
INTRODUCTION
Malignancies of the breast, along with colon cancer, cause the most frequent cancer-associated deaths in women. Although long-term survival has improved, 16.7% of breast cancer patients develop deadly metastatic disease [1]. Epithelial-to-mesenchymal transition (EMT) and mesenchymal-to-epithelial transition (MET) are principal tissue plasticity programs in embryogenesis, wound healing, fibrosis, cancer progression and metastasis [2]. This epithelial-mesenchymal plasticity (EMP) is a critical determinant of tumor invasion, stemness and chemoresistance of breast cancer cells, and its pivotal importance for disease outcome has been highlighted by numerous reports [3–9]. To further elucidate the molecular mechanisms that contribute to EMP and EMT, we used the well-characterized WAP-T mouse model that mimics the spontaneous development of mammary adenocarcinoma in humans [10–15]. In WAP-T mice, mammary glands develop normally. However, with the onset of lactation, the WAP promoter is specifically activated in epithelial cells of the mammary gland and drives expression of the simian virus 40 (SV40) large tumor antigen (LT), which compromises the tumor-suppressive activity of pRB and p53 [10, 16]. Invasive mammary carcinomas in WAP-T mice share features with human basal-like/triple-negative cancer [10, 13–15, 17]. Recently, we demonstrated that expression of mutant p53 in WAP-T tumors is mechanistically linked with EMP and enhanced metastasis [18, 19]. Interestingly, this aggressive tumor phenotype was inversely correlated with the expression of carcinoembryonic-related antigen cell adhesion molecule 1 (CEACAM1).
CEACAM1 is a member of the carcinoembryonic antigen (CEA) family in the superfamily of immunoglobulins. It is expressed on epithelial, endothelial or hematopoietic cells and its transcript undergoes extensive splicing, producing variants with or without a transmembrane domain, and long (CEACAM1-L) or short cytoplasmic tails (CEACAM1-S) [20]. CEACAM1 is involved in the regulation of metabolism, maintenance of tissue architecture and differentiation, tumor growth, innate immunity, and blood and lymphatic vessel growth and remodeling [21–25]. Recently, CEACAM1 has been identified as a crucial factor mediating the tolerance and exhaustion of immune cells through interaction of its variable (V)-like domain with that of T-cell immunoglobulin domain and mucin domain-3 (TIM-3) [20, 26–29]. Its role as a negative regulatory co-receptor also depends on an intact ITIM domain, which serves as a binding partner for Src family kinases and the Src-homology 2 domain-containing phosphatases (SHP) SHP-1 and SHP-2 ([20], and references therein). By means of CEACAM1-dependent scaffolding of signaling complexes, CEACAM1-L down-modulates the activity of receptor tyrosine kinases.
CEACAM1 expression levels are usually reciprocally connected to poor prognosis in many cancers including mammary carcinomas [30–36]. More specifically, CEACAM1 expression correlates with good prognosis in mammary carcinomas, whereas in melanomas, up-regulation of CEACAM1 is accompanied by poor overall survival. Beyond its prognostic value in cancers, CEACAM1 has often been referred to as a marker for tissue differentiation, especially in the context of mammary gland differentiation and mammary epithelial lumen formation [37–39]. Shively´s group has demonstrated that CEACAM1-S expression was mandatory to establish acinar lumen formation in vitro [40–42]. In addition, CEACAM1 expression was also shown to revert malignant mammary cells to a differentiated, lumen-forming phenotype in vitro [41]. Intriguingly, they identified a direct molecular interaction between the CEACAM1-L cytoplasmic domain and β-catenin in vitro. However, in vivo evidence to corroborate these data and to connect CEACAM1-L and Wnt signaling in breast cancer development is lacking so far.
Based on these observations, we hypothesized that CEACAM1-L could negatively modulate the Wnt/β-catenin signaling by retaining β-catenin at the cell membrane, analogous to the role of E-cadherin (CDH1) in vitro [38]. Activation of the canonical Wnt signaling pathway involves re-localization of β-catenin from the cell membrane to the nucleus, where it initiates the transcriptional program that induces EMT [43]. The present study reveals that CEACAM1-L expression reduces β-catenin phosphorylation at positions Y86, a post-translational modification known to sustain activity of the Wnt-pathway [44, 45]. Our data strongly support a CEACAM1-dependent repression of β-catenin-phosphorylation at Y86 based on recruitment of SHP- 2. We furthermore observed that CEACAM1-L not only serves as a membrane scaffold for β-catenin and SHP-2, but also promotes Wnt-pathway inhibitory phosphorylation at S33/S37/T41 [46]. Loss of CEACAM1 in WAP-T tumor cells produced increased canonical Wnt signaling and promoted cellular invasiveness in vitro, and importantly, dramatically enhanced the metastasis rate of mammary adenocarcinomas in CEACAM1null mice in vivo. Also, analyses of a publicly available database revealed that CEACAM1 expression is inversely correlated with survival of human mammary carcinoma patients.
Together, these results suggest that CEACAM1 acts as a gatekeeper for the maintenance of the epithelial phenotype in breast cancer cells in vitro and in vivo.
RESULTS
Mesenchymal markers are specifically up- regulated in CEACAM1low/- G-2 cells
In this study, we characterize CEACAM1-dependent effects on EMT in vitro and in vivo. For our in vitro studies, we used G-2 cells derived from primary mammary adenocarcinomas grown in WAP-T mice [12]. G-2 cells exhibit cancer stem cell-like properties and are composed of mixed epithelial and mesenchymal subpopulations in vitro, as shown in the phase contrast image in Figure 1A. These two cellular subsets are complementary and reciprocally switch between an epithelial-like (white dotted line) and a mesenchymal-like phenotype (black arrows), or vice versa [12]. As demonstrated by immune fluorescence (Figure 1B, 1C), the epithelial-like subpopulation co-expresses the epithelial markers CEACAM1 (red) and EpCAM (green) at the membrane (Figure 1B), whereas the mesenchymal-type cells are negative for CEACAM1 or EpCAM, but prominently express the mesenchymal marker Vimentin (green in Figure 1C), (Figure 1C). These observations suggest that an epithelial-like phenotype is connected to expression of CEACAM1.
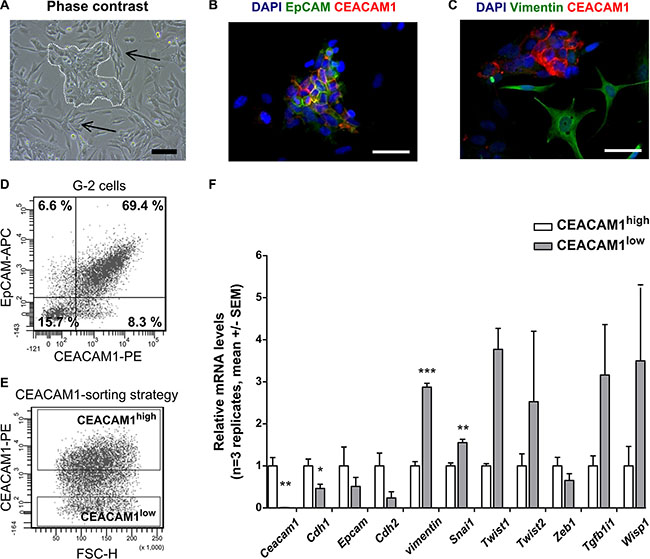
Figure 1: CEACAM1-expression and epithelial and mesenchymal properties of G-2 cells. (A–C) microscopic images depict the epithelial and mesenchymal morphology and marker expression in phase contrast (A) and fluorescence microscopy (B, C). In (A), the epithelial subpopulation of the G-2 cells exhibits a cobble-stone morphology in distinct cell colonies (white dotted line), whereas the mesenchymal fraction of the cell population (black arrows) exhibits extended, spindle-shaped cell bodies. (B, C) CEACAM1 expression (red) is shown in the epithelial cells, where it is co-localized with EpCAM (green) at the cellular membrane (B). In contrast, mesenchymal, Vimentin-expressing cells (C, green) do not express CEACAM1. Flow cytometric analyses for EpCAM and CEACAM1 expression (D, E) and sorting strategy for CEACAM1high and CEACAM1low G-2 cells (E) are shown. (F) the relative expression levels of characteristic epithelial and mesenchymal markers genes are compared after cell sorting according to (E) between CEACAM1highand CEACAM1low G-2 cells (Ceacam1, Cdh1, Epcam, Cdh2, vimentin, Snai1, Twist1, Twist2, Zeb1, Tgfb1i1, Wisp1). Expression data from CEACAM1high and CEACAM1low cells are shown in white and grey bars, respectively. Data shown here are representative of three independently repeated experiments in three replicates. Statistical analyses: Student´s t-test. Scale bars: A: 100 μm, B and C: 50 μm.
To further identify regulatory pathways that determine epithelial cell differentiation in a CEACAM1-dependent manner, we sorted CEACAM1high and CEACAM1low G-2 cells using fluorescence activated cell sorting (FACS) and analyzed their individual epithelial and mesenchymal gene signatures (Figure 1D–1F). As shown in Figure 1D, the majority of the G-2 cells co-express CEACAM1 and EpCAM at variable levels. Previous experiments demonstrated that abrogation of CEACAM1 expression in G-2 cells greatly interferes with their survival (F. Wegwitz, A.K. Horst, W. Deppert, unpublished observations). Therefore we compared enriched CEACAM1high with CEACAM1low G-2 populations (Figure 1E) with regards to their expression of EMT marker genes as in Lenfert et al. (Figure 1F) [18] We found that CEACAM1high cells expressed significantly higher RNA amounts of E-cadherin (Cdh1), and significantly lower RNA amounts of the EMT-drivers snail homolog 1 (Snai1) and vimentin compared to CEACAM1low G-2 cells (Figure 1F). In addition, up-regulation of the mesenchymal marker genes Twist-related protein 1 and 2 (Twist1, Twist2), Transforming growth factor beta 1-induced transcript 1 (Tgfb1i1) and Wnt-inducible signaling pathway protein 1 (Wisp1) was detected in CEACAM1low G-2 cells (Figure 1F).
CEACAM1 co-localizes and co-precipitates with β-catenin in murine G-2 cells
To ascertain if our hypothesis that CEACAM1 functions as a component of the EMT switch, we next analyzed whether E-cadherin, β-catenin and CEACAM1 interacted at the protein level. The interaction of human CEACAM1 with β-catenin has been demonstrated before in vitro: human CEACAM1-L contain a β-catenin binding motif in its cytoplasmic tail [38, 47]. This is missing in CEACAM1-S, but its cytoplasmic domain binds to Actin and protein kinase C. In Jurkat cells, CEACAM1-dependent regulation of Fas-mediated apoptosis depends on its interaction with β-catenin [48]. Similarly, in a model of colonic carcinogenesis (APC1638N/+:Ceacam1–/–mice), a correlation was found between the absence of CEACAM1 and β-catenin nuclear translocation [38, 49]. To verify the putative interaction between CEACAM1 and β-catenin in G-2 cells, we performed co-localization studies and co-immunoprecipitation with β-catenin. As shown in Figures 2A and 2B, CEACAM1-expressing cells exhibit a prominent membrane co-labeling of CEACAM1 and E-cadherin, or of CEACAM1 and β-catenin, respectively. Furthermore, we show in co-immunoprecipitation assays that CEACAM1 physically interacts with β-catenin in G-2 cells, whereas E-cadherin only showed binding with β-catenin (Figure 2C). These observations support our hypothesis that distinct ß-catenin fractions are maintained at the cell membrane by either interacting with E-cadherin or CEACAM1.
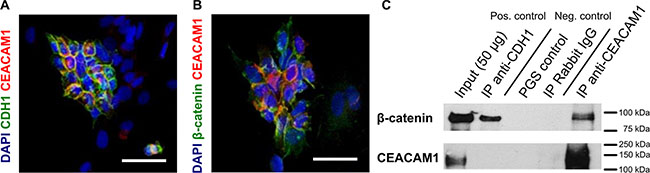
Figure 2: Co-localization and physical interaction of CEACAM1 with β-catenin. (A and B) co-localization of E-cadherin (green, A) or β-catenin (green, B) and CEACAM1 (red) is shown in the epithelial subpopulation of G-2 cells in immune fluorescence microscopy. (C) results from co-precipitation of CEACAM1 with β-catenin or CDH1 from 200 μg G-2 cell lysates are shown, followed by immunoblotting with anti β-catenin (upper panel) and anti-CEACAM1-antibodies (lower panel). Polyclonal anti-rabbit immunoglobulins or protein A/G sepharose (PAS/PGS) were used as controls. β-catenin co-precipitates with CEACAM1 and E-cadherin, whereas precipitation of E-cadherin does not reveal binding of CEACAM1. Scale bars: 50 μm.
Reduction of CEACAM1 expression facilitates EMT and Wnt signaling
As subcellular β-catenin localization and its phosphorylation determine Wnt signaling activity, we subsequently investigated whether β-catenin phosphorylation and Wnt signaling were connected to CEACAM1-L expression and could influence EMT. We asked whether different levels of CEACAM1 could modulate the activation of Wnt/β-catenin signaling and thereby affect the balance between the epithelial and mesenchymal phenotypes in G-2 cells. We thus generated variants with different levels of CEACAM1 expression using shRNA technology (Figure 3A and 3B; G-2scr, G-2shCC1#2 and G-2shCC1#3). With approximatively 50% CEACAM1+ cells, the cell line obtained after transduction with scrambled shRNA construct (G-2scr) closely resembled the G-2 parental cell line (Wegwitz, F., Deppert, W., unpublished observations and Figure 1). Furthermore, two stable cell lines with clear reduction in their CEACAM1+ fractions (24% and 11%, respectively) were selected (Figure 3B; middle and right panel). Interestingly, gradual reduction of CEACAM1 expression enlarged the proportion of mesenchymal-like cells (Figure 3A): epithelial-like cells are decreased in the G-2shCC1#2 population, and are no longer detectable in the G-2shCC1#3 population (Figure 3A, right upper panel). This indicates that a minimal proportion of CEACAM1-expressing cells, or minimal CEACAM1 levels are required for maintenance of the epithelial phenotype. In addition, intercellular adhesion appeared markedly reduced in the two CEACAM1low populations, where a cobblestone-like cellular morphology was largely absent (Figure 3A; G-2shCC1#2 and G-2shCC1#3), such that individual cells remained scattered and did not form epithelial-like colonies.
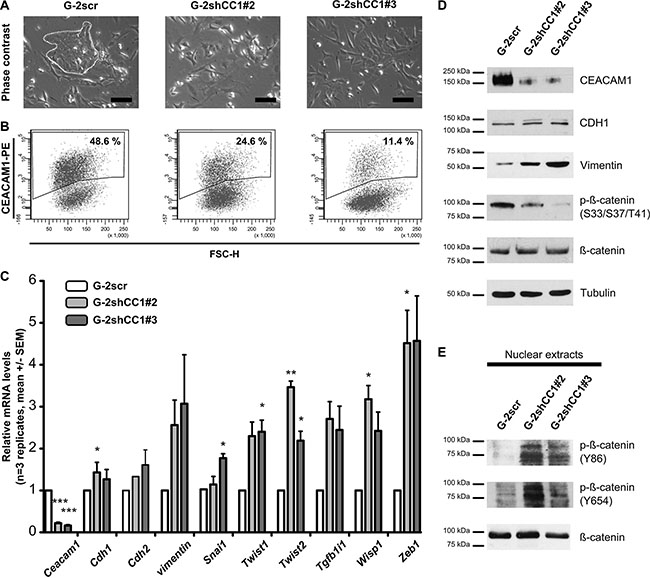
Figure 3: Analyses of EMT after down-regulation of CEACAM1 expression in G-2 cells. (A) CEACAM1 expression was down-regulated in stable anti-CEACAM1 shRNA –transfectants (clones G-2shCC1#2 and G-2shCC1#3); as a control, a cell clone stably expressing scrambled shRNAs was used (G-2scr). Whereas the epithelial phenotype is preserved in G-2scr cell colonies (left panel, white dotted line), the G-2shCC1#2 and G-2shCC1#3 populations exhibit a rather mesenchymal phenotype, also evident by poor intercellular adhesion and scattered cell growth (A, middle and right panel). Scale bars: 100 μm. (B) Dot plot histograms from flow cytometry analyses of the different G-2 populations are shown; they reveal maintenance of CEACAM1 expression (48.6%) in the G-2scr cells, and reduction of the fraction of CEACAM1-positive/CEACAM1high cells to 24.6% and 11.4% in the G-2shCC1#2 and G-2shCC1#3 clonal populations. (C) Comparative qRT-PCR expression analyses of key epithelial and mesenchymal marker genes (Ceacam1, Cdh1, Epcam, Cdh2, vimentin, Snai1, Twist1, Twist2, Zeb1, Tgfb1i1, Wisp1) in G-2scr, G-2shCC1#2 and G-2shCC1#3 cells. Data from G-2scr cells, G-2shCC1#2 and G-2shCC1#3 cells are shown in white, light grey and dark grey bars, respectively. Data sets are expressed as means ± SEM and were repeated three times independently in technical triplicates. Statistics: Student´s t-test. (D) Comparative, representative Western blot analyses of CEACAM1, CDH1, Vimentin, Tubulin, and phosphorylated at S33/S37/T41 in β-catenin in G-2scr, G-2shCC1#2 and G-2shCC1#3 cells. (E) Western blots of nuclear extracts showing tyrosine phosphorylation (Y86, Y654) of β-catenin in G-2scr, G-2shCC1#2 and G-2shCC1#3 cells.
To document CEACAM1-dependent regulation of the epithelial or mesenchymal phenotype, we performed RT-qPCR and analyzed the expression of key regulators of EMT: we first validated the down-regulation of Ceacam1 gene transcripts in the CEACAM1low G-2shCC1#2 and G-2shCC1#3 cell lines (Figure 3C). Strikingly, we observed an up-regulation of Tgfb1i1, Twist1, Twist2, vimentin, Wisp1 and Zeb1. Congruently, at the protein levels, this EMT-weighted signature is corroborated with reduction of CEACAM1 expression and increased concentration of Vimentin (Figure 3D). Interestingly, the levels of E-cadherin remained unaltered, pointing towards enhanced EMP rather than a complete EMT, where E-cadherin expression is frequently lost (Figure 3D). Furthermore, we demonstrate that increases in cellular plasticity in CEACAM1low cells coincide with a reduction of β-catenin phosphorylation on S33/S37/T41. These serine and threonine residues are subject to phosphorylation by glycogen synthase kinase 3β, and target β-catenin for poly-ubiquitinylation and degradation, and thus negatively regulate its transcriptional activity [46, 50, 51]. Contrary to phosphorylation on S33/S37/T41, β-catenin tyrosine phosphorylation on Y86 is enhanced in CEACAM1low cell populations exhibiting comparable total nuclear β-catenin levels (Figure 3E). In contrast to S33/S37/T41 phosphorylation, phosphorylation of this tyrosine residue stimulates the transcriptional activity of β-catenin [44, 45]. Taken together, these findings support our data that expression of CEACAM1 interferes with activation and nuclear translocation of β-catenin by inhibiting its phosphorylation at Y86.
CEACAM1 suppresses β-catenin transcriptional activity
To further corroborate our findings on the crosstalk between CEACAM1 and Wnt signaling, we performed luciferase-based β-catenin activity reporter assays using T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factor promoter constructs (TOP-FLASH); as a control, constructs harboring mutated TCF/LEF-binding site were used (FOP-FLASH). As previously reported in colon cancer cells [49], we found that reduction of CEACAM1 expression induced TCF/LEF dependent β-catenin transcriptional activity (Figure 4A). In agreement with our data presented above, functional investigations demonstrated that MatrigelTM cellular invasion was enhanced by approximately 2–3 -fold in cell clones with reduced CEACAM1 expression and increased β-catenin–inducible promoter activity (Figure 4B). These observations support our hypothesis that CEACAM1 expression blocks mesenchymal conversion of G-2 cells and thus impairs cellular motility.
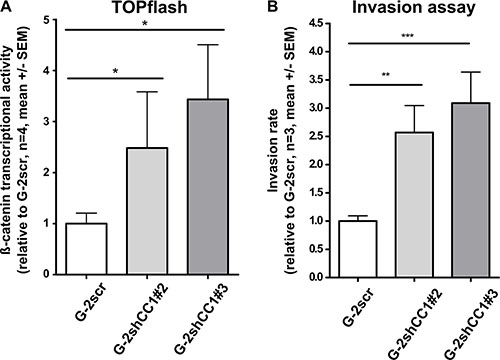
Figure 4: Comparison of β-catenin luciferase reporter activity and invasion potential of G-2 cells with normal or reduced CEACAM1 expression. (A) Relative activity of the β-catenin-inducible promoter is compared in Luciferase assays (TOPflash assay: β-catenin-inducible promoter with intact TCF/LEF binding sites) in G-2 cells with normal (G-2scr) or reduced (G-2shCC1#2 and G-2shCC1#3) CEACAM1 expression. As a control, G-2scr, G-2shCC1#2 and G-2shCC1#3 populations were transfected with a control plasmid containing mutant TCF/LEF binding sites in the β-catenin inducible promoter (TOPflash). TOPflash values were calibrated to FOPflash results and normalized to β-catenin-inducible promoter activity in G-2scr cells. Data sets are expressed as means ± SEM and were repeated three times in quadruplet analyses. (B) Comparison of invasive capacities of G-2scr, and G-2shCC1#2 and G-2shCC1#3 cells in a basal membrane (BME) invasion assay; data from G-2scr, and G-2shCC1#2 and G-2shCC1#3 cells are shown in white, light grey and dark grey bars, respectively. Data sets are expressed as means ± SEM and were repeated three times.
CEACAM1 overexpression in G-2 cells inhibits EMT-related gene expression
Our data above provide further evidence that EMT is facilitated when CEACAM1 expression is reduced. However, whether re-introduction or over-expression of CEACAM1 in epithelial cells inhibits EMT has not been studied so far. Therefore, we overexpressed CEACAM1 in G-2 cells and analyzed changes in their cellular morphology as well as the transcriptional activity and protein levels of EMT-related genes (Figure 5). We also assayed β-catenin transcriptional activity and its phosphorylation status in response to CEACAM1 overexpression. Phase contrast images depict that enhanced CEACAM1 expression imposes an epithelial-like phenotype on G-2 cells (Figure 5A). This clear change in cellular morphology is accompanied by a congruent change in the expression of E-cadherin, Epcam and Tgfb1i1, whereas the majority of EMT inducers, Wisp1, Twist1, Twist2 and Snai1 were down-regulated significantly (Figure 5B). Changes in expression of vimentin is only weak on RNA levels (Figure 5B), but protein levels of SNAI1 and Vimentin were significantly reduced in G-2 cells overexpressing CEACAM1 (Figure 5C). In addition, S33/S37/T41 phosphorylated forms of β-catenin were increased after enforced CEACAM1 expression (Figure 5C). In contrast, protein levels of E-cadherin and those of ZO-1, a gatekeeper of epithelial polarity, were only moderately increased, whereas Y86 phosphorylation was slightly decreased (Figure 5C). In line with these findings, transcriptional activity of β–catenin inversely correlated with CEACAM1 expression in G-2 cells (Figure 5D). The reduction of β–catenin transcriptional activity was even more pronounced when canonical Wnt signaling was activated by stimulation with WNT3a in CEACAM1 overexpressing G-2 cells (Supplementary Figure S1A).
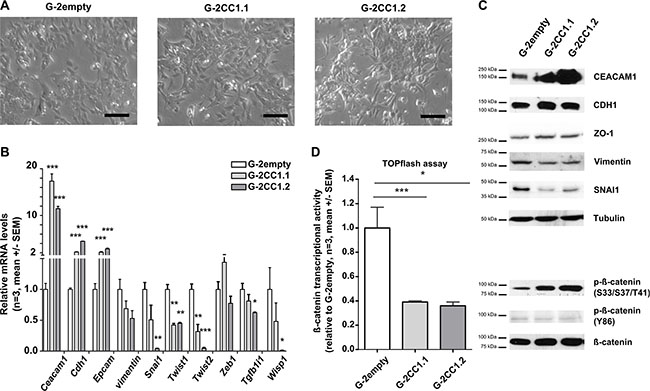
Figure 5: Overexpression of CEACAM1 in G-2 cells reduces the EMT phenotype. (A) Phase contrast microscopic images document maintenance of the epithelial phenotype in G-2 cells, as well as in G-2 cells overexpressing CEACAM1 (G-2CC1.1, middle and G-2CC1.2, right panel) Scale bars: 75 μm (B) Expression analyses of key epithelial and mesenchymal marker genes (Ceacam1, Cdh1, Epcam, Cdh2, vimentin, Snai1, Twist1, Twist2, Zeb1, Tgfb1i1, Wisp1) in G-2empty, G-2CC1.1 and G-2CC1.2 populations via qRT- PCR. Data from G-2empty, G-2CC1.1 and G-2CC1.2 cells are shown in white, light grey and dark grey bars, respectively. Data sets are expressed as means ± SEM and were repeated three times in triplicate analyses. (C) Western blot for proteins levels comparison of CEACAM1, CDH1, ZO-1, Vimentin, SNAI1, β-catenin and phosphorylated β-catenin; (S33/S37/T41 and Y86). Tubulin levels were used as loading controls. (D) Relative levels of β-catenin-inducible promoter activity assessed by TOPflash-Luciferase assays in G-2empty, G-2CC1.1 and G-2CC1.2 cells. TOPflash values were calibrated to FOPflash results and normalized to β-catenin-inducible promoter activity in G-2scr cells. Data sets are expressed as means ± SEM and were repeated three times in triplicate analyses.
SHP-2 binds to CEACAM1 and maintains the epithelial phenotype in G-2 cells
Since endothelial SHP-2 is known to regulate the recovery of adherent junctions through control of β-catenin phosphorylation, we tested whether SHP-2 and CEACAM1 could interact in CEACAM1-expressing G-2 cells [52]. Indeed, we were able to confirm a physical interaction of CEACAM1 and SHP-2 in pervanadate-treated G-2 cells (Figure 6A), but not in G-2 cells with reduced CEACAM1-levels (G-2shCC1#3). NSC-87877 has been described as small molecule specifically inhibiting phosphatase activity of SHP-2 as well as of SHP-1 [53]. Since SHP-1 protein levels were not detectable in G-2 cells (data not shown), we assumed that the effects of NSC-87877 pharmacological inhibition are specific to SHP-2 inhibition. To test whether SHP-2 activity was required to maintain the epithelial phenotype of G-2 cells, we treated G-2 cells with NSC-87877 (Figure 6B, 6C) and compared β-catenin phosphorylation (Figure 6B), cellular morphology (Figure 6C), and the epithelial and mesenchymal marker signatures (Figure 6D). We found that SHP-2 inhibition produced a mild increase in β-catenin Y86 phosphorylation (1.4× compared to control) after 24 hours, whereas phosphorylation of β-catenin inhibitory residues S33/S37/T41 was reduced (approximatively 0.74×) (Figure 6B). Accordingly, we observed increased Vimentin expression in G-2 cells treated with the SHP1/2 inhibitor (Figure 6B). Congruently, the proportion of mesenchymal-like cells increased over time under NSC-87877 treatment (Figure 6C; epithelial cell colonies are marked by white dotted lines, mesenchymal cells are indicated by black arrows). The importance of intact SHP-2 signaling for the maintenance of an epithelial phenotype in G-2 cells was corroborated by analyses of the gene expression profiles of EMT inducers (Figure 6D). We found that the expression of mesenchymal signature genes was strongly enhanced upon SHP-2 inhibition in G-2 cells. The majority of EMT markers and regulators, such as Vimentin, Snai1, Twist1, Zeb1, Tgf1i1 and Wisp1, were significantly upregulated after NSC-87877 treatment for 72 hrs, accompanied by significant down-regulation of epithelial marker genes, such as Epcam and E-cadherin (Figure 6D). Here, we demonstrate that CEACAM1 is critical for the maintenance of the epithelial phenotype in G-2 cells by regulating β-catenin activity through SHP-2-dependent de-phosphorylation of Y86, accompanied by increased phosphorylation on residues S33/S37/T41.
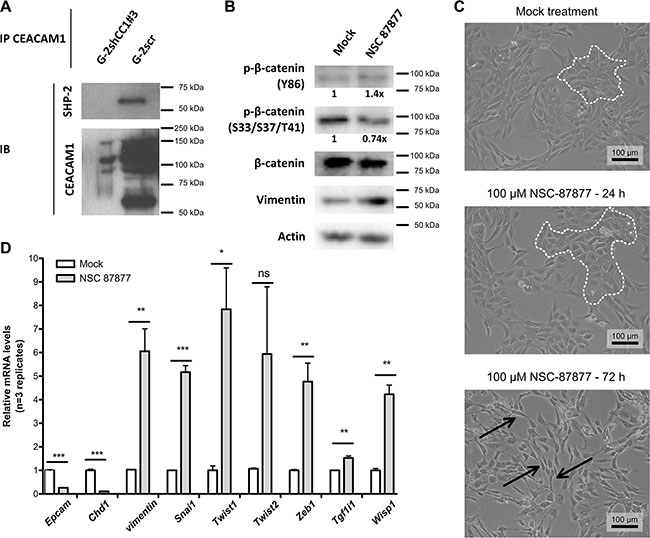
Figure 6: SHP-2 interacts with CEACAM1 and blocks EMT in G-2 cells. (A) Co-immunoprecipitation of SHP-2 with CEACAM1 in CEACAM1-expressing G-2scr cells, but not in G-2shCC1#3 with reduced CEACAM1 levels, demonstrates a physical interaction between SHP-2 and CEACAM1. (B) assessment of β-catenin phosphorylation by Western blot in cells without (mock) or with SHP-2 inhibitor treatment (NSC87877, 100 μM, 24 hrs): treated cells display moderately increased levels of β-catenin phosphorylation at Y86 as well as reduced β-catenin phosphorylation at S33/S37/T41; concomitantly, the expression level of Vimentin is increased; quantities were normalized relative to β-catenin levels. (C) Phase contrast images of CEACAM1-expressing G-2 cells treated with 100 μM SHP- 2 inhibitor NSC-87877 for 24 hrs or 72 hrs shows that mesenchymal transformation of the G-2 cells increases with time in comparison to mock treated cells; epithelial cell clusters are indicated by white dotted lines, mesenchymal cells with poor intercellular contacts and elongated cell bodies are indicated by black arrows; scale bar: 100 μm. (D) RT-qPCR-based gene expression analyses of EMT-signature marker genes in G-2 cells with (shaded bars) or without (white bars) SHP-2 inhibitor treatment (NSC-87877, 100 μM, 72 hrs). Note that the expression of Epcam and E-cadherin is significantly reduced cells with SHP-2 inhibition, whereas EMT drivers such as vimentin, Snai1, Twist1, Zeb1, Tgf1i1, and Wisp1 are elevated; Twist2 did not exhibit significant alteration of expression.
Enhanced activation of Wnt signaling and increased pulmonary metastasis in WAP-T/CEACAM1null mice
To translate these findings into an in vivo model, we crossbred WAP-T mice with CEACAM1-deficient mice and analyzed the spontaneously grown mammary tumors and their progression and metastasis [10, 54]. As shown in Figure 7A, WAP-T/CEACAM1null mice exhibit significantly higher rates of metastasis; in WAP-T mice, pulmonary metastases were found in approximately 8% of all mice examined. By contrast, pulmonary metastases were detected in approximately 40% of the tumor-bearing WAP-T/CEACAM1null mice (Figure 7A, 7B). The position of the tumor-bearing mammary gland did not affect metastasis incidence (data not shown). Surprisingly, in H&E staining, morphology of primary lesions was similar in CEACAM1 wild-type and deficient mice ([54] and data not shown). When pulmonary metastases were analyzed for large T-Antigen (LT) expression, we found that in the majority of the mice examined, metastases were LT-positive (Figure 7B, upper panel). In the lower panel, intra-arterial and inter-septic pulmonary metastases are depicted.
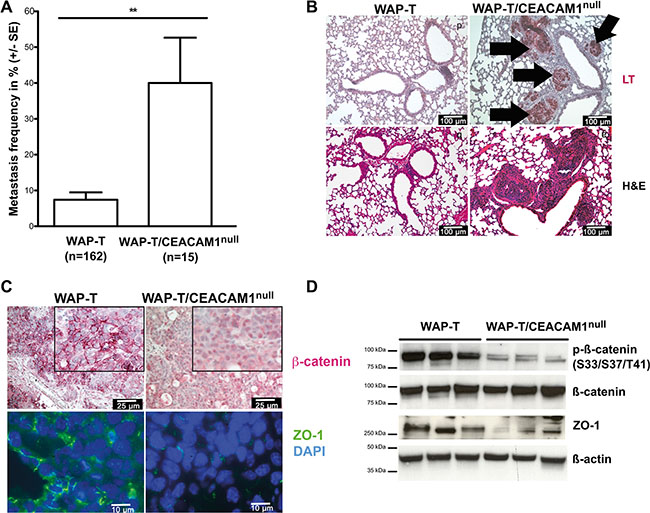
Figure 7: Loss of CEACAM1 in vivo enhances EMT and pulmonary metastasis of murine mammary adenocarcinomas. (A) Quantitative evaluation of pulmonary metastasis in WAP-T and WAP-T/CEACAM1null mice presents significant differences in metastastic frequencies. Data shown in percent ± standard error (B) Histological analyses of metastasis-free lung tissue and pulmonary metastases in WAP-T and WAP-T/CEACAM1null mice reveal LT-expressing metastases in WAP-T/CEACAM1null mice (upper panel, bold arrows point at clusters of intra-vascular and inter-septal metastases). These were identified in H&E stained tissue sections of tumor-bearing WAP-T mouse (lower panels), whereas no metastases were found in the depicted tissue sections of a tumor-bearing WAP-T mouse; (C) Immune histological analyses of expression and localization of β-catenin and ZO-1 in mammary tumors from WAP-T and WAP-T/CEACAM1null mice reveal differential localization of β-catenin (red staining) at cell membranes (WAP-T mice, inset enlarged) and in the nuclei (WAP-T/CEACAM1null mice; insets enlarged). In immune fluorescence, ZO-1 (shown in green) is localized at cellular junctions in tumors from the WAP-T mice, but is largely absent from CEACAM1null tumors. Nuclei were stained with DAPI (blue); scale bars are indicated (D) Analyses of protein expression levels of phosphorylated β-catenin (upper lane), and total β-catenin (second lane), as well as total ZO-1 protein in tumor lysates from WAP-T and WAP-T/CEACAM1null mice. Protein levels were normalized to β-Actin (lower lane). Note that phosphorylation of β-catenin is clearly reduced in tumors from WAP-T/CEACAM1null mice. Similarly, ZO-1 protein expression is reduced in WAP-T/CEACAM1null mice. Scale bars as indicated.
In immunostainings for β-catenin and Zonula occludens protein 1 (ZO-1) expression, we found that β-catenin exhibited a distinct staining pattern at the cell membrane in WAP-T tumors (Figure 7C, upper panel). In contrast, β-catenin had preferentially translocated in the tumor cell nuclei in tumors from WAP-T/CEACAM1null mice (Figure 7C, upper right panel). This observation corroborates our data that Wnt signaling is persistently activated in the absence of CEACAM1 (cf. Figures 3D, 3C, 3D and 4A). Furthermore, intercellular junctions were reduced in CEACAM1-negative tumors, as evidenced by reduction and focal loss of ZO-1 staining along cell membranes (Figure 7C, lower right panel). These data were confirmed in immunoblots, where a clear reduction of β-catenin phosphorylation at S33/S37/T41, and total reduction of ZO-1 levels were observed (Figure 7D).
Since loss of CEACAM1 represents a clear adverse prognosis parameter in WAP-T mouse mammary carcinomas, we took advantage of a publicly available database, www.klplot.com, to assess the impact of CEACAM1 expression levels on breast cancer patient survival [55, 56]. In line with our experimental results, CEACAM1 expression levels were inversely correlated with 10-yr survival rates of patients (Supplementary Figure S1B), irrespective of the particular tumor subtype (Supplementary Figure S1C–S1F).
In summary, our study demonstrates for the first time that loss of CEACAM1 expression facilitates EMT in murine mammary carcinoma in vitro and in vivo. Loss of CEACAM1 expression impairs negative modulation of Wnt signaling. Consequently, low CEACAM1 expression in breast cancer patients correlates with poorer outcome than high CEACAM1 expression.
DISCUSSION
Dichotomous roles have been assigned to CEACAM1 in tumor development and progression: on the one hand, its expression has been correlated to increased tumor invasion in melanoma, lung carcinoma, in carcinomas of the thyroid, liver and colon; in these specimens, CEACAM1 is up- regulated during progression of the disease and has been established as an independent prognostic marker [57, 58]. On the other hand, in early stages, CEACAM1-L is down-regulated in tumor cells of different organs including neoplastic lesions of the colon, stomach, bladder and prostate [59–65]. Similarly, CEACAM1 is strongly expressed on normal breast epithelial cells and is down-regulated in benign as well as malignant breast cancer [31, 66–69]. As CEACAM1 can act as an inhibitor of tumor cell growth of several cancers, e.g. colon and prostate cancer, it was identified as a putative tumor suppressor [57, 58].
Based on our previous findings that overexpression of mutant p53 increases mammary carcinoma cell aggressiveness and is associated with an EMT gene expression signature and reduction of CEACAM1-expression, we analyzed the specific molecular role of CEACAM1 in EMT and cancer progression [11, 18]. In the present report, we demonstrate that CEACAM1 is a critical determinant in the regulation of the EMT switch. Taking advantage of the WAP-T mammary carcinoma mouse model and of the G-2 cancer stem cell line derived from it, we demonstrate for the first time that expression of CEACAM1-L can control the tumor cell aggressiveness in vitro by reducing EMT gene expression and invasion capabilities, and in vivo by decreasing its metastatic propensity. So far, only the CEACAM1-S isoform has been studied to greater detail with respect to mammary cell differentiation. However, the molecular basis for the interaction between β-catenin and CEACAM1-L has been determined in vitro, see below [38].
CEACAM1 is an integral component of adhesion junctions, where it co-localizes with E-cadherin, and β-catenin [70]. In culture systems, CEACAM1 is present on WAP-T cells with epithelial but not mesenchymal phenotype. It co-localizes and physically interacts with β-catenin, as previously reported for cancer cell lines [38, 49]. In normal epithelial cells, the majority of β-catenin is localized at the plasma membrane, in close apposition with adhesion molecules (e.g. E-cadherin) and thus is presumed to contribute to the maintenance of epithelial cell polarity. The structural integrity of the β-catenin/ E-cadherin complex and its associated cell junction properties is regulated by protein kinase and phosphatase activity [46]. The first evidence for an in vivo connection between CEACAM1 expression and the regulation of Wnt signaling were reported in the compound APC1638N:Ceacam1–/– mice. Here, absence of CEACAM1 in murine colonic adenocarcinomas led to enhanced nuclear translocation of β-catenin. Additionally, CEACAM1 expression was required in murine colonic tumor cells to down-modulate activity of the β-catenin-inducible promoter, which is in agreement with our findings [49]. Similarly, in the colonic tumor cell lines CT26 and CT51, interaction of β-catenin with CEACAM1-L, but not CEACAM1-S, was confirmed [49].
Furthermore, we provide a causal relationship between specific expression of CEACAM1-L and changes in phosphorylation of β-catenin on S33/37/T41. Reduction of CEACAM1 levels in cultured tumor cells and in endogenous tumors from WAP-T/CEACAM1null mice was associated with decreased phosphorylation of β-catenin on S33/37/T41, a hallmark of activated Wnt/β-catenin pathway [51]. Phosphorylation of S33/37/T41 reduces β-catenin transcriptional activity by increasing affinity for E-cadherin and thereby its retention at the plasma membrane. On the other hand, unbound intracytoplasmic β-catenin is targeted for degradation by Skp, Cullin, F-box containing complex (SCF)-class E3-ubiquitin ligase [46, 71]. Importantly, this feature was associated with increased localization of β-catenin in the nucleus of the WAP-T tumor cells and consequently enhanced Wnt/β-catenin-pathway activity. Also, reduction of Ceacam1 expression levels was coupled to a clear increase of tyrosine phosphorylation of β-catenin at Y86. Few reports exist on the functional consequences of distal β-catenin tyrosine phosphorylation. However, Coluccia et al. state that phosphorylation on Y86 enhances nuclear translocation and transcriptional activity [72]. In line, overexpression of CEACAM1-L leads to increased S33/37/T41 phosphorylation levels, reduced Y86 phosphorylation levels, and the inhibition of β-catenin transcriptional activity, suggesting that CEACAM1-L expression impacts on expression/activity of specific kinases and phosphatases responsible for these events.
Because of its potential tumor suppressive properties, research has largely focused on the CEACAM1 isoform with long cytoplasmic tail containing two immunoreceptor tyrosine-based inhibition motifs (ITIMs). These ITIMs are phosphorylated upon activation of kinases of the Src familiy as well as receptor tyrosine kinases like epidermal growth factor (EGF)- or Insulin-receptor (IR) [57, 73–75]. Consequently, they harbor binding motifs for inhibitory phosphatases containing Src homology 2 domains (SH2-domains), especially SHP-1 and SHP-2 [82]. Importantly, phosphatase activity of SHP-1 and SHP-2 down-modulates Src- or Syk-kinase signaling and reduces the activation of the mitogen-activated protein kinase (MAPK) cascade [20]. Reduction in the juxtamembrane SHP-1/-2 signaling has been described in Ceacam1–/– mice, where CEACAM1-dependent tyrosine phosphatase recruitment leads to persistent or exacerbated receptor activation. As a consequence, persistent toll-receptor (TLR)-signaling, hyperactivation of platelet adhesion, inadequate activation of IL-2R, EGFR and IR, as well as endothelial nitric oxide synthase or matrix-metalloproteinase 9 (MMP9) secretion were observed [27, 73, 76, 77].
The consequences of canonical Wnt/β-catenin signaling in the absence of CEACAM1 have not been fully elucidated. Simoneau et al. reported that ectopic expression of SHP-1 but not its inactive variant could inhibit phosphorylation of β-catenin on Y86 [78]. Similarly, Lee et al. described the ability of SHP-2 to dephosphorylate β-catenin at Y654 [79]. Considering the ability of both SHP-1 and SHP- 2 to dephosphorylate different tyrosine residues of β-catenin, thereby decreasing canonical Wnt-pathway activity, we describe here for the first time that the interaction with CEACAM1-L and SHP-2 contributes significantly to the regulation of the β-catenin phosphorylation status: SHP-2 inhibition moderately enhances phosphorylation of β-catenin on Y86, but this increase was even more pronounced if G-2 cells exhibited partial loss of CEACAM1 expression. Once SHP-2 activity is compromised by small molecule inhibitors, or CEACAM1 is downregulated in G-2 cells, phosphorylation of β-catenin on its inactivating S33/S37/T41 residues is impaired. This could be due to the fact that in G-2 cells, mesenchymal and epithelial populations overlap and that additional regulatory pathways funnel into the regulatory network that determines the dichotomous β-catenin phosphorylation pattern. Further studies will have to be conducted to reveal additional CEACAM1 interaction partners that participate in the CEACAM1-SHP-2-β-catenin signaling scaffold.
The present report clearly points towards an involvement of CEACAM1 in the regulation of the EMT-phenotype of tumor cells through Wnt/β-catenin pathway which impacts on cancer progression and metastasis. Even though the exact molecular mechanism remains to be deciphered, our data identify CEACAM1 as a gatekeeper in EMT that provides novel insights into the regulation of cancer invasion and metastasis.
MATERIALS AND METHODS
Animals
All animals were housed under SPF conditions in the animal facility of the University Medical Center Hamburg-Eppendorf and approved by Hamburg’s Authority for Health and Environmental Protection (TVG 61/05; TVG 88/06). The model of prolactin-driven large T-Antigen (LT) expression in WAP-T mice (mice with prolactin-sensitive whey acidic protein-promoter driven expression of the large T-antigen) has been described [11, 54]. In brief, expression of the oncogenic LT is initiated during gestation, when lactation commences around gestational day 14. Following this induction, WAP-T mice undergo a sequence of dysplasia, DCIS, and adenocarcinoma formation within 8 months. Tumor growth, tumor histology, and metastasis were analyzed in both WAP-T and WAP-T/CEACAM1null (Ceacam1–/–) mice [54, 80].
Immunohistology
For histological analysis tumor and lung specimens were processed as previously described [12, 54].
Cell culture
The WAP-T cell line G-2 and derivatives were cultured in DMEM medium (PAA) supplemented with 10% FCS (PAA) in a humidified incubator at 37°C and 5% CO2. Generation of the G-2 cell line and its properties have been described previously [12]. Phase contrast pictures were obtained using a TS100 microscope (Nikon) equipped with a μEYE 1240ML-C-HQ camera (Imaging Development Systems).
Plasmid construction and lentiviral transduction of G-2 cells (shCC1 and CC1cDNA)
For the ectopic expression of Ceacam1-L in G-2 cells, Ceacam1 cDNA was extracted from the pcDNA-mCC1-4 via EcoRI-digestion and subsequently cloned into lentiviral vector LeGO-iG2 (using EcoRI restriction site), kindly provided by Dr. C. Stocking [81]. Sequences were confirmed by DNA sequencing (Seqlab, Germany). Production of lentiviral vectors and transduction of G-2 cells was performed as previously described [12]. Commercially available shRNA constructs against mouse Ceacam1 mRNA using the pGIPZ vectorsystem were purchased at Open Biosystems (# RMM4532). The production of the lentiviral particles was performed according to the manufacturer instructions. GFP positivity was used to enrich cells that received the vector via fluorescence-activated cell-sorting.
Flow cytometry
Flow cytometric analyses were performed on FACSCanto flow cytometer (BD Biosciences) and fluorescence-activated cell sorting was performed on a FACSAria cell sorter (BD Bioscience). Cells were stained as described [12] with a PE-labeled anti-CEACAM1 antibody (Biolegend, #134505) and/or an APC-labeled anti-EpCAM antibody (Biolegend, #118214). A complete list of primary antibodies is given in Supplementary Table S2.
Quantitative real-time PCR
RNA from cultivated cells was purified using the innuPREP RNA Mini kit (Analytik Jena). Conversion of RNA into cDNA and quantitative real-time PCRs were performed as described [12]. Hspa8 mRNA was used for normalization. Relative normalized transcript levels were calculated using the 2−ΔΔCT algorithm. Quality and specificity of all PCR primers was validated by melting curve analyses and standard curve fitting. The primers used in this study are detailed in the Supplementary Methods section (Supplementary Table S1).
Immunofluorescence staining
Immunofluorescence staining was performed as described previously [12] with the antibodies listed in Supplementary Table S2. Secondary antibodies used for immunofluorescence staining were appropriate DyLight®- or Alexa®Dye- conjugates obtained from Invitrogen/Molecular Probes or Dianova. Confocal microscopy was performed on a LSM 510 Meta (Zeiss). Conventional fluorescence microscopy was performed on a DMI6000B (Leica).
Western blot analysis
Cells or tumors were lysed with Laemmli lysis buffer. Protein concentration was estimated using the BCA Protein Assay (Thermo Scientific). Equal amounts of protein were separated by SDS-PAGE and transferred onto nitrocellulose (Hybond, Amersham). The membrane was blocked with 5% skim milk in TBS-T for at least 60 min at RT. Primary antibodies were used as detailed in Supplementary Table S2. All secondary antibodies conjugated with horseradish peroxidase (Dianova) were used in dilutions of 1:5000–1:10000 in 5% skim milk (TBS-T). For the extraction of the nuclei, trypsinized cells were resuspended in cold DMEM + 10% FCS and centrifuged for 5 min at 300×g, 4°C. Cells pellets were washed with ice cold PBS, resuspended in Nuclei-Buffer I (10 mM Tris-HCl pH 7.9; 10 mM KCl; 15 mM MgCl2) and incubated for 10 min on ice. After centifugation (1000×g/ 2 min/4°C), the nuclei were lysed in Laemmli lysis buffer supplemented with 1 μl Benzonase (Roche) and incubated for 5 min at 95°C. Protein concentration was estimated using the BCA Protein Assay (Thermo Scientific). All buffers used here were supplemented with protease (cOmplete mini EDTA free, Roche) and phosphatase inhibitors (2 mM Na3VO4 and 5 mM NaF). For protein quantification, signal acquisition and measurement was performed on a Gel Doc XR+ System (BioRad).
Immunoprecipitation
Cells were harvested in ice cold CHAPS buffer (100 mM Tris-HCl pH 7.4, 50 mM NaCl, 1 mM EDTA, 1% NP40, 0,1% CHAPS) supplemented 1 μl Benzonase as well as with protease and phosphatase inhibitors. Cell lysates were incubated on ice for 30 min and cell debris removed by centrifugation (12000×g/15 min/4°C). Protein concentration was determined by Bradford (Bio-Rad) assay; 200 μg of the protein lysates were used for immunoprecipitation. 10 μl protein G sepharose beads (GE Healthcare) were incubated at 4°C overnight with 2 μg anti-mouse CEACAM1 antibody CC1 (eBioscience) or anti-E-cadherin antibodies (Cell Signaling) as positive control. Precleaning was performed with 20 μl protein G sepharose at 4°C overnight. The precleaned lysates were then incubated with appropriate antibody-coupled protein G sepharose beads over night at 4°C. Co-precipitation of SHP-2 with CEACAM1 was performed after pervanadate treatment of G-2 cells using biotinylated anti-CEACAM1 antibodies (eBioscience), streptaviding sepharose (ThermoFisher) and anti-SHP-2 antibody (Cell Signaling Technology), as described in [82]. A complete list of primary antibodies is given in Supplementary Table S2.
Invasion assay
Cells were harvested and kept for at least 3 hrs in serum-free DMEM. 2.5 × 104 cells in medium without FCS were transferred onto basement membrane coated inserts (CultreCoat® Medium BME Cell Invasion Assay, Trevigen). The lower chamber was filled with DMEM supplemented with 10% FCS as chemo-attractant, and the assay was incubated for 72 hrs at 37°C. Cells attached to the bottom of the inserts were fixed with 4% PFA and stained with DAPI. Cells were counted under a fluorescence microscope (Leica DMLB) at 10× magnification. Experiments were performed three times in triplicates.
Luciferase assay
1 × 105 cells in 12-wells were transfected with Renilla Luciferase (pGL4.73, 100 ng) and Firefly Luciferase (either M50 Super 8× TOPFlash or FOPFlash vector, 2 μg, kindly provided by Dr. Hanna Taipaleenmäki) using PEI (25 kD, 4 μl per well of a 1 μg/μl solution). Afterward, the cells were incubated at 37°C for 24 hrs before luciferase activity was measured using the Dual-Luciferase® Reporter Assay (Promega) in a GloMax 20/20 (Promega). Calculation of sample signal was performed as following:

Subsequently, samples were normalized on values of G-2scr or G-2empty respectively.
SHP-2 inhbition with NSC-87877
Parental G-2 cells were seeded in 6-wells (1.5 × 105 per well) and grown for 48 hrs in DMEM medium with 10% FCS. The culture medium was then replaced by DMEM/10% FCS supplemented with 100 μM NSC-87877 (Merck/Millipore, #565851, stock solution: 20 mM in H2O) or with appropriate H2O-volume as mock-control. The cells were finally allowed to grow for 24 hrs, 48 hrs or 72 hrs and harvested for subsequent analyses.
Software and statistical analyses
Numeric data were first entered in Excel (Office 2013, Microsoft) and processed with GraphPad PRISM 4.0 (GraphPad Software) for generation of the graphs as well as for statistical analyses. For the results of the qPCR, invasion- and luciferase-assays, we performed Student´s t-tests. Fisher´s exact test was employed to test the significance of the different metastasis rate in the mice. Standard error of these binominal datasets were calculated as following: SEp = sqrt [p (1–p) / n]. p values: *p ≥ 0.05; **p ≥ 0,01; ***p ≥ 0.001. Figures were generated with Inkscape (www.inkscape.org) and Photoshop (Adobe). Fluorescence and phase contrast pictures were processed with ImageJ. Confocal pictures were processed in Imaris 5.0 (Bitplane).
ACKNOWLEDGMENTS
We would like to thank Prof. J. Wienands and Dr. D. Dittmann for their kind scientific support during the revision of this manuscript. We also gratefully acknowledge excellent technical assistance by Annette Preuss, Renke Brixél, Krimhild Scheike, Christa Reinhold, Gundula Pilnitz-Stolze, and the staff of the animal facility at the Heinrich-Pette-Institute and the University Medical Center Hamburg-Eppendorf.
CONFLICTS OF INTEREST
The authors declare that they have no financial disclosure or conflicts of interest.
FUNDING
This project was funded by the Erich und Gertrud Roggenbuck-Stiftung to FW (grant “Modulation of tumor cell dissemination and metastasis in a mouse model for breast cancer by targeting EMT-associated genes”) as well as by the German Research Foundation (DFG) in the Research Priority Programme “SPP1190: The tumor-vessel-interface” to WD, CW, and AKH (grant DE212/24-1).
REFERENCES
1. Altobelli E, Lattanzi A. Breast cancer in European Union: An update of screening programmes as of March 2014 (Review). International journal of oncology. 2014; 45:1785–1792.
2. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nature reviews Molecular cell biology. 2014; 15:178–196.
3. Bastid J. EMT in carcinoma progression and dissemination: facts, unanswered questions, and clinical considerations. Cancer Metastasis Rev. 2012; 31:277–283.
4. Bonnomet A, Syne L, Brysse A, Feyereisen E, Thompson EW, Noel A, Foidart JM, Birembaut P, Polette M, Gilles C. A dynamic in vivo model of epithelial-to-mesenchymal transitions in circulating tumor cells and metastases of breast cancer. Oncogene. 2012; 31:3741–3753.
5. Pinto CA, Widodo E, Waltham M, Thompson EW. Breast cancer stem cells and epithelial mesenchymal plasticity - Implications for chemoresistance. Cancer letters. 2013; 341:56–62.
6. Jannasch K, Wegwitz F, Lenfert E, Maenz C, Deppert W, Alves F. Chemotherapy of WAP-T mouse mammary carcinomas aggravates tumor phenotype and enhances tumor cell dissemination. Int J Cancer. 2015; 137:25–36.
7. Tiligada E. Chemotherapy: induction of stress responses. Endocrine-related cancer. 2006; 13:S115–124.
8. Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, Andre S, Piccart M, Campone M, Brain E, Macgrogan G, Petit T, Jassem J, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nature medicine. 2009; 15:68–74.
9. Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, Chen ZQ, Liu XP, Xu ZD. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clinical cancer research. 2009; 15:2657–2665.
10. Schulze-Garg C, Lohler J, Gocht A, Deppert W. A transgenic mouse model for the ductal carcinoma in situ (DCIS) of the mammary gland. Oncogene. 2000; 19:1028–1037.
11. Heinlein C, Krepulat F, Lohler J, Speidel D, Deppert W, Tolstonog GV. Mutant p53(R270H) gain of function phenotype in a mouse model for oncogene-induced mammary carcinogenesis. Int J Cancer. 2008; 122:1701–1709.
12. Wegwitz F, Kluth MA, Manz C, Otto B, Gruner K, Heinlein C, Kuhl M, Warnecke G, Schumacher U, Deppert W, Tolstonog GV. Tumorigenic WAP-T mouse mammary carcinoma cells: a model for a self-reproducing homeostatic cancer cell system. PLoS One. 2010; 5:e12103.
13. Jannasch K, Dullin C, Heinlein C, Krepulat F, Wegwitz F, Deppert W, Alves F. Detection of different tumor growth kinetics in single transgenic mice with oncogene-induced mammary carcinomas by flat-panel volume computed tomography. Int J Cancer. 2009; 125:62–70.
14. Otto B, Gruner K, Heinlein C, Wegwitz F, Nollau P, Ylstra B, Pantel K, Schumacher U, Baumbusch LO, Martin-Subero JI, Siebert R, Wagener C, Streichert T, et al. Low- grade and high-grade mammary carcinomas in WAP-T transgenic mice are independent entities distinguished by Met expression. Int J Cancer. 2013; 132:1300–1310.
15. Otto B, Streichert T, Wegwitz F, Gevensleben H, Klatschke K, Wagener C, Deppert W, Tolstonog GV. Transcription factors link mouse WAP-T mammary tumors with human breast cancer. Int J Cancer. 2013; 132:1311–1322.
16. Pipas JM. SV40: Cell transformation and tumorigenesis. Virology. 2009; 384:294–303.
17. Hirner H, Gunes C, Bischof J, Wolff S, Grothey A, Kuhl M, Oswald F, Wegwitz F, Bosl MR, Trauzold A, Henne-Bruns D, Peifer C, Leithauser F, et al. Impaired CK1 delta activity attenuates SV40-induced cellular transformation in vitro and mouse mammary carcinogenesis in vivo. PLoS One. 2012; 7:e29709.
18. Lenfert E, Maenz C, Heinlein C, Jannasch K, Schumacher U, Pantel K, Tolstonog GV, Deppert W, Wegwitz F. Mutant p53 promotes epithelial-mesenchymal plasticity and enhances metastasis in mammary carcinomas of WAP-T mice. Int J Cancer. 2015; 136:E521–533.
19. Maenz C, Lenfert E, Pantel K, Schumacher U, Deppert W, Wegwitz F. Epithelial-mesenchymal plasticity is a decisive feature for the metastatic outgrowth of disseminated WAP-T mouse mammary carcinoma cells. BMC Cancer. 2015; 15:178.
20. Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nature reviews Immunology. 2006; 6:433–446.
21. Horst AK, Ito WD, Dabelstein J, Schumacher U, Sander H, Turbide C, Brummer J, Meinertz T, Beauchemin N, Wagener C. Carcinoembryonic antigen-related cell adhesion molecule 1 modulates vascular remodeling in vitro and in vivo. J Clin Invest. 2006; 116:1596–1605.
22. Ludewig P, Flachsbarth K, Wegscheid C, Tiegs G, Richard DG, Wagener DC, Bartsch DU, Horst AK. CEACAM1 confers resistance towards oxygen-induced vessel damage in a mouse model of retinopathy of prematurity. Investigative ophthalmology & visual science. 2014.
23. Najjar SM. Regulation of insulin action by CEACAM1. Trends Endocrinol Metab. 2002; 13:240–245.
24. Fiset A, Xu E, Bergeron S, Marette A, Pelletier G, Siminovitch KA, Olivier M, Beauchemin N, Faure RL. Compartmentalized CDK2 is connected with SHP-1 and beta-catenin and regulates insulin internalization. Cell Signal. 2011; 23:911–919.
25. Poy MN, Yang Y, Rezaei K, Fernstrom MA, Lee AD, Kido Y, Erickson SK, Najjar SM. CEACAM1 regulates insulin clearance in liver. Nat Genet. 2002; 30:270–276.
26. Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen BS, Melum E, Pertel T, Clayton KL, Raab M, Chen Q, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2014.
27. Chen D, Iijima H, Nagaishi T, Nakajima A, Russell S, Raychowdhury R, Morales V, Rudd CE, Utku N, Blumberg RS. Carcinoembryonic antigen-related cellular adhesion molecule 1 isoforms alternatively inhibit and costimulate human T cell function. J Immunol. 2004; 172:3535–3543.
28. Chen Z, Chen L, Qiao SW, Nagaishi T, Blumberg RS. Carcinoembryonic antigen-related cell adhesion molecule 1 inhibits proximal TCR signaling by targeting ZAP-70. J Immunol. 2008; 180:6085–6093.
29. Hosomi S, Chen Z, Baker K, Chen L, Huang YH, Olszak T, Zeissig S, Wang JH, Mandelboim O, Beauchemin N, Lanier LL, Blumberg RS. CEACAM1 on activated NK cells inhibits NKG2D-mediated cytolytic function and signaling. European journal of immunology. 2013; 43:2473–2483.
30. Tsukada M, Wakai T, Matsuda Y, Korita PV, Shirai Y, Ajioka Y, Hatakeyama K, Kubota M. Loss of carcinoembryonic antigen-related cell adhesion molecule 1 expression predicts metachronous pulmonary metastasis and poor survival in patients with hepatoblastoma. J Pediatr Surg. 2009; 44:1522–1528.
31. Wang JL, Sun SZ, Qu X, Liu WJ, Wang YY, Lv CX, Sun JZ, Ma R. Clinicopathological significance of CEACAM1 gene expression in breast cancer. Chin J Physiol. 2011; 54:332–338.
32. Shinozuka K, Uzawa K, Fushimi K, Yamano Y, Shiiba M, Bukawa H, Yokoe H, Tanzawa H. Downregulation of carcinoembryonic antigen-related cell adhesion molecule 1 in oral squamous cell carcinoma: correlation with tumor progression and poor prognosis. Oncology. 2009; 76:387–397.
33. Cruz PV, Wakai T, Shirai Y, Yokoyama N, Hatakeyama K. Loss of carcinoembryonic antigen-related cell adhesion molecule 1 expression is an adverse prognostic factor in hepatocellular carcinoma. Cancer. 2005; 104:354–360.
34. Laack E. Expression of CEACAM1 in Adenocarcinoma of the Lung: A Factor of Independent Prognostic Significance. Journal of Clinical Oncology. 2002; 20:4279–4284.
35. Thom I, Schult-Kronefeld O, Burkholder I, Schuch G, Andritzky B, Kastendieck H, Edler L, Wagener C, Bokemeyer C, Schumacher U, Laack E. Expression of CEACAM-1 in pulmonary adenocarcinomas and their metastases. Anticancer Res. 2009; 29:249–254.
36. Thies A, Moll I, Berger J, Wagener C, Brummer J, Schulze HJ, Brunner G, Schumacher U. CEACAM1 expression in cutaneous malignant melanoma predicts the development of metastatic disease. J Clin Oncol. 2002; 20:2530–2536.
37. Nguyen T, Chen CJ, Shively JE. Phosphorylation of CEACAM1 molecule by calmodulin kinase IID in a three-dimensional model of mammary gland lumen formation. The Journal of biological chemistry. 2014; 289:2934–2945.
38. Jin L, Li Y, Chen CJ, Sherman MA, Le K, Shively JE. Direct interaction of tumor suppressor CEACAM1 with beta catenin: identification of key residues in the long cytoplasmic domain. Exp Biol Med (Maywood). 2008; 233:849–859.
39. Yokoyama S, Chen CJ, Nguyen T, Shively JE. Role of CEACAM1 isoforms in an in vivo model of mammary morphogenesis: mutational analysis of the cytoplasmic domain of CEACAM1–4S reveals key residues involved in lumen formation. Oncogene. 2007; 26:7637–7646.
40. Kirshner J, Schumann D, Shively JE. CEACAM1, a cell-cell adhesion molecule, directly associates with annexin II in a three-dimensional model of mammary morphogenesis. The Journal of biological chemistry. 2003; 278:50338–50345.
41. Kirshner J, Chen CJ, Liu P, Huang J, Shively JE. CEACAM1–4S, a cell-cell adhesion molecule, mediates apoptosis and reverts mammary carcinoma cells to a normal morphogenic phenotype in a 3D culture. Proc Natl Acad Sci USA. 2003; 100:521–526.
42. Chen CJ, Kirshner J, Sherman MA, Hu W, Nguyen T, Shively JE. Mutation analysis of the short cytoplasmic domain of the cell-cell adhesion molecule CEACAM1 identifies residues that orchestrate actin binding and lumen formation. The Journal of biological chemistry. 2007; 282:5749–5760.
43. Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. Journal of mammary gland biology and neoplasia. 2010; 15:117–134.
44. Kajiguchi T, Katsumi A, Tanizaki R, Kiyoi H, Naoe T. Y654 of beta-catenin is essential for FLT3/ITD-related tyrosine phosphorylation and nuclear localization of beta-catenin. European journal of haematology. 2012; 88:314–320.
45. Xi Y, Wei Y, Sennino B, Ulsamer A, Kwan I, Brumwell AN, Tan K, Aghi MK, McDonald DM, Jablons DM, Chapman HA. Identification of pY654-beta-catenin as a critical co-factor in hypoxia-inducible factor-1alpha signaling and tumor responses to hypoxia. Oncogene. 2013; 32:5048–5057.
46. Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. The EMBO journal. 2012; 31:2714–2736.
47. Li C, Chen CJ, Shively JE. Mutational analysis of the cytoplasmic domain of CEACAM1–4L in humanized mammary glands reveals key residues involved in lumen formation: stimulation by Thr-457 and inhibition by Ser-461. Experimental cell research. 2009; 315:1225–1233.
48. Li Y, Shively JE. CEACAM1 regulates Fas-mediated apoptosis in Jurkat T-cells via its interaction with beta-catenin. Experimental cell research. 2013; 319:1061–1072.
49. Leung N, Turbide C, Balachandra B, Marcus V, Beauchemin N. Intestinal tumor progression is promoted by decreased apoptosis and dysregulated Wnt signaling in Ceacam1-/- mice. Oncogene. 2008; 27:4943–4953.
50. Zhu G, Wang Y, Huang B, Liang J, Ding Y, Xu A, Wu W. A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene. 2012; 31:1001–1012.
51. Sadot E, Conacci-Sorrell M, Zhurinsky J, Shnizer D, Lando Z, Zharhary D, Kam Z, Ben-Ze’ev A, Geiger B. Regulation of S33/S37 phosphorylated beta-catenin in normal and transformed cells. Journal of cell science. 2002; 115:2771–2780.
52. Timmerman I, Hoogenboezem M, Bennett AM, Geerts D, Hordijk PL, van Buul JD. The tyrosine phosphatase SHP2 regulates recovery of endothelial adherens junctions through control of beta-catenin phosphorylation. Mol Biol Cell. 2012; 23:4212–4225.
53. Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, Sebti SM, Lawrence NJ, Wu J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Molecular pharmacology. 2006; 70:562–570.
54. Gerstel D, Wegwitz F, Jannasch K, Ludewig P, Scheike K, Alves F, Beauchemin N, Deppert W, Wagener C, Horst AK. CEACAM1 creates a pro-angiogenic tumor microenvironment that supports tumor vessel maturation. Oncogene. 2011; 30:4275–4288.
55. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q, Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast cancer research and treatment. 2010; 123:725–731.
56. Gyorffy B, Surowiak P, Budczies J, Lanczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One. 2013; 8:e82241.
57. Beauchemin N, Arabzadeh A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013; 32:643–671.
58. Fiori V, Magnani M, Cianfriglia M. The expression and modulation of CEACAM1 and tumor cell transformation. Annali dell’Istituto superiore di sanita. 2012; 48:161–171.
59. Tilki D, Irmak S, Oliveira-Ferrer L, Hauschild J, Miethe K, Atakaya H, Hammerer P, Friedrich MG, Schuch G, Galalae R, Stief CG, Kilic E, Huland H, et al. CEA-related cell adhesion molecule-1 is involved in angiogenic switch in prostate cancer. Oncogene. 2006; 25:4965–4974.
60. Oliveira-Ferrer L, Tilki D, Ziegeler G, Hauschild J, Loges S, Irmak S, Kilic E, Huland H, Friedrich M, Ergun S. Dual role of carcinoembryonic antigen-related cell adhesion molecule 1 in angiogenesis and invasion of human urinary bladder cancer. Cancer Res. 2004; 64:8932–8938.
61. Zhou CJ, Liu B, Zhu KX, Zhang QH, Zhang TG, Xu WH, Wang HB, Yu WH, Qu YD, Wang HJ, Wu HL, Sun SZ, Guo JQ. The different expression of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) and possible roles in gastric carcinomas. Pathol Res Pract. 2009; 205:483–489.
62. Bamberger AM, Riethdorf L, Nollau P, Naumann M, Erdmann I, Gotze J, Brummer J, Schulte HM, Wagener C, Loning T. Dysregulated expression of CD66a (BGP, C-CAM), an adhesion molecule of the CEA family, in endometrial cancer. Am J Pathol. 1998; 152:1401–1406.
63. Riethdorf L, Lisboa BW, Henkel U, Naumann M, Wagener C, Loning T. Differential expression of CD66a (BGP), a cell adhesion molecule of the carcinoembryonic antigen family, in benign, premalignant, and malignant lesions of the human mammary gland. J Histochem Cytochem. 1997; 45:957–963.
64. Luo W, Tapolsky M, Earley K, Wood CG, Wilson DR, Logothetis CJ, Lin SH. Tumor-suppressive activity of CD66a in prostate cancer. Cancer Gene Ther. 1999; 6:313–321.
65. Lin SH, Pu YS. Function and therapeutic implication of C-CAM cell-adhesion molecule in prostate cancer. Semin Oncol. 1999; 26:227–233.
66. Huang J, Hardy JD, Sun Y, Shively JE. Essential role of biliary glycoprotein (CD66a) in morphogenesis of the human mammary epithelial cell line MCF10F. Journal of cell science. 1999; 112:4193–4205.
67. Huang J, Simpson JF, Glackin C, Riethorf L, Wagener C, Shively JE. Expression of biliary glycoprotein (CD66a) in normal and malignant breast epithelial cells. Anticancer Res. 1998; 18:3203–3212.
68. Liu Q, Yang YM, Zhang QH, Zhang TG, Zhou Q, Zhou CJ. Inhibitor of differentiation is overexpressed with progression of benign to malignant lesions and related with carcinoembryonic antigen-related cell adhesion molecule 1 distribution in mammary glands. Ann Diagn Pathol. 2011; 15:30–36.
69. Neumaier M, Paululat S, Chan A, Matthaes P, Wagener C. Biliary glycoprotein, a potential human cell adhesion molecule, is down-regulated in colorectal carcinomas. Proc Natl Acad Sci USA. 1993; 90:10744–10748.
70. Sundberg U, Beauchemin N, Obrink B. The cytoplasmic domain of CEACAM1-L controls its lateral localization and the organization of desmosomes in polarized epithelial cells. Journal of cell science. 2004; 117:1091–1104.
71. Cadigan KM, Peifer M. Wnt signaling from development to disease: insights from model systems. Cold Spring Harbor perspectives in biology. 2009; 1:a002881.
72. Coluccia AM, Vacca A, Dunach M, Mologni L, Redaelli S, Bustos VH, Benati D, Pinna LA, Gambacorti-Passerini C. Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through its tyrosine phosphorylation. The EMBO journal. 2007; 26:1456–1466.
73. Abou-Rjaily GA, Lee SJ, May D, Al-Share QY, Deangelis AM, Ruch RJ, Neumaier M, Kalthoff H, Lin SH, Najjar SM. CEACAM1 modulates epidermal growth factor receptor—mediated cell proliferation. J Clin Invest. 2004; 114:944–952.
74. Brummer J, Neumaier M, Gopfert C, Wagener C. Association of pp60c-src with biliary glycoprotein (CD66a), an adhesion molecule of the carcinoembryonic antigen family downregulated in colorectal carcinomas. Oncogene. 1995; 11:1649–1655.
75. Najjar SM, Blakesley VA, Li Calzi S, Kato H, LeRoith D, Choice CV. Differential phosphorylation of pp120 by insulin and insulin-like growth factor-1 receptors: role for the C-terminal domain of the beta-subunit. Biochemistry. 1997; 36:6827–6834.
76. Lu R, Pan H, Shively JE. CEACAM1 negatively regulates IL-1beta production in LPS activated neutrophils by recruiting SHP-1 to a SYK-TLR4-CEACAM1 complex. PLoS pathogens. 2012; 8:e1002597.
77. Nouvion AL, Oubaha M, Leblanc S, Davis EC, Jastrow H, Kammerer R, Breton V, Turbide C, Ergun S, Gratton JP, Beauchemin N. CEACAM1: a key regulator of vascular permeability. Journal of cell science. 2010; 123:4221–4230.
78. Simoneau M, Coulombe G, Vandal G, Vezina A, Rivard N. SHP-1 inhibits beta-catenin function by inducing its degradation and interfering with its association with TATA-binding protein. Cell Signal. 2011; 23:269–279.
79. Lee SH, Peng IF, Ng YG, Yanagisawa M, Bamji SX, Elia LP, Balsamo J, Lilien J, Anastasiadis PZ, Ullian EM, Reichardt LF. Synapses are regulated by the cytoplasmic tyrosine kinase Fer in a pathway mediated by p120catenin, Fer, SHP-2, and beta-catenin. The Journal of cell biology. 2008; 183:893–908.
80. Bickert T, Marshall RP, Zhang Z, Ludewig P, Binder M, Klinke A, Rottbauer W, Amling M, Wagener C, Ito WD, Horst AK. Acceleration of collateral development by carcinoembryonic antigen-related cell adhesion molecule 1 expression on CD11b/(+)Gr-1(+) myeloid cells--brief report. Arterioscler Thromb Vasc Biol. 2012; 32:2566–2568.
81. Weber K, Bartsch U, Stocking C, Fehse B. A multicolor panel of novel lentiviral “gene ontology” (LeGO) vectors for functional gene analysis. Molecular therapy. 2008; 16:698–706.
82. Huber M, Izzi L, Grondin P, Houde C, Kunath T, Veillette A, Beauchemin N. The carboxyl-terminal region of biliary glycoprotein controls its tyrosine phosphorylation and association with protein-tyrosine phosphatases SHP-1 and SHP-2 in epithelial cells. J Biol Chem. 1999; 274:335–344.