
INTRODUCTION
Brain tumors are the most common pediatric solid tumors and the leading cause of cancer mortality in children. High-dose ionizing radiation and rare inherited syndromes are the only established risk factors for brain tumors, causing a small proportion of cases [1]. Thus, brain tumors are considered as multifactorial disorders resulting from progressive accumulation of genetic and epigenetic alterations in concert with environmental exposures. It has been suggested that genetic polymorphisms in four main pathways including DNA repair, cell cycle, metabolism, and inflammation play an important role in brain carcinogenesis [2]. Therefore, candidate gene-association studies of adult brain tumors have mainly focused on these four hypothesized pathways to identify genetic susceptibility factors for adult brain tumors [3–9].
Whereas a considerable number of candidate gene-association studies are available on adult brain tumors, very few and small genetic studies have been performed on brain tumors in children and adolescents [10–13], due to difficulties in collecting a sufficient number of DNA samples. Hence, the role of genetic polymorphisms in pediatric brain tumor (PBT) etiology is largely unknown. However, some studies have found similar genetic mutation patterns for adult and pediatric brain tumor progression within specific histological types [14–18]. These similarities in prognostic factors provide an important starting point for identifying candidate genetic risk factors for PBTs; that might be similar to those involved in adult brain tumorigenesis [19]. Moreover, in our previous genetic association study of PBT risk, we concluded that single nucleotide polymorphisms (SNPs) identified by genome-wide association studies (GWAS) on adult glioma might also be associated with the risk of brain tumors in children [20].
The aim of this study, which represents the largest series of PBT cases to date, was to determine whether the SNPs identified by candidate gene-association studies on adult brain tumors, involved in the four main pathways discussed above, are also related to pediatric brain tumor risk.
RESULTS
We successfully genotyped 63 SNPs in 245 cases and 489 controls. The distributions of allele frequencies in 3 SNPs (rs4444903, rs9288516, and rs61754966) were not in agreement with HWE (p < 0.001) and therefore these SNPs were dropped from analyses. As shown in Table 1, the age and sex distributions were similar in cases and controls.
Table 1: Characteristics of cases and controls
Characteristics |
Cases |
Astrocytomas |
Non-astrocytomas |
Controls |
|---|---|---|---|---|
No. of participants |
245 |
134 |
111 |
489 |
Sex |
||||
Males |
136 (56%) |
74 (55%) |
62 (56%) |
261 (53%) |
Females |
109 (44%) |
60 (45%) |
49 (44%) |
228 (47%) |
Age-group |
||||
7–9 years old |
48 (20%) |
28 (21%) |
20 (18%) |
112 (23%) |
10–14 years old |
108 (44%) |
60 (45%) |
48 (43%) |
219 (45%) |
15–19 years old |
89 (36%) |
46 (34%) |
43 (39%) |
158 (32%) |
Country |
||||
Sweden |
106 (43%) |
48 (36%) |
58 (52%) |
174 (36%) |
Norway |
24 (10%) |
15 (11%) |
9 (8%) |
62 (13%) |
Denmark |
62 (25%) |
37 (28%) |
25 (23%) |
134 (27%) |
Switzerland |
53 (22%) |
34 (25%) |
19 (17%) |
119(24%) |
Type of tumor |
||||
Astrocytoma (IIIb) |
134 (55%) |
|||
Pilocytic astrocytoma |
93 |
|||
Supependymal giant cell astrocytoma |
5 |
|||
Pleomorphic xanthoastrocytoma |
4 |
|||
Diffuse astrocytoma |
13 |
|||
Anaplastic astrocytoma |
11 |
|||
Fibrillary astrocytoma |
2 |
|||
Glioblastoma |
5 |
|||
Giant cell glioblastoma |
1 |
|||
Other gliomas (IIId) |
20 (8%) |
|||
Malignant glioma |
11 |
|||
Oligoastrocytoma |
2 |
|||
Oligodendroglioma |
6 |
|||
Anaplastic oligodendroglioma |
1 |
|||
Ependymoma (IIIa) |
19 (8%) |
|||
Subependymoma |
2 |
|||
Choroid plexus papilloma |
4 |
|||
Choroid plexus caricinoma |
1 |
|||
Ependymoma |
7 |
|||
Papillary ependymoma |
1 |
|||
Anaplastic ependymoma |
4 |
|||
Intracranial embryonal tumors (IIIc) |
7 (3%) |
|||
CNS primitive neuroectodermal tumor |
6 |
|||
Neuroepithelioma |
1 |
|||
Other specified intracranial neoplasms (IIIe) |
49 (20%) |
|||
Germinoma |
7 |
|||
Yolk sac tumor |
1 |
|||
Teratoma, mature |
1 |
|||
Haemangioblastoma |
1 |
|||
Desmoplastic infantile ganglioglioma |
2 |
|||
Dysembryoplastic neuroepithelial tumor |
6 |
|||
Ganglioglioma |
26 |
|||
Anaplastic ganglioglioma |
1 |
|||
Centrol neurocytoma |
3 |
|||
Neurilemoma |
1 |
|||
Unspecified intracranial neoplasm (IIIf) |
16 (6%) |
a: Restricted to ICD-O-3 location C71, subclassified according to WHO histological subclassification, 2007; patients with neurofibromatosis and tuberous sclerosis were excluded.
As Table 2 illustrates, the A alleles of EGFR rs730437 (ORDOM 0.59 [95% CI 0.42-0.83], p = 0.002) and EGFR rs11506105 (ORDOM 0.71 [95% CI 0.51–0.98], p = 0.036) involved in cell cycle pathway were associated with decreased susceptibility to PBTs, whereas the A allele of ERCC1 rs3212986 (ORDOM 1.53 [95% CI 1.11–2.09], p = 0.009) involved in DNA repair pathway was associated with an increased risk of these tumors. Moreover, the interactions between these SNPs and covariates including age, sex and country were not significant.
Table 2: Summary results for SNPs associated with pediatric brain tumors
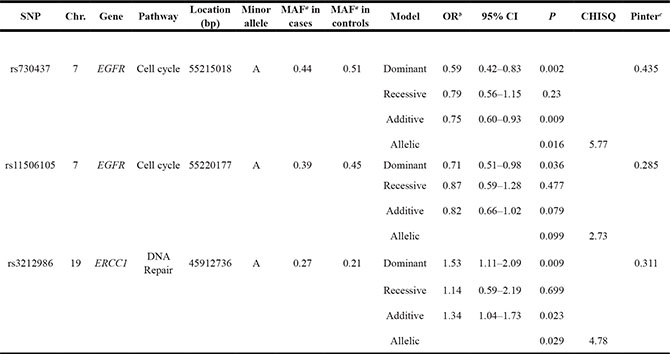
a: MAF=Minor Allele Frequency b: OR adjusted for age, sex, and country c: P value for interactions between SNPs and demographic variables including age, sex and country.
The stratified analyses of two histological subtypes indicated that the protective effect of EGFR rs730437 remained significant in both patients with astrocytoma and non-astrocytoma tumor subtypes (pDOM=0.018 and pDOM=0.014, respectively), whereas the risk effect of ERCC1 rs3212986 was more evident in patients with astrocytoma subtype (pDOM=0.002). Moreover, a decreased risk of astrocytoma subtype associated with the C alleles of CHAF1A rs243341 and rs2992 as well as the T allele of XRCC1 rs25487 involved in DNA repair pathway was detected (pDOM=0.040, pDOM=0.049, and pDOM=0.033, respectively). In addition, the stratified analyses showed an increased risk of non-astrocytoma tumor subtype associated with the C alleles of EGFR rs9642393, EME1 rs12450550, and ATM rs170548, and the T allele of GLTSCR1 rs1035938 (pREC=0.021, pREC=0.001, pDOM=0.041, and pREC=0.027, respectively) as well as a decreased risk of this subtype associated with the A allele of XRCC4 rs7721416 and the C allele of XRCC4 rs2662242 (DNA repair pathway) (pREC=0.032 and pREC=0.024, respectively) (Tables 3 and 4).
Table 3: Summary results for SNPs associated with astrocytoma subtype
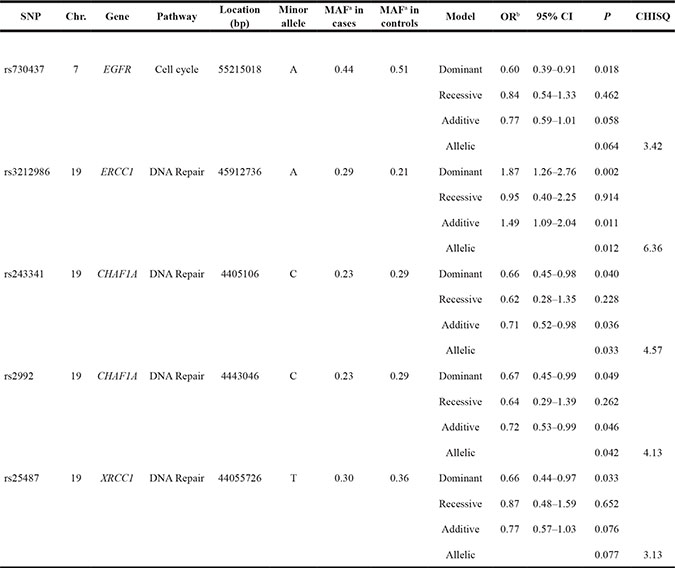
a: MAF=Minor Allele Frequency b: OR adjusted for age, sex, and country.
Table 4: Summary results for SNPs associated with non-astrocytoma subtype
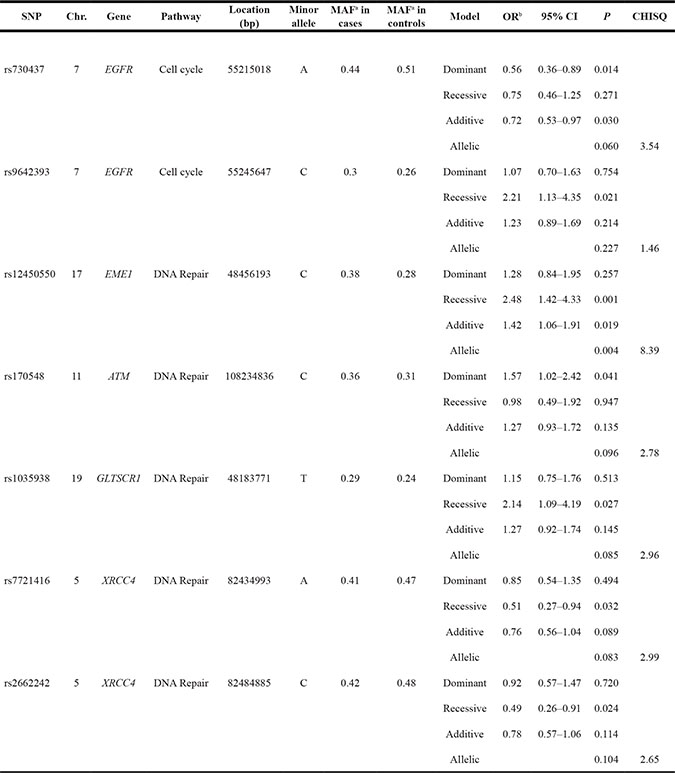
a: MAF=Minor Allele Frequency b: OR adjusted for age, sex, and country.
Non-significant findings as well as the significant findings with wide confidence intervals are shown in the online appendix Tables S1–S3.
Strong LD (D´ ≥ 0.95) was observed between four genotyped SNPs in EGFR (rs730437, rs11506105, rs4947986, and rs3752651) in which five haplotypes with frequency of > 1% were detected. In the EGFR block, the distribution of haplotypes was suggestively different between PBT patients and controls (χ2 = 8.1, df = 4, p = 0.089) and the most common haplotype (CGGT) had a significant risk effect compared with the other haplotypes combined (OR 1.31 [95% CI 1.05–1.64], p = 0.017) (Table 5). In the astrocytoma subgroup, the CHAF1A block (rs243341, rs105038, rs243356, and rs2992) with three haplotypes with frequency of >1% was found; however, the distribution of haplotypes in this haploblock was not significantly different between patients and controls (χ2 = 2.95, df = 2, p = 0.229). Moreover, in the non-astrocytoma subgroup, two haploblocks including ATM (rs228599, rs664143, rs170548, rs3092993, and rs3092992) and XRCC4 (rs7721416 and rs2662242) with, respectively, five and three haplotypes with frequency of > 1% were observed; nevertheless, in none of these haploblocks, the distribution of haplotypes was significantly different between patients and controls (χ2 = 1.92, df = 4, p = 0.751 and χ2 = 2.98, df = 2, p = 0.226, respectively).
Table 5: Haplotype analysis of SNPs in EGFR
SNPs |
Haplotype |
Frequency |
ORa |
95% CI |
P |
|---|---|---|---|---|---|
EGFR: rs730437, rs11506105, rs4947986, rs3752651 |
AAAC |
0.21 |
0.92 |
0.69–1.23 |
0.569 |
AAAT |
0.08 |
0.70 |
0.44–1.11 |
0.126 |
|
AAGT |
0.14 |
0.82 |
0.59–1.15 |
0.247 |
|
AGGT |
0.06 |
0.69 |
0.41–1.15 |
0.153 |
|
CGGT |
0.51 |
1.31 |
1.05–1.64 |
0.017 |
a: Odds ratio for haplotype compared with all other haplotypes adjusted for age, sex, and country.
As described above, 248 testing procedures were performed. When the Bonferroni correction is applied, the reference p value is 0.0002 for an experiment-wide significance level of 0.05, and 0.0004 for a significance level of 0.10; none of the reported associations met these limits. As summarized in Table S4, no significant SNP-SNP interactions were observed (p < 0.001).
DISCUSSION
This study suggests that SNPs involved in cell cycle and DNA repair pathways previously linked with brain tumors in adults are also associated with pediatric brain tumorigenesis. The results indicate that the A alleles of EGFR rs730437 and rs11506105 may decrease susceptibility to PBTs, whereas the A allele of ERCC1 rs3212986 may increase the risk of these tumors. Moreover, the C alleles of CHAF1A rs243341 and rs2992 as well as the T allele of XRCC1 rs25487 were associated with a decreased risk of astrocytoma subtype. In addition, an increased risk of non-astrocytoma tumor subtype associated with the C alleles of EGFR rs9642393, EME1 rs12450550, and ATM rs170548, and the T allele of GLTSCR1 rs1035938 as well as a decreased risk of this subtype associated with the A allele of XRCC4 rs7721416 and the C allele of XRCC4 rs2662242 were detected.
Genetic and epigenetic aberrations involved in cell cycle and DNA repair pathways have been proposed to play a role in PBT pathogenesis and progression [21–25]. Overexpression of the epidermal growth factor receptor (EGFR), which plays an important role in cell growth and development through the cell cycle pathway, is shown to be common and correlated with tumor grade in pediatric brain tumors [26, 27] . The function of genetic variants of EGFR is still unclear; however, intronic variations of EGFR found in this study may affect EGFR expression and play a role in pediatric brain tumorigenesis.
DNA repair is an important mechanism to maintain genomic stability and its functional failure may lead to carcinogenesis. Somatic variations of DNA repair genes have been identified in PBTs [28–30]; however, no study has investigated the role of DNA repair gene variations in PBT etiology. In this study, we examined the association between PBT risk and 37 SNPs in 17 DNA repair genes, shown to be associated with risk of adult brain tumors. Of these, 9 SNPs in 7 DNA repair genes (ERCC1, CHAF1A, XRCC1,EME1, ATM, GLTSCR1, and XRCC4) were also associated with susceptibility to PBTs. A few studies have previously indicated that DNA repair gene alterations may increase the predisposition to cancer formation in children [31, 32]. Moreover, these variations might predict the tumor drug sensitivity and the treatment outcome [33].
The ERCC1 gene encodes excision repair cross-complementing group 1 which is involved in DNA nucleotide excision repair (NER) pathway [34]. The result of association between the A allele of ERCC1 rs3212986 and increased risk of PBTs is in line with our finding in a meta-analysis of this polymorphism in adult brain tumors [3]. XRCC1 which encodes an enzyme named X-ray cross-complementing group 1, plays an important role in base excision repair pathway (BER) [35]. Chromatin assembly factor 1, subunit A (CHAF1A) is involved in DNA mismatch repair (MMR) during the correction of DNA replication errors [36]. XRCC4 encodes X-ray repair complementing defective repair in Chinese hamster cells 4 that functions in the repair of DNA double-strand breaks (DSBs) produced by ionizing radiation and restriction enzymes [37].
EME1 encodes essential meiotic structure-specific endonuclease 1 which forms an endonuclease complex with methyl methanesulfonate-sensitive UV-sensitive 81 protein (MUS81) and plays an important role in DNA repair and maintaining of genome integrity [38]. ATM encodes a protein kinase which is a member of phosphatidyl inositol 3-kinase-like kinase (PIKK) family. ATM is a key regulator of cell cycle checkpoint signaling pathways which responds to DNA strand breaks by inducing cell-cycle arrest and hence facilitates DNA repair [39]. GLTSCR1 stands for glioma tumor suppressor candidate region 1 and GLTSCR1 rs1035938 polymorphism alters a CpG site within the 5′ CpG island of the gene. This germ-line alteration might affect the transcription of GLTSCR1 and other candi-date genes in the region [40].
The present study was conducted based on the largest series of pediatric brain tumor cases to date with the purpose to investigate the association between genetic polymorphisms along with four main pathways hypothesized to be involved in brain tumorigenesis and PBT risk. Although investigating the effect of environmental risk factors was not the aim of our study, the fact that we did not take into consideration the possible gene-environment interactions is a limitation of this work. Moreover, due to shortage of large enough pediatric patient materials with access to DNA samples, we were not able to replicate our findings. Therefore, additional studies are necessary to validate these results, explore the mechanisms through which these genetic polymorphisms influence cancer susceptibility, and to investigate gene-environment interactions underling risk of PBTs.
Since this study describes the association between genetic variations and risk of pediatric brain tumors, it provides an important starting point for understanding the mechanisms behind the PBT etiology that in turn might lead to identifying clinically meaningful genetic risk and protective factors and eventually cancer prevention and treatment.
In conclusion, the present study indicates that while the minor alleles of polymorphisms in ERCC1 may increase the risk of PBTs, the minor alleles of SNPs in EGFR are associated with decreased susceptibility to these tumors. Furthermore, polymorphisms in CHAF1A and XRCC1 are associated with the risk of astrocytoma, whereas SNPs in EME1, ATM, GLTSCR1, and XRCC4 are associated with susceptibility to non-astrocytoma subtypes. Therefore, genetic polymorphisms in cell cycle and DNA repair pathways associated with susceptibility to adult brain tumors seem also to be associated with PBT risk suggesting that pediatric and adult brain tumors might share similar etiological pathways.
MATERIALS AND METHODS
Study subjects and procedures
This population-based case-control study is based on the Cefalo study, a large international study of brain tumors in children and adolescents conducted in Sweden, Denmark, Norway, and Switzerland. Details of the study methods have been described previously [41]. Briefly, all children aged 7–19 years during the period between 1 April 2004 and 31 August 2008, diagnosed with a primary intracranial brain tumor defined according to the International Classification of Childhood Cancer, third edition (ICCC-3), group III [42], restricted to ICD-O-3 location C71, were considered as case subjects. Cases were subclassified according to the fourth edition of the World Health Organization (WHO) classification of tumors of the central nervous system [43]. Participants with neurofibromatosis or tuberous sclerosis were excluded from analyses. Medulloblastoma cases were excluded here since they will be included in a separate study. Controls were randomly selected from the population registers and matched to the cases by age, sex, and geographic region. A total of 352 (82%) cases and 646 (71%) controls participated in the interviews. The study was approved by the National data protection boards and ethical committees in all participating countries, and written informed consent was obtained from all participants and/or their parents.
Saliva collection and DNA extraction were performed using the Oragene self-collection kit (DNA Genotek, Ottawa, ON, Canada) and the DNA yield was quantitated using PicoGreen (Invitrogen, Carlsbad, CA, USA), according to the standard protocols. This study, in total, included saliva DNA of 245 cases and 489 controls. Distribution of diagnostic subtypes among cases were 134 astrocytoma, 19 ependymoma, 7 intracranial embryonal tumors, 20 other gliomas, 49 other specified intracranial neoplasms, and 16 with unspecified intracranial neoplasms (Table 1).
Selection of candidate SNPs and genotyping
The PubMed database was searched (up to December 2012) using combinations of the following terms: ‘brain tumor’, ‘single nucleotide polymorphism’, ‘association’, ‘gene’, ‘risk’, ‘case control’, ‘susceptibility’, and ‘polymorphism’ to identify all the published peer-reviewed candidate gene-association studies of brain tumors. All the statistically significant SNPs reported by childhood brain tumor genetic association studies [11–13] as well as the SNPs reported by at least two candidate gene-association studies as being statistically associated with the risk of adult brain tumors [3–9, 44–49] were selected for genotyping.
Of 68 selected SNPs, 63 SNPs were satisfactorily genotyped (success rate ≥ 80%). Genotyping was performed at the Mutation Analysis core Facility (MAF), Clinical Research Centre, Huddinge University Hospital, Stockholm, Sweden, with staff blinded to sample status, using the Sequenom iPlex Gold platform with matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry. The average success rate was 97% and the concordance rate for duplicate genotyping was 100%.
Statistical analyses
The χ2 goodness-of-fit test was used to examine the consistency of allele frequencies with Hardy-Weinberg equilibrium (HWE) among the controls and p < 0.001 was considered statistically significant. Odds ratios (ORs) and corresponding 95% confidence intervals (CIs) were calculated using unconditional logistic regression modelling to evaluate the association between each SNP and the risk of PBT based on the Cochran–Armitage trend test of additivity (trend) as well as dominant (DOM) and recessive (REC) models, with adjustment for age, sex, and country. We applied the χ2 test to compare the allelic frequencies of the genotyped SNPs between cases and controls as well as the Wald test to evaluate the significance of interactions between SNPs and demographic variables including age, sex and country. SNP-SNP interactions were investigated for pairwise combinations of all the SNPs involved in the same pathway and p < 0.001 was set as the significance level of interaction analyses. Stratified analyses were performed by astrocytoma alone and the other tumor types combined (including ependymoma, intracranial embryonal tumors (except medulloblastoma), other gliomas, other specified intracranial neoplasms, and unspecified intracranial neoplasm); in order to increase the statistical power. To measure the linkage disequilibrium (LD) between the genotyped SNPs, D´ was calculated. Haploblocks were defined based on the default LD block parameters in Haploview v4.2. Haplotype analyses were performed for the haplotype blocks harboring the SNPs that were found to be associated with PBTs. Haplotypes with a frequency > 1% were considered in the blocks with different haplotype distribution between cases and controls (p < 0.1). Although selection of SNPs for the analyses was based on a priori knowledge from candidate gene-association studies on adults, the possibility of false-positive findings was considered by providing the reference p value for an experiment-wide significance with Bonferroni correction (0.0002 for an experiment-wide significance level of 0.05, and 0.0004 for a significance level of 0.10). The analyses were carried out using PLINK v1.07 [50] and SAS statistical software version 9.3 (SAS Institute, Inc., Cary, NC, USA).
ACKNOWLEDGMENTS
We gratefully acknowledge collaboration with and support from clinicians and other hospital staff in all countries, as well as assistance from national and local cancer registers with identification of patients. We also acknowledge the skillful work of the research nurses, interviewers, and research assistants in all countries. Finally, we acknowledge all those with whom we have collaborated previously within the CEFALO study. Please see Aydin et al. for an exhaustive list [41].
URLs
PLINK: http://pngu.mgh.harvard.edu/~purcell/plink/
SAS: http://www.sas.com/
CONFLICTS OF INTEREST
The authors have declared no conflicts of interest.
GRANT SUPPORT
The Swedish part of the CEFALO study was supported by grants from the Swedish Council for Working Life and Social Research [2004–0504, 2007–0224]; the Swedish Research Council [K2008-70X-15366-04-3]; the Swedish Cancer Society [09 0666]; the Swedish Childhood Cancer Society [PROJ06/050, PROJ09/086]; and the Swedish Radiation Protection Authority [SSI P 1572]. The Danish CEFALO study was supported by the Danish Strategic Research Council [2103-05-0006, 2064-04-0010]. The Swiss part of the CEFALO study was supported by the Swiss Federal Office of Public Health [05.001626]; the Swiss Research Foundation on Mobile Communication [A2006.18]; and the Swiss National Science Foundation [PDFMP3_122873]. The Norwegian CEFALO study was supported by the Research Council of Norway [175163/V40].
REFERENCES
1. Pollack IF, Jakacki RI. Childhood brain tumors: epidemiology, current management and future directions. Nat Rev Neurol. 2011; 7:495–506.
2. Gu J, Liu Y, Kyritsis AP, Bondy ML. Molecular epidemiology of primary brain tumors. Neurotherapeutics. 2009; 6:427–435.
3. Adel Fahmideh M, Schwartzbaum J, Frumento P, Feychting M. Association between DNA repair gene polymorphisms and risk of glioma: a systematic review and meta-analysis. Neuro Oncol. 2014; 16:807–814.
4. Rajaraman P, Wang SS, Rothman N, Brown MM, Black PM, Fine HA, Loeffler JS, Selker RG, Shapiro WR, Chanock SJ, Inskip PD. Polymorphisms in apoptosis and cell cycle control genes and risk of brain tumors in adults. Cancer Epidemiol Biomarkers Prev. 2007; 16:1655–1661.
5. Andersson U, Schwartzbaum J, Wiklund F, Sjostrom S, Liu Y, Tsavachidis S, Ahlbom A, Auvinen A, Collatz-Laier H, Feychting M, Johansen C, Kiuru A, Lonn S, et al. A comprehensive study of the association between the EGFR and ERBB2 genes and glioma risk. Acta Oncol. 2010; 49:767–775.
6. Schwartzbaum JA, Xiao Y, Liu Y, Tsavachidis S, Berger MS, Bondy ML, Chang JS, Chang SM, Decker PA, Ding B, Hepworth SJ, Houlston RS, Hosking FJ, et al. Inherited variation in immune genes and pathways and glioblastoma risk. Carcinogenesis. 2010; 31:1770–1777.
7. Amirian E, Liu Y, Scheurer ME, El-Zein R, Gilbert MR, Bondy ML. Genetic variants in inflammation pathway genes and asthma in glioma susceptibility. Neuro Oncol. 2010; 12:444–452.
8. Rajaraman P, Hutchinson A, Rothman N, Black PM, Fine HA, Loeffler JS, Selker RG, Shapiro WR, Linet MS, Inskip PD. Oxidative response gene polymorphisms and risk of adult brain tumors. Neuro Oncol. 2008; 10:709–715.
9. De Roos AJ, Rothman N, Brown M, Bell DA, Pittman GS, Shapiro WR, Selker RG, Fine HA, Black PM, Inskip PD. Variation in genes relevant to aromatic hydrocarbon metabolism and the risk of adult brain tumors. Neuro Oncol. 2006; 8:145–155.
10. Jeon S, Han S, Lee K, Choi J, Park SK, Park AK, Ahn HS, Shin HY, Kang HJ, Koo HH, Seo JJ, Choi JE, Kim H, et al. Genetic variants of AICDA/CASP14 associated with childhood brain tumor. Genet Mol Res. 2013; 12:2024–2031.
11. Searles Nielsen S, McKean-Cowdin R, Farin FM, Holly EA, Preston-Martin S, Mueller BA. Childhood brain tumors, residential insecticide exposure, and pesticide metabolism genes. Environ Health Perspect. 2010; 118:144–149.
12. Sirachainan N, Wongruangsri S, Kajanachumpol S, Pakakasama S, Visudtibhan A, Nuchprayoon I, Lusawat A, Phudhicharoenrat S, Shuangshoti S, Hongeng S. Folate pathway genetic polymorphisms and susceptibility of central nervous system tumors in Thai children. Cancer Epidemiol Biomarkers Prev. 2008; 32:72–78.
13. Salnikova LE, Zelinskaya NI, Belopolskaya OB, Aslanyan MM, Rubanovich AV. Association study of xenobiotic detoxication and repair genes with malignant brain tumors in children. Acta naturae. 2010; 2:58–65.
14. Dubuc AM, Northcott PA, Mack S, Witt H, Pfister S, Taylor MD. The genetics of pediatric brain tumors. Curr Neurol Neurosci Rep. 2010; 10:215–223.
15. Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009; 361:1173–1178.
16. Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Lyons-Weiler MA, LaFramboise WA, Burger PC, Brat DJ, Rosenblum MK, Holmes EJ, Zhou T, Jakacki RI, Children’s Oncology G. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2011; 27:87–94.
17. Broderick DK, Di C, Parrett TJ, Samuels YR, Cummins JM, McLendon RE, Fults DW, Velculescu VE, Bigner DD, Yan H. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004; 64:5048–5050.
18. Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, Ahmadi R, Thieme B, Joos S, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008; 118:1739–1749.
19. Gilbertson RJ, Gutmann DH. Tumorigenesis in the brain: location, location, location. Cancer Res. 2007; 67:5579–5582.
20. Adel Fahmideh M, Lavebratt C, Schuz J, Roosli M, Tynes T, Grotzer MA, Johansen C, Kuehni CE, Lannering B, Prochazka M, Schmidt LS, Feychting M. CCDC26, CDKN2BAS, RTEL1 and TERT Polymorphisms in pediatric brain tumor susceptibility. Carcinogenesis. 2015.
21. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O’Kelly M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010; 17:98–110.
22. Sie M, den Dunnen WF, Hoving EW, de Bont ES. Anti-angiogenic therapy in pediatric brain tumors: an effective strategy? Crit Rev Oncol Hematol. 2014; 89:418–432.
23. Nageswara Rao AA, Scafidi J, Wells EM, Packer RJ. Biologically targeted therapeutics in pediatric brain tumors. Pediatr Neurol. 2012; 46:203–211.
24. Pollack IF. Pediatric Brain Tumors: Application of Stratifi cation Criteria to Refine Patient Management. Vol. 2. Pediatric Cancer: Springer. 2012.
25. Donson AM, Addo-Yobo SO, Handler MH, Gore L, Foreman NK. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatr Blood Cancer. 2007; 48:403–407.
26. Karajannis M, Allen JC, Newcomb EW. Treatment of pediatric brain tumors. J Cell Physiol. 2008; 217:584–589.
27. Pfister S, Hartmann C, Korshunov A. Histology and molecular pathology of pediatric brain tumors. J Child Neurol. 2009; 24:1375–1386.
28. Zarghooni M, Bartels U, Lee E, Buczkowicz P, Morrison A, Huang A, Bouffet E, Hawkins C. Whole-genome profiling of pediatric diffuse intrinsic pontine gliomas highlights platelet-derived growth factor receptor alpha and poly (ADP-ribose) polymerase as potential therapeutic targets. J Clin Oncol. 2010; 28:1337–1344.
29. Agnihotri S, Burrell K, Buczkowicz P, Remke M, Golbourn B, Chornenkyy Y, Gajadhar A, Fernandez NA, Clarke ID, Barszczyk MS, Pajovic S, Ternamian C, Head R, et al. ATM regulates 3-methylpurine-DNA glycosylase and promotes therapeutic resistance to alkylating agents. Cancer Discov. 2014; 4:1198–1213.
30. Kim JH, Huse JT, Huang Y, Lyden D, Greenfield JP. Molecular diagnostics in paediatric glial tumours. Lancet Oncol. 2013; 14:e19–27.
31. Rube CE, Fricke A, Schneider R, Simon K, Kuhne M, Fleckenstein J, Graber S, Graf N, Rube C. DNA repair alterations in children with pediatric malignancies: novel opportunities to identify patients at risk for high-grade toxicities. Int J Radiat Oncol Biol Phys. 2010; 78:359–369.
32. Poley JW, Wagner A, Hoogmans MM, Menko FH, Tops C, Kros JM, Reddingius RE, Meijers-Heijboer H, Kuipers EJ, Dinjens WN, Rotterdam Initiative on Gastrointestinal Hereditary T. Biallelic germline mutations of mismatch-repair genes: a possible cause for multiple pediatric malignancies. Cancer. 2007; 109:2349–2356.
33. Kennedy RD, D’Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006; 24:3799–3808.
34. Wood RD. Nucleotide excision repair in mammalian cells. J Biol Chem. 1997; 272:23465–23468.
35. Taylor RM, Thistlethwaite A, Caldecott KW. Central role for the XRCC1 BRCT I domain in mammalian DNA single-strand break repair. Mol Cell Biol. 2002; 22:2556–2563.
36. Kim JA, Haber JE. Chromatin assembly factors Asf1 and CAF-1 have overlapping roles in deactivating the DNA damage checkpoint when DNA repair is complete. Proc Natl Acad Sci USA. 2009; 106:1151–1156.
37. Terasawa M, Shinohara A, Shinohara M. Canonical non-homologous end joining in mitosis induces genome instability and is suppressed by M-phase-specific phosphorylation of XRCC4. PLoS Genet. 2014; 10:e1004563.
38. Abraham J, Lemmers B, Hande MP, Moynahan ME, Chahwan C, Ciccia A, Essers J, Hanada K, Chahwan R, Khaw AK, McPherson P, Shehabeldin A, Laister R, et al. Eme1 is involved in DNA damage processing and maintenance of genomic stability in mammalian cells. EMBO J. 2003; 22:6137–6147.
39. Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol. 2006; 8:37–45.
40. Yang P, Kollmeyer TM, Buckner K, Bamlet W, Ballman KV, Jenkins RB. Polymorphisms in GLTSCR1 and ERCC2 are associated with the development of oligodendrogliomas. Cancer. 2005; 103:2363–2372.
41. Aydin D, Feychting M, Schuz J, Tynes T, Andersen TV, Schmidt LS, Poulsen AH, Johansen C, Prochazka M, Lannering B, Klaeboe L, Eggen T, Jenni D, et al. Mobile phone use and brain tumors in children and adolescents. J Natl Cancer Inst. 2011; 103:1264–1276.
42. Steliarova-Foucher E, Stiller C, Lacour B, Kaatsch P. International Classification of Childhood Cancer, third edition. Cancer. 2005; 103:1457–1467.
43. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007; 114:97–109.
44. Bethke L, Webb E, Murray A, Schoemaker M, Feychting M, Lonn S, Ahlbom A, Malmer B, Henriksson R, Auvinen A, Kiuru A, Salminen T, Johansen C, et al. Functional polymorphisms in folate metabolism genes influence the risk of meningioma and glioma. Cancer Epidemiol Biomarkers Prev. 2008; 17:1195–1202.
45. De Roos AJ, Rothman N, Inskip PD, Linet MS, Shapiro WR, Selker RG, Fine HA, Black PM, Pittman GS, Bell DA. Genetic polymorphisms in GSTM1, -P1, -T1, and CYP2E1 and the risk of adult brain tumors. Cancer Epidemiol Biomarkers Prev. 2003; 12:14–22.
46. Schwartzbaum JA, Ahlbom A, Lonn S, Malmer B, Wigertz A, Auvinen A, Brookes AJ, Collatz Christensen H, Henriksson R, Johansen C, Salminen T, Schoemaker MJ, Swerdlow AJ, et al. An international case-control study of interleukin-4Ralpha, interleukin-13, and cyclooxygenase-2 polymorphisms and glioblastoma risk. Cancer Epidemiol Biomarkers Prev. 2007; 16:2448–2454.
47. Schwartzbaum J, Ahlbom A, Malmer B, Lonn S, Brookes AJ, Doss H, Debinski W, Henriksson R, Feychting M. Polymorphisms associated with asthma are inversely related to glioblastoma multiforme. Cancer Res. 2005; 65:6459–6465.
48. Ruan Z, Zhao Y, Yan L, Chen H, Fan W, Chen J, Wu Q, Qian J, Zhang T, Zhou K, Mao Y, Zhou L, Huang Y, et al. Single nucleotide polymorphisms in IL-4Ra, IL-13 and STAT6 genes occurs in brain glioma. Front Biosci (Elite Ed). 2011; 3:33–45.
49. Rajaraman P, Brenner AV, Butler MA, Wang SS, Pfeiffer RM, Ruder AM, Linet MS, Yeager M, Wang Z, Orr N, Fine HA, Kwon D, Thomas G, et al. Common variation in genes related to innate immunity and risk of adult glioma. Cancer Epidemiol Biomarkers Prev. 2009; 18:1651–1658.
50. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81:559–575