
INTRODUCTION
Leukemia is the most common hematologic malignancy, with 54,270 new cases in the USA in 2015, representing a 14.4% increase over 2012. The most common forms of leukemia are acute myeloid leukemia (AML) representing 40% of all cases, followed by chronic lymphocytic leukemia (CLL) and chronic myeloid leukemia (CML) [1-3].
Treatment strategies for myeloid leukemia include chemotherapy, radiotherapy and immunotherapy, with current research focusing on antibody-based targeted immunotherapy because of its versatility and advantages in terms of production, regulatory compliance and pharmaceutical logistics [4]. Several molecules overexpressed on the surface of AML cells have been investigated as targets for antibody-based therapy, including CD33, CD47, C-type lectin, CD64, CD123 and most recently CD89 [5-10].
CD33 is the most common target for AML therapy. The antibody-drug conjugate (ADC) gemtuzumab ozogamicin targets this molecule, and showed promising efficacy as a monotherapy (overall response rate of ~30%). However, is use as a supplement to conventional chemotherapy achieved no survival advantage but caused higher toxicity in phase III trials, leading to the withdrawal of approval in 2010 [11, 12]. Alternative targeting agents include immunotoxins (ITs) and targeted fully human cytolytic fusion proteins (hCFPs). In both cases, selectivity is achieved by an antibody derivative such as a single chain variable fragment (scFv). Conventional ITs use bacterial toxins to induce apoptosis, such as truncated Pseudomonas aeroginosa exotoxin A (ETA’) [13], whereas hCFPs are equipped with human pro-apoptotic effectors such as granzyme B, granzyme M or angiogenin [8, 14, 15]. The main disadvantages of ITs are the immunogenicity of the non-human effectors and off-target effects such as hepatotoxicity and vascular leakage, which have limited their performance in clinical trials [16]. In contrast, hCFPs are fully human constructs and the risk of immunogenicity is much lower [17].
We previously reported the identification of a novel cytolytic effector protein known as microtubule-associated protein tau (MAP) which promotes the assembly of the mitotic spindle [18-20]. MAP binds to microtubules via microtubule-binding repeats and enhances the stability of microtubule polymers [21-23]. Native MAP is regulated by phosphorylation at two sites to induce its dissociation and allow microtubule disassembly after mitosis, so the cytotoxic version of MAP was mutated to remove these sites (S156A and S204A) [18]. Consequently, the modified MAP binds irreversibly to microtubules and interrupts their normally dynamic behavior, ultimately leading to the induction of apoptosis [24].
The modified MAP protein kills proliferating EGFR+ cancer cells, EpCAM+ carcinoma cells, AChR+ rhabdomyosarcoma cells and CD30+ lymphoma cells when fused to appropriate targeting components [18-20, 25]. Most recently, the CD64-targeting construct H22(scFv)-MAP was shown to eliminate pro-inflammatory M1 macrophages but not the anti-inflammatory M2 population following intradermal administration to the chronically inflamed skin of transgenic mice expressing human CD64 [26]. The latter study also revealed that H22(scFv)-MAP kills rapidly-proliferating pro-leukemic/monocytic HL-60 cells in a dose-dependent manner, indicating a potential for anti-leukemic activity.
We therefore tested H22(scFv)-MAP against primary cells isolated from seven untreated leukemia patients. Our ex vivo data confirmed the targeted pro-apoptotic activity of H22(scFv)-MAP and suggest it should be developed further as an immunotherapeutic candidate for the treatment of leukemia.
RESULTS
Phenotyping primary cells from leukemia patients
Double staining for CD33 and CD64 indicated that blood samples from both AML M4 patients, the AML M4/M5 patient and two of the three CMML patients yielded ~70% CD33+ CD64+ blasts (Table 1). The other CMML sample contained 53% CD33+ CD64+ blasts, whereas the sample from the AML M5 patient yielded only 20% CD33+ CD64+ blasts (Figure 1A).
Table 1: Characteristics of the leukemia patients
Parameter |
Number |
||||||
|---|---|---|---|---|---|---|---|
Patient designation |
AML M4 (I) |
AML M4 (II) |
AML M4/M5 |
AML M5 |
CMML (I) |
CMML (II) |
CMML (III) |
Age |
51 |
78 |
81 |
74 |
91 |
79 |
n.i. |
Gender |
Female |
Male |
Male |
Male |
Female |
Male |
n.i. |
FAB subtype |
AML M4 (AMML) |
AML M4 (AMML) |
AML M4/M5 |
AML M5 |
CMML-2 |
CMML-2 |
CMML-2 |
WBC (G/L) |
80.7 |
88.7 |
113.4 |
104.9 |
105.7 |
4.9 |
n.i. |
% Monocytes |
3 |
1 |
44 |
6 |
9 |
23.6 |
n.i. |
Hemoglobin (g/L) |
91 |
116 |
10 |
83 |
98 |
119 |
n.i. |
Platelet count (G/L) |
28 |
45 |
134 |
128 |
64 |
258 |
n.i. |
Previous treatment |
No |
No |
No |
No |
No |
No |
No |
CD64+ CD33+ (%) |
70 |
73 |
71 |
20 |
69 |
74 |
53 |
CD64+ cells (%) |
70 |
73 |
73 |
23 |
70 |
74 |
53 |
FAB = French-American-British classification; n. i. = no information.
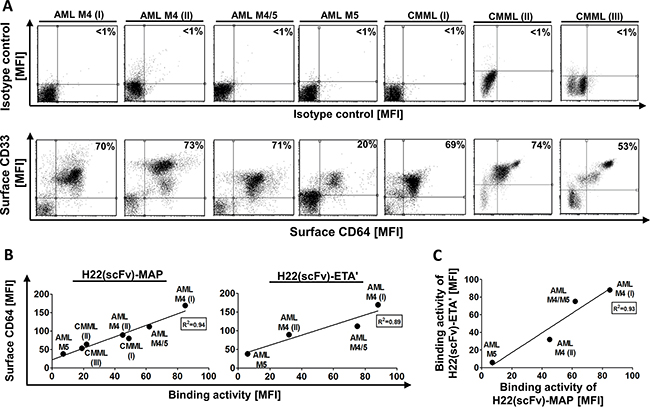
Figure 1: Characterization of the CD64+ leukemic blasts and H22(scFv)-MAP binding. Blood samples were obtained from seven untreated patients diagnosed with different forms of leukemia (Table 1). A. The proportion of primary CD33+ CD64+ leukemic blasts from seven patients is compared directly to the isotype control. Percentiles indicate the proportion of double positive cells from all measured events. B. The ability of purified H22(scFv)-MAP to bind the leukemic cells in vitro was tested by flow cytometry. The binding activity of H22(scFv)-MAP (left) and H22(scFv)-ETA’ (right) correlate to the estimated CD64+ level. C. Comparative binding activities of both CD64-targeting fusion proteins. Abbreviations: MFI, mean fluorescence intensity.
Binding of H22(scFv)-MAP to CD64+ target cells
H22(scFv)-MAP bound to CD64+ blasts from both AML M4 patients, the AML M4/M5 patient and all three CMML patients (Supplementary Figure S1A). H22(scFv)-ETA’ showed an almost identical binding profile, but was only tested against the samples from AML patients (Supplementary Figure S1B). Both constructs showed very low binding activity to the leukemic blasts from the AML M5 patient even though as stated above the sample contained ~20% CD64+ CD33+ cells. As expected, the detection antibody alone and a CD30-specific non-binding control (Mock-MAP) did not bind to the isolated leukemic blasts. The binding activity of H22(scFv)-MAP and H22(scFv)-ETA correlated to the surface expression levels of CD64 on the blasts (Figure 1B) and there was also a strong correlation in binding properties when these constructs were compared (Figure 1C).
H22(scFv)-MAP induces apoptosis specifically in leukemic blasts
Having confirmed the specific binding of H22(scFv)-MAP to CD64+ leukemic blasts, we tested the CD64-specific cytotoxicity of H22(scFv)-MAP, Mock-MAP and H22(scFv)-ETA’ by staining with Annexin V-eGFP (AV) and propidium iodide (PI) to detect cells undergoing apoptosis. We have shown in previous studies that 200 nM is a sufficient dose of hCFP or IT to achieve the receptor-specific induction of apoptosis within 12 h [7, 8, 14, 15]. We therefore used the same dose for the constructs tested in this study. First we ascertained the background frequency of apoptosis in each sample using a vehicle control, i.e. the buffer formulation but no active fusion protein. In most samples, 5–15% of cells underwent apoptosis spontaneously, but the frequency was higher for the AML M4/M5 (~32%), AML M5 (57%) and CMML (I) (44%) samples (Figure 2). Importantly, the background frequency of apoptosis did not increase in the presence of Mock-MAP, confirming that MAP is not cytotoxic in the absence of specific binding. In contrast, the proportion of early-apoptotic (AV+/PI–) cells increased substantially in six of the samples when the leukemic cells were treated with H22(scFv)-MAP (Figure 3A). The AML M5 blasts exhibited the highest level of background cell death, which in combination with low CD64 profile resulted in no response to H22(scFv)-ETA’ or H22(scFv)-MAP treatment (Figure 2A). The pro-apoptotic effect of H22(scFv)-ETA’ correlated to that of H22(scFv)-MAP in the samples from the four AML patients (Figure 3C). Interestingly, there was a correlation between the binding activity and cytotoxicity of H22(scFv)-MAP in the AML samples, but in the CMML samples there was an inverse relationship between binding activity and cytotoxicity (Figure 3B).
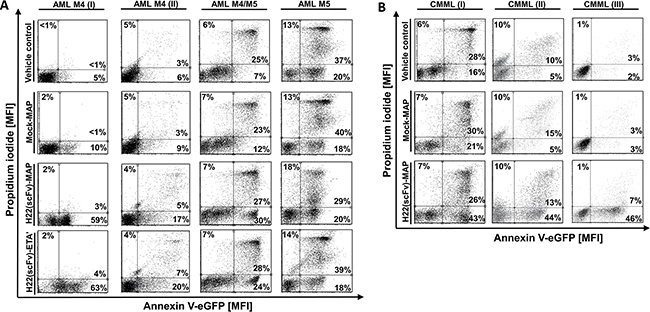
Figure 2: H22(scFv)-MAP induces apoptosis in CD64+ leukemic blasts ex vivo. Leukemic blasts isolated from four AML patients A. and three CMML patients B. were treated ex vivo with 200 nM H22(scFv)-ETA’, H22(scFv)-MAP or Mock-MAP for 12 h. The leukemic cells were stained with Annexin V–eGFP and PI. Dot blots (ungated) show the specific pro-apoptotic effect of the H22(scFv)-MAP compared to Mock-MAP and H22(scFv)-ETA’. The experiment was carried out in duplicate.
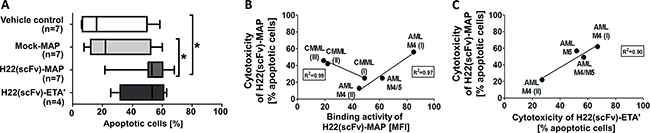
Figure 3: H22(scFv)-MAP is cytotoxic towards CD64+ leukemic blasts ex vivo. A. Quantitative analysis of the dot blots in Figure 2 representing the proportion of cells undergoing apoptosis (the sum of early (Annexin V+/PI-) and late (Annexin V+/PI+) apoptotic cells) in the different experimental groups. Values are presented as box and whiskers plots with the interquartile range and minimum/maximum values indicated by the error bars. Each measurement was taken in duplicate. Statistical analysis was carried out using the non-parametric one-way ANOVA test (Kruskal-Wallis test by ranks): *p ≤ 0.05. B. Cytotoxicity of H22(scFv)-MAP plotted against its binding activity on leukemic blasts. C. Correlation between the cytotoxicity of the CD64-targeting fusion proteins. Abbreviations: MFI, mean fluorescence intensity.
H22(scFv)-MAP was safer than paclitaxel against primary PBMCs and TAMs
Peripheral blood mononuclear cells (PBMCs) from healthy blood donors were used to investigate nonspecific toxicity of H22(scFv)-MAP. Figure 4A confirms that neither Mock-MAP nor H22(scFv)-MAP are cytotoxic towards freshly-isolated PBMCs, whereas paclitaxel is potently cytotoxic at same concentration of 200 nM (dot blots in Supplementary Figure S2). The isolated PBMCs were then polarized to different macrophage phenotypes: M0, M1, M2, and tumor-associated macrophages (TAMs) (Supplementary Figure S4). The TAMs expressed much lower levels of CD64 than the other phenotypes (Figure 4B), resulting in weak H22(scFv)-MAP binding activity (Supplementary Figure S1C). Accordingly, CD64low TAMs showed strong resistance towards H22(scFv)-MAP treatment, whereas paclitaxel induced apoptosis in all the healthy cells we tested (Figures 4C and 4D).
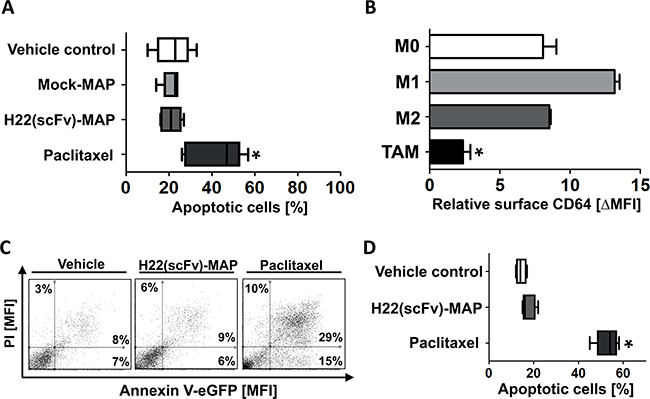
Figure 4: H22(scFv)-MAP shown no cytotoxicity towards healthy PMBC-derived cells. A. Quantitative analysis of the proportion of healthy PMBC-derived cells undergoing apoptosis (the sum of early (Annexin V+/PI-) and late (Annexin V+/PI+) apoptotic cells) in the different experimental groups. Dot blots are shown in Supplementary Figure S2. B. TAMs express significantly lower amounts of CD64. The levels of CD64 on differently polarized, healthy PBMC-derived macrophages (n=3 for M0/1/2 and n=5 for TAM) were assessed by flow cytometry. C. Representative dot blots (ungated) confirm that TAMs are resistant to the CD64-directed MAP hCFP but not to the nonspecific cytostatic agent paclitaxel. D. Quantification of TAM apoptosis (n=5 PBMC donors) in the different experimental groups. Values are presented as box and whiskers plots with the interquartile range and minimum/maximum values indicated by the error bars. Each measurement was taken in duplicate. Statistical analysis was carried out using the non-parametric one-way ANOVA test (Kruskal-Wallis test by ranks): *p ≤ 0.05.
DISCUSSION
Our experiments showed that H22(scFv)-MAP is suitable for the targeted elimination of CD64+ leukemic blasts from patients with different leukemic subtypes. The advantages of CD64 as a target for the delivery of cytotoxic effector molecules include its rapid internalization, which reduces the likelihood of off-target effects during hCFP therapy [8, 27, 28]. The therapeutic potential of H22(scFv)-MAP was recently demonstrated against inflammatory CD64high macrophages (M1 subtype) in the context of chronic cutaneous inflammation [26]. The M1 population is thought to be beneficial in a tumor environment (including solid tumors and circulating tumor cells) but this environment is overwhelmed by anti-inflammatory signals, and type-II cytokines (e.g. IL-4, IL-10 and IL-13) tend to dominate (Supplementary Figure S3) [29-32]. The anti-inflammatory cytokines inhibit the M1 phenotype (CD64high) and induce alternative polarized macrophages (CD64low M2/TAM subtype), which according to our observations are resistant to H22(scFv)-MAP (Figure 4) [26, 30, 33]. It is therefore anticipated that the homeostasis of the healthy CD64+ myeloid-derived cell population is spared by hCFP treatment because cells strictly representing the M1 subtype are sensitive [26, 33]. Furthermore, the co-expression of CD64 and CD33 by 50–70% of the leukemic blasts in most of our samples confirms their malignant nature. The binding of H22(scFv)-MAP in most samples demonstrated the efficient recognition of leukemic blasts. The exception was AML M5, which had only ~20% CD64+ CD33+ blasts, and this sample also failed to respond to the ex vivo treatment with H22(scFv)-MAP. Interestingly, the cytotoxic potency of the hCFP correlated directly with the abundance of CD64+ in AML blasts, but there was an inverse correlation in the CMML blasts. This may reflect the distinct maturation stages of the acute and chronic myeloid blasts [34], which could result in the CD64-delivered cargo undergoing different forms of intracellular processing. Notably, our data suggest that H22(scFv)-MAP has the ability to kill both dim and bright CD64+ cells.
Cytostatic agents (e.g. paclitaxel) that disrupt microtubule dynamics are already used for chemotherapy against diverse forms of cancer [35-37]. These act in a similar way to MAP by stabilizing polymerized microtubules, blocking mitosis and ultimately leading to cell cycle arrest and apoptosis [38]. However, in line with our observations, the nonspecific delivery of taxanes causes off-target effects, including life-threatening toxicity towards healthy cells [39]. This was overcome by developing microtubule-blocking ADCs such as the FDA-approved trastuzumab emtansine (anti-HER2) and brentuximab vedotin (anti-CD30) [40-42]. Using the rationale for targeted microtubule-blocking therapy, H22(scFv)-MAP demonstrated the ability to bind CD64+ primary cancer cells specifically and kill them efficiently. Furthermore, no untargeted cytotoxicity was observed by treating the same cells with a non-binding Mock-MAP control. Our ex vivo data revealed that six of seven leukemia patients responded to the CD64-directed hCFP and its cytotoxic efficacy was similar to that of hCFPs based on granzymes or angiogenin [8, 14, 15].
In summary, H22(scFv)-MAP is an efficacious immunotherapeutic candidate that could be developed as a post-remission treatment for CD64+ hematological diseases, or combined with standard chemotherapy to improve prognosis. The development of novel immunotherapeutic strategies based on hCFPs is necessary because the FDA has withdrawn its approval for gemtuzumab ozogamicin, the only ADC approved for the treatment of AML, due to its lack of efficacy and the off-target toxicity revealed during phase III clinical trials [43].
MATERIALS AND METHODS
Patent samples and cell cultivation
Peripheral blood was collected from either untreated leukemia patients (diagnosed according to FAB classifications and clinical parameters as shown in Table 1) or healthy individuals after written informed consent was received and with the approval of the Clinical Research Ethics Board of the University of Aachen. PBMCs were isolated by density gradient centrifugation using Biocoll separating solution (BioChrom GmbH, Berlin, Germany). After three washes in phosphate buffered saline (PBS), the cells were cultured in standard RPMI 1640 medium (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) inactivated human serum (BioChrom), 50 μg/ml penicillin and 100 μg/ml streptomycin (Gibco, Thermo Fisher Scientific) at 37°C, 5% CO2 and 100% humidity.
Production of the recombinant H22(scFv)-MAP hCFP
Recombinant H22(scFv)-MAP was expressed in Escherichia coli BL21 (DE3) cells using the protocol for periplasmic stress expression in the presence of compatible solutes as previously described [26]. Briefly, bacteria were grown after transformation to an optical density (OD) of 1.6 followed by stress induction with 500 mM D-sorbitol, 10 mM betaine monohydrate and 4% (w/w) NaCl. After incubating for 30 min at 26°C with agitation (180 rpm), protein expression was induced by adding 2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). Bacteria were harvested 18 h after induction by centrifugation (4000 x g, 10 min, 4°C) and frozen at –80°C overnight. The frozen pellet was resuspended in preparation buffer (75 mM Tris-HCl, 300 mM NaCl, 5 mM DTT, 10 mM EDTA, 10% (v/w) glycerol, pH 8.0) containing a complete protease inhibitor cocktail (Roche, Penzberg, Germany) at 4°C and sonicated five times for 60 s at 200 W. Cell debris was removed by centrifugation (24,000 x g, 20 min, 4°C) and EDTA was removed by dialysis against PBS (pH 7.4) at 4°C overnight. H22(scFv)-MAP was purified by immobilized metal-ion affinity chromatography (IMAC) on an Äkta Purifier System (GE Healthcare, Little Chalfont, UK) using IMAC Sepharose 6 Fast Flow as resin and an XK16-20 column (GE Healthcare). Preparative size exclusion chromatography (SEC) was carried out on an Äkta Purifier System (GE Healthcare) using an XK16-70 column packed with SuperdexTM 75 (GE Healthcare), with PBS as the mobile phase (pH 7.4) at a flow rate of 1.0 ml/min. The purity of H22(scFv)-MAP was determined by SDS-PAGE followed by staining with Coomassie Brilliant Blue and its identify was confirmed by western blotting. The protein concentration was determined by densitometric analysis of stained gels using AIDA software and was verified using the Bradford assay (Bio-Rad, Munich, Germany). Bovine serum albumin standards were used for calibration.
Analysis of cell phenotypes
The phenotypes of the isolated cells were determined using labeled antibodies and flow cytometry. Double staining for CD33 and CD64 was carried out on leukemic material, and monostaining for CD14, CD64, CD163, CD200R, CD206, CD273, and CD301 was carried out on healthy material. Briefly, we incubated 2 × 105 cells with 125 ng of anti-human CD33[P67.6]-APC, anti-human CD206[15-2]-PE, anti-human CD301[H037G3]-APC, anti-human CD163[GHI/61]-PE, anti-human CD273[MIH18]-PE, anti-human CD200R[OX-108]-PE, anti-human CD14[63D3]-PE, or anti-human CD64[10.1]-PE (all from eBioscience, Frankfurt, Germany) in 1% (v/v) human antibody-binding serum (BioChrom) for 30 min on ice in the dark, followed by washing with PBS. The corresponding mouse IgG1[MOPC-21] antibody (Biolegend, San Diego, CA, USA) with either a phycoerythrin (PE) or allophycocyanin (APC) label was used as the corresponding isotype control. Finally, the cells were washed twice with PBS and analyzed on a FACSCalibur flow cytometer (Becton Dickinson, Heidelberg, Germany) with Flowing Software v2.5.1.
Binding analysis by flow cytometry
The cell-binding activity of Mock-MAP (non-binding control), H22(scFv)-ETA’ and H22(scFv)-MAP was analyzed by flow cytometry. We incubated 2 x 105 cells with 200 ng of each construct in PBS (pH 7.4) containing 1% (v/v) human antibody-blocking serum for 30 min on ice followed by washing with PBS. Because each construct contained a His6-tag, fluorescence staining was carried out using 100 ng of an anti-His5 Alexa Fluor 488 antibody (Qiagen, Hilden, Germany) for 30 min on ice in the dark. Finally, the cells were washed twice with PBS and analyzed on a FACSCalibur flow cytometer.
Apoptosis assay
An Annexin V/PI assay was used to determine the pro-apoptotic effect of H22(scFv)-MAP. We incubated 5 x 105 cells per ml with 200 nM H22(scFv)-MAP or 200 nM H22(scFv)-ETA’ or 200 nM paclitaxel (Sigma Aldrich, Steinheim, Germany) in a 24-well plate (Greiner Bio-One GmbH, Frickenhausen, Germany) for 12 h at 37°C, 5% CO2 and 100% humidity. We used the anti-CD30 construct Ki4-MAP (200 nM) as the non-binding Mock-MAP control [20]. After incubation, the cells were washed twice with Annexin V buffer (pH 7.4) and stained with Annexin V–eGPF (produced by Anh-Tuan Pham, Fraunhofer IME, Aachen) in Annexin V binding buffer (10 mM HEPES/NaOH, pH 7.4), 140 mM NaCl, 2.5 mM CaCl2) for 30 min at room temperature. Finally, the cells were washed carefully as described above, resuspended in Annexin V buffer containing 10 μg/ml PI and analyzed by flow cytometry using a FACSCalibur flow cytometer.
In vitro polarization of macrophages
Freshly-isolated PBMCs were cultured at a concentration of 5 x 105 cells per ml for 48 h in complete medium supplemented with the appropriate polarization stimulus. Medium only was used for non-polarized M0 cells, 100 U/ml human IFNγ (Peprotech, Hamburg, Germany) was used for M1 cells, and 20 ng/ml IL-4 (Peprotech) was used for M2 cells. Sterile serum from patient AML M4 (I) was used to induce the TAM phenotype. The added serum amount (60 μl/ml) resulted in a final concentration of 20 ng/ml IL-4. No serum-mediated toxicity was observed in the TAM culture. Thereafter, macrophages were used for phenotyping or targeting experiments.
ACKNOWLEDGMENTS
The authors thank Reinhard Rosinke and Anh-Tuan Pham (both from Fraunhofer IME in Aachen) for assistance with the production of recombinant H22(scFv)-MAP, H22(scFv)-ETA’ and Annexin-eGFP, Dr Richard M. Twyman for critically reading the manuscript, and Fanny Frenzel (University Hospital, Aachen) for providing blood parameters for the leukemia patients. C.S. was supported by a fellowship from the German José Carreras Leukemia Foundation (grant no. DJCLS F 14/05). M.W was supported by the RWTH scholarship of Young Researchers at RWTH Aachen University (RFwN). R.M. was supported by a scholarship from the Jürgen-Manchot-Foundation.
CONFLICTS OF INTEREST
DH, RM, TT, and SB are co-inventors on a patent describing H22(scFv)-MAP, which is assigned to Fraunhofer IME. The authors otherwise declare no conflicts of interest.
GRANT SUPPORT
This work was funded by the Fraunhofer project “The markets of the future – SKINHEAL“.
Authors’ Contributions
Conception and design: DH, RM, TT, RFi, SB
Acquisition of data: DH, RM, CC, GG, EG, LH, MA, DK, MW, EJ, TB
Work involving patient-derived material: EJ, TB, RF, CS, RFe
Analysis and interpretation of data: RM, DH, CC, SB
Writing, reviewing, and/or revision of the manuscript: RM, DH, CS, RFi, SB, TT, MA
Study supervision: SB, TT.
REFERENCES
1. Siegel R, Naishadham D and Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62:10-29.
2. Siegel R, Ma J, Zou Z and Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014; 64:9-29.
3. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65:5-29.
4. Grosso DA, Hess RC and Weiss MA. Immunotherapy in acute myeloid leukemia. Cancer. 2015; 121:2689-2704.
5. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, Jr., van Rooijen N and Weissman IL. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009; 138:286-299.
6. Bakker AB, van den Oudenrijn S, Bakker AQ, Feller N, van Meijer M, Bia JA, Jongeneelen MA, Visser TJ, Bijl N, Geuijen CA, Marissen WE, Radosevic K, Throsby M, et al. C-type lectin-like molecule-1: a novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004; 64:8443-8450.
7. Mladenov R, Hristodorov D, Cremer C, Hein L, Kreutzer F, Stroisch T, Niesen J, Brehm H, Blume T, Brummendorf TH, Jost E, Thepen T, Fischer R, Stockmeyer B, Barth S and Stein C. The Fc-alpha receptor is a new target antigen for immunotherapy of myeloid leukemia. Int J Cancer. 2015; 137:2729-2738.
8. Schiffer S, Rosinke R, Jost E, Hehmann-Titt G, Huhn M, Melmer G, Barth S and Thepen T. Targeted ex vivo reduction of CD64-positive monocytes in chronic myelomonocytic leukemia and acute myelomonocytic leukemia using human granzyme B-based cytolytic fusion proteins. Int J Cancer. 2014; 135:1497-1508.
9. Stein C, Kellner C, Kugler M, Reiff N, Mentz K, Schwenkert M, Stockmeyer B, Mackensen A and Fey GH. Novel conjugates of single-chain Fv antibody fragments specific for stem cell antigen CD123 mediate potent death of acute myeloid leukaemia cells. Br J Haematol. 2010; 148:879-889.
10. Thol F and Schlenk RF. Gemtuzumab ozogamicin in acute myeloid leukemia revisited. Expert Opin Biol Ther. 2014; 14:1185-1195.
11. Cowan AJ, Laszlo GS, Estey EH and Walter RB. Antibody-based therapy of acute myeloid leukemia with gemtuzumab ozogamicin. Front Biosci (Landmark Ed). 2013; 18:1311-1334.
12. Ehninger A, Kramer M, Rollig C, Thiede C, Bornhauser M, von Bonin M, Wermke M, Feldmann A, Bachmann M, Ehninger G and Oelschlagel U. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood cancer journal. 2014; 4:e218.
13. Tur MK, Huhn M, Jost E, Thepen T, Brummendorf TH and Barth S. In vivo efficacy of the recombinant anti-CD64 immunotoxin H22(scFv)-ETA’ in a human acute myeloid leukemia xenograft tumor model. Int J Cancer. 2011; 129:1277-1282.
14. Cremer C, Braun H, Mladenov R, Schenke L, Cong X, Jost E, Brummendorf TH, Fischer R, Carloni P, Barth S and Nachreiner T. Novel angiogenin mutants with increased cytotoxicity enhance the depletion of pro-inflammatory macrophages and leukemia cells ex vivo. Cancer Immunol Immunother. 2015.
15. Schiffer S, Letzian S, Jost E, Mladenov R, Hristodorov D, Huhn M, Fischer R, Barth S and Thepen T. Granzyme M as a novel effector molecule for human cytolytic fusion proteins: CD64-specific cytotoxicity of Gm-H22(scFv) against leukemic cells. Cancer Lett. 2013; 341:178-185.
16. Alewine C, Hassan R and Pastan I. Advances in anticancer immunotoxin therapy. Oncologist. 2015; 20:176-185.
17. Harding FA, Stickler MM, Razo J and DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010; 2:256-265.
18. Hristodorov D, Mladenov R, Pardo A, Pham AT, Huhn M, Fischer R, Thepen T and Barth S. Microtubule-associated protein tau facilitates the targeted killing of proliferating cancer cells in vitro and in a xenograft mouse tumour model in vivo. Br J Cancer. 2013; 109:1570-1578.
19. Hristodorov D, Amoury M, Mladenov R, Niesen J, Arens K, Berges N, Hein L, Di Fiore S, Pham AT, Huhn M, Helfrich W, Fischer R, Thepen T, et al. EpCAM-selective elimination of carcinoma cells by a novel MAP-based cytolytic fusion protein. Mol Cancer Ther. 2014; 13:2194-2202.
20. Hristodorov D, Nordlohne J, Mladenov R, Huhn M, Fischer R, Thepen T and Barth S. Human microtubule-associated protein tau mediates targeted killing of CD30(+) lymphoma cells in vitro and inhibits tumour growth in vivo. Br J Haematol. 2014; 164:251-257.
21. Jho YS, Zhulina EB, Kim MW and Pincus PA. Monte carlo simulations of tau proteins: effect of phosphorylation. Biophys J. 2010; 99:2387-2397.
22. Weingarten MD, Lockwood AH, Hwo SY and Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975; 72:1858-1862.
23. Al-Bassam J, Ozer RS, Safer D, Halpain S and Milligan RA. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J Cell Biol. 2002; 157:1187-1196.
24. Gan PP, McCarroll JA, Po’uha ST, Kamath K, Jordan MA and Kavallaris M. Microtubule dynamics, mitotic arrest, and apoptosis: drug-induced differential effects of betaIII-tubulin. Mol Cancer Ther. 2010; 9:1339-1348.
25. Brehm H, Hristodorov D, Pardo A, Mladenov R, Niesen J, Fischer R, Tur MK and Barth S. Targeted killing of rhabdomyosarcoma cells by a MAP-based human cytolytic fusion protein. Cancer Lett. 2015; 365:149-155.
26. Hristodorov D, Mladenov R, Fischer R, Barth S and Thepen T. Fully human MAP-fusion protein selectively targets and eliminates proliferating CD64 M1 macrophages. Immunol Cell Biol. 2016.
27. Tur MK, Huhn M, Thepen T, Stocker M, Krohn R, Vogel S, Jost E, Osieka R, van de Winkel JG, Fischer R, Finnern R and Barth S. Recombinant CD64-specific single chain immunotoxin exhibits specific cytotoxicity against acute myeloid leukemia cells. Cancer Res. 2003; 63:8414-8419.
28. Zhong RK, van de Winkel JG, Thepen T, Schultz LD and Ball ED. Cytotoxicity of anti-CD64-ricin a chain immunotoxin against human acute myeloid leukemia cells in vitro and in SCID mice. Journal of hematotherapy & stem cell research. 2001; 10:95-105.
29. Biswas SK, Sica A and Lewis CE. Plasticity of macrophage function during tumor progression: regulation by distinct molecular mechanisms. J Immunol. 2008; 180:2011-2017.
30. Martinez FO and Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014; 6:13.
31. Sica A and Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012; 122:787-795.
32. Sanchez-Correa B, Bergua JM, Campos C, Gayoso I, Arcos MJ, Banas H, Morgado S, Casado JG, Solana R and Tarazona R. Cytokine profiles in acute myeloid leukemia patients at diagnosis: survival is inversely correlated with IL-6 and directly correlated with IL-10 levels. Cytokine. 2013; 61:885-891.
33. Hristodorov D, Mladenov R, von Felbert V, Huhn M, Fischer R, Barth S and Thepen T. Targeting CD64 mediates elimination of M1 but not M2 macrophages in vitro and in cutaneous inflammation in mice and patient biopsies. MAbs. 2015; 7:853-862.
34. Murati A, Brecqueville M, Devillier R, Mozziconacci MJ, Gelsi-Boyer V and Birnbaum D. Myeloid malignancies: mutations, models and management. BMC Cancer. 2012; 12:304.
35. Crown J and O’Leary M. The taxanes: an update. Lancet. 2000; 355:1176-1178.
36. King KM, Lupichuk S, Baig L, Webster M, Basi S, Whyte D and Rix S. Optimal use of taxanes in metastatic breast cancer. Curr Oncol. 2009; 16:8-20.
37. Norris B, Pritchard KI, James K, Myles J, Bennett K, Marlin S, Skillings J, Findlay B, Vandenberg T, Goss P, Latreille J, Rudinskas L, Lofters W, et al. Phase III comparative study of vinorelbine combined with doxorubicin versus doxorubicin alone in disseminated metastatic/recurrent breast cancer: National Cancer Institute of Canada Clinical Trials Group Study MA8. J Clin Oncol. 2000; 18:2385-2394.
38. Fitzpatrick JM and de Wit R. Taxane mechanisms of action: potential implications for treatment sequencing in metastatic castration-resistant prostate cancer. European urology. 2014; 65:1198-1204.
39. Wang GS, Yang KY and Perng RP. Life-threatening hypersensitivity pneumonitis induced by docetaxel (taxotere). Br J Cancer. 2001; 85:1247-1250.
40. Muller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K, Savic S, Harbeck N, Nitz U, Gluz O, von Bergwelt-Baildon M, Kreipe H, et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Transl Med. 2015; 7:315ra188.
41. Klute K, Nackos E, Tasaki S, Nguyen DP, Bander NH and Tagawa ST. Microtubule inhibitor-based antibody-drug conjugates for cancer therapy. Onco Targets Ther. 2014; 7:2227-2236.
42. Guillemard V and Saragovi HU. Taxane-antibody conjugates afford potent cytotoxicity, enhanced solubility, and tumor target selectivity. Cancer Res. 2001; 61:694-699.
43. Rowe JM and Lowenberg B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood. 2013; 121:4838-4841.