
INTRODUCTION
The Polycomb group protein complexes alter methylation of histone tails and play a critical role during development by compacting chromatin and repressing transcription of differentiation-associated genes. Among the best studied chromatin regulators is enhancer of zeste homolog 2 (EZH2), the catalytic subunit of the polycomb repressor complex 2 (PRC2). EZH2 contains a Suvar 3-9 Enhancer of zeste Trithorax (SET) domain and functions as a histone methyltransferase (HMT) for the well characterized repressive mark histone H3 lysine 27 trimethylation (H3K27me3) [1]. Deregulation and function-altering mutations of chromatin modifiers [2-4] have been widely reported in cancer, including in ovarian cancer (OC), but their involvement and regulation in tumorigenesis are complex and remain not fully understood [5].
The concept that the epigenome may buffer DNA from environmental insults is an area of emerging interest. In cancer cells, environmental stimuli have been shown to induce changes in chromatin [6], but the mechanisms underlying chromatin remodeling in response to environmental cues is unclear. One of the key regulatory factors in the tumor microenvironment is the transforming growth factor β1 (TGF-β), which has been widely implicated in cancer progression [7]. TGF-β exerts both tumor suppressor and oncogenic activities in cancer [8] and while its effects are mediated transcriptionally, the cytokine has recently been shown to alter chromatin organization [9]. In this study, we sought to determine whether chromatin modifications imparted by EZH2 play a role in the response of OC cells to TGF-β.
In OC, TGF-β is abundantly secreted in the peritoneal environment by cancer and stromal cells [10, 11] and induces cancer cell invasiveness, epithelial to mesenchymal transition (EMT), and peritoneal metastasis [8, 12, 13]. We and others have shown that TGF-β signaling is active in OC [11, 12] inducing EMT, which permits break-down of cell junctions and loss of cell polarity allowing epithelial cells to become migratory and invasive [14]. While the genetic events triggered by the cytokine, such as activation of the E-cadherin repressors, Twist, ZEB1/2 and SNAI1/2 are well delineated [15], it is not clear whether TGF-β has additional effects on chromatin conformation and if such changes contribute to gene regulation linked to the EMT program.
To understand whether the cellular response to TGF-β involves epigenetic alterations, we recently completed a methylome analysis and demonstrated that TGF-β induced changes in DNA methylation that directly affected gene transcription [16]. DNA methylation is strongly correlated with polycomb repression [17, 18], prompting us to further dissect how the PRC2 complex responds to TGF-β and whether the EMT process is regulated by removal of repressive histone marks. By using a TGF-β-inducible EMT model in OC cells, we show that EZH2 is required for maintaining the epithelial characteristics of cancer cells and that erasure of the H3K27me3 marks catalyzed by EZH2 permits transition to a mesenchymal state. We also report that ZEB2, an important regulator of the late phases of EMT, is directly repressed by H3K27me3 and that removal of this transcriptional break is required for sustaining the mesenchymal phenotype induced by TGF-β. In all, our data support that EZH2 functions as repressor of a key EMT inducer preserving an epithelial phenotype and that TGF-β-induced gene regulation involves not only a well-defined molecular signaling pathway, but also chromatin modifications that appear to be necessary during the later phases of EMT enabling the maintenance of a mesenchymal phenotype.
RESULTS
Kinetics of TGF-β-induced EMT in OC cells
TGF-β-induced EMT [16] in SKOV3 cells was confirmed by examining changes in cellular shape from epithelial to a mesenchymal morphology (Figure 1A-1I, -1II), a decrease in protein levels of E-cadherin (Figure 1B), and an increase of expression levels of the mesenchymal marker, vimentin (Figure 1C). The morphological changes became evident at 24-48 hours and progressed during TGF-β stimulation up to 120 hours. Downregulation of E-cadherin (CDH1 gene) during EMT is induced by a well described transcriptional machinery including the repressors SNAI1, SNAI2 (Slug), TWIST, ZEB1, ZEB2, and E47 [19-23]. To understand the dynamic involvement of these transcription factors in TGF-β-induced EMT in OC cells, we studied their expression levels in a time course experiment. We observed an increase in mRNA levels of SNAI1, SNAI2, ZEB1 and ZEB2 at 12 hours, and a slight decrease and plateau from 48-120 hours. In contrast, mRNA levels of SNAI2 and ZEB2 were significantly increased (approximately 20-fold) at later time-points (120 h) (Figure 1D-1G). These data suggest that in this model, several transcription factors including SNAI1, SNAI2, ZEB1 and ZEB2, initiate EMT, while maintenance of the mesenchymal phenotype depends mostly on SNAI2 and ZEB2.
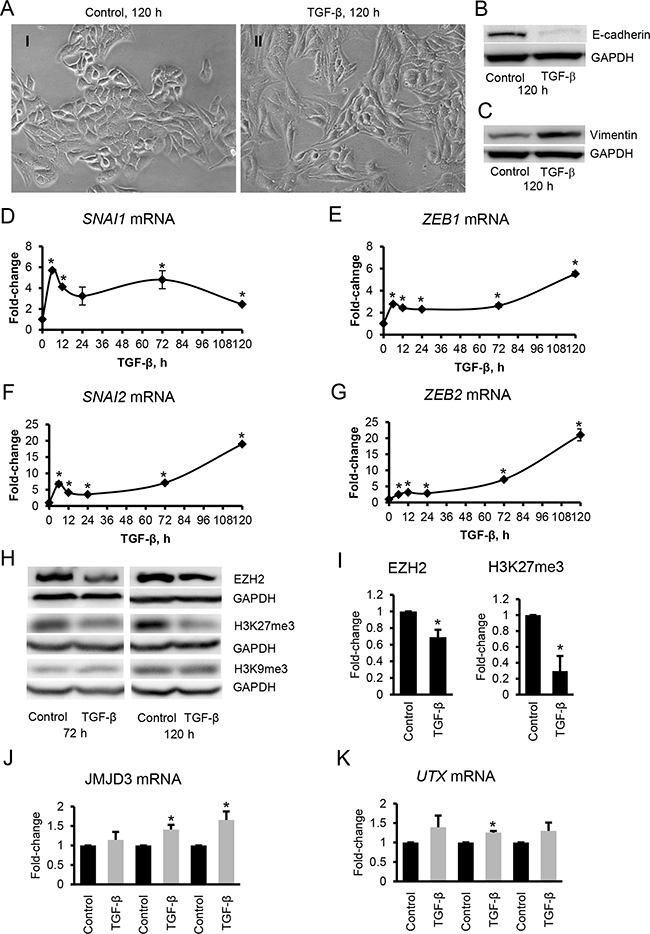
Figure 1: TGF-β induces EMT and changes in EZH2 and H3K27me3 in ovarian cancer cells. A. Phase contrast microscopy identifies morphological changes induced in SKOV3 cells by TGF-β (5 ng/mL) at 120 hours (magnification: 200X). B, C. Western blotting for E-cadherin (B) and vimentin (C) in SKOV3 cells treated with TGF-β (5 ng/mL) for 120 hours. D-G. Relative changes in mRNA expression levels of the EMT inducers SNAI1, SNAI2, ZEB1 and ZEB2 in SKOV3 cells treated with TGF-β (5 ng/mL) for 6, 12, 24, 72, and 120 hours. mRNA expression levels were measured by qRT-PCR and normalized to GAPDH. Values are means ± SE (n=3). *Indicate significant difference (P<0.05) relative to control. H. Western blotting for EZH2, H3K27me3, and H3K9me3 in SKOV3 cells treated with TGF-β (5 ng/mL) for 72 and 120 hours. I. Quantification of EZH2 (n=5) and H3K27me3 (n=3) levels in SKOV3 cells treated with TGF-β (5 ng/mL) for 120 hours. Intensity of protein bands measured by densitometry was normalized to GAPDH levels. Bars represent means ± SE, *indicate P<0.05 relative to control. J, K. qRT-PCR measures mRNA expression levels of JMJD3 and UTX in SKOV3 cells treated with TGF-β (5 ng/mL) for the indicated times. Expression levels were normalized to GAPDH. Bars represent means ± SE (n=3). *Denote significant differences relative to control (P<0.05).
Histone H3 trimethylation during TGF-β-induced EMT in OC cells
Because of observed differences in timing of transcriptional factor engagement by TGF-β, we hypothesized that the delayed overexpression of SNAI2 and ZEB2 could be regulated through epigenetic mechanisms [24]. To determine whether TGF-β stimulation is associated with changes in histone methylation, we measured the total levels of the H3K27me3 by western blotting. H3K27me3 levels decreased significantly 72 hours after TGF-β treatment and remained repressed at 120 hours (Figure 1H and 1I). This was accompanied by a modest decrease in protein levels of the methyltransferase EZH2 (Figure 1H and 1I). In contrast, levels of H3K9me3, another repressive mark not mediated by EZH2, were not altered by stimulation with TGF-β (Figure 1H). Interestingly, TGF-β did not alter EZH2 protein levels in untransformed ovarian surface epithelial (IOSE) cells (Supplementary Figure S1) suggesting that effects may be restricted to transformed cells. Additionally, TGF-β stimulation had no effect on EZH2 mRNA levels in SKOV3 cells (Supplementary Figure S2).
Because the H3K27me3 levels are influenced by both H3K27 methylation as well as by demethylation, we also examined the expression of the specific H3K27 demethylases UTX (also known as KDM6A) and JMJD3 (also known as KDM6B). Stimulation with TGF-β increased (P<0.05) the mRNA expression levels of JMJD3 at 72 and 120 hours (Figure 1K). TGF-β also increased (P<0.05) UTX expression (72 h) but to a lesser extent than JMJD3 (Figure 1J). These results support that TGF-β induces a decrease in the total amount of the repressive mark H3K27me3 by decreasing EZH2 protein level and by increasing expression of H3K27 demethylases.
Inhibition of EZH2 enzymatic activity induces mesenchymal characteristics in OC cells
To determine whether the process of EMT involves events regulated by H3K27me3, we used the enzymatic inhibitor GSK126 and siRNA-mediated knock down. GSK126 is a specific S-adenosyl-methionine-competitive inhibitor of the EZH2 methyltransferase activity [25]. SKOV3 cells treated with GSK126 alone for 120 hours exhibited a change in shape from cobblestone to a mesenchymal morphology (Figure 2A-2I vs -2III). The morphological changes induced by GSK126 were similar to those induced by TGF-β (Figure 2A-2II). The combination of TGF-β plus GSK126 potently induced mesenchymal characteristics (Figure 2A-2IV). H3K27me3 levels were decreased by GSK126, TGF-β, or the combination treatment (Figure 2B), and EZH2 protein levels were decreased by TGF-β and were not affected by GSK126 (Supplementary Figure S3), in agreement with previous reports [25]. The mesenchymal morphology induced by GSK126 and by TGF-β correlated with an increased expression of vimentin (Figure 2C) and N-cadherin (Supplementary Figure S4). Interestingly however, the levels of E-cadherin did not change after GSK126 treatment (Figure 2D), suggesting that the enzymatic activity of EZH2 is not implicated in the regulation of E-cadherin expression.
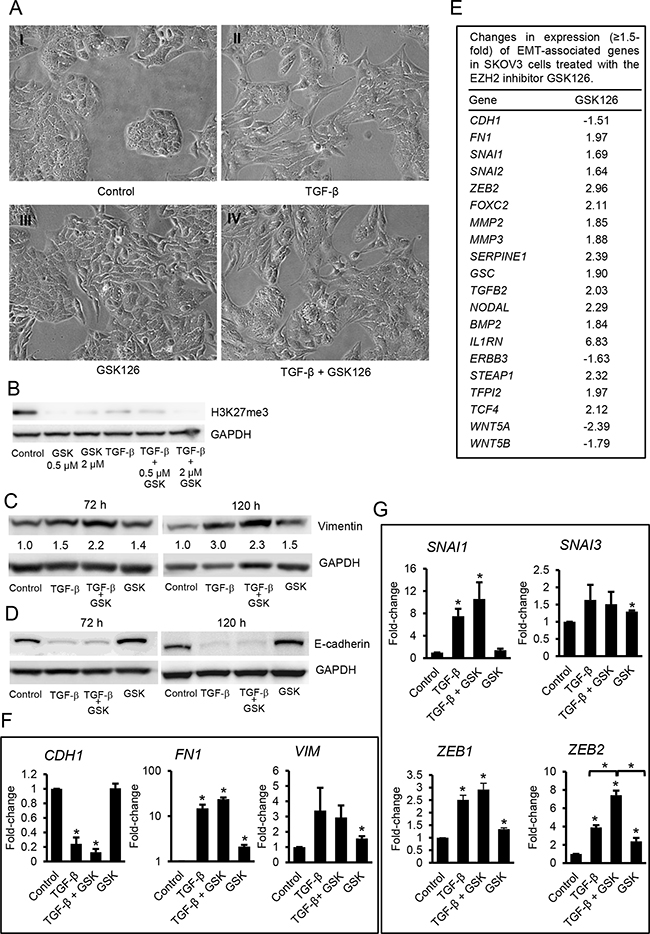
Figure 2: Effects of GSK126 on ovarian cancer cells. A. Phase contrast microscopy identifies morphological changes induced in SKOV3 cells by diluent (control, A-I), TGF-β (5 ng/mL, picture A-II), GSK126 (2 µM, picture A-III), or the combination (picture A-IV) for 120 hours (magnification: 200X). B. Western blotting for H3K27me3 in SKOV3 cells treated with TGF-β (5 ng/mL), GSK126 (2 µM or 0.5 µM), or the combination for 120 hours. C, D. Effects of TGF-β (5 ng/mL), GSK126, and the combination on Vimentin (C) and E-cadherin (D) protein expression levels as measured by western blotting at 72 and 120 hours. Densitometric values relative to GADH are marked. E. List of EMT-associated genes and fold-change (≥ 1.5) in response to treatment with GSK126 (2 µM for 120 hours) measured by RT2 Profiler PCR arrays for EMT. mRNA expression levels were normalized to the average expression of five housekeeping genes (ACTB, B2M, GAPDH, HPRT1 and RPLP0). F, G. Validation using qRT-PCR of mRNA expression levels for (F) EMT markers CDH1 (E-cadherin), FN1 (fibronectin-1), and VIM (vimentin), and (G) EMT-associated transcription factors SNAI1, SNAI3, ZEB1, and ZEB2 in SKOV3 cells treated with TGF-β (5 ng/mL), GSK126 (2 µM), or the combination for 120 hours. GAPDH was used for normalization. Bars represent means ± SE (n=3). *Indicate significant differences compared to controls or between values linked by brackets (P<0.05).
To further explore the involvement of EZH2 and H3K27me3 during the process of EMT, we used an RT-PCR array to determine changes in expression of 84 known EMT-associated genes in SKOV3 cells treated with 2 μM GSK126 for 120 hours. Twenty genes showed ≥1.5-fold change in cells treated with GSK126 including EMT inducers and mesenchymal markers (Figure 2E). Quantitative RT-PCR subsequently confirmed that EZH2 inhibition significantly increased (P<0.05) expression levels of the mesenchymal markers FN1 (fibronectin 1) and VIM (vimentin) (Figure 2F) and the EMT inducers SNAI3, ZEB1, and ZEB2 (Figure 2G). In most cases, the levels of up-regulation induced by GSK126 were less than those induced by TGF-β. However, the upregulation of ZEB2 induced by TFG-β and by GSK126 were comparable, and the combination of TGF-β and GSK126 had an additive effect (P<0.01) on ZEB2 expression compared to each agent alone (Figure 2G).
Next, we examined the induction of EMT in a different OC cell line. EMT was induced in Kuramochi cells by TGF-β after prolonged treatment (up to 14 days) as indicated by subtle changes in morphology to elongated shapes (Figure 3A-3I vs 3A-3II), a decrease of E-cadherin protein levels and an increase in vimentin expression (Figure 3B). Kuramochi cells treated with GSK126, or TGF-β, alone or in combination for 96 hours, showed a similar mesenchymal-like morphology (Figure 3C). GSK126 treatment increased (P<0.05) expression of the mesenchymal markers FN1 and VIM (Figure 3D) and EMT inducer ZEB2 (Figure 3E). The effect of combining TGF-β plus GSK126 on ZEB2 expression was greater (P=0.01) than TGF-β alone (Figure 3E), similar to the responses observed in SKOV3 cells.
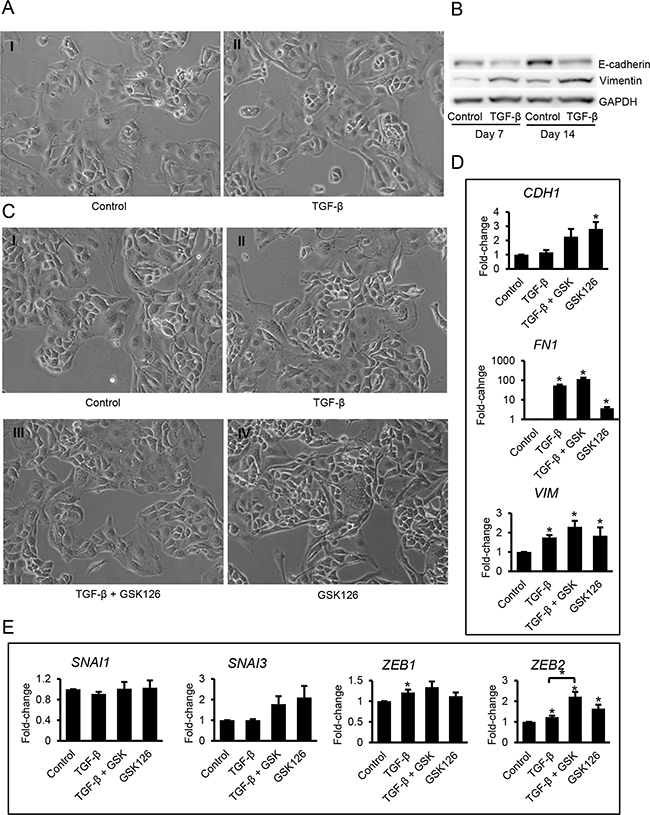
Figure 3: Effects of TGF-β and GSK126 on Kuramochi ovarian cancer cells. A. Phase contrast microscopy identifies morphological changes induced in Kuramochi cells by TGF-β (5 ng/ml) at 7 days (magnification: 200X). B. Western blotting for E-cadherin and vimentin in lysates of Kuramochi cells treated with TGF-β (5 ng/mL) for 7 and 14 days. C. Morphology of Kuramochi cells treated with vehicle (C-I), TGF-β (5 ng/mL, C-II), TGF-β (5 ng/mL) plus GSK126 (2 μM, C-III), or GSK126 (2 μM, C-IV) for 7 days (magnification: 200X). D, E. Relative mRNA expression levels (means ± SE, n=3) of CDH1 (E-cadherin), FN1 (fibronectin) and VIN (Vimentin) (D) and EMT-associated transcription factors SNAI1, SNAI3, ZEB1, and ZEB2 (E) measured by qRT-PCR in Kuramochi cells treated as described in C for 96 hours. mRNA expression levels were normalized to GAPDH. Bars represent means ± SE. *Indicate significant differences relative to control or between bars linked by brackets (P<0.05).
EZH2 knockdown induces a mesenchymal phenotype in SKOV3 cells
To further examine the effects of EZH2 inhibition on EMT we used siRNA targeting EZH2 (siEZH2). Western blot confirmed efficient EZH2 knockdown by siEZH2 compared with scrambled siRNA (siControl, Figure 4B). SKOV3 cells transfected with siControl and treated with TGF-β displayed a mesenchymal morphology indicative of EMT (Figure 4A-4I vs. -4III). SKOV3 cells transfected with siEZH2 and either treated or untreated with TGF-β also exhibited a mesenchymal morphology (Figures 4A-4II and 4A-4IV), a decrease in protein levels of E-cadherin (Figure 4C), an increase in vimentin (Figure 4D and 4E), and an increase in migratory properties (transwell migration, Figure 4F) consistent with induction of EMT.
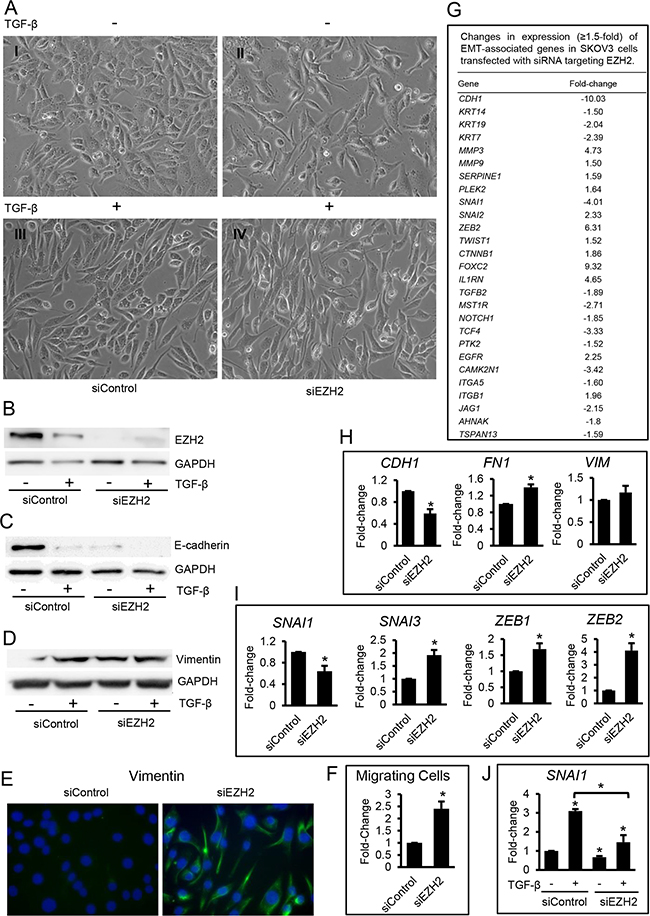
Figure 4: Effects of EZH2 knock down by siRNA in ovarian cancer cells. A. Morphology of SKOV3 cells transfected with control siRNA (siControl, A-I and A-III) or siRNA targeting EZH2 (siEZH2, A-II and A-IV), and treated with control vehicle (AI and II) or TGF-β (5 ng/mL-AII and IV) for 48 hours (magnification: 200X). B, C, D. Western blotting of EZH2 (B), E-cadherin (C), and vimentin (D) in SKOV3 cell lysates treated as described in A. E. Immunofluorescent staining of vimentin (green) in SKOV3 cells transfected with control siRNA (siControl) or siRNA targeting EZH2 (siEZH2) (magnification: 400X). Nuclei show blue DAPI staining. F. Transwell migration of SKOV3 cells treated as described in E. G. List of EMT-associated genes and fold-change (≥1.5) in SKOV3 cells transfected with siRNA targeting EZH2 or control siRNA, as measured by RT2 Profiler PCR arrays for EMT. mRNA expression levels were normalized to the average expression of five housekeeping genes (ACTB, B2M, GAPDH, HPRT1 and RPLP0). H, I. Validation using qRT-PCR of mRNA expression levels for (H) EMT markers CDH1 (E-cadherin), FN1 (fibronectin-1), and VIM (vimentin), and (I) EMT-associated transcription factors SNAI1, SNAI3, ZEB1, and ZEB2 in SKOV3 cells transfected with siRNA targeting EZH2 (siEZH2) or control siRNA (siControl). GAPDH was used for normalization. J. qRT-PCR measurements of SNAI1 mRNA expression levels in SKOV3 cells transfected with control siRNA (siControl) or siRNA targeting EZH2 (siEZH2) and then treated with vehicle or TGF-β (5 ng/mL) for 48 hours. mRNA expression levels were normalized to GAPDH. Bars represent means ± SE (n=3). *Indicate significant differences compared to controls or between bracket-linked means (P<0.05).
The expression of EMT-associated genes in SKOV3 cells transfected with siRNA targeting EZH2 was next examined by using a quantitative RT-PCR EMT array. Interestingly, more EMT associated genes were affected by EZH2 knockdown compared to enzymatic inhibition. Specifically, EZH2 knockdown caused an increase in mRNA expression levels for the EMT inducers SNAI2, ZEB2, TWIST1, and of genes with known roles in mesenchymal cell function (MMP3, MMP9, SERPINE1, and PLEK2), and a decrease of the epithelial markers CDH1, KRT14, KRT19, and KRT7 (Figure 4G). The effects of EZH2 knockdown on EMT induction were confirmed by qRT-PCR which showed a decrease in CDH1 (Figure 4H), and increases in FN1 (Figure 4H) and the EMT inducers ZEB1, ZEB2, and SNAI3, (Figure 4I). Taken together with the previous data obtained using the enzymatic inhibitor GSK126, these results support that inhibition of EZH2 induces EMT in OC cells. Consequently, relatively high EZH2 expression levels (and likely activity) appear necessary for maintaining an epithelial phenotype.
Interestingly, the only transcriptional regulator linked to EMT, which was suppressed, rather than upregulated by EZH2 knockdown was SNAI1 (Figure 4I). It is possible that SNAI1 is not necessary for EMT induction in this model and that its functions are redundant with those of other TGF-β-inducible transcription factors. This is consistent with results observed with the use of GSK126 which induced mesenchymal cell characteristics without causing any changes in SNAI1 expression (Figure 2G and 3E). Repression of SNAI1 mRNA levels induced by EZH2 knockdown could also indicate that EZH2 directly regulates its expression. To examine this possibility, we determined the effect of EZH2 knockdown on TGF-β-induced SNAI1 upregulation in SKOV3 cells. Indeed, we observed TGF-β-induced upregulation of SNAI1 in cells expressing normal levels of EZH2 but no upregulation in cells transfected with siEZH2 and treated with TGF-β (Figure 4J), indicating that EZH2 is required for TGF-β-induced SNAI1 upregulation.
TGF-β decreases EZH2 binding to and H3K27me3 marking of the ZEB2 promoter
Among the EMT inducers, ZEB2 which appears to be important to the late stages of EMT (Figure 1G) was the most responsive based on fold-change to EZH2 inhibition by GSK126 (Figure 2F) or knockdown (Figure 4I). To test its involvement relative to EZH2 in TGF-β-induced EMT, SKOV3 cells transfected with control or siRNA targeting EZH2 were treated with TGF-β for 48 hours. TGF-β notably upregulated (~17-fold) ZEB2 expression in SKOV3 cells transfected with siEZH2 relative to cells transfected with control siRNA (P<0.05, Figure 5A). These results are consistent with the ZEB2 response observed in cells treated with TGF-β and the EZH2 inhibitor GSK126 (Figure 2F), suggesting that the ZEB2 promoter may be marked by the H3K27me3 repressive mark, and could become more responsive to TGF-β upon removal of the histone mark. To determine whether TGF-β alters EZH2 binding to and H3K27me3 marking of the ZEB2 promoter, we used ChIP with antibodies against EZH2 and H3K27me3. We examined within the -700 to +300 sequence relative to the transcription start site (TSS) of the ZEB2 promoter, two regions containing SMAD binding sites (SBS, minimal consensus sequence AGAC) designated SBS1 (-103 to -256) and SBS2 (-428 to -585) (Figure 5B). Pull down of the region containing the SBS1 using EZH2 and H3K27me3 antibodies was not affected by TGF-β (Figure 5B). However, TGF-β decreased (P<0.05) EZH2 binding and the amount of H3K27me3 in the region containing the SBS2 (Figure 5D). As a control, SKOV3 cells treated with the EZH2 inhibitor GSK126 displayed decreased (P<0.05) levels of H3K27me3 marking in the two examined regions (Figure 5C and 5D). These results demonstrate that TGF-β decreases H3K27me3 repressive marking in specific regions of the ZEB2 promoter, which may enable more efficient binding of TGF-β induced SMADs (or other transcription factors) that, in turn, regulate ZEB2 expression.
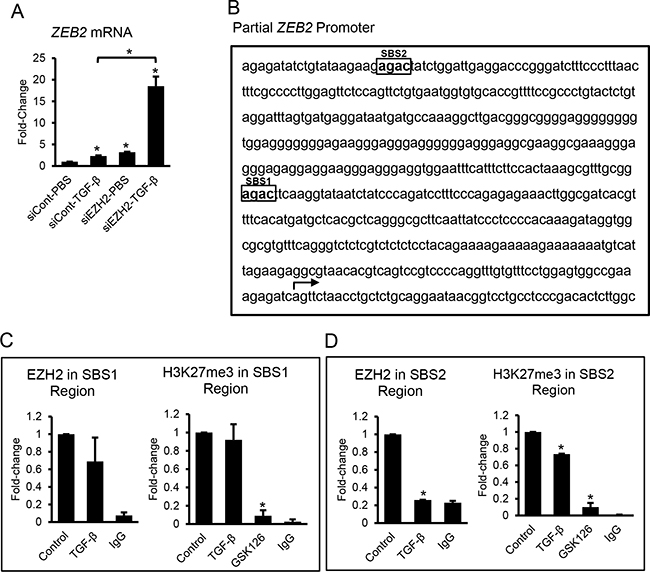
Figure 5: TGF-β and EZH2 regulation of ZEB2 expression. A. qRT-PCR measures ZEB2 mRNA expression levels (means ± SE, n=3) in SKOV3 cells transfected with control siRNA (siControl) or siRNA targeting EZH2 (siEZH2) and treated with vehicle (control) or TGF-β for 48 hours. mRNA expression levels were normalized to GAPDH. Bars represent means ± SE (n=3). *Indicate significant differences compared to controls or between bracket-linked values (P<0.05). B. Partial sequence of the ZEB2 promoter illustrates two SMAD binding sites (designated as SBS1 and SBS2). The transcription start site (TSS) is marked by an arrow. C, D. Chromosome immunoprecipitation measures effects of TGF-β on EZH2 and H3K27me3 binding to the ZEB2 promoter containing the SBS1 (C) and SBS2 (D). GSK126 decreases H3K27me3 binding to the same regions (positive control). Input levels were used for normalization. Bars represent means ± SE (n=2). *Indicate significant differences relative to control (P<0.05).
Inhibition of H3K27 demethylases suppresses TGF-B-induced EMT
Stability of the H3K27me3 chromatin repressive marking depends on both the activity of the HMT EZH2 and of the H3K27 demethylases JMJD3 and UTX [26]. To examine the involvement of the demethylases JMJD3 and UTX, which we found to be modestly upregulated by TGF-β (Figure 1J and 1K) during the process of EMT, we used the small molecule inhibitor GSK-J4 which is enzymatically converted into GSK-J1 upon entering the cytosol and blocks both JMJD3 and UTX [27]. We observed that SKOV3 cells treated with GSK-J4 for 96 hours maintained an epithelial cobblestone appearance (Figure 6A-6IV) similar to that of control-treated cells (Figure 6A-6I). The mesenchymal phenotype induced by TGF-β (Figure 6A-6II) was prevented by concomitant treatment with GSK-J4 (Figure 6A-6III). GSK-J4 also inhibited (P<0.05) TGF-β-induced upregulation of the mesenchymal markers, FN1 (fibronectin) and VIM (vimentin, Figure 6B), but did not alter the effects of TGF-β on CDH1 (Figure 6B). These data add to the concept that H3K27me3 mediated repression of mesenchymal characteristics is responsive to TGF-β treatment. In contrast, the epithelial marker E-cadherin appears less affected by modulation of this histone mark in response to TGF-β. Furthermore, the E-cadherin repressors SNAI1, ZEB1, and ZEB2, which are potently induced by TGF-β, were only modestly affected by GSK-J4 (only SNAI1) and the demethylase inhibitor did not significantly alter their response to TGF-β (Figure 6C). In all, these results support that inhibition of JMJD3 and UTX by GSK-J4 partially suppresses TGF-β-induced EMT by preventing upregulation of mesenchymal markers.
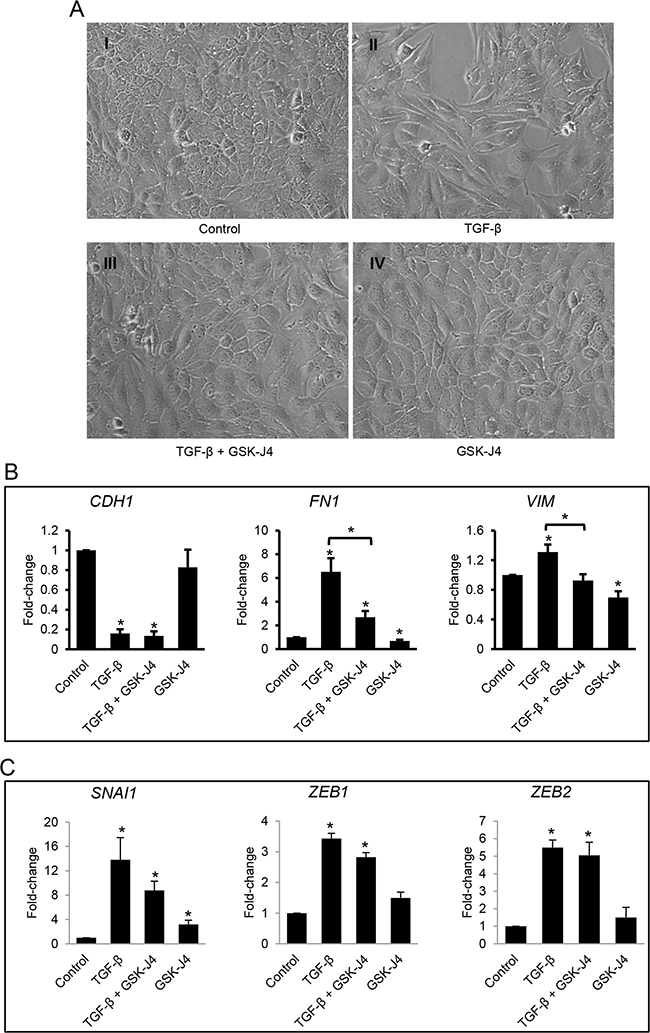
Figure 6: Effects of the JMJD3 and UTX inhibitor GSK-J4 on TGF-β-induced EMT in ovarian cancer cells. A. Morphology accessed by phase contrast microscopy of SKOV3 cells treated with vehicle (A-I), TGF-β (5 ng/mL, A-II), TGF-β (5 ng/mL) plus GSK-J4 (4 μM, A-III), or GSK-J4 (4 μM, panel A-IV) for 96 hours (magnification: 200X). B, C. qRT-PCR measurements of mRNA expression levels for (B) EMT markers CDH1, FN1, and VIM, and (C) EMT-associated transcription factors SNAI1, ZEB1, and ZEB2 in SKOV3 cells treated as described in A. GAPDH was used for normalization. Values are means ± SE (n=3). *Indicate significant differences relative to control or between bracket-linked values (P<0.05).
DISCUSSION
Epithelial-to-mesenchymal transition is a cellular process involved in the early phases of development and in cancer metastasis. Recent data support that regulation of this “plastic” process involves not only genetic factors, but also epigenetic remodeling, including alterations in DNA methylation and effects of histone modifiers on chromatin assembly [16, 28]. The Polycomb (PcG) and the Trithorax groups (TrxG) of proteins function as epigenetic repressors or activators respectively, being implicated in regulation of gene transcription during development [29]. It has been established that their loss of function impacts the fate of embryonic stem cells and their differentiation into the three germ layers [30]. In the present study we describe a novel mechanism by which EMT is sustained in OC cells through loss of the repressive chromatin mark H3K27me3 at the promoter of transcription factors involved in the induction of mesenchymal characteristics. Our data point to ZEB2 as a transcriptional regulator activated late during the transition, whose promoter loses the H3K27me3 histone mark in response to TGF-β. Mesenchymal characteristics of OC cells are enhanced by both EZH2 knockdown or by enzymatic inhibitors, suggesting that EZH2 is required for maintaining the epithelial cellular characteristics in this system.
Consistent with our findings, a recent report described the methyltransferase activity of EZH2 as a regulator of mesenchymal to epithelial transition (MET) during human pluripotent cell reprogramming. In this model, EZH2 inhibitors prevented Oct4/Sox2/Klf4/c-Myc induced transformation of fibroblasts into iPSCs [31]. Likewise, it has been shown that inhibition of EZH2 augments the expression of transgelin/smooth muscle-22α, a mesenchymal marker, in response to TGF-β in endothelial cells [32]. Loss of EZH2 in the lung epithelium resulted in defective epithelial layer differentiation and alveoli formation [33]. Lastly, conditional overexpression of EZH2 using the MMTV-LTR model in the mammary gland caused epithelial hyperplasia through activation of the Wnt/β-catenin pathway [34], while conditional EZH2 knock-out delayed the outgrowth of the mammary epithelium [35]. In all, like our data, these studies support that EZH2 functions as a guardian of epithelial cell fate and perhaps, as a break against TGF-β-induced transition to mesenchymal features.
The increased expression of EZH2 in ovarian tumors [36] could explain the reported relative resistance of OC cells to EMT [37, 38], or their preference towards MET, reverting to an epithelial phenotype immediately after dissemination from the primary tumor site. The transition between EMT and MET may be particularly relevant to OC, where dissemination occurs intraperitoneally and cells are required to adopt an epithelial phenotype and to adhere to the mesothelium shortly after relocating to distant sites. It is possible that EZH2 facilitates this process during tumor metastasis, tilting the balance in favor of MET.
On the other hand, the findings that EZH2 enhances the epithelial characteristics of OC cells argue against the concept that this HMT is an oncogene associated with cancer metastasis [36, 39]. EZH2 was previously shown to repress E-cadherin in breast, pancreatic and prostate cancer cells [40, 41]. Cooperation between Snail1 and EZH2 in the repression of E-cadherin was also observed during neural crest development [42]. In this regard, our findings are discrepant, as we show that E-cadherin expression is suppressed by EZH2 knockdown in OC cells. Interestingly, we also observed that the EZH2 inhibitor GSK126 did not alter E-cadherin expression, suggesting that in our model, E-cadherin regulation by EZH2 is independent of its HMTase function. Polycomb-independent regulatory functions of EZH2 have been also recognized in other cancer models, where the protein cooperates with other transcription factors (such as the androgen receptor or NF-κB) to exert oncogenic properties [43, 44]. Our findings likely reflect EZH2’s distinct cell context-dependent functions, which have been previously recognized [43]. Indeed, other studies have proposed a tumor suppressor role for EZH2, as loss-of-function mutations associated with decreased H3K27me3 levels were noted in patients with myelodysplastic syndromes and myeloproliferative neoplasms (MDS/MPN) [45, 46], and deletion of EZH2 was sufficient to induce MDS/MPN-like disease in mice [47].
We attribute the EMT blocking role of EZH2 to its repressive effects on several transcriptional regulators, and in particular on ZEB2. This is the first report demonstrating that ZEB2 is a direct EZH2 target whose promoter, which is repressed by the H3K27me3 mark, responds to TGF-β stimulation. We show that blockade of EZH2 by using either siRNA mediated knockdown or pharmacological inhibitors leads to de-repression of ZEB2 and drives OC cells through EMT. Polycomb-mediated regulation of the ZEB1 promoter has been recently implicated in the conversion of breast cancer non-stem to stem cells [48] and loss of H3K27me3 at the poised promoter of ZEB1 was functionally linked to activation of the stemness regulatory transcriptional machinery.
Our results also show that TGF-β stimulation is associated with a decrease of the H3K27me3 repressive mark, which appears to occur late during the EMT process. Consistent with our findings, Ramadoss, et al. [49] have shown a decrease of the H3K27me3 mark in response to TGF-β in a mammary epithelial system, which was attributed to upregulation of the demethylase JMJD3. We also observed in our OC cell model upregulated demethylase levels in response to TGF-β. However, the decrease of the repressive chromatin mark in our system appears to be related to both increased levels of demethylases as well as decreased EZH2 activity. Epigenetic reprogramming including a global reduction of the H3K9me2 marked heterochromatin involving LOCKS regions and an increase in H3K4Me3 associated euchromatin was described in hepatocytes exposed to TGF-β [9]. In monocytes conditioned by TGF-β, global mapping of histone marks showed that a larger number of differentiation associated genes was marked by H3K4me3, while significantly fewer genes were marked by H3K27me3 in response to the cytokine [50]. Collectively these data support that epigenomic modifications occur during EMT, are most likely cell context-dependent, and participate in the fine tuning of cellular transformation and plasticity with its acquired new functions.
Although EZH2 has been considered a potential cancer therapeutic target [51, 52], our findings also point to potentially unexpected consequences of EZH2 inhibition leading to de-repression not only of tumor suppressor genes that can block cancer growth, but also of pro-tumorigenic factors which can unleash unanticipated aggressive cancer behavior. The difficulty manipulating epigenomically-directed therapies is linked to their “imprecise targeting” and ability to globally reprogram the oncogenic machinery. We posit that an in-depth analysis of both potential benefits as well as adverse consequences must be a priori completed and understood. Efforts to design pathway specific epigenomic blocking strategies that avoid non-specific off-target effects are critically needed.
MATERIALS AND METHODS
Cell culture and treatments
SKOV3 cells were obtained from the American Type Culture Collection (ATCC). Kuramochi cells were from the Japanese Cancer Research Resources Bank. The immortalized ovarian surface epithelial (IOSE) cells were kindly provided by Dr. N. Auersperg from the University of British Columbia. SKOV3 and IOSE cells were maintained in media containing 1:1 MCDB 105 (Sigma-Aldrich, St. Louis, MO) and M199 (Cellgro, Tewksbury, MA). Kuramochi cells were cultured in RPMI 1640. Media were supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution. Cells were treated with TGF-β1 (hereafter referred to as TGF-β, R&D Systems, Minneapolis, MN), EZH2 inhibitor GSK126 (Xcess Biosciences, Inc., San Diego, CA), UTX and JMJD3 inhibitor GSK-J4 (Sigma-Aldrich), or combinations of TGF-β with inhibitors at doses and durations as indicated.
Cell transfection
Transient knockdown of EZH2 was performed by transfecting SKOV3 cells with a pool of 3 target-specific small interfering RNA (siRNA) (catalogue#: sc-35312, Santa Cruz Biotechnology, Inc. Dallas, TX) in the presence of the DreamFect Gold transfection reagent (OZ Biosciences, San Diego, CA). Scrambled siRNA (Santa Cruz Biotechnologies, Inc.) served as control.
Migration assay
Cell migration was evaluated using transwell inserts (8 µm pore size) in a 24-well plate (Corning Inc., Corning NY). Ten thousand cells suspended in serum-free medium were seeded in the upper compartment, and medium containing 20% FBS was added to the lower compartment as attractant. Cells were cultured for 24 hours under standard conditions. Non-migrating cells were removed with a cotton swab, and remaining migrating cells were fixed with 4% paraformaldehyde, stained with 1% crystal violet, and counted using a light microscope.
RNA isolation and real-time quantitative RT-PCR (qRT-PCR)
Total RNA was extracted with the RNA Stat-60 reagent (Tel-Test, Inc., Friendswood, TX). An iScript cDNA Synthesis kit (Bio-Rad Laboratories, Hercules CA) was used for reverse transcription of RNA, and iTaq Universal SYBR Green Supermix (Bio-Rad) was used for quantitative PCR (qPCR) amplification of cDNAs following procedures from the manufacturers. Relative changes in mRNA expression levels were calculated by the 2-ΔΔCT method using GAPDH for normalization. The sequences of primers used for qRT-PCR are presented in Supplementary Table S1. The human EMT RT2 Profiler PCR Array was purchased from SA Bioscience (Frederick, MD) and used with an ABI Prism 7900 HT Real-Time PCR system (Applied Biosystems, Foster City, CA), according to the manufacturer’s instructions. Array data analysis was performed based on the 2-ΔΔCT method with normalization of the raw data to the housekeeping genes included in the array using a Microsoft Excel algorithm provided by the manufacturer. Fold-changes were calculated for each gene in treated cells compared to controls. All experiments were repeated at least three times in independent conditions.
Western blotting
Proteins were resolved by SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes (Millipore, Billerica, MA). Membranes were blocked and then probed with antibodies against the proteins of interest. Antibodies against vimentin (# 3932), E-cadherin (#3195), EZH2 (#3147), and tri-methyl-histone H3 (Lys27) (H3K27me3, #9756) were from Cell Signaling Technology, tri-methyl-histone H3 (Lys9) (H3K9me3, ab8898) from Abcam Inc. (Cambridge, MA), and GAPDH (#H86504M) from Meridian Life Sciences, Inc. (Memphis, TN). Membranes were incubated with HRP-conjugated secondary antibody, followed by detection of antigen-antibody complexes using an enhanced chemiluminescent substrate (SuperSignal West Pico, Thermo Scientific, Waltham, MA). Digital images of blots were captured by a luminescent image analyzer (ImageQuant LAS 4000, GE Healthcare Life Sciences, Piscataway, NJ). Densitometric analysis of protein bands was performed with ImageJ 1.48 software (National Institutes of Health of the USA). Experiments were repeated at least three times.
Immunofluorescence
Cells grown in 8-well culture slides (Millicell EZ slide, Millipore) were fixed with 4% paraformaldehyde for 30 min, and blocked/permeabilized with PBS containing 5% normal goat serum and 0.3% Triton X-100 for 60 minutes. Thereafter, cells were incubated with primary antibody overnight at 4°C, and then with Alexa Fluor 488-labeled anti-rabbit IgG (H+L) for 2 hours. Slides were coverslipped using Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA) and subsequently examined and photographed with a fluorescence microscope fitted with a digital camera.
Chromosome immunoprecipitation (ChIP)
ChIP assays were performed using a kit (#17-295, Millipore) following the manufacturer’s protocol. Briefly, chromatin was cross-linked with 1% formaldehyde for 10 minutes at 37°C. Cells were treated with SDS lysis buffer followed by sonication (5 pulses of 10 seconds each at 30 seconds interval using Fisher Scientific Model 100 set at 3). Protein-chromatin complexes were immunoprecipitated with antibodies against EZH2 (#5246, Cell Signaling Technology, Danvers, MA), H3K27me3 (#9756, Cell Signaling Technology), or IgG (negative control). Complexes were separated by incubation with salmon sperm DNA/protein A agarose slurry, and then the chromatin was eluted and the cross-links reversed. DNA was purified with a Chromatin IP DNA Purification kit (Active Motif, Carlsbad, CA). The primers used for amplification of specific regions of the ZEB2 promoter by qPCR are described in Supplementary Table S2. Chromosome immunoprecipitation experiments were repeated at least twice.
Statistical analysis
Data were analyzed using the Student’s t-test. Differences were considered statistically significant when P<0.05.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This work was supported by the National Cancer Institute (CA182832 to DM and KN) and the V Foundation (to DM and KN).
REFERENCES
1. Cao R and Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Current opinion in genetics & development. 2004; 14:155-164.
2. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, Leiserson MD, Miller CA, Welch JS, Walter MJ, Wendl MC, Ley TJ, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013; 502:333-339.
3. Herviou L, Cavalli G, Cartron G, Klein B and Moreaux J. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget. 2016; 7:2284-2296. doi: 10.18632/oncotarget.6198.
4. Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R and Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013; 14:765-780.
5. Jiang T, Wang Y, Zhou F, Gao G, Ren S and Zhou C. Prognostic value of high EZH2 expression in patients with different types of cancer: a systematic review with meta-analysis. Oncotarget. 2015; 7:4584-97. doi: 10.18632/oncotarget.6612.
6. O’Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW, Clements EG, Cai Y, Van Neste L, Easwaran H, Casero RA, Sears CL and Baylin SB. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer cell. 2011; 20:606-619.
7. Papageorgis P. TGFbeta Signaling in Tumor Initiation, Epithelial-to-Mesenchymal Transition, and Metastasis. Journal of oncology. 2015; 2015:587193.
8. Akhurst RJ and Derynck R. TGF-beta signaling in cancer--a double-edged sword. Trends in cell biology. 2001; 11:S44-51.
9. McDonald OG, Wu H, Timp W, Doi A and Feinberg AP. Genome-scale epigenetic reprogramming during epithelial-to-mesenchymal transition. Nature structural & molecular biology. 2011; 18:867-874.
10. Abendstein B, Stadlmann S, Knabbe C, Buck M, Muller-Holzner E, Zeimet AG, Marth C, Obrist P, Krugmann J and Offner FA. Regulation of transforming growth factor-beta secretion by human peritoneal mesothelial and ovarian carcinoma cells. Cytokine. 2000; 12:1115-1119.
11. Cao L, Shao M, Schilder J, Guise T, Mohammad KS and Matei D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene. 2012; 31:2521-2534.
12. Do TV, Kubba LA, Du H, Sturgis CD and Woodruff TK. Transforming growth factor-beta1, transforming growth factor-beta2, and transforming growth factor-beta3 enhance ovarian cancer metastatic potential by inducing a Smad3-dependent epithelial-to-mesenchymal transition. Molecular cancer research. 2008; 6:695-705.
13. Heldin CH, Vanlandewijck M and Moustakas A. Regulation of EMT by TGFbeta in cancer. FEBS letters. 2012; 586:1959-1970.
14. Thiery JP, Acloque H, Huang RY and Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139:871-890.
15. Syed V. TGF-beta Signaling in Cancer. Journal of cellular biochemistry. 2016.
16. Cardenas H, Vieth E, Lee J, Segar M, Liu Y, Nephew KP and Matei D. TGF-beta induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics. 2014; 9:1461-1472.
17. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y and Fuks F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006; 439:871-874.
18. Ning X, Shi Z, Liu X, Zhang A, Han L, Jiang K, Kang C and Zhang Q. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer letters. 2015; 359:198-205.
19. Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002; 2:442-454.
20. Nagafuchi A, Shirayoshi Y, Okazaki K, Yasuda K and Takeichi M. Transformation of cell adhesion properties by exogenously introduced E-cadherin cDNA. Nature. 1987; 329:341-343.
21. Comijn J, Berx G, Vermassen P, Verschueren K, van Grunsven L, Bruyneel E, Mareel M, Huylebroeck D and van Roy F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Molecular cell. 2001; 7:1267-1278.
22. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004; 117:927-939.
23. Moreno-Bueno G, Cubillo E, Sarrio D, Peinado H, Rodriguez-Pinilla SM, Villa S, Bolos V, Jorda M, Fabra A, Portillo F, Palacios J and Cano A. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer research. 2006; 66:9543-9556.
24. Diaz-Martin J, Diaz-Lopez A, Moreno-Bueno G, Castilla MA, Rosa-Rosa JM, Cano A and Palacios J. A core microRNA signature associated with inducers of the epithelial-to-mesenchymal transition. The Journal of pathology. 2014; 232:319-329.
25. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, LaFrance LV, Mellinger M, Duquenne C, Tian X, Kruger RG, McHugh CF, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012; 492:108-112.
26. Hong S, Cho YW, Yu LR, Yu H, Veenstra TD and Ge K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A. 2007; 104:18439-18444.
27. Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H, Eberhard D, Hutchinson S, Jones E, Katso R, Leveridge M, Mander PK, et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature. 2012; 488:404-408.
28. Wang Y and Shang Y. Epigenetic control of epithelial-to-mesenchymal transition and cancer metastasis. Experimental cell research. 2013; 319:160-169.
29. Geisler SJ and Paro R. Trithorax and Polycomb group-dependent regulation: a tale of opposing activities. Development (Cambridge, England). 2015; 142:2876-2887.
30. Harikumar A and Meshorer E. Chromatin remodeling and bivalent histone modifications in embryonic stem cells. EMBO reports. 2015; 16:1609-1619.
31. Rao RA, Dhele N, Cheemadan S, Ketkar A, Jayandharan GR, Palakodeti D and Rampalli S. Ezh2 mediated H3K27me3 activity facilitates somatic transition during human pluripotent reprogramming. Scientific reports. 2015; 5:8229.
32. Maleszewska M, Gjaltema RA, Krenning G and Harmsen MC. Enhancer of zeste homolog-2 (EZH2) methyltransferase regulates transgelin/smooth muscle-22alpha expression in endothelial cells in response to interleukin-1beta and transforming growth factor-beta2. Cellular signalling. 2015; 27:1589-1596.
33. Galvis LA, Holik AZ, Short KM, Pasquet J, Lun AT, Blewitt ME, Smyth IM, Ritchie ME and Asselin-Labat ML. Repression of Igf1 expression by Ezh2 prevents basal cell differentiation in the developing lung. Development (Cambridge, England). 2015; 142:1458-1469.
34. Li X, Gonzalez ME, Toy K, Filzen T, Merajver SD and Kleer CG. Targeted overexpression of EZH2 in the mammary gland disrupts ductal morphogenesis and causes epithelial hyperplasia. The American journal of pathology. 2009; 175:1246-1254.
35. Michalak EM, Nacerddine K, Pietersen A, Beuger V, Pawlitzky I, Cornelissen-Steijger P, Wientjens E, Tanger E, Seibler J, van Lohuizen M and Jonkers J. Polycomb group gene Ezh2 regulates mammary gland morphogenesis and maintains the luminal progenitor pool. Stem cells (Dayton, Ohio). 2013; 31:1910-1920.
36. Rao ZY, Cai MY, Yang GF, He LR, Mai SJ, Hua WF, Liao YJ, Deng HX, Chen YC, Guan XY, Zeng YX, Kung HF and Xie D. EZH2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of TGF-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis. 2010; 31:1576-1583.
37. Sundfeldt K. Cell-cell adhesion in the normal ovary and ovarian tumors of epithelial origin; an exception to the rule. Molecular and cellular endocrinology. 2003; 202:89-96.
38. Gao J, Zhu Y, Nilsson M and Sundfeldt K. TGF-beta isoforms induce EMT independent migration of ovarian cancer cells. Cancer cell international. 2014; 14:72.
39. Li H, Cai Q, Godwin AK and Zhang R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Molecular cancer research. 2010; 8:1610-1618.
40. Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, Kleer CG, Varambally S and Chinnaiyan AM. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008; 27:7274-7284.
41. Han T, Jiao F, Hu H, Yuan C, Wang L, Jin ZL, Song WF and Wang LW. EZH2 promotes cell migration and invasion but not alters cell proliferation by suppressing E-cadherin, partly through association with MALAT-1 in pancreatic cancer. Oncotarget. 2016; 7:11194-207. doi: 10.18632/oncotarget.7156.
42. Tien CL, Jones A, Wang H, Gerigk M, Nozell S and Chang C. Snail2/Slug cooperates with Polycomb repressive complex 2 (PRC2) to regulate neural crest development. Development (Cambridge, England). 2015; 142:722-731.
43. Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, Liou YC and Yu Q. Context-specific regulation of NF-kappaB target gene expression by EZH2 in breast cancers. Molecular cell. 2011; 43:798-810.
44. Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, Gregory RI, Morrissey C, Vessella RL, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science (New York, NY). 2012; 338:1465-1469.
45. Nikoloski G, Langemeijer SMC, Kuiper RP, Knops R, Massop M, Tonnissen E, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA and Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nature genetics. 2010; 42:665-667.
46. Volkel P, Dupret B, Le Bourhis X and Angrand PO. Diverse involvement of EZH2 in cancer epigenetics. American journal of translational research. 2015; 7:175-193.
47. Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, Sanada M, Miyagi S, Saraya A, Kamio A, Nagae G, Nakaseko C, Yokote K, Shimoda K, Koseki H, Suzuki Y, et al. Concurrent loss of Ezh2 and Tet2 cooperates in the pathogenesis of myelodysplastic disorders. The Journal of experimental medicine. 2013; 210:2627-2639.
48. Chaffer CL, Marjanovic ND, Lee T, Bell G, Kleer CG, Reinhardt F, D’Alessio AC, Young RA and Weinberg RA. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 2013; 154:61-74.
49. Ramadoss S, Chen X and Wang CY. Histone demethylase KDM6B promotes epithelial-mesenchymal transition. The Journal of biological chemistry. 2012; 287:44508-44517.
50. Huang Y, Min S, Lui Y, Sun J, Su X, Liu Y, Zhang Y, Han D, Che Y, Zhao C, Ma B and Yang R. Global mapping of H3K4me3 and H3K27me3 reveals chromatin state-based regulation of human monocyte-derived dendritic cells in different environments. Genes and immunity. 2012; 13:311-320.
51. Agarwal P, Alzrigat M, Parraga AA, Enroth S, Singh U, Ungerstedt J, Osterborg A, Brown PJ, Ma A, Jin J, Nilsson K, Oberg F, Kalushkova A and Jernberg-Wiklund H. Genome-wide profiling of histone H3 lysine 27 and lysine 4 trimethylation in multiple myeloma reveals the importance of Polycomb gene targeting and highlights EZH2 as a potential therapeutic target. Oncotarget. 2016; 7:6809-23. doi: 10.18632/oncotarget.6843.
52. Baker T, Nerle S, Pritchard J, Zhao B, Rivera VM, Garner A and Gonzalvez F. Acquisition of a single EZH2 D1 domain mutation confers acquired resistance to EZH2-targeted inhibitors. Oncotarget. 2015; 6:32646-32655.