control.
Table 2d: Differentially expressed miRNA that target genes important for proliferative control – miRNA upregulated by PAX3-FOXO1 relative to empty vector.
PROLIFERATIVE CONTROL |
|||||
miR |
Target |
Gene Function |
V vs. PF |
P3 vs PF |
V vs P3 |
*31-3p |
ERBB2 E2F1 |
HER2 growth factor receptor Cell cycle regulatory transcription factor |
+4.19 |
+2.04 |
+2.05 |
*196a-5p |
CDKN1B HMGA2 ING5 |
Cyclin dependent kinase inhibitor Promotes myoblast proliferation Inhibitor of growth – p53 pathway |
+24.39 |
+19.33 |
|
*301a-3p |
E2F2 E2F7 ERBB4 JARID2 MAPK1 MDM4 PTEN |
Promotes cell cycle progression Promotes cell cycle progression HER4 – growth factor receptor Inhibits proliferation Promotes proliferation Promotes proliferation Inhibits proliferation |
+3.07 |
+2.33 |
|
*16-5p |
Wnt3a CCND1 Mdm4 Jun CCNE1 G2E3 FGF7 FGF2 |
Promotes proliferation Cylcin D1 – G1 progression Promotes proliferation Transcription factor Cyclin E1 – G1/S transition G2/M-specific E3 Ubiquitin ligase Growth factor – pro-proliferative Growth factor – pro-proliferative |
+2.07 |
+2.56 |
|
*222-3p |
CDKN1B |
Cyclin-dependent kinase inhibitor |
+2.78 |
+7.02 |
-2.52 |
*155-5p |
Jarid2 Myb FGF7 Fos |
Inhibits proliferation Regulates proliferation Fibroblast growth factor ligand Transcription factor |
+2.18 |
+5.54 |
-2.53 |
*221-3p |
CDKN1B |
Cyclin dependent kinase inhibitor (p57 |
+2.10 |
+6.97 |
-3.31 |
92b-5p |
CDKN1C |
Cyclin dependent kinase inhibitor (p16) |
+3.75 |
-2.77 |
|
*30c-2-5p |
HDAC4 MAPKBP1 ATF1 KRAS IRS2 IGF1R FOXO3 |
Promotes proliferation Promotes proliferation Promotes proliferation Proto-oncogene – proliferative Insulin signaling pathway Insulin receptor – proliferative Promotes proliferation |
+3.55 |
-2.32 |
|
181c-3p |
E2F7 |
Promotes cell cycle |
+2.74 |
-3.77 |
|
*30a-5p |
Egfr RUNX2 SOS IGF1R |
Growth factor receptor – proliferative Inhibits cellular proliferation Promotes cellular proliferation Growth factor receptor |
+2.44 |
-2.50 |
|
99a-5p |
FGFR3 IGF1R FOXO4 |
Growth factor receptor Growth factor receptor Proliferative transcription factor |
+2.05 |
-2.45 |
|
*Let-7g-5p |
Myc IGFBP1 CDKN2A |
Proliferative transcription factor Inhibits IGF-dependent proliferation Cyclin dependent kinase inhibitor (p16) |
+2.04 |
-2.15 |
|
*130b-3p |
MET MAPK1 JARID2 E2F2 E2F7 ERBB4 PTEN MDM4 |
Hepatocyte growth factor receptor Kinase – promotes proliferation Inhibits proliferation Promotes cell cycle progression Promotes cell cycle progression HER4 – growth factor receptor Inhibits proliferation Promotes proliferation |
+3.39 |
+2.20 |
|
*206-3p |
HDAC4 Spry1 Id1 TIMP3 |
Promotes proliferation Antagonist of the FGF pathway Promotes proliferation Inhibits proliferation |
+2.42 |
+4.49 |
|
*15b-5p |
CCNE2 CCND1 MYB |
Cyclin E – G1/S phase progression Cyclin D – G1/S phase progression Promotes proliferation |
+2.86 |
||
*30b-5p |
CCNE2 |
Cyclin E – G1/S phase progression |
+1.98 |
||
The microRNA, target gene name, function, and fold change for each miRNA are listed; downregulated microRNA indicated by negative numbers, upregulated miRNA indicated by positive numbers. Genes and functions listed in normal font indicate validated targets; genes and functions listed in italics indicate genes highly predicted to be targets of the indicated microRNA. Comparisons are made between empty vector and PAX3-FOXO1 (V vs. PF), PAX3 and PAX3-FOXO1 (P3 vs. PF) or empty vector and PAX3 (V vs. P3) and the *indicates miRNA that target genes involved with the promotion of aneuploidy and in proliferative control.
Further, we observed an increased number of disrupted chromosomal structures in cells stably expressing PAX3-FOXO1 relative to the negative control cells (vector and PAX3). We noted 16 cells (15.5%) with sister chromatid dissociation (Figure 1C, right panel, solid arrows), 17 cells (16.5%) with telomere association, and 4 cells (3.9%) with double minutes (Table 1B) with only two of these cells having the presence of both sister chromatid dissociation and telomere association. Although some chromosomal disruptions were observed in the control cells, these events were minimal (incidence <4%) and did not include the presence of double minutes (Table 3B).
We previously published that PAX3-FOXO1 is phosphorylated at three independent sites [27, 28, 40] and phosphorylation contributes to ARMS oncogenic phenotypes in an in vitro cellular model [26]. To determine how phosphorylation at these sites affects PAX3-FOXO1-dependent changes in chromosome number and structure, we utilized mutants in which each individual site was mutated to a phospho-incompetent alanine (S201A, S205A, or S209A) [26]. A Western blot analysis of mouse primary myoblasts stably transduced with these mutants demonstrates that all mutants were expressed to levels equivalent to that of wild-type PAX3-FOXO1 (Figure 1B).
Cytogenetic analysis demonstrated that similar to wild-type PAX3-FOXO1, a majority of cells stably expressing S201A [57/64 cells - 89.1%], S205A [62/69 cells - 89.8%], and S209A [55/57 cells - 96.5%] had chromosome numbers <2N or >2N to <4N with an increase in the number of hypertetraploid cells (>4N) for all three samples (Table 3A). We also observed similar chromosomal abnormalities for each of the phopsho-incompetent mutants relative to wild-type PAX3-FOXO1 (Figure 1D); however, the individual phosphorylation events seem to impact these chromosomal aberrations differently (Table 3B). While all three mutants have a similar percentage of cells containing sister chromatid dissociation relative to wild-type PAX3-FOXO1, the expression of S209A increased the number of individual events within each cell, with some cells having over 60 dissociations. Although both S201A and S205A increased the percentage of cells with telomere association, loss of phosphorylation at S205 or S209 decreased the number of individual events within each cell relative to PAX3-FOXO1. Finally, double minutes were present in a similar percentage of cells expressing the fusion protein as either wild type or mutant. However, cells stably expressing the S205A mutant had an increased number of double minutes per cell relative to the wild-type fusion protein. Taken together, these results are the first to demonstrate that the presence of an oncogenic fusion protein, PAX3-FOXO1, is sufficient to promote aneuploidy and disrupt chromosome structure and that phosphorylation contributes to this state.
Table 3A: Quantification of PAX3-FOXO1-dependent aneuploidy on cells stably transduced with empty vector (Vector), PAX3, PAX3-FOXO1, or the PAX3-FOXO1 phosphomutants (S201A, S205A, and S209A). .
Sample |
Chromosome Number |
|||||
Hypodiploid |
Diploid |
Hyperdiploid/ Hypotetraploid |
Tetraploid |
Hypertetraploid |
||
Number cells analyzed |
<2N (<40) |
2N (40) |
>2N to <4N (41 – 79) |
4N (80) |
>4N (>80) |
|
Vector |
62 |
7 (11.3%) |
25 (40.3%) |
7 (11.3%) |
23 (37.1%) |
0 (0.0%) |
PAX3 |
50 |
2 (4.0%) |
34 (68.0%) |
2 (4.0%) |
12 (24.0%) |
0 (0.0%) |
PAX3-FOXO1 |
103 |
51 (49.5%) |
6 (5.8%) |
42 (40.8%) |
4 (3.9 %) |
0 (0.0%) |
S201A |
64 |
29 (45.3%) |
5 (7.8%) |
27 (42.2%) |
2 (3.1%) |
1 (1.5%) |
S205A |
69 |
13 (18.8%) |
3 (4.3%) |
45 (65.2%) |
4 (5.8%) |
4 (5.8%) |
S209A |
57 |
18 (31.6%) |
2 (3.5%) |
29 (50.8%) |
0 (0.0%) |
8 (14.0%) |
The chromosome number for individual cells was determined and classified as hypodiploid (<2N), diploid (2N), hyperdiploid/hypotetrapoid (>2N to <4N), tetraploid (4N) or hypertetraploid (>4N)
Table 3B: Quantification of PAX3-FOXO1-dependent chromosomal structural abnormalities.
Sample |
Number cells analyzed |
Sister Chromatid Dissociation |
Telomere Association |
Double Minutes |
|||
Number of Cells |
Range of Events/Cell |
Number of Cells |
Range of Events/Cell |
Number of Cells |
Range of Events/Cell |
||
Vector |
62 |
1 (1.6%) |
2 |
2 (3.2%) |
2 |
0 (0.0%) |
0 |
PAX3 |
50 |
2 (4.0%) |
2 |
2 (4.0%) |
2 |
0 (0.0%) |
0 |
PAX3-FOXO1 |
103 |
16 (15.5%) |
1 – 5 |
17 (16.5%) |
1 – 16 |
4 (3.9%) |
5 – 20 |
S201A |
64 |
6 (9.4%) |
2 – 6 |
21 (32.8%) |
2 – 18 |
4 (6.3%) |
6 – 12 |
S205A |
69 |
9 (13.0%) |
1 – 3 |
20 (28.9%) |
1 – 4 |
5 (7.2%) |
1 – 62 |
S209A |
57 |
7 (12.3%) |
5 – 63 |
6 (10.5%) |
1 – 3 |
4 (7.0%) |
5 – 40 |
Metaphase chromosome analysis on cells stably transduced with empty vector (Vector), PAX3, PAX3-FOXO1, or the PAX3-FOXO1 phosphomutants (S201A, S205A, or S209A). The number of cells containing the indicated structural abnormality along with a range of the number of occurrences of the abnormality within each cell is listed.
PAX3-FOXO1 overrides cell cycle inhibition to enhance cellular proliferation
One of the initial cellular responses to aneuploidy is an attenuation of proliferation [41]. An examination of our comparative transcriptomic analysis revealed that of the 846 differentially expressed mRNA, 93 (nearly 11%) have biological functions important for cellular proliferation (Table 1), twelve of which have previously described PAX3-FOXO1 binding sites in their proximal promoter (Table 1, carat) [29], and 30 had altered gene expression levels consistent with changes seen in human tumor samples [30-33] (Table 1, pound sign). We also found 13 of the downregulated and 9 of the upregulated miRNAs are experimentally validated on miRTarBase to target mRNA whose biological function is important for proliferative control. Further, 15 of the downregulated and 9 of the upregulated miRNAs are highly predicted to target growth regulatory genes (Table 2), based on their predicted efficacy of targeting or probability of conserved targeting, as described above.
Consistent with the predicted cellular response to the aneuploid state, we found that of the 93 alternatively expressed mRNA, 23 are cell cycle regulatory genes and include decreases in cyclins and their related cyclin dependent kinases and increases in the expression of cyclin dependent kinase inhibitors (Table 1). Further, we found 11 of the downregulated and 12 of the upregulated miRNAs also affect cell cycle regulatory proteins in a manner consistent with the attenuation of growth. To determine how these changes translate into effects on cellular growth, we determined doubling times of primary myoblasts stably transduced with empty vector, PAX3, or PAX3-FOXO1. We found the doubling time of primary myoblasts transduced with empty vector to be approximately 35 hours and that the stable expression of PAX3 enhanced the growth rate by reducing the doubling time to approximately 20 hours (Figure 2). Primary myoblasts stably expressing PAX3-FOXO1 also had a reduced doubling time of approximately 20 hours (Figure 2), a result that seems to be in contrast to the presence of aneuploidy in these cells and the observed changes in cell cycle regulatory mRNA and miRNA. Further, we determined proliferation rates on primary myoblasts stably expressing the PAX3-FOXO1 phospho-mutants. We found that although cells expressing S201A have an enhanced proliferation rate relative to the negative control, the rate is significantly slower than for cells stably expressing wild-type PAX3-FOXO1 (Figure 2). The stable expression of S205A and S209A resulted in proliferation rates that were indistinguishable from the empty vector negative control.
To better understand this result, we examined our mRNA and miRNA comparative transcriptomic analyses for alterations in mRNA and miRNAs whose biological functions may affect growth independent of cell cycle regulatory proteins. We found that the expression of PAX3-FOXO1 is sufficient to alter the mRNA for multiple growth factors (14 genes) and growth factor receptors (18 genes) in a manner that is in direct alignment with our observed increased proliferation rate. These include the growth factor receptors c-MET [22], and IGF1R [20] (both direct targets of PAX3-FOXO1), FGFR4, Erbb3 (HER3), IL6ST, and the receptor NOTCH2, which in certain cell types enhances proliferation. We also found the increased expression of many of the associated growth factors including IGF2, IGFBP3, and IGFBP5 and several proliferative transcription factors (17 genes), including CREB3, FOXO4, HOXB9, and Myc (Table 1). Further, most of the miRNA have validated or predicted targets that are growth factor receptors, growth factors, or proliferative transcription factors (Table 2). Interestingly, 18 of the downregulated and 13 of the upregulated miRNAs target genes important for both aneuploidy and the regulation of proliferative control. Taken together, these results demonstrate that the expression of an oncogene is sufficient to override aneuploid-dependent attenuation of growth by globally altering the expression of growth factor related mRNA and miRNA.
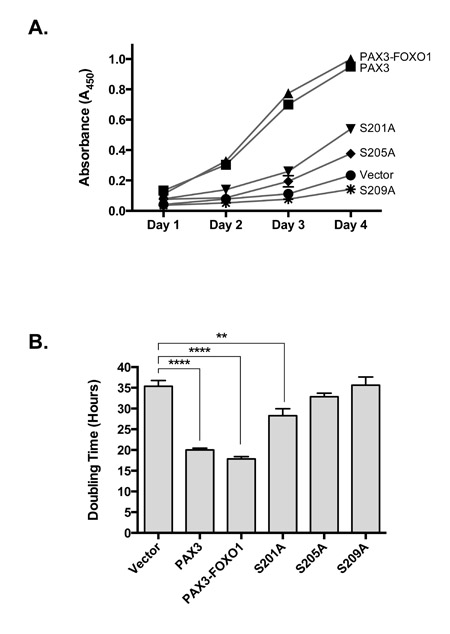
Figure 2: Stably transduced primary myoblasts (vector, PAX3, wild-type PAX3-FOXO1, or PAX3-FOXO1 phosphomutant) were plated, grown for four days, growth determined using the CCK-8 cell counting kit and plotted as a function of time (A) from which doubling times were determined, as described in the Materials and Methods. Error bars represent the standard deviation from three independent determinations and P-values were computed using non-parametric two-way ANOVA analyses comparing each treatment condition to the empty vector negative control. (**P = 0.005, ****P < 0.0001).
Phosphorylation contributes to PAX3-FOXO1-dependent differential gene expression
To determine how phosphorylation of PAX3-FOXO1 affects the expression of genes important for maintaining chromosome number and structure, we performed a qRT-PCR analysis on a subset of genes with fusion protein-dependent altered expression. We tested genes in chromosome segregation (AurkA), chromosome cohesion (BUB1, Cep250, and Sgol1), and cytokinesis (COROC1, Myh10, and Prc1). We observed changes in gene expression consistent with our mRNA deep sequencing results, in which the presence of PAX3-FOXO1 promotes a significant decrease in the expression of all seven genes (Figure 3A). Further, with the exception of Myh10 and Cep250, we found inhibiting the phosphorylation of PAX3-FOXO1 at Ser201 or Ser205 corrected these decreases, with gene expression levels equivalent to the empty vector or PAX3 controls (Figure 3A). However, loss of phosphorylation at Ser209 was unable to correct these decreases and in fact promoted a further 2-fold decrease in the expression of Prc1.
A qRT-PCR analysis on a subset of proliferation genes, including cell cycle regulatory genes (CDKN1C, CDK1, and Cyclin D1) and growth factor genes (Erbb3, IGF2, and IGFBP5) once again showed PAX3-FOXO1-dependent changes consistent with our mRNA deep sequencing results (Figure 3B). We found the stable expression of the phospho-incompetent mutants also increased (CDKN1C) or decreased (CDK1 and Cyclin D1) cell cycle regulatory gene expression relative to the negative control. However, with the exception of CDK1 in cells expressing the S209A mutant, these changes were much less severe relative to wild-type PAX3-FOXO1. With respect to growth factor related genes, we found that the S209A mutant reduced the expression of all three genes to levels indistinguishable from the negative control. Further, the expression of S201A and S205A also altered the expression of IGF2 and IGFBP5 that trended toward background levels, but had little effect on the expression of Erbb3.
We performed a similar quantitative analysis on a select set of miRNAs, chosen for their ability to target genes important for the promotion of aneuploidy and for the regulation of proliferation. We found changes in miRNA expression consistent with our deep sequencing results (Figure 3C). As with our mRNA results, we found that inhibiting phosphorylation of PAX3-FOXO1 at Ser201 or Ser205 returned miRNA expression levels to that of the empty vector or PAX3 controls (Figure 3C). While loss of phosphorylation at Ser209 was able to correct the altered miRNA expression for miR30c-2-5p, S209A had no effect on the altered expression of miR130b-3p. Interestingly, the expression of S209A resulted in a 2- or 4-fold increase in the expression of miR-196a-5p and miR301a-3p, respectively, relative to wild-type PAX3-FOXO1 (Figure 3C). Taken together, our results demonstrate that phosphorylation contributes to the altered expression of genes and miRNAs important for the PAX3-FOXO1-dependent promotion of aneuploidy, aberrant chromosome structure, and regulation of growth.
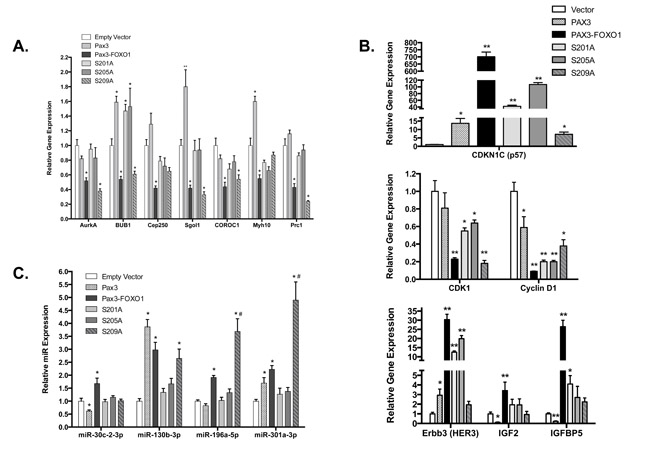
Figure 3: Quantitative RT-PCR analysis for aneuploidy mRNA (A), proliferation mRNA (B) and miRNA (C). Total RNA was isolated from stably transduced cells [empty vector (white), PAX3 (stippled), PAX3-FOXO1 (black), or the phosphomutants S201A (light gray), S205A (medium gray), or S209A (hashed gray). Quantitative RT-PCR was performed using primers specific for the indicated mRNA A. and B. or microRNA C., as described in the Materials and Methods. Error bars represent the standard deviation from three independent determinations and P-values were computed using non-parametric two-way ANOVA analyses. The asterisk indicates statistical comparisons between empty vector and each sample (*P = 0.001, **P < 0.0001). The hash-tap indicates comparisons between wild-type PAX3-FOXO1 and the sample (#P = 0.001).
DISCUSSION
Aneuploidy is common in solid tumors, which given the enhanced proliferative activity of transformed or malignant cells, suggests that these cells have acquired mechanisms to overcome proliferative defects associated with this state. In this report we demonstrate for the first time that the presence of an oncogenic protein, PAX3-FOXO1, is sufficient to serve as a driving mutation to initiate a cascade of changes in mRNA and miRNA whose biological functions are important for maintaining proper chromosome number and structure and proliferative control (Tables 1 and 2). We show that these changes translate into biological effects, since cells expressing PAX3-FOXO1 are primarily aneuploid (Table 3A), have altered chromosome structure (Table 3B), and have compensated to override the anti-proliferative responses to aneuploidy in physiologically relevant primary myoblasts (Figure 2). Consistent with our previous work [26], we demonstrate that inhibiting PAX3-FOXO1 phosphorylation reverts many of the gene expression changes to background levels (Figure 3), which translate into biological effects by altering the extent of chromosome structural abnormalities (Figure 1 and Table 3B) and enhanced proliferation rates (Figure 2). Given that these changes result explicitly from the expression and phosphorylation of PAX3-FOXO1, we conclude that the acquisition of the oncogene is the initiating driver mutation in the development of ARMS.
Because the aneuploid state is detrimental to a cell, growth retardation is one of the first cellular responses [42, 43]. Therefore, in order for muscle cells to develop into ARMS, the cells containing the fusion protein must overcome this proliferative defect. Consistent with the cell’s normal response to the aneuploid state, we saw changes in cell cycle regulatory genes that would be expected to inhibit cellular proliferation. However, we also found that the presence of the fusion protein enhances proliferation, which seems in direct contrast to altered cell cycle regulatory genes. This apparent contradiction can be explained by the fact that we also observed significant increases in the expression of growth factor related genes and proliferative transcription factors, several of which are known direct targets of PAX3-FOXO1 [20, 44]. Taken together, these results support the idea that the oncogenic fusion protein serves a second role as a driving mutation: it promotes the increased expression of growth factor related genes, either directly or indirectly through downstream effects, that are capable of negating the inhibitory changes in cell cycle regulatory proteins to override the growth retarding effects of the aneuploid state.
Based on the evidence presented here, we propose the following model by which PAX3-FOXO1 serves as the initial driving mutation in the development of ARMS (Figure 4). In this model myogenic cells in situ randomly and somatically acquire the t(2;13)(q35;q14) translocation, an event which is mimicked in our in vitro model system through the stable transduction of primary myoblasts. The subsequent expression of PAX3-FOXO1, maintained over repeated cellular duplications either in situ or in vitro, will bind to select promoters and regulatory sequences, thereby directly altering the expression of a subset of genes (Table 1, carats). These alterations could then initiate a cascade of indirect downstream events resulting in a global alteration of gene regulatory networks that ultimately reprogram proliferating myoblasts. This reprogramming results in the disruption of multiple aspects that lead to aneuploidy, including maintenance of the segregation machinery, regulation of chromosome segregation, promotion of chromosome cohesion and condensation, and insuring proper progression through mitosis.
In response to the aneuploid state, the cells attempt to halt proliferation, which is evidenced through our observed changes in the expression of cell cycle regulatory genes and miRNAs, and which most likely result from the aneuploid state and not from direct involvement of the fusion protein. However, PAX3-FOXO1 directly induces the expression of multiple and redundant proliferative genes, including growth factors, growth factor receptors, and proliferative transcription factors (Table 2, carats), thereby overriding aneuploidy-induced proliferation defects. Finally, the cells must be able to acquire a tolerance to the aneuploid state, most likely by affecting genes important in p53 regulation, in order to promote ARMS progression.
We reported that PAX3-FOXO1 is phosphorylated at Ser201 by GSK3β and small molecule inhibitors of this enzyme not only reduce phosphorylation at this site but also attenuate ARMS tumor phenotypes [26]. Consistent with this work, and adding to our present model, we found that phosphorylation contributes to the PAX3-FOXO1-dependent changes in gene and miRNA expression leading to the acquisition and persistence of aneuploidy. We found that although inhibiting phosphorylation of PAX3-FOXO1 at Ser201 and Ser205 did not affect aneuploidy, it altered the severity of chromosomal structural abnormalities (Figure 1 and Table 3B) and blocked the enhanced proliferation of primary myoblasts (Figure 2). We found phosphorylation contributes to the altered expression of multiple genes and miRNAs, which was evidenced by the reversion of gene expression to levels equivalent to the negative control in cells stably expressing phospho-incompetent mutants (Figure 3). Therefore, our results provide additional evidence to support the role post-translational modifications make in regulating the contributions of an oncogene to the development of a solid tumor. Further, these results identify this event, the specific phosphorylation of PAX3-FOXO1, as a viable biological target for therapy development (Figure 4). Interestingly, inhibition of phosphorylation at Ser209 enhanced the severity of sister chromatid dissociation (Figure 1D and Table 3B), and had proliferation rates indistinguishable from the negative control (Figure 1). Although we previously reported undetectable levels of phosphorylation of PAX3-FOXO1 at Ser209 in this system [28], the results presented here would suggest that this event does occur at greatly reduced levels and below the limits of detection.
The results presented here are important for directing the development of new therapies for the treatment of ARMS (Figure 4) and provide implications for how others may approach the development of therapies for solid tumors. Many present experimental therapies for ARMS, and other solid tumors, focus on inhibiting a single gene or a single pathway genetically located downstream of the oncogene. However, these therapies have not proven effective in Phase I or Phase II clinical trials for ARMS [45-48]. These outcomes are not surprising given the global nature of mRNA and miRNA alterations seen in cells that express PAX3-FOXO1. As such, therapies targeting a single gene product or pathway would be expected to have limited efficacy since multiple genes with similar biological functions could easily compensate.
Therefore, we propose the development of a multi-faceted regimen that targets genetic or biological characteristics identified from our work. We published that small molecules such as LiCl attenuate ARMS tumor phenotypes [26]. Others demonstrated that compounds that interfere with growth factor dependent proliferation, such as Cixutumumab [48], or the survival of aneuploid cells, such as chloroquine [49], might be effective as new treatments for human solid tumors. Therefore, we propose a combination therapy that minimally utilizes these three drugs for the treatment of ARMS. This regimen is expected to target both the driver mutation, with LiCl or other GSK3beta inhibitors preventing the phosphorylation of PAX3-FOXO1 at Ser201 [26] thereby inhibiting altered gene expression and the biological consequences of this driver mutation, with chloroquine killing aneuploid cells while Cixutumumab removes the growth-factor-dependent proliferative advantage. This regimen is expected to be specific for ARMS tumor cells, since all targets derive from the presence of the unique genetic mutation, have minimal negative effects since all proposed drugs are proven safe in human subjects, and would inhibit three molecular events essential for the viability of ARMS tumor cells.
Finally, the paradigm created here has implications that may be applied to other tumor models. Studies are being published with more frequency that utilize deep-sequencing strategies to examine global gene and miRNA expression changes resulting from the presence of an oncogene or the process of malignant transformation for examples see [50-53]. Often a single gene, miRNA, or miRNA locus is selected from these global studies for more in depth investigation to utilize as a specific target for therapy development. However, given that our results demonstrate that individual tumor phenotypes (e.g., induction of aneuploidy) arise from alterations in multiple biological aspects of this phenotype (e.g., mitotic progression, segregation machinery, etc.), it would be expected that a cell would be capable of compensating for the therapeutic inhibition of a single gene or miRNA. Therefore, as illustrated in the present study, deep sequencing studies should be utilized to discover how global gene regulatory alterations affect biological phenotypes and then use this knowledge to develop a multi-faceted therapeutic approach to target specific aspects of multiple different tumor phenotypes that are essential for the progression of the disease.
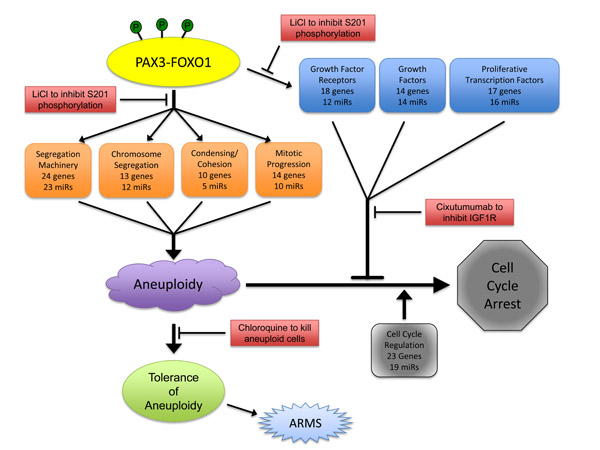
Figure 4: Model describing the role of PAX3-FOXO1 in the development of ARMS and how it informs potential therapy development.
MATERIALS AND METHODS
Primary cells and culture conditions
Mouse primary myoblasts were isolated from 2 - 4 day old C57/Bl6 mice as previously described [40]. Cells were grown as previously described [25-28, 40] and were passage-matched to prevent possible differences due to passage conditions.
Stable transduction of primary myoblasts
Mouse primary myoblasts were stably transduced as previously described [24, 40] with the MSCV-IRES-puromycin empty vector, vector containing FLAG epitope-tagged Pax3 (FLAG-Pax3), FLAG-PAX3-FOXO1, or FLAG-PAX3-FOXO1 in which each of the previously identified phosphorylation sites [27, 28] were mutated to the phospho-incompetent alanine (S201A, S205A, or S209A) [27, 28]. Three days post-transduction, cells were selected using puromycin, as previously described [28]. The stably transduced cells were harvested and pooled from three independent transductions to create a single population that express each construct.
Western blot analysis
Stably transduced cells were grown to 80% confluency, harvested, and total cell extracts made, as previously described [26-28, 40]. Equal amounts of total cell lysates (12μg) were separated by 8% SDS-PAGE and analyzed by Western blot analysis using antibodies specific for Pax3 [54] as previously described [27, 28].
Metaphase chromosome analysis
Transduced primary myoblasts were grown until logarithmic phase growth (approximately 70 - 80% confluency). The cells were then treated with Colcemeid (100ng/ml), harvested, and prepared for metaphase chromosome analysis, as previously described [55]. Metaphase images were captured using an Applied Imaging Model ER-3339 cooled CCD camera (Applied Spectral Imaging) mounted on top of a Nikon Eclipse E400 with CytoVision version 3.1 image-capture software (Applied Spectral Imaging).
RNA extraction, cDNA library construction, and cDNA deep sequencing
Primary myoblasts stably expressing empty vector, FLAG-PAX3, or FLAG-PAX3-FOXO1 were grown to approximately 80% confluency. Total RNA was isolated using the miRNeasy mini kit (Qiagen), allowing for the isolation of RNA <30 bp in length, according to the manufacturer’s specifications. Poly-A+ mRNA or miRNA were isolated from 4μg total RNA, to generate the respective cDNA libraries, both using the Illumina sample preparation kits according to the manufacturer’s specifications. The cDNA libraries were provided a unique index identifier, allowing the clustering of several samples into a single sequencing lane, and deep sequencing analyses were performed in triplicate from three independent cell growth, RNA isolation, and cDNA library constructions.
mRNA-seq data analysis
The raw data was groomed and trimmed for quality of read using online Galaxy analysis (https://usegalaxy.org), resulting in 40 - 41 high quality base pair reads for each sequence with between 4 - 6 million independent reads for each sample. The sequences were mapped to the mouse genome using Tophat analysis, transcripts were assembled using the Cufflinks program, and individual replicates were merged into a single file using Cuffmerge. The resulting transcript reads were normalized using Fragments Per Kilobase of transcript per Million mapped reads (FPKM) analysis, which normalizes each identified sequence for the length of the identified transcript and the volume of the total read yield from each run. Differential expression was determined from these normalized values using the Cuffdiff program, which not only compares differential expression of the merged files between sets but also utilizes the sequence results from the three independent determinations within each set to assign statistical significance to the differential expression.
miRNA-seq data analysis
Raw fastq sequences were obtained from the Illumina Genome Analyzer II using the “Demultiplex” algorithm in the CASAVA 1.8.2 software (Illumina) that allows the identification of individual samples by “index sequences” contained within the adapters and introduced during the adapter ligation and amplification of the samples. miRNAKey, a software package used for analyzing small RNA data obtained from deep sequencing experiments, was used for data analysis at default settings. The pipeline clips the Illumina 3’ adaptor sequences from the reads, maps the clipped reads to miRBase and uses the Seq-EM algorithm to estimate the distribution of multiply mapped reads across the observed miRNAs. Sequences less than 16 bases after adaptor clipping were removed. The read counts obtained were then used for differential expression analysis between control and experimental samples using EBSeq from the R package with a False Discovery Rate (FDR) of 5%. We used the default ‘Median Normalization’ in EBSeq to make the counts comparable across samples.
Proliferation assay
The proliferation rate of cells stably expressing empty vector, FLAG-PAX3, FLAG-PAX3-FOXO1, or the phospho-incompetent mutants was assessed using the CCK-8 colorimetric assay kit, according to the manufacturer’s specifications (Cell Counting Kit-8, Dojindo Molecular Technologies) and as previously described [26].
qRT-PCR
Total RNA was extracted from cells stably expressing empty vector, FLAG-PAX3, FLAG-PAX3-FOXO1, or the FLAG-PAX3-FOXO1 phospho-mutants. Equal amounts of isolated RNA (100ng) were used for cDNA synthesis using the iScript cDNA synthesis kit (Bio-Rad) for mRNA and the Taqman miRNA reverse transcription kit (Applied Biosystems) for miRNA. The qRT-PCR was performed on the resulting cDNA using the CFX96 Touch™ Real-Time PCR Detection System using commercially available primer/probe sets and the Applied Biosystems Universal Master Mix, according to the manufacturer’s specifications. All results were normalized for GAPDH (mRNA) or the U6 small nuclear RNA for miRNA and reported as fold expression relative to the results obtained for cells stably transduced with the empty vector.
ACKNOWLEDGMENTS
All deep sequencing was performed in the LCRC Translational Genomics Core facility.
CONFLICTs OF INTEREST
The authors have no conflicts of interest related to the work described in this manuscript.
FINANCIAL SUPPORT
Funding for this project is from the National Cancer Institute grant R01CA138656, the Louisiana State University School of Medicine Research Enhancement Bridge Grant, and the Louisiana Cancer Research Consortium (LCRC) (ADH). JZ has been partially supported by grants from the National Institute of General Medicine Sciences (NIGMS) grants P20GM103501, subproject #2, P30GM114732, and U54GM104940-01, and the National Institute on Minority Health and Health Disparities (NIMHD) grants P20MD004817, and U54MD006176-01.
REFERENCES
1. Weaver BA and Cleveland DW. Does aneuploidy cause cancer? Curr Opin Cell Biol. 2006; 18:658-667.
2. Gonzalez-Loyola A, Fernandez-Miranda G, Trakala M, Partida D, Samejima K, Ogawa H, Canamero M, de Martino A, Martinez-Ramirez A, de Carcer G, Perez de Castro I, Earnshaw WC and Malumbres M. Aurora B overexpression causes aneuploidy and p21Cip1 repression during tumor development. Mol Cell Biol. 2015.
3. Perez de Castro I and Malumbres M. Mitotic Stress and Chromosomal Instability in Cancer: The Case for TPX2. Genes Cancer. 2012; 3:721-730. doi: 10.1177/1947601912473306.
4. Zhang N, Ge G, Meyer R, Sethi S, Basu D, Pradhan S, Zhao YJ, Li XN, Cai WW, El-Naggar AK, Baladandayuthapani V, Kittrell FS, Rao PH, Medina D and Pati D. Overexpression of Separase induces aneuploidy and mammary tumorigenesis. Proc Natl Acad Sci U S A. 2008; 105:13033-13038.
5. Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW and Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007; 11:9-23.
6. Cui Y, Borysova MK, Johnson JO and Guadagno TM. Oncogenic B-Raf(V600E) induces spindle abnormalities, supernumerary centrosomes, and aneuploidy in human melanocytic cells. Cancer Res. 2010; 70:675-684.
7. De Keersmaecker K, Real PJ, Gatta GD, Palomero T, Sulis ML, Tosello V, Van Vlierberghe P, Barnes K, Castillo M, Sole X, Hadler M, Lenz J, Aplan PD, et al. The TLX1 oncogene drives aneuploidy in T cell transformation. Nature medicine. 2010; 16:1321-1327.
8. Thompson SL and Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010; 188:369-381.
9. Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE and Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008; 322:703-709.
10. Santaguida S and Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nat Rev Mol Cell Biol. 2015; 16:473-485.
11. Goldstein M, Meller I, Issakov J and Orr-Urtreger A. Novel genes implicated in embryonal, alveolar, and pleomorphic rhabdomyosarcoma: a cytogenetic and molecular analysis of primary tumors. Neoplasia. 2006; 8:332-343.
12. Horn RC, Jr. and Enterline HT. Rhabdomyosarcoma: a clinicopathological study and classification of 39 cases. Cancer. 1958; 11:181-199.
13. Shapiro DN, Sublett JE, Li B, Downing JR and Naeve CW. Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res. 1993; 53:5108-5112.
14. Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ, 3rd, Emanuel BS, Rovera G and Barr FG. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993; 5:230-235.
15. Chen Z, Coffin CM, Smith LM, Issa B, Arndt S, Shepard R, Brothman L, Stratton J, Brothman AR and Zhou H. Cytogenetic-clinicopathologic correlations in rhabdomyosarcoma: a report of five cases. Cancer Genet Cytogenet. 2001; 131:31-36.
16. Kullendorff CM, Donner M, Mertens F and Mandahl N. Chromosomal aberrations in a consecutive series of childhood rhabdomyosarcoma. Med Pediatr Oncol. 1998; 30:156-159.
17. Gordon T, McManus A, Anderson J, Min T, Swansbury J, Pritchard-Jones K, Shipley J, United kingdom Children’s Cancer Study G and United Kingdom Cancer Cytogenetics G. Cytogenetic abnormalities in 42 rhabdomyosarcoma: a United Kingdom Cancer Cytogenetics Group Study. Med Pediatr Oncol. 2001; 36:259-267.
18. Gil-Benso R, Lopez-Gines C, Carda C, Lopez-Guerrero JA, Ferrer J, Pellin-Perez A and Llombart-Bosch A. Cytogenetic and molecular findings related to rhabdomyosarcoma. An analysis of seven cases. Cancer Genet Cytogenet. 2003; 144:125-133.
19. Fredericks WJ, Galili N, Mukhopadhyay S, Rovera G, Bennicelli J, Barr FG and Rauscher FJ, 3rd. The PAX3-FKHR fusion protein created by the t(2;13) translocation in alveolar rhabdomyosarcomas is a more potent transcriptional activator than PAX3. Mol Cell Biol. 1995; 15:1522-1535.
20. Ayalon D, Glaser T and Werner H. Transcriptional regulation of IGF-I receptor gene expression by the PAX3-FKHR oncoprotein. Growth Horm IGF Res. 2001; 11:289-297.
21. Epstein JA, Song B, Lakkis M and Wang C. Tumor-specific PAX3-FKHR transcription factor, but not PAX3, activates the platelet-derived growth factor alpha receptor. Mol Cell Biol. 1998; 18:4118-4130.
22. Ginsberg JP, Davis RJ, Bennicelli JL, Nauta LE and Barr FG. Up-regulation of MET but not neural cell adhesion molecule expression by the PAX3-FKHR fusion protein in alveolar rhabdomyosarcoma. Cancer Res. 1998; 58:3542-3546.
23. Roeb W, Boyer A, Cavenee WK and Arden KC. PAX3-FOXO1 controls expression of the p57Kip2 cell-cycle regulator through degradation of EGR1. Proc Natl Acad Sci U S A. 2007; 104:18085-18090.
24. Hollenbach AD, Sublett JE, McPherson CJ and Grosveld G. The Pax3-FKHR oncoprotein is unresponsive to the Pax3-associated repressor hDaxx. Embo J. 1999; 18:3702-3711.
25. Miller PJ and Hollenbach AD. The oncogenic fusion protein Pax3-FKHR has a greater post-translational stability relative to Pax3 during early myogenesis. Biochim Biophys Acta. 2007; 1770:1450-1458.
26. Loupe JM, Miller PJ, Ruffin DR, Stark MW and Hollenbach AD. Inhibiting phosphorylation of the oncogenic PAX3-FOXO1 reduces alveolar rhabdomyosarcoma phenotypes identifying novel therapy options. Oncogenesis. 2015; 4:e145.
27. Dietz KN, Miller PJ and Hollenbach AD. Phosphorylation of serine 205 by the protein kinase CK2 persists on Pax3-FOXO1, but not Pax3, throughout early myogenic differentiation. Biochemistry. 2009; 48:11786-11795.
28. Dietz KN, Miller PJ, Iyengar AS, Loupe JM and Hollenbach AD. Identification of serines 201 and 209 as sites of Pax3 phosphorylation and the altered phosphorylation status of Pax3-FOXO1 during early myogenic differentiation. Int J Biochem Cell Biol. 2011; 43:936-945.
29. Cao L, Yu Y, Bilke S, Walker RL, Mayeenuddin LH, Azorsa DO, Yang F, Pineda M, Helman LJ and Meltzer PS. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 70:6497-6508.
30. Lae M, Ahn EH, Mercado GE, Chuai S, Edgar M, Pawel BR, Olshen A, Barr FG and Ladanyi M. Global gene expression profiling of PAX-FKHR fusion-positive alveolar and PAX-FKHR fusion-negative embryonal rhabdomyosarcomas. The Journal of pathology. 2007; 212:143-151.
31. Ebauer M, Wachtel M, Niggli FK and Schafer BW. Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene. 2007; 26:7267-7281.
32. Davicioni E, Finckenstein FG, Shahbazian V, Buckley JD, Triche TJ and Anderson MJ. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006; 66:6936-6946.
33. De Pitta C, Tombolan L, Albiero G, Sartori F, Romualdi C, Jurman G, Carli M, Furlanello C, Lanfranchi G and Rosolen A. Gene expression profiling identifies potential relevant genes in alveolar rhabdomyosarcoma pathogenesis and discriminates PAX3-FKHR positive and negative tumors. Int J Cancer. 2006; 118:2772-2781.
34. Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH, Chu CF, Huang HY, Lin CM, Ho SY, Jian TY, Lin FM, Chang TH, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014; 42:D78-85.
35. Garcia DM, Baek D, Shin C, Bell GW, Grimson A and Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nature structural & molecular biology. 2011; 18:1139-1146.
36. Grimson A, Farh KK, Johnston WK, Garrett-Engele P, Lim LP and Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007; 27:91-105.
37. Friedman RC, Farh KK, Burge CB and Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009; 19:92-105.
38. Lu J and Clark AG. Impact of microRNA regulation on variation in human gene expression. Genome Res. 2012; 22:1243-1254.
39. Petersen A, Stewenius Y, Bjorkqvist M and Gisselsson D. Euploidy in somatic cells from R6/2 transgenic Huntington’s disease mice. BMC Cell Biol. 2005; 6:34.
40. Miller PJ, Dietz KN and Hollenbach AD. Identification of serine 205 as a site of phosphorylation on Pax3 in proliferating but not differentiating primary myoblasts. Protein Sci. 2008; 17:1979-1986.
41. Gordon DJ, Resio B and Pellman D. Causes and consequences of aneuploidy in cancer. Nature reviews Genetics. 2012; 13:189-203.
42. Compton DA. Mechanisms of aneuploidy. Curr Opin Cell Biol. 2011; 23:109-113.
43. Torres EM, Williams BR and Amon A. Aneuploidy: cells losing their balance. Genetics. 2008; 179:737-746.
44. Epstein JA, Shapiro DN, Cheng J, Lam PY and Maas RL. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc Natl Acad Sci U S A. 1996; 93:4213-4218.
45. Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, Chugh R, Ladanyi M, Grippo JF, Dall G, Staddon AP, Chawla SP, Maki RG, Araujo DM, et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer. 2014; 120:2448-2456.
46. Geoerger B, Kieran MW, Grupp S, Perek D, Clancy J, Krygowski M, Ananthakrishnan R, Boni JP, Berkenblit A and Spunt SL. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur J Cancer. 2012; 48:253-262.
47. Bagatell R, Norris R, Ingle AM, Ahern C, Voss S, Fox E, Little AR, Weigel BJ, Adamson PC and Blaney S. Phase 1 trial of temsirolimus in combination with irinotecan and temozolomide in children, adolescents and young adults with relapsed or refractory solid tumors: a Children’s Oncology Group Study. Pediatric blood & cancer. 2014; 61:833-839.
48. Weigel B, Malempati S, Reid JM, Voss SD, Cho SY, Chen HX, Krailo M, Villaluna D, Adamson PC and Blaney SM. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: a report from the Children’s Oncology Group. Pediatric blood & cancer. 2014; 61:452-456.
49. Tang YC, Williams BR, Siegel JJ and Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell. 2011; 144:499-512.
50. Saito M, Shimada Y, Shiraishi K, Sakamoto H, Tsuta K, Totsuka H, Chiku S, Ichikawa H, Kato M, Watanabe S, Yoshida T, Yokota J and Kohno T. Development of lung adenocarcinomas with exclusive dependence on oncogene fusions. Cancer Res. 2015; 75:2264-2271.
51. Nagaraja AK, Creighton CJ, Yu Z, Zhu H, Gunaratne PH, Reid JG, Olokpa E, Itamochi H, Ueno NT, Hawkins SM, Anderson ML and Matzuk MM. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol Endocrinol. 2010; 24:447-463.
52. Zender L, Villanueva A, Tovar V, Sia D, Chiang DY and Llovet JM. Cancer gene discovery in hepatocellular carcinoma. J Hepatol. 2010; 52:921-929.
53. Honegger A, Schilling D, Bastian S, Sponagel J, Kuryshev V, Sultmann H, Scheffner M, Hoppe-Seyler K and Hoppe-Seyler F. Dependence of intracellular and exosomal microRNAs on viral E6/E7 oncogene expression in HPV-positive tumor cells. PLoS pathogens. 2015; 11:e1004712.
54. Lam PY, Sublett JE, Hollenbach AD and Roussel MF. The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol Cell Biol. 1999; 19:594-601.
55. Howe B, Umrigar A and Tsien F. Chromosome preparation from cultured cells. Journal of visualized experiments. 2014; :e50203.