
INTRODUCTION
Cisplatin (cis-diamminedichloroplatinum(II), cDDP), is one of the most potent and widely used chemotherapeutic agents. cDDP has been successfully applied in treating many different tumor types, including head and neck, lung, testis, ovarian, cervix and breast [1, 2]. cDDP induces formation of intra- and interstrand crosslinks, which may induce DNA single and double strand breaks (SSBs and DSBs) in replicating cells [3, 4]. The accumulation of unrepaired DNA lesions, particularly DSBs, can lead to cell death. In mammalian cells, DSBs are repaired by mainly two major pathways: the classical non-homologous end joining (c-NHEJ) and the homologous recombination (HR). HR is mainly active in S- and G2-phase, ensures accurate repair by using the undamaged sister chromatid as a template and involves, among others, BRCA2 and RAD51 [5]. Mild hyperthermia (HT), 1 h at 41–42.5°C, can disrupt this pathway temporarily by inducing degradation of BRCA2 and preventing the recruitment of RAD51 to DSBs [6]. c-NHEJ is active in all cell cycle phases, rejoins the break ends without the requirement for homologous template and is therefore considered to be more error prone [7–9]. Besides inducing DNA breaks, cDDP has also been shown to disrupt c-NHEJ [10–13].
The combination of cDDP and HT has been shown to be an effective clinical modality and is the standard treatment for previously irradiated patients with recurrent cervical cancer in the Netherlands [14]. Nevertheless, commonly used dose of cDDP can be very toxic for a substantial percentage of patients due to sometimes irreversible neural toxicity (hearing loss, numbness and tingling of the extremities) and kidney failure [15, 16]. Kidney failure is particularly dreaded in women with advanced cervical cancer in whom kidney function is already impaired by renal obstruction due to the tumor. Therefore, a large number of patients would benefit it the required dose of cDDP could be reduced without compromising tumor control.
Tumor resistance to cDDP plus HT may not only be explained by an insufficient cDDP dose, but also by the existence of a third DNA-repair pathway, so-called back-up NHEJ (b-NHEJ), which becomes active when either or both c-NHEJ and HR are impaired [17]. Poly-(ADP-ribose)-polymerase1 (PARP1) is essential for functioning of b-NHEJ [18, 19], and at least partly explains the accumulation of DSBs. Additional benefits of this approach may come from the observations that PARP1 plays a crucial role in regulating replication fork progression. Collapsed replication forks caused by a PARP1-i are converted to DSBs and require HR for repair [20]. In HR deficient cells, including cells harboring inactivation mutations in BRCA1 or BRCA2 and cells suffering from a HT-induced BRCA2 degradation, such lesions become highly cytotoxic in what can be considered a form of synthetic lethality [6, 20–24]. Importantly, PARP1 inhibitors have already been in multiple clinical trials in BRCA negative breast and ovarian cancers, and generally show favorable clinical profile [25, 26].
Here we set out to test, in vitro, whether the efficacy of cDDP+HT modality can benefit from the inhibition of PARP1. We focused on two interrelated aspects of such therapeutic strategy. First, we investigated whether PARP1-i can enhance the cytotoxicity of the standard cDDP+HT regimen. Second, we asked whether addition of PARP1-i can allow significant reduction of the overall cDDP dose, while maintaining the cytotoxic potential of the treatment. This is a clinically relevant question, particularly in the case that inhibition of PARP1 does not significantly alter the efficacy of HT when combined with standard cDDP doses, due to relatively high cytotoxicity of the two modality approach. Given that the concentrations of cDDP in necrotic or poorly vascularized tumor areas are likely low, reducing the cDDP dose required for efficient cell killing by co-administering PARP1-i may allow maintaining local tumor control while limiting the systemic side effects associated with standard cDDP concentrations.
RESULTS
Mild hyperthermia induces cell cycle arrest, apoptosis and inhibits homologous recombination
To determine the effect of HT on R1, SiHa and HeLa cells, we first measured changes in cell cycle distribution and induction of apoptosis. In the cell cycle analysis (Figure 1A) a G2-arrest was observed 16 h after treating cells for 1 h with 42°C. This effect was moderate for R1 cells and more pronounced for SiHa and HeLa cells. Flow cytometric analysis of DNA content showed a 20% increase in apoptosis for all cell lines (Figure 1B). Next, We measured the effects of HT on HR activity by quantifying accumulation of HR factor RAD51 on alpha-particle induced DSBs. Consistently with previously published results, HT treatment temporarily abrogated accumulation of RAD51 on DSB sites in all cell lines (Figure 1C), confirming inactivation of HR.
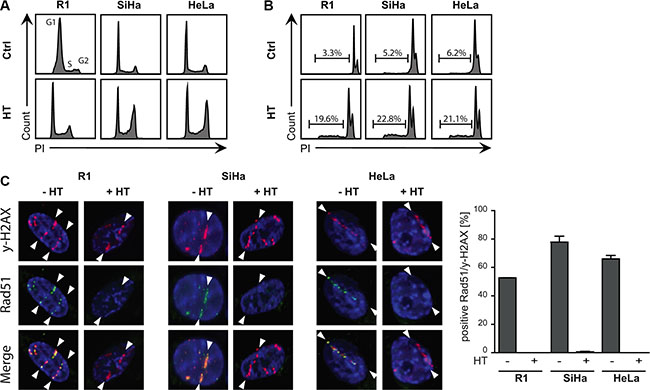
Figure 1: Sensitivity of cells to hyperthermia. (A) Cell cycle analysis were determined via FACS analysis after BrdU incorporation. A G2-arrest is observed after HT treatment. (B) Apoptosis levels were studied using the Nicoletti assay. HT induced apoptosis in all cell lines. (C). Representative pictures of co-localization of γ-H2AX and RAD51 foci on α-irradiation tracks in untreated cells and after HT treatment. RAD51 is no longer detected 30 min after HT, indicating that HR is not active. The bar graph with the standard error of the mean shows the mean of at least three independent experiments. For each condition more than 300 cells were analyzed.
PARP1-i sensitizes cells moderately to combinational treatment of cDDP with hyperthermia
Having confirmed that HR is inhibited by HT in the used cell lines, we set out to determine the effects of PARP1 inhibition on the cytotoxicity of cDDP+HT treatment. To this end, clonogenic survival assays were conducted as schematically shown. Clonogenic assays were conducted to investigate the effect of the different treatments on cell survival Figure 2A. In Figure 2B, percentages of survival are normalized to the untreated samples. We observed a ~50% decrease in cell survival after 1 h cDDP (5 μM) treatment alone. Hyperthermia as a monotherapy was less effective than treatment with a PARP1-i. Combining the PARP1-i with cDDP caused a slight decrease in cell survival, compared to samples treated with cDDP only. The combination of cDDP and HT was highly cytotoxic and killed > 85% of cells, while the combination of hyperthermia and PARP1-i was less effective. The triple treatment with cDDP, PARP1-i and HT resulted in a slightly lower cell survival than the double treatment of cDDP and hyperthermia, but this difference was not statistically significant.
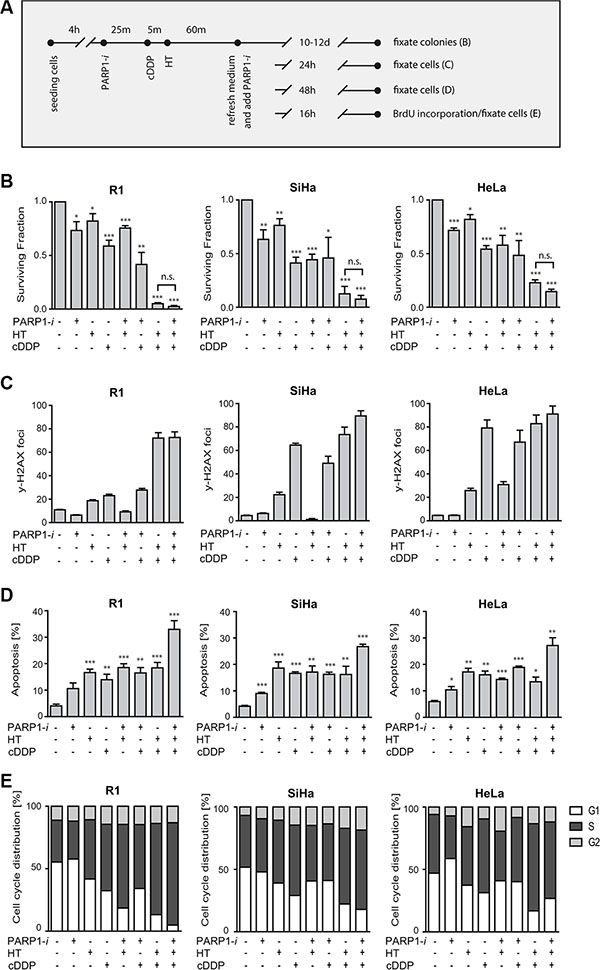
Figure 2: Effects of PARP1-i (100 μM NU1025/continuously), HT (42°C/1 h), cDDP (5 μM/1 h). (A) Overview of treatment schemes for different experiments represented in B–E. (B) Clonogenic assays were conducted in order to study the cell survival after 10–12 days post-treatment. No significant differences were found between HT+cDDP and cDDP + HT + PARP1-i. (R1: p = 0.10, SiHa: p = 0.12, HeLa: p = 0.10). (C) DNA DSBs were analyzed using the γ-H2AX assay. The induction of DSBs in SiHa and HeLa after cDDP treatment is 3-4 times higher than in R1 cells. Nonetheless, all three cells lines show around 70–80 DSBs after cDDP+HT or cDDP+HT+PARP1-i. (D) The Nicoletti assay was performed to observe apoptosis levels after different treatments. The triple combination treatment gave the highest levels of apoptosis. (E) Cell cycle was determined via FACS analysis after BrdU incorporation. cDDP+HT and cDDP+HT+PARP1-i show a slight increase in cells in S-phase. Graph bars represent mean of at least three experiments with standard error of the mean. Asterisks indicate the significant differences compared to the untreated sample (ctrl), this was tested using the non-parametric Mann-Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001.
To further explore the effects of various treatments, the induction of DSBs was quantified by counting γ-H2AX foci (Figure 2C). In R1 cells, cDDP treatment induced around 20 γ-H2AX foci, while in SiHa and HeLa a 3–4 fold more γ-H2AX foci were found 24 h after treatment with cDDP. Treatment with hyperthermia and cDDP induced a similar amount of DSBs (70–90) as the triple modality. The number of detected DSBs was relatively low after the single treatment with PARP1-i or hyperthermia. The combination of the PARP1-i with hyperthermia resulted in a slight increase in the number of DSBs in HeLa cells, but not in SiHa and R1 cells.
DNA content analysis was performed 48 h after the various treatments to test whether apoptosis is responsible for the observed induction of cell death (Figure 2D). All three cell lines showed > 20% of apoptotic cells after the single and double modalities, while after the triple treatment the induction of apoptosis increased significantly (up to 25–30%).
Cell cycle distribution was not significantly affected 16 h after treatments, except for the combination of hyperthermia and cDDP and the triple combination, which caused a ~30–55% increase in accumulation of cells in S-phase (Figure 2E). These results indicate that addition of PARP1-i moderately sensitizes cells to the combinational cDDP+HT treatment, mainly by increasing apoptosis.
Cell death and cell cycle arrest are prominent in cells treated with PARP1-i, cDDP and hyperthermia
To study the cell cycle distribution in more detail, we followed cells using time-lapse microscopy for 96 h, starting immediately after the various treatments. In Figure 3A representative images of the cells are shown at 0 h (left column) and 96 h (right column) after the end of the indicated treatments. SiHa and HeLa cells ceased to grow rapidly after any treatment containing cDDP. However, thermochemotherapy alone (cDDP+HT) as well as the triple treatment were the most toxic for these cells (last two rows of pictures). The combination treatments had similar effects on the R1 cells, although they appeared to be less sensitive to cDDP alone. After 96 h, in cDDP+HT or cDDP+HT+PARP1-i treatments, most of the R1 cells died, while the SiHa and HeLa cells interrupted their cell cycle progression (Figure 3C). Cells that did not divide for over 50 h, but did not die, were regarded to be in ‘cell cycle arrest’. Nevertheless, the cell cycle time was unaffected in the surviving cells under the different treatment conditions, both in the first and second generation post-treatment, except for cells treated with cDDP+HT and the triple combination, where the few surviving cells were all in cell cycle arrest (Figure 3B).
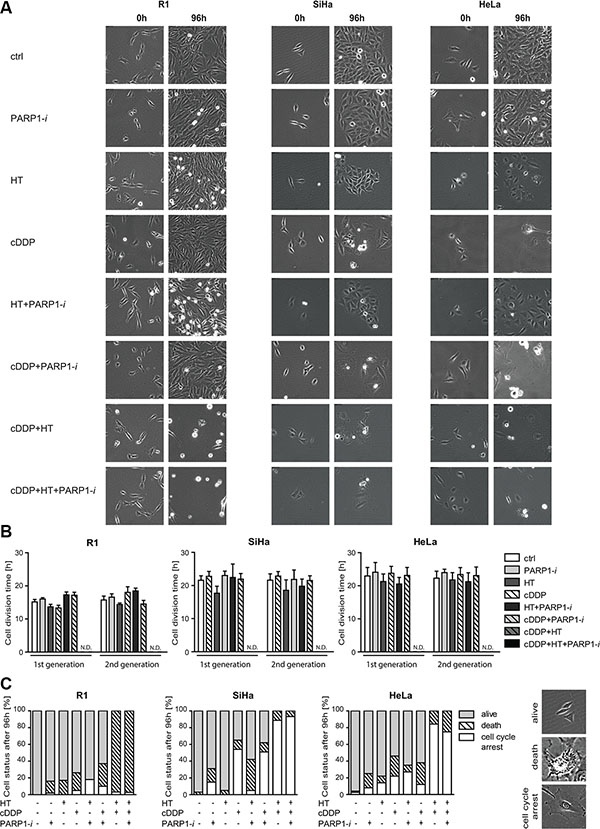
Figure 3: Time-lapse microscopy analysis 0 and 96 h after combination treatments with PARP1-i (100 μM NU1025/continuously), HT (42°C/1 h), cDDP (5 μM/1 h). (A) Representative pictures of cells directly after treatment (0 h) and four days after treatment (96 h). (B) Cell division times of two generations post treatment. Cells that did not divide for over 50 h, but did not die, were regarded to be in ‘cell cycle arrest’. R1 cells have a shorter cell division time than SiHa and HeLa cells. But no significant differences were found after any of the treatments compared to the untreated cells. Graph bars represent mean with standard error of the mean of three independent experiments with the standard error of the mean of three independent experiments. (C) Graph bars with means represent cell status 96 h after treatment with examples pictures of each cell status on the right. Each condition has been performed in triplicate.
Adding PARP1-i to cDDP+HT treatment allows lowering cDDP dose while maintaining similar levels of cytotoxicity
The effects PARP1-i could be masked by the relatively high cytotoxicity of the cDDP+HT combination treatment. To test this hypothesis, we compared the effectiveness of the commonly used dose of cDDP concentration (5 μM for 1 h; cDDP/C) to a tenfold lower cDDP concentration (0.5 μM for 1 h; cDDP/L). We found similar levels of cell death (Figure 4A) in cells treated with either cDDP/L+HT+PARP1-i or cDDP/C+HT (R1: p = 0.10; SiHa: p = 0.86; HeLa: p = 0.22). Time-lapse microscope analysis confirmed these results at 96 h after treatment (Figure 4B). Moreover, clonogenic survival analysis showed that cDDP/L+HT+PARP1-i had similar cytotoxic effects as the combination of cDDP/C+HT (Figure 4C). No significant differences were found in R1, SiHa and HeLa cells (p = 0.10, p = 0.70 and p = 0.20 respectively). These results demonstrate that using a PARP1-i could allow lowering cDDP dosage without significantly affecting the treatment efficacy. This might be of great importance for patients who do not tolerate the standard concentration of cDDP, due to toxicity.
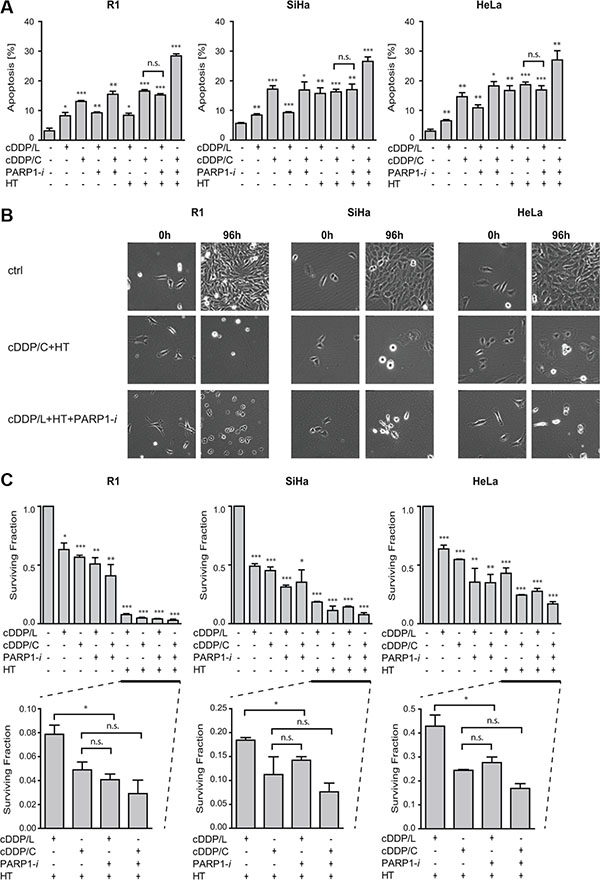
Figure 4: Addition of PARP1-i (100 μM NU1025/continuously) to the commonly used dose of cDDP (5 μM/1 h; cDDP/C) and HT (42°C/1 h) permits lowering of cDDP concentration (0.5 μM/1 h; cDDP/L). (A) To study apoptosis levels, the Nicoletti assay was performed 48 h after different treatments. No significant differences were observed between cDDP/L+HT+PARP1-i and cDDP/C+HT (R1: p = 0.10, SiHa: p = 0.86, HeLa: p = 0.22). (B) Time-lapse microscopy was performed 0–96 h after treatments. Pictures represent cells directly after treatment (0 h) and at the end of the analysis (96 h). (C) Clonogenic assays were conducted to study the effect 10-12 days after treatments. No significant differences were observed between cDDP/L+HT+PARP1-i and cDDP/C+HT (R1: p = 0.10, SiHa: p = 0.70, HeLa: p = 0.20). Graph bars show mean of at least three independent experiments with the standard error of the mean. Asterisks indicate the significant differences compared to the untreated sample (ctrl), tested using the non-parametric Mann-Whitney test. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
Our results show that the combination of a standard cDDP-dose plus HT already yields a high tumor kill. This agrees with clinical studies in women with recurrent cervical cancer [27]. Although, the addition of PARP1-i in an experimental setting only gives a minor enhancement of this effect, a moderate increase in cell death was observed in all cases. Moreover, at a ten-fold-lower cDDP concentration plus HT, the loss of tumorcidal effect can be compensated by PARP1 inhibition. These findings are consistent both in terms of cell surviving fraction, γ-H2AX foci staining and apoptosis. The advantage of the low-dose cDDP+HT+PARP1-i treatment, may be a lower cDDP related toxicity in patients. Our speculation would be that inhibition of PARP1-i does not cause an induction of DNA double strand breaks, but interferes with the b-NHEJ to prevent repair of double strand breaks [18, 19].
In the Netherlands, combinational treatment of cDDP and hyperthermia is one of the standard treatments options for women with recurrent cervical cancer after previous radiotherapy [14]. Previously irradiated tumors may have a lower perfusion caused by the irradiation inducing hypoxia, and hypoxic tumors are less sensitive to ionizing radiation [28–30]. Clinical trials have shown that HT is an effective chemosensitizer for cDDP [31, 32] that increases cDDP uptake in tumor cells and thus enhances the induction of DNA damage [33]. Additionally, as described earlier, hyperthermia also temporality inhibits one of the major DNA repair pathways, HR, by downregulation of BRCA2 [34, 35], explaining the rational for thermochemotherapy. However, one of the limiting effects of cDDP is nephrotoxicity.
Our results may have important clinical implications, as they suggest the possibility to diminish cDDP related toxicity, without reducing the desired cytotoxic effect. Therefore, combining a low dose of cDDP with HT and a PARP1-i may be a promising approach to increase tumor control whilst reducing systematic cDDP-toxicity, particularly in women with (recurrent) cervical cancer. The triple combination might be interesting for the treatment of other tumor types as well, because the treatment will also target tumors with wild-type BRCA status [36, 37] and it may also be an option for patients in whom radiotherapy is contra-indicated.
To assess if not only chemotherapy, but also hyperthermia dose can be reduced, it is also relevant to further elaborate whether reduction in time at 42°C or the use of a lower temperature combined with chemotherapy and a PARP1-i can cause comparable outcome. This is of clinical interest, since it is not always possible to achieve 42°C for 1 h at the tumor site. If the addition of a PARP1-i can be used to compensate for hyperthermia treatment at a shorter time or at a lower temperature, this may improve treatment outcome.
In several clinical trials the effect of high- and low-dose cDDP has been compared for squamous head and neck carcinoma [38–40] and cervical carcinoma [41], in which these studies report acceptable toxicity of low-dose cDDP in radiochemotherapy without significant differences in response rate between high- and low-dose cDDP.
Another way to decrease the severe nephrotoxicity caused by cDDP is to administer high-volume hydration in cDDP-based chemotherapy. This has been tested in clinical trials in patients with lung cancer [42, 43] and resulted in reduction of renal toxicity after high-volume hydration, while renal function was significantly worse after low-volume hydration. These effects might be related to osmolality, and both serum and urinary concentrations of cDDP [43, 44]. Likewise, as applying low-cDDP dose, a decrease in nephrotoxicity is achieved when administering high-volume hydration in patients.
Our results provide rational for adding PARP1-i to the standard cDDP+HT therapy, because this could slightly increase overall treatment efficacy, potentially allow reducing of cDDP concentration. It could also increase efficacy in tumor area that are poorly accessible for cDDP and it could increase efficacy in patient that are subjected to high-volume hydration. In vivo experiments need to be performed to confirm these findings.
MATERIALS AND METHODS
Cell lines
The rat rhabdomyosarcoma cells (R1) were grown in MEM and the cervical carcinoma cells (SiHa and HeLa) were grown in EMEM. The R1 cell line was created in our own laboratory [44]. SiHa and HeLa cell lines were obtained from the American Type Culture Collection (ATCC). All media contain 25 mM Hepes (Gibco-BRL life technologies, Breda, The Netherlands) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 2 mM glutamine. Cells were maintained at 37°C in an incubator with humidified air supplemented with respectively 2% and 5% CO2. The cell division time during exponential growth of R1 cells is approximately 16 h and of the cervical cancer cells 24 h.
Chemical agents
Cells were treated with an optimized concentration [33, 45, 46] of 5 μM for 1 h cisplatin (cDDP; Platosin®, Pharmachemie B.V., Haarlem, The Netherlands). For experiments in which low and commonly used cisplatin concentrations were compared, cells were treated with either 0.5 μM (cDDP/L) or the standard dose of 5 μM (cDDP/C) for 1 h, respectively. The 0.5 μM dose was determined by performing a titration curve on clonogenic assays with concentrations between 0 and 5.0 μM cDDP dose combined with PARP1-i – and found that a 0.5 μM dose was the lowest dose of cDDP where the combination had an effect comparable to the effect of the standard concentration cDDP alone. The medium was refreshed directly after. To inhibit the b-NHEJ and SSB repair, 100 μM of PARP1-i (NU1025, Tocris Bioscience, Bristol, UK) was added 30 min prior to hyperthermia. After refreshing the medium, PARP1-i was re-added till the end of the experiment in the appropriate concentration.
Hyperthermia
Incubation of the cells was performed by partially submerging the culture dishes in a thermostatically controlled water bath (Lauda aqualine AL12, Beun de Ronde, Abcoude, The Netherlands) for 1 h at 42°C. Temperature was checked in parallel dishes and the desired temperature (± 0.1°C) was reached in approximately 5 min. The atmosphere of the water bath was adjustable. R1 cells were heated in a 2% CO2/98% air atmosphere and SiHa and HeLa cells were heated in a 5% CO2/95% air atmosphere with an air inflow of 2 L/min.
Immunohistochemistry
Cells were plated 24 h prior to treatment on sterile coverslips or 1.8 μm mylar membrane dishes. Cells on coverslips were treated with cDDP, PARP1-i and/or HT, while cells on mylar were treated with only hyperthermia and directly followed by irradiation with α-particles for 1 min. Next, cells were fixed with 2% paraformaldehyde after 15 min, 30 min (α-particles) or 24 h. After washing with PBS and incubated for 30 min with TNBS (PBS containing 0.1% Triton X-100 and 1% FCS), cells were stained with γ-H2AX (Millipore, dilution 1:100 in TNBS) and RAD51 antibody (dilution 1:25 in TNBS) as described by Aten et al. [47]. Finally, vectashield with DAPI (Life technologies, USA) was dropped at the slide and the coverslip is turned upside down on the slide, for scoring under the fluorescence microscope.
Clonogenic assays
Cell survival was studied on R1, SiHa and HeLa cells using different treatment combinations; hyperthermia (1 h at 42°C), cDDP (cDDP/C: 5 μM for approximately 1 h, cDDP/L: 0.5 μM for approximately 1 h), PARP1-i (100 μM, continuously). Clonogenic assays were conducted as described by Franken et al. [48]. Cells were plated before treatment into 6-well culture plates (Costar, USA). Dishes were placed in an incubator with 5% CO2 at 37°C until sufficiently large clones were formed. Afterwards the medium is removed and cells were washed with PBS. A mixture of 6.0% glutaraldehyde (2–3 ml) and 0.5% crystal violet was added for at least 30 min at room temperature. Next, plates were washed with water and dried in normal air at room temperature. Colonies were counted under a light microscope [49]. Surviving fractions were calculated by dividing the plating efficiency of treated cells by that of control cells [50].
Apoptosis assay
The Nicoletti assay [51] was used to study apoptosis in all cell lines. At 48 h after treatment cells were collected and pellets were resuspended in Nicoletti buffer (0.1% w/v Sodium citrate, 0.1% v/v Triton X-100 in demi water, pH 7.4). Analyses were made using flow cytometry (FACS Canto, BD Biosciences, USA).
Cell cycle analysis
Cell cycle distribution was analyzed using the thymidine analogue 5-Bromo-2′-deoxy-uridine (BrdU, Sigma Aldrich, USA) that can incorporate into S-phase cells (60 min at 37°C). BrdU was added 16 h after treatment and 1 h prior to fixation in 2 ml PBS and 6 ml of 100% ethanol. The pellet was resuspended in pepsin-HCl (0.4 mg/ml, 0.1 N HCl) and cells were stored 30 min at room temperature. Cells were washed with PBT (0.5% Tween-20, Sigma Aldrich USA in 0.5 l PBS). The pellet was resuspended in HCl (2 N, Merck) and cells were incubated 30 min at 37°C. 100 μl primary antibody rat-anti-BrdU (Abcam, UK) in PBTb (1% bovine serum albumin, Sigma, in PBT) was added 1 h at 37ºC. After washing, 100 μl IgG goat-anti-rat FITC (Abcam, UK) in PBTg (1% normal goat serum, Dako, USA, in PBT) was added and left for 1 h at 37ºC. Final, PI (Sigma-Aldrich, USA) was added, cell suspensions were vortexed and directly measured using flow cytometry (FACS Canto, BD Biosciences, USA).
Time-lapse microscopy
Cells were grown in 6-wells plates for 24 h, before treatments. Dishes were placed under a confocal microscope (Leica PS2, The Netherlands) with a temperature and CO2/air controlled chamber and cells were imaged for 96 h. A minimum of fifty cells were analyzed per condition.
Statistics
All represented in vitro data are means ± standard error of the mean of at least three independent experiments. Cell survival and apoptosis were analyzed using SPSS (Chicago, IL, USA) statistical software using a non-parametric Mann-Whitney test.
ACKNOWLEDGMENTS
This work was supported by the Dutch Cancer Society (UVA 2012-5540).
CONFLICTS OF INTEREST
None.
REFERENCES
1. Hartmann JT, Fels LM, Knop S, Stolt H, Kanz L, Bokemeyer C. A randomized trial comparing the nephrotoxicity of cisplatin/ifosfamide-based combination chemotherapy with or without amifostine in patients with solid tumors. Invest New Drugs. 2000; 18:281–289.
2. Prestayko AW, D’Aoust JC, Issell BF, Crooke ST. Cisplatin (cis-diamminedichloroplatinum II). Cancer Treat Rev. 1979; 6:17–39.
3. Poklar N, Pilch DS, Lippard SJ, Redding EA, Dunham SU, Breslauer KJ. Influence of cisplatin intrastrand crosslinking on the conformation, thermal stability, and energetics of a 20-mer DNA duplex. Proc Natl Acad Sci USA. 1996; 93:7606–7611.
4. Rudd GN, Hartley JA, Souhami RL. Persistence of cisplatin-induced DNA interstrand crosslinking in peripheral blood mononuclear cells from elderly and young individuals. Cancer Chemother Pharmacol. 1995; 35:323–326.
5. Jasin M, Rothstein R. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol. 2013; 5:a012740.
6. Krawczyk PM, Eppink B, Essers J, Stap J, Rodermond H, Odijk H, Zelensky A, van Bree C, Stalpers LJ, Buist MR, Soullie T, Rens J, Verhagen HJ, et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proc Natl Acad Sci USA. 2011; 108:9851–9856.
7. Essers J, van Steeg H, de Wit J, Swagemakers SM, Vermeij M, Hoeijmakers JH, Kanaar R. Homologous and non-homologous recombination differentially affect DNA damage repair in mice. EMBO J. 2000; 19:1703–1710.
8. Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000; 19:3398–3407.
9. Fleck O, Nielsen O. DNA repair. J Cell Sci. 2004; 117(Pt 4):515–517.
10. Diggle CP, Bentley J, Knowles MA, Kiltie AE. Inhibition of double-strand break non-homologous end-joining by cisplatin adducts in human cell extracts. Nucleic Acids Res. 2005; 33:2531–2539.
11. Myint WK, Ng C, Raaphorst GP. Examining the non-homologous repair process following cisplatin and radiation treatments. Int J Radiat Biol. 2002; 78:417–424.
12. Turchi JJ, Henkels KM, Zhou Y. Cisplatin-DNA adducts inhibit translocation of the Ku subunits of DNA-PK. Nucleic Acids Res. 2000; 28:4634–4641.
13. Boeckman HJ, Trego KS, Turchi JJ. Cisplatin sensitizes cancer cells to ionizing radiation via inhibition of nonhomologous end joining. Mol Cancer Res. 2005; 3:277–285.
14. Heijkoop ST, van Doorn HC, Stalpers LJ, Boere IA, van der Velden J, Franckena M, Westermann AM. Results of concurrent chemotherapy and hyperthermia in patients with recurrent cervical cancer after previous chemoradiation. Int J Hyperthermia. 2014; 30:6–10.
15. Sastry J, Kellie SJ. Severe neurotoxicity, ototoxicity and nephrotoxicity following high-dose cisplatin and amifostine. Pediatr Hematol Oncol. 2005; 22:441–445.
16. Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol. 2003; 23:460–464.
17. Iliakis G, Wu W, Wang M. DNA double strand break repair inhibition as a cause of heat radiosensitization: re-evaluation considering backup pathways of NHEJ. Int J Hyperthermia. 2008; 24:17–29.
18. Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004; 279:55117–55126.
19. Wang M, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006; 34:6170–6182.
20. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917.
21. Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005; 434:917–921.
22. Dedes KJ, Wilkerson PM, Wetterskog D, Weigelt B, Ashworth A, Reis-Filho JS. Synthetic lethality of PARP inhibition in cancers lacking BRCA1 and BRCA2 mutations. Cell Cycle. 2011; 10:1192–1199.
23. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011; 5:387–393.
24. Eppink B, Krawczyk PM, Stap J, Kanaar R. Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. Int J Hyperthermia. 2012; 28:509–517.
25. Schreiber V, Illuzzi G, Heberle E, Dantzer F. [From poly(ADP-ribose) discovery to PARP inhibitors in cancer therapy]. Bull Cancer. 2015; 102:863–873.
26. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010; 10:293–301.
27. Franckena M, De Wit R, Ansink AC, Notenboom A, Canters RA, Fatehi D, Van Rhoon GC, Van Der Zee J. Weekly systemic cisplatin plus locoregional hyperthermia: an effective treatment for patients with recurrent cervical carcinoma in a previously irradiated area. Int J Hyperthermia. 2007; 23:443–450.
28. Rockwell S, Dobrucki IT, Kim EY, Marrison ST, Vu VT. Hypoxia and radiation therapy: past history, ongoing research, and future promise. Curr Mol Med. 2009; 9:442–458.
29. Oldenborg S, Griesdoorn V, van Os R, Kusumanto YH, Oei BS, Venselaar JL, Zum Vorde Sive Vording PJ, Heymans MW, Kolff MW, Rasch CR, Crezee H, van Tienhoven G. Reirradiation and hyperthermia for irresectable locoregional recurrent breast cancer in previously irradiated area: Size matters. Radiother Oncol. 2015; 117:223–228.
30. Vaupel P, Briest S, Hockel M. Hypoxia in breast cancer: pathogenesis, characterization and biological/therapeutic implications. Wien Med Wochenschr. 2002; 152:334–342.
31. Westermann A, Mella O, Van Der Zee J, Jones EL, Van Der Steen-Banasik E, Koper P, Uitterhoeve AL, De Wit R, Van Der Velden J, Burger C, Schem BC, Van Der Wilt C, Dahl O, et al. Long-term survival data of triple modality treatment of stage IIB-III-IVA cervical cancer with the combination of radiotherapy, chemotherapy and hyperthermia - an update. Int J Hyperthermia. 2012; 28:549–553.
32. Westermann AM, Jones EL, Schem BC, van der Steen-Banasik EM, Koper P, Mella O, Uitterhoeve AL, de Wit R, van der Velden J, Burger C, van der Wilt CL, Dahl O, Prosnitz LR, et al. First results of triple-modality treatment combining radiotherapy, chemotherapy, and hyperthermia for the treatment of patients with stage IIB, III, and IVA cervical carcinoma. Cancer. 2005; 104:763–770.
33. Bergs JW, Haveman J, Ten Cate R, Medema JP, Franken NA, Van Bree C. Effect of 41 degrees C and 43 degrees C on cisplatin radiosensitization in two human carcinoma cell lines with different sensitivities for cisplatin. Oncol Rep. 2007; 18:219–226.
34. Genet SC, Fujii Y, Maeda J, Kaneko M, Genet MD, Miyagawa K, Kato TA. Hyperthermia inhibits homologous recombination repair and sensitizes cells to ionizing radiation in a time- and temperature-dependent manner. J Cell Physiol. 2013; 228:1473–1481.
35. Oei AL, Vriend LE, Crezee J, Franken NA, Krawczyk PM. Effects of hyperthermia on DNA repair pathways: one treatment to inhibit them all. Radiat Oncol. 2015; 10:165.
36. Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009; 361:123–134.
37. Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmana J, Mitchell G, Fried G, Stemmer SM, Hubert A, Rosengarten O, Steiner M, Loman N, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015; 33:244–250.
38. Veronesi A, Zagonel V, Tirelli U, Galligioni E, Tumolo S, Barzan L, Lorenzini M, Comoretto R, Grigoletto E. High-dose versus low-dose cisplatin in advanced head and neck squamous carcinoma: a randomized study. J Clin Oncol. 1985; 3:1105–1108.
39. Hoebers FJ, Heemsbergen W, Balm AJ, van Zanten M, Schornagel JH, Rasch CR. Concurrent chemoradiation with daily low dose cisplatin for advanced stage head and neck carcinoma. Radiother Oncol. 2007; 85:42–47.
40. Jeremic B, Milicic B. Influence of low-dose daily cisplatin on the distant metastasis-free survival of patients with locally advanced nonmetastatic head and neck cancer treated with radiation therapy. Radiother Oncol. 2008; 87:201–203.
41. Mitsuhashi A, Uno T, Usui H, Nishikimi K, Yamamoto N, Watanabe M, Tate S, Hirashiki K, Kato K, Yamazawa K, Shozu M. Daily low-dose cisplatin-based concurrent chemoradiotherapy in patients with uterine cervical cancer with emphasis on elderly patients: a phase 2 trial. Int J Gynecol Cancer. 2013; 23:1453–1458.
42. Horinouchi H, Kubota K, Itani H, Taniyama TK, Nakamichi S, Wakui H, Kanda S, Nokihara H, Yamamoto N, Sekine I, Tamura T. Short hydration in chemotherapy containing cisplatin (>/=75 mg/m2) for patients with lung cancer: a prospective study. Jpn J Clin Oncol. 2013; 43:1105–1109.
43. Oka T, Kimura T, Suzumura T, Yoshimoto N, Nakai T, Yamamoto N, Matsuura K, Mitsuoka S, Yoshimura N, Kudoh S, Hirata K. Magnesium supplementation and high volume hydration reduce the renal toxicity caused by cisplatin-based chemotherapy in patients with lung cancer: a toxicity study. BMC Pharmacol Toxicol. 2014; 15:70.
44. Reinhold HS. A cell dispersion technique for use in quantitative transplantation studies with solid tumours. Eur J Cancer. 1965; 1:67–71.
45. Wang Q, Zheng XL, Yang L, Shi F, Gao LB, Zhong YJ, Sun H, He F, Lin Y, Wang X. Reactive oxygen species-mediated apoptosis contributes to chemosensitization effect of saikosaponins on cisplatin-induced cytotoxicity in cancer cells. J Exp Clin Cancer Res. 2010; 29:159.
46. Koivusalo R, Krausz E, Ruotsalainen P, Helenius H, Hietanen S. Chemoradiation of cervical cancer cells: targeting human papillomavirus E6 and p53 leads to either augmented or attenuated apoptosis depending on the platinum carrier ligand. Cancer Res. 2002; 62:7364–7371.
47. Aten JA, Stap J, Krawczyk PM, van Oven CH, Hoebe RA, Essers J, Kanaar R. Dynamics of DNA double-strand breaks revealed by clustering of damaged chromosome domains. Science. 2004; 303:92–95.
48. Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006; 1:2315–2319.
49. Bergs JW, Franken NA, ten Cate R, van Bree C, Haveman J. Effects of cisplatin and gamma-irradiation on cell survival, the induction of chromosomal aberrations and apoptosis in SW-1573 cells. Mutat Res. 2006; 594:148–154.
50. van Bree C, Franken NA, Snel FA, Haveman J, Bakker PJ. Wild-type p53-function is not required for hyperthermia-enhanced cytotoxicity of cisplatin. Int J Hyperthermia. 2001; 17:337–346.
51. Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006; 1:1458–1461.