
INTRODUCTION
The International Agency for Research on Cancer defined Helicobacter pylori (H. pylori) as a class I carcinogen [1] as H. pylori caused gastric carcinogenesis [2] and the eradication of H. pylori prevented metachronous gastric cancer (MGC) after endoscopic resection of early gastric cancer [3]. During carcinogenesis, H. pylori provoked gastric inflammation, oxidative stress, and several harmful events including genetic and epigenetic pathways [4], after which its eradication can be a solution for prevention. However, in intervention trials studying gastric cancer prevention, H. pylori eradication did not prevent MGC in patients undergoing endoscopic submucosal dissection, have yielded rather disappointing results [5]. Therefore, non-microbial dietary intervention has been considered asan alternate eradication method or as a provision of surrounding break up to either remove mutagenic inflammation leading to cancer progression [6–9].
Generally, strategies to reduce the occurrence of gastric cancer, include improvement of sanitation, high intake of fresh fruits, safe food-preservation methods, and avoidance of smoking [10]. Additionally, mitigating the chronic inflammatory response associated with infectious disease has been recommended [11], including non-microbial dietary intervention or supplementation with phytoceuticals with the hope that these approaches may be an effective way of preventing cancer through long-term control of gastric inflammation [12]. Increased consumption of fatty fish or fish oil supplements containing anti-inflammatory ω-3 PUFAs is also an intriguing intervention since ω-3 PUFA has been shown to have a therapeutic role in inflammatory diseases such as rheumatoidarthritis, inflammatory bowel disease, asthma, cardiovascular, and neurodegenerative diseases [13–15] and various cancers by reducing the level of AA-derived eicosanoids and inflammatory cytokines, (including Interleukin (IL)-1, IL-2, IL-6, IL-8, Interferon (IFN)-γ,and Leukotriene B4) and Tumor necrosis factor-α, promoting anti-inflammatory activities [16, 17]. In spite of these achievements, the expected clinical impact of ω-3 PUFAs has been reduced, perhaps because applying ω-3 PUFA-containing diet scan bring considerable variations between the experimental groups [18, 19], resulting from an in consistent intake and uncertain purity of ω-3 PUFAs.
Fat-1 transgenic (Fat-1 TG) mice are capable of producing ω-3 fatty acids from ω-6 type fatty acids because of the transgenic over-expression of n-3 desaturase lead to abundant ω-3 PUFAs with reduced levels of ω-6 fatty acids in their organs and tissues without a dietary n-3 supply [20]. Here we used these mice to explore the effect of ω-3 PUFAs, especially the exact effect of ω-3 PUFAs on H. pylori associated gastric lesions, something that has never been studied before. We hypothesize that Fat-1 TG mice may be conferred a chemo-preventive benefit through anti-inflammatory or anti-mutagenic actions of ω-3 PUFAs. Using H. pylori infected mice, our study can provide important preclinical evidence that ω-3 PUFAs are efficient in attenuating H. pylori-associated gastritis and preventing gastric tumors and reveal molecular insights for the use of dietary ω-3 PUFAs in chemoprevention of H. pylori-associated gastric cancer.
RESULTS
Attenuated gastric inflammation in Fat-1 TG mice compared to WT mice at 16 weeks after H. pylori infection
After16 weeks of H. pylori infection, there were no significant changes on gross lesions of resected stomachs of H. pylori-infected WT and Fat-1 TG mice (see Supplementary Figure S1). However, the gastric mucosa of WT mice were thicker than Fat-1 TG mice and this thickening was accompanied with intense infiltrations of inflammatory cells in sub-mucosal and mucosal layers. Compared to the non-infected group, marked inflammatory cells were observed in the sub-mucosa and mucosa in the stomachs of H. pylori-infected WT mice (Figure 1B), whereas these inflammatory cell infiltrations were significantly decreased in Fat-1 TG mice after H. pylori infection (P < 0.05, Figure 1C). Therefore, we compared the expression levels of inflammatory mediators between H. pylori-infected WT and Fat-1 TG mice. As seen in Figure 1D, the expression of cytokines known to be increasingly expressed at H. pylori-infected gastritis including Vascular endothelial growth factor (VEGF), Cyclooxygenase (Cox)-2, IL-1β, IL-8, INF-γ, and IL-6, [21], were all significantly increased in H. pylori-infected WT compared to non-infected vehicle control group (P < 0.001). However, their expression levels were all significantly decreased in H. pylori-infected Fat-1 TG mice (P < 0.01). Since the sources of these inflammatory cytokines are macrophages and monocytes, we tested for macrophages using F4/80 immunohistochemical staining. As seen in Figure 1E, F4/80 expressions were significantly increased after H. pylori infection in WT mice, but significantly decreased in Fat-1 TG mice (P < 0.05).
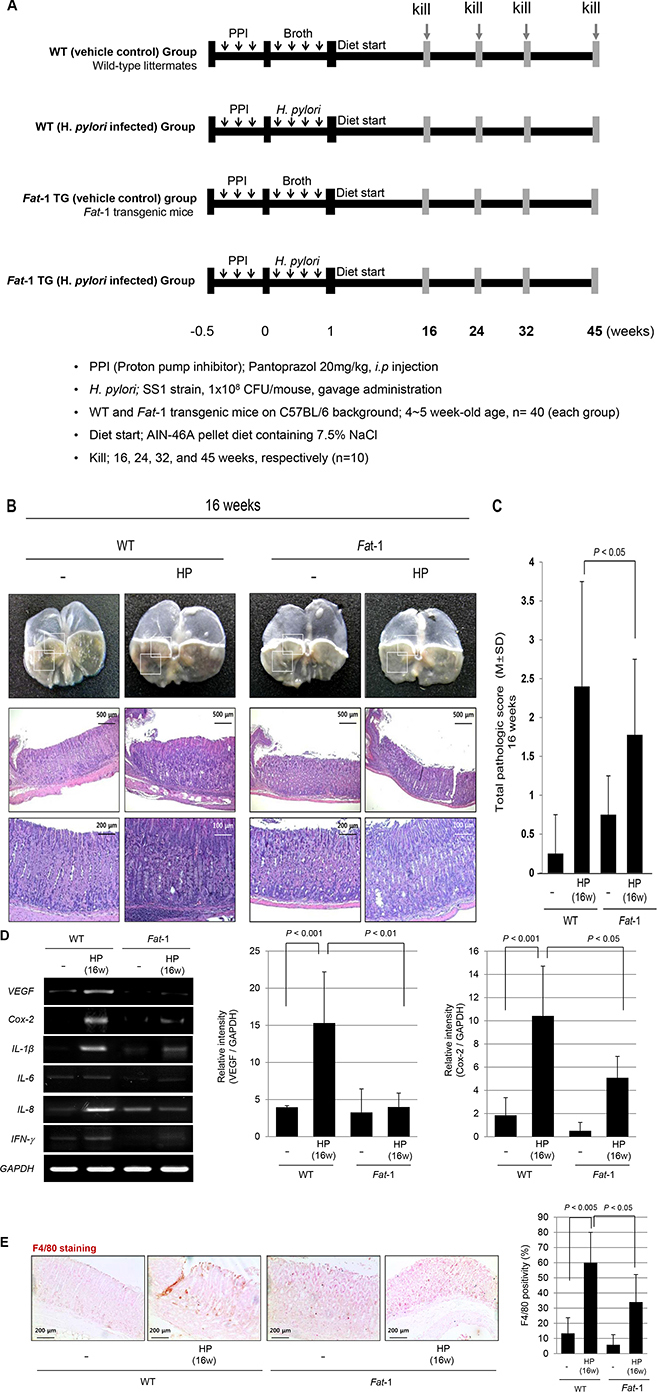
Figure 1: The influence of Fat-1 mice on H. pylori-initiated, high salt diet-promoted gastric tumorigenesis model at 16 weeks. (A) Experimental scheme for H. pylori-associated gastritis model. The mice were grouped into four groups; non-infected normal in WT mice (n = 40), H. pylori-infected WT mice (n = 40), non-infected normal in Fat-1 TG mice (n = 40), and H. pylori-infected Fat-1 TG mice (n = 40), which were serially sacrificed at 16, 24, 32 and 45 weeks after H. pylori-initiated, high salt diet-promoted gastric tumorigenesis model. (B) Gross lesions and total pathological score according to group. H. pylori infection followed with high salt diet leads to some erosion, erythematous gastric mucosa at 16 weeks (box indicated the portion of pathologic analysis). At 16 weeks, H. pylori infections lead to significantly increased inflammatory cells infiltration at submucos and mucosa on body and antrum area and mucosal proliferations in WT mice, while these changes were significantly ameliorated in H. pylori-infected Fat-1 TG mice. (H&E stain, magnification, × 40, 100 and 200). (C) Total pathological scores among groups. (D) Expression of VEGF, Cox-2, IL-1β, IFN-γ, and IL-6 mRNA is increased in H. pylori-infected WT mice group. On RT-PCR analysis for mucosal Cox-2 mRNA expression, Cox-2 was significantly increased after H. pylori infection (P < 0.001), but their expressions were significantly decreased in Fat-1 TG mice group (P < 0.05). The VEGF was also significantly increased after H. pylori infection (P < 0.001), but their expressions were significantly decreased in the Fat-1 mice group (P < 0.05). (E) The immunohistochemical changes of F4/80 denoting macrophage infiltrations according to groups (Magnification, × 100). H. pylori infection led to significant increased positivity in WT mice (P < 0.005), but significantly attenuated in Fat-1 TG mice (P < 0.05).
Mitigated chronic atrophic gastritis in Fat-1 TG mice compared to WT mice at 24 weeks after H. pylori infection
Contrary to gross lesions at 16 weeks, H. pylori infection lead to significant gross changes at 24 weeks, as seen in Figure 2A and Supplementary Figure S2; portions of the gastric mucosa had a nodular and granular appearance, while the remaining portion of stomach looked thin and transparent. Instead, the gross lesions of H. pylori-infected Fat-1 TG mice were not changed compared to non-infected WT or Fat-1 TG mice even at 24 weeks. Some mice administered only HSD without H. pylori infection showed some mass-like lesion on gross observation (see Supplementary Figure 2A), whereas none were observed in HSD-administered Fat-1 TG mice. Almost all H. pylori-infected WT mice showed multiple nodular lesions accompanied by a thin surrounding gastric wall, whereas no significant changes were noted in Fat-1 TG mice at 24weeks (see Supplementary Figure 2A and 2B). On pathological analysis, gastritis cysticaprofunda, disappearance of parietal cells, and profuse inflammatory cell infiltrations on submucosa and mucosa were noted at H. pylori-infected WT. As seen in Figure 2A, there were no prominent changes in H. pylori-infected Fat-1 TG mice, only mild gastritis and focal erosive changes were observed. The pathological scoring between H. pylori-infected WT and Fat-1 TG mice was differed significantly at 24 weeks (P < 0.05, Figure 2B). Since there were significant differences in inflammatory activities, proliferative status, and angiogenic activities between WT and Fat-1 TG mice at 24 weeks of H. pylori infection (Figure 2B), we had measured the expression levels of the inflammatory mediators Cox-2 and IL-1β and angiogenic growth factors VEGF and Platelet-derived growth factor (PDGF); there were significant differences between WT and Fat-1 TG mice (Figure 2C). Further evaluation of COX-2 and Prostaglandin dehydrogenase (PDGH) was done by Western blots, COX-2 levels were significantly increased in WT mice after H. pylori infection (P < 0.05), but not in Fat-1 TG mice. The expression of 15-PGDH was significantly lower in H. pylori-infected WT mice at 24 weeks (P < 0.001), but significantly preserved at Fat-1 TG mice (P < 0.05, Figure 2D). In order to compare the angiogenic activities, we performed immunohistochemical staining with Cluster of differentiation (CD)31 endothelial antibody. As seen in Figure 2E, H. pylori infection led to a significant increase of CD31 in gastric mucosa (P < 0.001), but these expressions were not increased in H. pylori-infected Fat-1 TG mice. Though the expression of CD31 was higher in Fat-1 TG mice compared to WT, there was no statistical significance. In order to screen further changes in molecular events between groups, we performed protein array analysis. As seen in Figure 2F, H. pylori infection in WT mice led to significant increases in IL-1α, endothelin-1, amphiregulin, and Fibroblast growth factor (FGF) (P < 0.01), however, no significant changes were observed in Fat-1 TG mice, suggesting that the ischemic and proliferative conditions in H. pylori-infected WT mice were significantly relieved in H. pylori-infected Fat-1 TG mice.
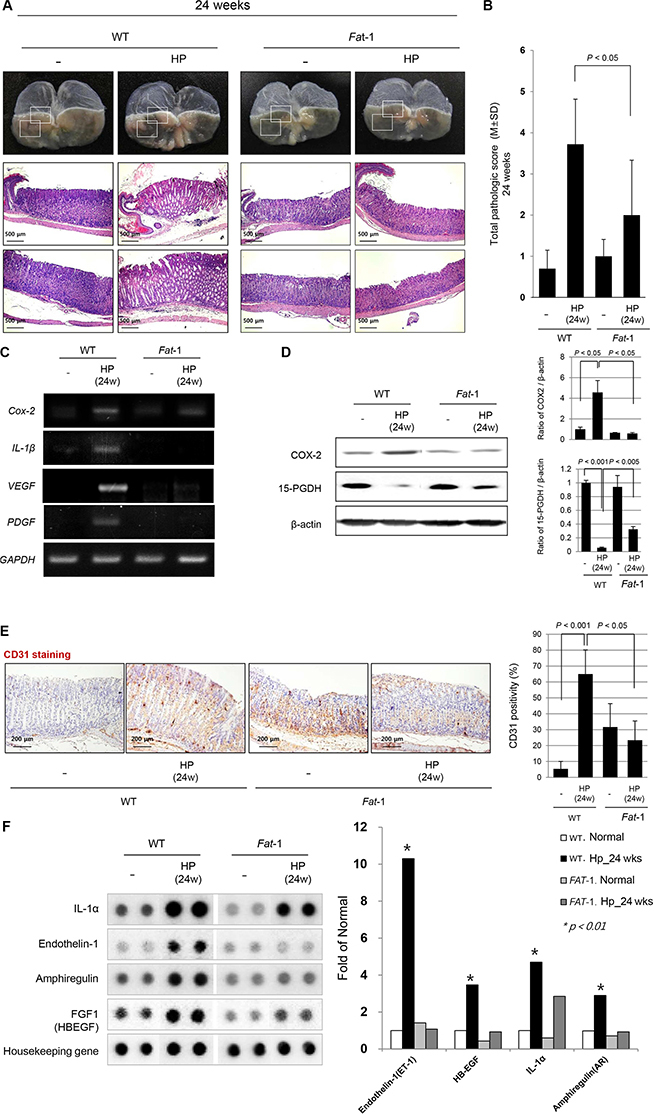
Figure 2: Mitigated chronic atrophic gastritis in Fat-1 TG mice compared to WT mice at 24 weeks after H. pylori infection. (A) H. pylori infection in WT mice lead to significant gross changes at 24 weeks. Some portion of gastric mucosa looks like nodular and granular appearance, while the other portion of stomach looks thin and transparent. However, the gross lesions of H. pylori-infected Fat-1 TG mice were not changed compared to non-infected WT or Fat-1 TG mice even at 24 weeks. Almost all H. pylori-infected WT mice showed multiple nodular lesions accompanied with thin surrounding gastric wall, whereas no significant changes were noted in Fat-1 TG mice at 24 weeks (H&E stain, magnification, × 40, 100). (B) Pathological evaluation showed gastritis cystica profunda, disappearance of parietal cells, and profuse inflammatory cell infiltrations on submucosa and mucosa at H. pylori-infected WT mice. On the other hand, there were no prominent changes in H. pylori-infected Fat-1 TG mice, only showing mild gastritis and focal erosive changes. The pathological scoring between H. pylori-infected WT and Fat-1 TG mice were significantly differed at 24 weeks (P < 0.05). (C) Expressions of Cox-2, IL-1β, VEGF, and PDGF mRNA were significantly increased after H. pylori infection in WT mice, but much lesser in Fat-1 TG mice even after H. pylori infection at 24 weeks. (D) Western blot analysis for COX-2 and 15-PGDH. The expressions of COX-2 were significantly increased after H. pylori infected in WT mice group, but their expressions were significantly decreased in the H. pylori-infected Fat-1 TG mice group (P < 0.05). H. pylori infection in WT mice significantly cancelled the expressions of 15-PGDH at 24 weeks (P < 0.001), but 15-PGDH expressions were significantly preserved in Fat-1 TG mice, though decreased compared to non-infected group (P < 0.005). (E) The immunohistochemical changes of endothelial marker CD31 denoting angiogenic activity according to groups (Magnification, × 100). H. pylori infection led to significant increased positivity in WT mice (P < 0.001), but CD31 immunostaining was significantly attenuated in Fat-1 TG mice (P < 0.05). (F) Cytokine protein array. H. pylori infection in WT mice showed significantly increased expressions of IL-1α, endothelin-1, amphiregulin, FGF1 (P < 0.01), but never in H. pylori-infected Fat-1 TG mice, suggesting ischemic condition and proliferative activities were relived in Fat-1 TG mice even under H. pylori infection.
Increased H. pylori-induced tumorigeneis in WT mice, but not in Fat-1TG mice observed at 32 weeks
Our model of H. pylori infection followed with 7.5% high salt diet provoked significant tumorigenesis after 32 weeks of H. pylori infection on gross observation (see Supplementary Figure S3A). As seen in Figure 3A, there were multiple ulcerated tumors on the stomach accompanied with several nodules, but very thin stomach walls at 32 weeks, whereas still no significant changes in gross appearance were noted in H. pylori-infected Fat-1 TG mice except that small sized nodular lesions were noted in 30% Fat-1 TG mice (see Supplementary Figure S4A and S4B). On pathological findings, gastric adenoma and chronic atrophic gastritis was prominent pathology observed at WT mice. On pathological scoring between groups, significant differences in pathological scores were seen at 32 weeks after H. pylori infection (Figure 3B). In order to elucidate the underlying molecular changes of these inflammatory and tumorigenic changes between groups, we performed RT-PCR to quantify the levels of inflammatory and angiogenic growth factors including Cox-2, IL-1β, IL-6, VEGF, and PDGF mRNA (Figure 3C) and immunohistochemical staining for CD31 (Figure 3E). As noted at 24 weeks, there were significant increases in the mRNA levels of Cox-2, IL-1β, IL-6, VEGF, PDGF and expressions of CD31 in H. pylori-infected WT mice, however no changes in Fat-1 TG mice at 32 weeks. The expression level of 15-PGDH was significantly decreased in H. pylori-infected WT mice (P < 0.005), and significantly preserved in Fat-1 TG mice. The expression levels of COX-2 and VEGF were similar, there were significant decreases in the oncogene COX-2 and VEGF, and significant increases in the expression of the tumor suppressive 15-PGDH in Fat-1 TG mice compared to WT mice (Figure 3D).
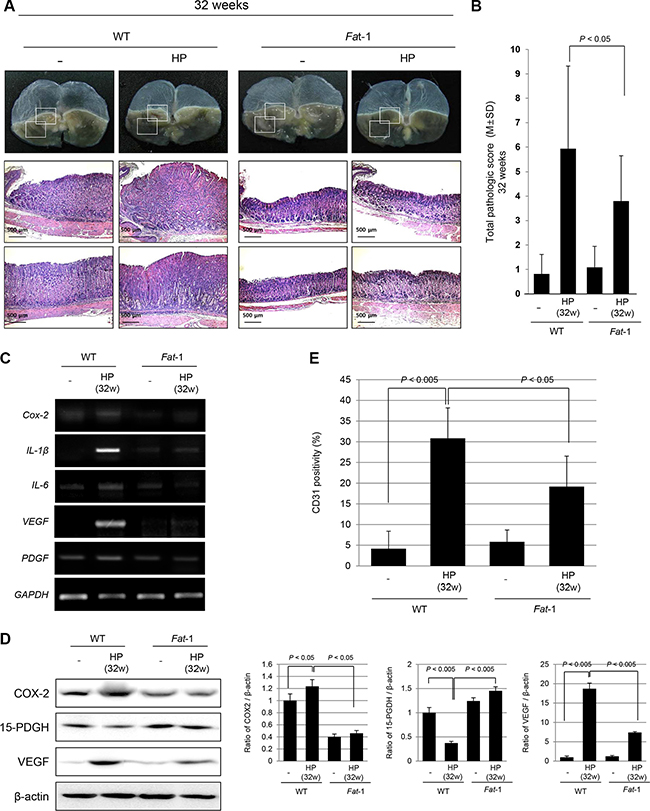
Figure 3: Significantly attenuated tumorigenesis in Fat-1 TG mice at 32 weeks of H. pylori infection. (A) Current model of H. pylori infection followed with 7.5% high salt diet provoked significant tumorigenesis after 32 weeks of H. pylori infection on gross observation. There were multiple ulcerated tumors on the stomach accompanied with several nodules, but very thin gastric wall, transparently looking. However, no significant changes in gross appearance were noted in H. pylori-infected Fat-1 TG mice except small sized nodular lesions in 30% Fat-1 TG mice. Pathological findings in H. pylori-infected WT mice at 32 weeks showed gastric adenoma and chronic atrophic gastritis (H&E stain, Magnification, × 40). (B) Pathological scoring between groups showed significant differences in pathological scores between WT and Fat-1 TG mice (P < 0.05). (C–D) RT-PCR for inflammatory and angiogenic growth factors and immunohistochemical staining for CD31 were done. There were significant increases in Cox-2, IL-1β, IL-6, VEGF, PDGF mRNA and CD31 in H. pylori-infected WT mice, but significantly decreased in Fat-1 TG mice. (D) Western blots for COX-2, 15-PGDH, and VEGF according to group. The expressions of 15-PGDH were significantly decreased in H. pylori-infected WT mice (P < 0.005), while COX-2 expressions were significantly increased in WT mice at 32 weeks of H. pylori infection. On the other hand, the expressions of tumor suppressive 15-PGDH were significantly preserved in H. pylori-infected Fat-1 TG mice (P < 0.005). Just like COX-2, there were significant decrements in VEGF expressions in Fat-1 TG mice compared to WT mice.
Long-term (45 weeks) preventive effects of ω-3 PUFA-producing Fat-1 TG mice against H. pylori-induced gastric carcinogenesis
We maintained our model up to 45 weeks after H. pylori infection to compare the chemo-preventive effects of ω-3 PUFAs. As seen in Figure 4A and Supplementary Figure S4A, all H. pylori-infected mice had developed variable sized gastric tumors, whereas only 30% of Fat-1 TG mice developed gastric tumors and those were smaller than those in WT mice (see Supplementary Figure S4B). When reviewing the gross and pathological appearance, some tumors were shown to be more invasive and larger in size in WT mice compared to Fat-1 TG mice (Figure 4A and 4B). As seen in Figure 4C, 1:1 mounting view of whole stomach of WT – H. pylori group showed moderately-differentiated adenocarcinoma, by which there was significant difference in tumorigenesis between WT and Fat-1 TG mice at 45 weeks after H. pylori infection. The expression of important inflammatory and angiogenic growth factors, including Cox-2, IL-1β, IL-6, and PDGF mRNA are presented in Figure 4D and were significantly increased in H. pylori-infected WT, but not Fat-1 TG mice at 45 weeks. The protein expressions of COX-2 and VEGF were noted in similar way. Explaining the significant differences in tumorigenesis between groups, proliferative activities were measured. β-catenin, Cyclin-dependent kinase (CDK)4, phosphorylated Protein kinase B (PKB or AKT) (Figure 4E), CD26, Heparin-binding EGF-like growth factor (HB-EGF), Insulin-like growth factor-binding protein (IGFBP)3 (Figure 4F), Bromodeoxyuridine (BrdU), and Ki-67 (Figure 4G) were included in these measurements reflecting H. pylori-associated proliferative activities. As seen in Figure 4E, 4F, and 4G, H. pylori infection was associated with significant increases in β-catenin, CDK4, Akt activation, Ki-67 as well as BrdU incorporation, whereas these proliferative markers were not significantly different from the non-infected Fat-1 TG mice (Figure 4E and 4G). At this stage, we also conducted protein arrays to identify proteins whose expressiosn are significantly different between WT and Fat-1 TG mice. As seen in Figure 4F, CD26, FGF1 (HB-EGF), and IGFBP3 were significantly increased in H. pylori-infected WT, but not changed in Fat-1 TG mice.
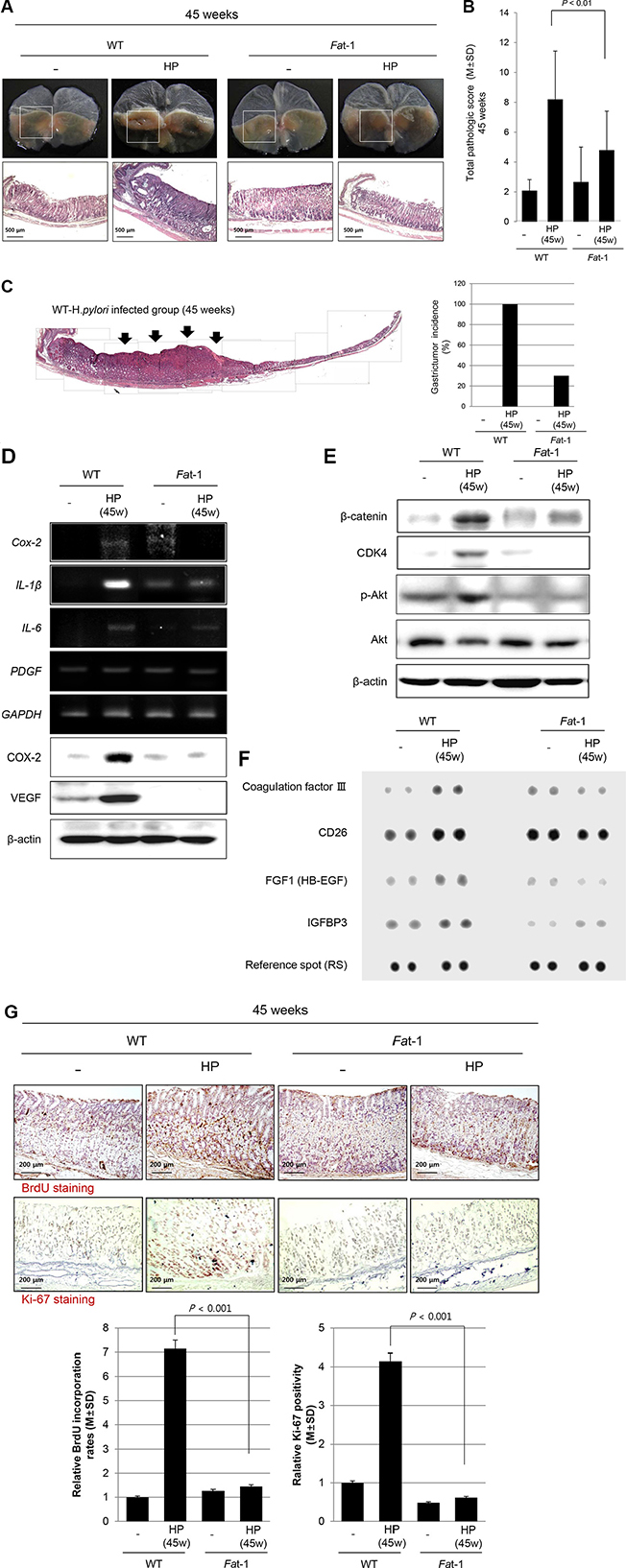
Figure 4: Long-term efficacy of ω-3 PUFA on H. pylori-induced gastric tumorigenesis at 45 weeks. (A) All of H. pylori-infected mice had developed variable sized gastric tumors, whereas only 30% mice developed gastric tumors in Fat-1 TG mice, smaller in size than WT mice (H&E stain, Magnification, x 40). (B–C) The mean pathological scoring was significantly decreased in Fat-1 TG mice compared to WT mice (P < 0.01). 1:1 mounting view of whole stomach of WT - H. pylori group showed moderately-differentiated adenocarcinoma, by which there was significant difference in tumorigenesis between WT and Fat-1 TG mice at 45 weeks after H. pylori infection. (D) Measuring the expressions of Cox-2, IL-1β, IL-6, and PDGF mRNA between groups, H. pylori-infected WT showed significantly increased expressions, but significantly attenuated at Fat-1 TG mice. (E) Western blots for β-catenin, CDK4, phosphorylated-Akt, and total Akt according to group. (F) Protein array. (G) Immunohistochemical staining for BrdU and ki-67. All of figure E, F, and G consistently suggested H. pylori infection for 45 weeks led to significantly increased proliferative actions in WT mice, but these mucosal proliferative mechanisms were significantly decreased in Fat-1 TG mice (P < 0.001, Magnification × 100). The stomachs were stained for BrdU and Ki67, the number of positive cells was represented as mean ± SD.
Dose of exogenous ω-3 PUFAs showing similar lipid profiles of stomach as seen in Fat-1 TG mice
Though the current experiment investigated the exact role of ω-3 PUFAs against H. pylori infection by using Fat-1 TG mice generating ω-3 PUFAs in the stomach after being fed ω-6 PUFAs rich diets, we tested how much exogenous ω-3 PUFAs is required to achieve the above protection from H. pylori infection. We administered various doses of ω-3 PUFAs (0.5 g/60 kg to 10 g/60 kg) through oral feeding and compared the eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) levels in the stomach. As seen in Figure 5A and 5B, ω-3 PUFAs more than 0.5 g/60 kg showed similar patterns in Fat-1 TG mice because the ratio of ω-6 PUFAs/ω-3 PUFAs was noted in mice fed more than 0.5 g/60 kg ω-3 PUFAs (Figure 5B).
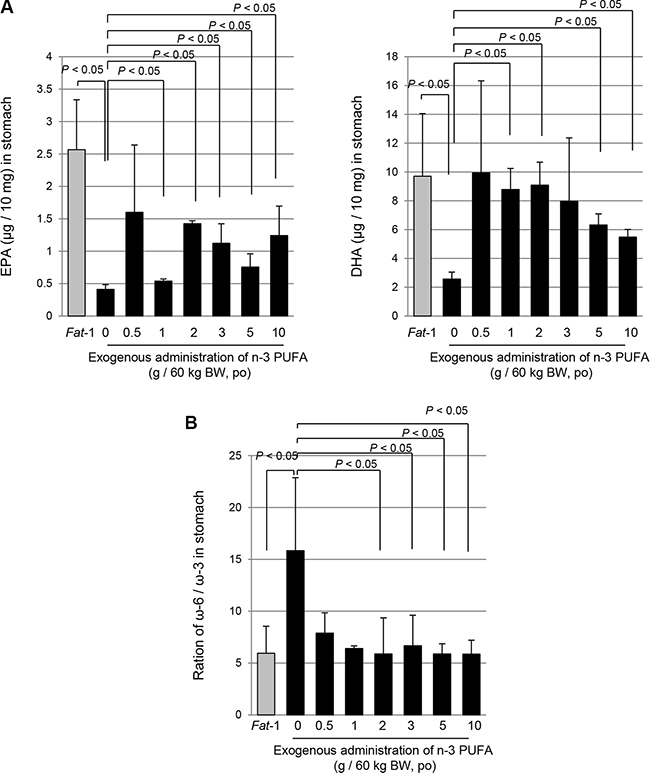
Figure 5: The comparison of concentration of DHA, EPA and ratio of ω-6 and ω-3 PUFAs; between WT mice following administration of exogenous ω-3 PUFA and endogenous ω-3 generated within Fat-1 TG mice. (A–B) The DHA and EPA were extracted from stomach of Fat-1 TG mice and WT mice administrated with ω-3 dose dependently and analyzed by LC/MS/MS as described in Materials and Methods.
DISCUSSION
In the current experiment, ω-3PUFAs attenuated H. pylori-associated gastric inflammation, rejuvenated H. pylori-induced CAG, and prevented H. pylori-associated gastric tumorigenesis in Fat-1 TG mice. In terms of cancer prevention, concerted mechanisms including COX-2 inhibition, 15-PGDH preservation, regulation of proliferation, and mitigated tumorigenic angiogenesis were revealed. Through measuring real levels of lipid profiles in the stomach after exogenous administration of ω-3 PUFAs, we found that dietary intake of more than 0.5 g/60 kg ω-PUFAs could achieve similar lipid profile as seen in Fat-1 TG mice. To our knowledge, this might be the first study demonstrating the inhibitory effect of ω-3PUFAs on H. pylori-induced inflammation and gastric tumorigenesis, signifying the future potential of ω-3 PUFAs to ameliorate either H. pylori-associated CAG or gastric cancer through dietary intervention in the clinic.
Under the same H. pylori infection conditions, the expression of inflammatory and angiogenic growth factors were significantly reduced in Fat-1 TG mice compared to WT (Figure 1D). This suggests that ω-3 PUFAs were capable of suppressing the main inflammatory mechanism and providing an efficient gastro-protection against H. pylori infection beyond inhibitory actions on H. pylori colonization. However, since several in vitro and in vivo studies showed that ω-3 PUFAs could inhibit H. pylori growth in vitro and its colonization in the gastric mucosa of mice in vivo [22, 23], we have investigated the possibility of inferior colonization or bactericidal effects due to ω-3 PUFAs synthesized in Fat-1 TG mice. Similar colonization statuses as measured using direct culture, rapid urease test, e.g., CLO test, and Giemsa staining were observed in WT and TG mice up to 8 weeks of colonization. To avoid reduced bacterial colonization in Fat-1 TG mice, we administered the proton pump inhibitor, pantoprazole, before H. pylori inoculation to enhance their colonization rates followed with thrice administration of H. pylori cultures, and following this protocol, we did not find any differences in H. pylori colonization between WT and Fat-1 TG mice. Therefore, we could rule out that the cancer prevention outcome was not related with lowered colonization in Fat-1 TG mice, even though an in vitro study showed some bacterostatic effects of EPA or DHA.
To determine the molecular mechanisms of the ω-3PUFAs-mediated decrease in H. pylori-associated inflammation 16 to 24 weeks after H. pylori infection, we first looked at inflammatory mediators and angiogenic growth factors. Since the increased expression of inflammatory mediators such as COX-2, IL-1β, IL-6, IFN-γ, and IL-8 were one of core pathogenic mechanisms in H. pylori-associated gastritis [24, 25] and these inflammatory responses are thought to be one of the core processes involved in gastric carcinogenesis [26, 27], we have focused on to the serial changes of COX-2-Prostaglandin E2 (PGE2) pathway [28]. As seen in Figures 1, 2, and 3, these inflammatory mediators were significantly decreased in Fat-1 TG mice compared WT mice. Both ω-6 and ω-3 PUFAs are precursors of potent lipid mediators termed eicosanoids, eicosanoids derived from ω-6 PUFAs have pro-inflammatory and immune-active functions, whereas eicosanoids derived from ω-3 PUFAs have anti-inflammatory properties [13, 29]. This is why the increased intake of ω-3 PUFAs led to a decrease in the risk of many chronic inflammation-based diseases including arthritis, diabetes, obesity, cardiovascular diseases, inflammation, and cancer [30, 31]. COX can generate anti-inflammatory mediators from ω-3 PUFAs [32] including: i) eletrophilicoxo-derivatives (EFOX), a peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist that mostly transduces the beneficial anti-inflammatory effects of DHA, ii) COX inhibitor, iii) aspirin and iv) ω-3 PUFAs-derived lipid autacoids termed resolvins and protectins [33]. Similarly, even though we have published the cancer-preventive effects of the non-steroidal anti-inflammatory drugs (NSAIDs) nimesulide and celecoxib [34] and multiple case-control cohort randomized control trial (RCT), a pooled analysis, and meta-analyses also suggest a preventive effect of aspirin or NSAIDs on the development of non-cardia gastric cancer [35]. The adverse effects associated with the use of NSAIDs may lead to poor adherence as chemo-preventive agents and lead to other harmful effects on the kidney and cardiovascular system. Therefore, it should be emphasized that ω-3 PUFAs raises low toxicological constraints.
Secondarily, the observation that at 24 weeks, the preservation of 15-PGDH—a PG degrading enzyme—accompanied with attenuated COX-2, VEGF, PDGF, and CD31 expression in Fat-1 TG mice compared to WT mice strongly explained the cancer-preventive actions of ω-3 PUFAs [36]. Since 15-PGDH may function as a tumor suppressor through antagonizing oncogenic action of COX-2, 15-PGDH has been found to be down-regulated and a contributor to elevated levels of PGE2 in most tumors, as seen in our H. pylori-associated gastric carcinogenesis model. Since the expression of 15-PGDH and COX-2 appears to be regulated reciprocally in cancer cells, up-regulation of 15-PGDH can be either down-regulated by transcriptional repressors or the attenuation of enzymatic turnover [37]. From our study, significant down-regulation was noted in WT mice after H. pylori infection, but preserved or elevated expression levels of 15-PGDH were observed in Fat-1 TG mice. We documented that ω-3 PUFAs significantly up-regulated 15-PGDH to prevent tumorigenesis relevant to intestinal polyposis [38]. Regarding the changes of decreased 15-PGDH following H. pylori infection [39], the decreased expression of 15-PGDH were reversed with successful H. pylori eradication, in which suppressed 15-PGDH expression levels were associated with Toll like receptor-4 and Myeloid differentiation primary response gene 88 expressions, phospho-Extracellular signal-regulated kinases1/2, and EGF receptor (EGFR)-Snail [40].
Thirdly, considering findings from 32 to 45 weeks in Fat-1 TG mice after H. pylori infection (Figure 3 and Figure 4), co-mechanisms regulating abnormal mucosal growth and considerable rejuvenation were orchestrated to prevent H. pylori-gastric carcinogenesis. As much as the above mentioned tumor suppressive 15-PGDH and sustained inhibition of oncogenic inflammatory mediators, significant control of abnormal mucosal proliferation was observed in Fat-1 TG mice. The proliferation of raised cells is a typical observation in either pre-neoplasia or neoplasia because cells in the proliferative stage are vulnerable to somatic mutations, especially frequently observed in inflammation-prevailed microenvironment conditions. Among the well-known mechanisms of H. pylori promoted gastric carcinogenesis, are abnormal mucosal proliferation [41, 42], promoted epithelial mesenchymal transition [43] via abnormal Akt or Wnt/β-catenin or sonic hedgehog oncogenic activation [44–46], aberrant promoter methylation in tumor suppressor genes, abnormal expression of microRNAs, and abnormal apoptosis [47] after the stage of CAG. As seen in Figure 2F, the expression of IL-1α, amphiregulin, and FGF1 were significantly increased after 24 weeks of H. pylori infection and the expression of β-catenin, CDK4, phosphorylated-Akt, CD26, coagulation factor III, IGFBP3 were significantly increased after 45 weeks of H. pylori infection. Among these factors,CD26, a surface dipeptidylpeptidase IV—a multifunctional protein with intrinsic peptidase activity that inactivates or degrades some bioactive peptides [48]. Due to its multiple functions, CD26/Dipeptidyl peptidase 4 has been shown to be related to the tumor-development process, but it remains unclear if it is a tumor suppressor or a marker of malignancy. However, all of these expressions relevant to abnormal mucosal growth were all mitigated in Fat-1 TG mice. Significantly, we observed that some of these biomarkers were already used for cancer risk of H. pylori-associated gastric cancer except for CD26 [49–53]; ω-3 PUFAs can be an effective agent to counteract biomarkers suggestive of cancer risk.
Fourthly, although they were not studied in this study, additional mechanisms of ω-3 PUFAs-mediated chemoprevention include its role as a structural component providing the optimal function of cellular membranes including: i) membrane fluidity, ii) enzyme activity, iii) balanced ω-6:ω-3 PUFAs production and gene expressions, suggesting that increasing the amount of ω-3 PUFAs consumed at the population level may be a possible potential health benefit; increased intake of ω-3 PUFAs result in prevention of chronic diseases and reduction in the burdening of the health care. From our study, we also investigated why a number of ω-3 PUFAs–containing dietary supplements have shown on the market claiming to protect against the development of a variety of conditions including cancer, but meta-analysis did not provide results of evidence based medicine to suggest a significant association between ω-3 PUFAs and cancer incidence [54]. In the current study, we have compared the gastric levels of ω-3 PUFAs after administering several dose of ω-3 PUFAs (Figure 5) and found that intake more than 0.5 g/60 kg resulted in a similar ω-6/ω-3 PUFAs ratio in the stomach. Our results support the use of dietary ω-3PUFAs in H. pylori-infected patients as a safe prophylactic/preventive strategy before stepping into irreversible condition. Clinical trials to document the chemo-preventive effects of ω-3 PUFAs have mostly focused on colorectal polyps; Higurashi T et al. [55] performed a double blind, placebo-controlled RCT to explore the effects of EPA against colorectal aberrant crypt foci (ACF) and highlighted the suppressive effect of 2.7g EPA/day on the formation of ACF and Hull MA et al. [56], after their SeAFOod trials, The seafood Polyp Prevention Trial, concluded that EPA can prevent polyp formation.
Lastly, we have checked microbiota changes in WT and Fat-1 TG mice during 24 and 32 weeks through 454 pyrosequencing measurement and found significant changes between WT and Fat-1 TG mice (data not shown). These significant changes in fecal microbiota in Fat-1 TG mice might come from the following two possibilities: i) the direct influence of ω-3 PUFAs on microbiota change or ii) changes reflecting mitigated conditions of H. pylori-induced chronic atrophic gastritis by fatty acid produced in Fat-1 TG mice. Conclusively, our findings suggest that he increased abundance of ω-3PUFAs in tissue significantly reduced gastric inflammation and tumorigenesis in Fat-1 mice. Clinical trials to document the effects of exogenous administration of more than 0.5 g/60 kg high purity ω-3 PUFAs in chronic H. pylori infection should be conducted to determine the benefit of the use of ω-3 PUFAs in chemo-preventive strategies for H. pylori-induced atrophic gastric disease.
MATERIALS AND METHODS
H. pylori-infected mice model
Animals
Five-week-old male C57BL/6 mice (WT mice) were purchased from Orient (Seoul, Korea) and Fat-1 transgenic (Fat-1 TG) mice were provided kindly from Dr. Jing X. Kang (Boston, MA). They were housed in a cage maintained at 23°C in a 12 h/12 h light/dark cycle under specific pathogen-free conditions (n = 160). We divided four groups; 1) WT mice as vehicle control group 2) WT mice as H. pylori-infected group 3) Fat-1 mice as vehicle control group 4) Fat-1 mice as H. pylori-infected group. First, all groups were given intraperitoneal (i.p.) injections of proton pump inhibitors (PPIs, pantoprazole, 20 mg/kg; Amore-Pacific Pharma) three times per week to facilitate H. pylori colonization through lowered gastric acidity. And then, each mouse was intragastrically inoculated with a suspension of H. pylori containing 109 CFUs/mL or with an equal volume (0.1 mL) of clean TS broth using gastric intubation needles. The H. pylori-infected mice were fed a special pellet diet based on AIN-46A containing 7.5% NaCl (high salt diet, Biogenomics, Seongnam, Korea) for 45 weeks (Figure 1A) to promote H. pylori-induced carcinogenic process in all infected animals [57]. And randomized groups of mice (n = 10) sacrificed at 16, 24, 32 and 45 weeks post H. pylori infection, respectively. The stomachs of mice were opened along the greater curvature and washed with ice cold PBS. The numbers of either erosions/ulcers or protruded nodule/mass were determined under the magnified photographs (see Supplementary Figures S1–S4). Stomachs were isolated and subjected to a histologic examination, ELISA, Western blotting, and RT-PCR. All animal studies were carried out in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of CHA University CHA Cancer Institute after IRB approval.
Gross lesion index
After sacrificing the mice, the isolated stomachs were open along the greater curvature and washed in ice-cold saline. To investigate the degree of gross mucosal pathology, the mucosal sides of the stomachs were photographed using a digital camera and part of the mucosa was immediately fixed with 10% formalin solution. The gross damage of the gastric mucosa was assessed by three gastroenterologists, who were blinded to the treatments, using a gross ulcer index [58]. All the gross photographs were displayed in Supplementary Figures and tumorous lesion was depicted with white arrow.
Index of histopathologic injury
For histopathological analysis, the stomach were fixed in 10% neutralized buffered formalin, processing using the standard method and embedded in paraffin. Sections of 4 μm thickness were then stained with hematoxylin and eosin [59]. The glandular mucosae of corpus and antrum were examined histologically. The pathological changes of H. pylori-infection, such as inflammatory cells infiltration, erosive lesions, ulceration, dysplasia, adenoma formation (precancerous lesion), were graded by three gastroenterologists, who were blinded to the group, using an index of histologic injury defined [60]. In this study, inflammation was defined as grade the infiltration of inflammatory cells, 0: none, 1: under the lamina propria, 2: half of mucosa 3: until the epithelial gland layer (all mucosa). The erosion was defined as proportion of erosive lesion, 0: none, 1: loss of epithelial gland layer (1/3 proportion), 2: two-three portion of mucosa (2/3 proportion) 3: all mucosa (3/3 proportion)
BrdU staining for assessing mucosal proliferation
To estimate the rate of proliferation that is increased during carcinogenesis, we injected BrdU before sacrifice, and performed immunohistochemical analysis with anti-BrdU antibody.
Immunohistochemical staining
Immunohistochemistry was performed on replicate sections of mouse gastric tissues. After deparaffinization were dewaxed and rehydrated with graded alcohol, and boiled three times in 100 mM Tris buffered saline (pH 6) with 5% urea in an 850 W microwave oven for 5 min each. And then cooling in water for 15 min and washed in PBS, and slides were incubated overnight with the primary antibody at 4°C. Antibodies: F4/80 (1:500; eBioscience, San Diego, CA) or CD31 (1:300; Dako, Santa Clara, CA) or Ki-67 (1:300; Santa cruz, Santa Cruz, CA) in the presence of 1.0% bovine serum albumin respectively. Slides incubated with secondary antibody (1:300) for 1 h at room temperature, and then with 40-6- diamidino-2-phenylindole (DAPI, 100 ng/ml) for 1 min at room temperature. And finally the slides were counterstained with hematoxylin (Sigma-Aldrich).
RT-PCR
Total RNA was isolated using the Trizol (Invitrogen, Carlsbad, CA). Trizol was added to 1.5 ml tube, which were then incubated 10 min at 4°C and gently mixed with 100 μl chloroform (Merck, Rahway, NJ). After incubation for 10 min in ice, samples were centrifuged at 10,000 g for 30 min. Supernatants were extracted and mixed with 200 μl isopropanol (Merck), and mixtures were incubated at 4°C for 1 h. After centrifuging at 13,000 g for 30 min, pellets were washed with 70% (v/v) ethanol. After allowing the ethanol to evaporate completely, pellets were dissolved in 40 μl diethylene pyrocarbonate-treated water (Invitrogen Life Technologies). cDNA was prepared using reverse transcriptase originating from Murine-Moloney leukemia virus (Promega, Madison, WI), according to the manufacturer’s instructions. The polymerase chain reaction (PCR) was performed over 25 cycles of: 94°C for 20 s, 58.5 for 30 s, and 72°C for 45 s. Oligonucleotide primers were purchased from Bioneer (Daejeon, Korea). Oligonucleotide primers were as follows; for VEGF, sense 5′-GAA GCT ACT GCC GTC CGA TT-3′ and antisense 5′-TCC TCT TCC TTC ATG TCA GGC-3′, for COX-2, sense 5′-GAA ATG GCT GCA GAG TTG AA-3′ and antisense 5′-TCA TCT AGT CTG GAG TGG GA-3′, for PDGF, sense 5′-ACG TCA TGT TAC GGC TTC CT-3′ and antisense 5′-CAG TGT GAC TGT GTC TCC CC-3′, for IL-1β, sense 5′-CAG GCT CCG AGA TGA ACA ACA AAA-3′ and antisense 5′-TGG GGA ACT CTG CAG ACT CAA ACT-3′, for IL-8, sense 5′-GGG GCT TTG CCG TGC AAT AA-3′ and antisense 5′-GCA CAG GGT TGA GCC AAA A-3′, for IFN-γ, sense 5′-ACA ATG AAC GCT ACA CAC TG-3′ and antisense 5′-TCA AAC TTG GCA ATA CTC AT-3′, for IL-6, sense 5′-AAG AGA CTT CCA GCC AGT TG-3′ and antisense 5′-TGG ATG GTC TTG GTC CTT AG-3′, and for GAPDH, sense 5′-GGT GCT GAG TAT GTC GTG GA-3′ and antisense 5′-TTC AGC TCT GGG ATG ACC TT-3′.
Western blotting
Cells were harvested and lysed in lysis buffer (Cell signaling Technology) containing 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma Aldrich St. Louis, MO). After 30 min of incubation, samples were centrifuged at 12,000 g for 15 min 4°C. The supernatants were then collected and protein quantification was carried out with a Bio-Rad protein assay. Equal amounts soluble protein (30 μg) were denaturated by heating at 100°C for 3 minutes. Proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride membranes. The membranes were blocked in 5% BSA in PBST for 30 min. And then, the membranes probed initially with specific primary antibody, washed, incubated with peroxidase-conjugated secondary antibodies, and rewashed. The protein bands were detected by chemiluminescence (Supersignal, Pierce) exposure on chemiluminescence system (GE Healthcare, Buckinghamshire, UK). The general procedure for Western blot analysis of cultured mouse gastric mucosal cells was similar to the procedures described above. Antibodies used in the current study were COX-2, purchased from Thermo and β-actin, VEGF, β-catenin, purchased from Santa Cruz and CDK4, phospho-Akt, Akt, purchased from cell Signaling Technology and 15-PGDH, purchased from Cayman.
Cytokine protein array
Cytokine protein array was performed using Mouse Cytokine Antibody Array 3 (4 membrane arrays) with Accessories, for simultaneous detection of 54 cytokines related proteins in 2 samples from R&D systems (Minneapolis, MN). After blocking the array membranes for 30 minutes, the membranes were incubated with 1 ml of serum at room temperature for 2 hours. After washing with buffer, we added primary biotin-conjugated antibody to each membrane, for incubation at room temperature for 2 hours. After washing with buffer and addition of horseradish peroxidase-conjugated streptavidin to each membrane, we exposed them to detection buffer, using a luminescent image analyzer system (LAS-4000, Fuji Film; Tokyo, Japan). Density was expressed as the percentage of the detected value from the sample versus the background result, using a gelpro32 program (Media Cybernetics, Rockville, MD).
Gas chromatography for measuring lipid profiles in the stomach
Fatty acid profiles were analyzed using gas chromatography as described previously [19]. Briefly, 1 cm of mice tails (in order to perform the phenotyping of mice) or blocks of colon tissue (5 × 5 mm) were grounded to powder under liquid nitrogen. Samples were then subjected to extraction of total lipids and fatty acid methylation by heating at 100°C for 1 h under 14% boron trifluoride (BF3)–methanol reagent (Sigma, St. Louis, MO) and hexane (Sigma). Fatty acid methyl esters were analysed by gas chromatography using a fully automated 6890 N Network GC System (Agilent Technologies) equipped with a flame-ionization detector. Peaks of resolved fatty acids were identified by comparison with fatty acid standards (Nu-chek-Prep), and area percentage for all resolved peaks was analysed using GC ChemStation Software (Agilent Technologies, Santa Clara, CA) [61].
Statistical analyses
The data are presented as means ± standard deviations (S.D.). The data were analyzed by 1-WAY ANOVA, and the statistical significance between groups was determined by Student t test. Statistical significance was accepted at P < 0.05.
Abbreviations
H. pylori, Helicobactor pylori; ω-3 PUFAs, Omega-3-polyunsaturated-fatty acids; WT, Wild-type; TG, Transgenic; COX, Cyclooxygenase; PGDH, Prostaglandin dehydrogenase; 15-PGDH, Hydroxyprostaglandin dehydrogenase; MGC, Metachronous gastric cancer; IL, Interleukin; IFN, Interferon; VEGF, Vascular endothelial growth factor; PDGF, Platelet-derived growth factor; CD, Cluster of differentiation; FGF, Fibroblast growth factor; CDK, Cyclin-dependent kinase; AKT, Protein kinase B; HB-EGF, Heparin-binding EGF-like growth factor; IGFBP, Insulin-like growth factor-binding protein; BrdU, Bromodeoxyuridine; PGE2, Prostaglandin E2; RCT, Randomized control trial.
ACKNOWLEDGMENTS
We thank Dr. Eun-Hee Kim for technical assistance.
CONFLICTS OF INTEREST
No conflicts of interest.
FINANCIAL SUPPORT
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2014R1A1A2058732 to JM Park), by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry and Fisheries (IPET) through High Value-added Food Technology Development Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (116015-03-1-CG000), and also by Japanese Society for Promotion of Science (to T Azuma).
REFERENCES
1. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994; 61:1–241.
2. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001; 345:784–789.
3. Fukase K, Kato M, Kikuchi S, Inoue K, Uemura N, Okamoto S, Terao S, Amagai K, Hayashi S, Asaka M, Japan Gast Study G. Effect of eradication of Helicobacter pylori on incidence of metachronous gastric carcinoma after endoscopic resection of early gastric cancer: an open-label, randomised controlled trial. Lancet. 2008; 372:392–397.
4. Zhang W, Lu H, Graham DY. An Update on Helicobacter pylori as the Cause of Gastric Cancer. Gastrointestinal tumors. 2014; 1:155–165.
5. Kuipers EJ, Sipponen P. Helicobacter pylori eradication for the prevention of gastric cancer. Helicobacter. 2006; 11:52–57.
6. Lee HJ, Park JM, Han YM, Gil HK, Kim J, Chang JY, Jeong M, Go EJ, Hahm KB. The role of chronic inflammation in the development of gastrointestinal cancers: reviewing cancer prevention with natural anti-inflammatory intervention. Expert review of gastroenterology & hepatology. 2016; 10:129–139.
7. Park JM, Jeong M, Kim EH, Han YM, Kwon SH, Hahm KB. Omega-3 Polyunsaturated Fatty Acids Intake to Regulate Helicobacter pylori-Associated Gastric Diseases as Nonantimicrobial Dietary Approach. BioMed research international. 2015; 2015:712363.
8. Jeong M, Park JM, Han YM, Park KY, Lee DH, Yoo JH, Cho JY, Hahm KB. Dietary prevention of Helicobacter pylori-associated gastric cancer with kimchi. Oncotarget. 2015; 6:29513–29526. doi: 10.18632/oncotarget.4897.
9. Han YM, Park JM, Jeong M, Yoo JH, Kim WH, Shin SP, Ko WJ, Hahm KB. Dietary, non-microbial intervention to prevent Helicobacter pylori-associated gastric diseases. Annals of translational medicine. 2015; 3:122.
10. Hartgrink HH, Jansen EP, van Grieken NC, van de Velde CJ. Gastric cancer. Lancet. 2009; 374:477–490.
11. Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002; 420:860–867.
12. Park SH, Kangwan N, Park JM, Kim EH, Hahm KB. Non-microbial approach for Helicobacter pylori as faster track to prevent gastric cancer than simple eradication. World journal of gastroenterology. 2013; 19:8986–8995.
13. Calder PC. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. The American journal of clinical nutrition. 2006; 83:1505S–1519S.
14. Calder PC. Dietary modification of inflammation with lipids. The Proceedings of the Nutrition Society. 2002; 61:345–358.
15. Simopoulos AP. Omega-3 fatty acids in inflammation and autoimmune diseases. Journal of the American College of Nutrition. 2002; 21:495–505.
16. Yates CM, Calder PC, Ed Rainger G. Pharmacology and therapeutics of omega-3 polyunsaturated fatty acids in chronic inflammatory disease. Pharmacology & therapeutics. 2014; 141:272–282.
17. Kremer JM. n-3 fatty acid supplements in rheumatoid arthritis. The American journal of clinical nutrition. 2000; 71:349S–351S.
18. Riediger ND, Othman RA, Suh M, Moghadasian MH. A systemic review of the roles of n-3 fatty acids in health and disease. Journal of the American Dietetic Association. 2009; 109:668–679.
19. Kang JX, Wang J. A simplified method for analysis of polyunsaturated fatty acids. BMC biochemistry. 2005; 6:5.
20. Kang JX. Fat-1 transgenic mice: a new model for omega-3 research. Prostaglandins, leukotrienes, and essential fatty acids. 2007; 77:263–267.
21. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. Journal of immunology research. 2014; 2014:149185.
22. Thompson L, Cockayne A, Spiller RC. Inhibitory effect of polyunsaturated fatty acids on the growth of Helicobacter pylori: a possible explanation of the effect of diet on peptic ulceration. Gut. 1994; 35:1557–1561.
23. Correia M, Michel V, Matos AA, Carvalho P, Oliveira MJ, Ferreira RM, Dillies MA, Huerre M, Seruca R, Figueiredo C, Machado JC, Touati E. Docosahexaenoic acid inhibits Helicobacter pylori growth in vitro and mice gastric mucosa colonization. PloS one. 2012; 7:e35072.
24. Nam KT, Oh SY, Ahn B, Kim YB, Jang DD, Yang KH, Hahm KB, Kim DY. Decreased Helicobacter pylori associated gastric carcinogenesis in mice lacking inducible nitric oxide synthase. Gut. 2004; 53: 1250–1255.
25. Yeo M, Kim DK, Han SU, Lee JE, Kim YB, Cho YK, Kim JH, Cho SW, Hahm KB. Novel action of gastric proton pump inhibitor on suppression of Helicobacter pylori induced angiogenesis. Gut. 2006; 55:26–33.
26. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008; 454:436–444.
27. Kovaleva IE, Grivennikov SI, Luzikov VN. On the effect of cholesterol on the fate of CYP11A1 imported into yeast mitochondria in vivo. Biochemistry Biokhimiia. 2000; 65:1206–1211.
28. Wang D, Dubois RN. Eicosanoids and cancer. Nature reviews Cancer. 2010; 10:181–193.
29. Wall R, Ross RP, Fitzgerald GF, Stanton C. Fatty acids from fish: the anti-inflammatory potential of long-chain omega-3 fatty acids. Nutrition reviews. 2010; 68:280–289.
30. Abete P, Testa G, Galizia G, Della-Morte D, Cacciatore F, Rengo F. PUFA for human health: diet or supplementation? Current pharmaceutical design. 2009; 15:4186–4190.
31. Chapkin RS, McMurray DN, Lupton JR. Colon cancer, fatty acids and anti-inflammatory compounds. Current opinion in gastroenterology. 2007; 23:48–54.
32. Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, Rudolph V, Freeman BA, Schopfer FJ. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nature chemical biology. 2010; 6:433–441.
33. Masoodi M, Kuda O, Rossmeisl M, Flachs P, Kopecky J. Lipid signaling in adipose tissue: Connecting inflammation & metabolism. Biochimica et biophysica acta. 2015; 1851:503–518.
34. Ulrich CM, Bigler J, Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nature reviews Cancer. 2006; 6:130–140.
35. Tan VP, Wong BC. Gastric cancer chemoprevention: the current evidence. Gastroenterology clinics of North America. 2013; 42:299–316.
36. Na HK, Park JM, Lee HG, Lee HN, Myung SJ, Surh YJ. 15-Hydroxyprostaglandin dehydrogenase as a novel molecular target for cancer chemoprevention and therapy. Biochemical pharmacology. 2011; 82:1352–1360.
37. Tai HH. Prostaglandin catabolic enzymes as tumor suppressors. Cancer metastasis reviews. 2011; 30:409–417.
38. Han YM, Park JM, Cha JY, Jeong M, Go EJ, Hahm KB. Endogenous conversion of omega-6 to omega-3 polyunsaturated fatty acids in fat-1 mice attenuated intestinal polyposis by either inhibiting COX-2/beta-catenin signaling or activating 15-PGDH/ IL-18. International journal of cancer. 2016; 138: 2247–2256.
39. Park JM, Park SH, Hong KS, Han YM, Jang SH, Kim EH, Hahm KB. Special licorice extracts containing lowered glycyrrhizin and enhanced licochalcone A prevented Helicobacter pylori-initiated, salt diet-promoted gastric tumorigenesis. Helicobacter. 2014; 19:221–236.
40. Thiel A, Ganesan A, Mrena J, Junnila S, Nykanen A, Hemmes A, Tai HH, Monni O, Kokkola A, Haglund C, Petrova TV, Ristimaki A. 15-hydroxyprostaglandin dehydrogenase is down-regulated in gastric cancer. Clinical cancer research. 2009; 15:4572–4580.
41. Nishizawa T, Suzuki H. Gastric Carcinogenesis and Underlying Molecular Mechanisms: Helicobacter pylori and Novel Targeted Therapy. BioMed research international. 2015; 2015:794378.
42. Ding SZ, Goldberg JB, Hatakeyama M. Helicobacter pylori infection, oncogenic pathways and epigenetic mechanisms in gastric carcinogenesis. Future oncology. 2010; 6:851–862.
43. Yu H, Zeng J, Liang X, Wang W, Zhou Y, Sun Y, Liu S, Li W, Chen C, Jia J. Helicobacter pylori promotes epithelial-mesenchymal transition in gastric cancer by downregulating programmed cell death protein 4 (PDCD4). PloS one. 2014; 9:e105306.
44. Sasaki T, Yamashita Y, Kuniyasu H. AKT plays a crucial role in gastric cancer. Oncology letters. 2015; 10:607–611.
45. Song X, Xin N, Wang W, Zhao C. Wnt/beta-catenin, an oncogenic pathway targeted by H. pylori in gastric carcinogenesis. Oncotarget. 2015; 6:35579–35588. doi: 10.18632/oncotarget.5758.
46. Donnelly JM, Chawla A, Houghton J, Zavros Y. Sonic hedgehog mediates the proliferation and recruitment of transformed mesenchymal stem cells to the stomach. PloS one. 2013; 8:e75225.
47. Xia HH, Talley NJ. Apoptosis in gastric epithelium induced by Helicobacter pylori infection: implications in gastric carcinogenesis. The American journal of gastroenterology. 2001; 96:16–26.
48. Beckenkamp A, Davies S, Willig JB, Buffon A. DPPIV/CD26: a tumor suppressor or a marker of malignancy? Tumour biology. 2016; 37:7059–7073.
49. Takaishi S, Wang TC. Gene expression profiling in a mouse model of Helicobacter-induced gastric cancer. Cancer science. 2007; 98:284–293.
50. Busiello I, Acquaviva R, Di Popolo A, Blanchard TG, Ricci V, Romano M, Zarrilli R. Helicobacter pylori gamma-glutamyltranspeptidase upregulates COX-2 and EGF-related peptide expression in human gastric cells. Cellular microbiology. 2004; 6: 255–267.
51. Tuccillo C, Manzo BA, Nardone G, D’Argenio G, Rocco A, Di Popolo A, Della VN, Staibano S, De Rosa G, Ricci V, Del Vecchio BC, Zarrilli R, Romano M. Up-regulation of heparin binding epidermal growth factor-like growth factor and amphiregulin expression in Helicobacter pylori-infected human gastric mucosa. Digestive and liver disease. 2002; 34:498–505.
52. Pham TM, Fujino Y, Kikuchi S, Tamakoshi A, Yatsuya H, Matsuda S, Yoshimura T, Group JS. A nested case-control study of stomach cancer and serum insulin-like growth factor (IGF)-1, IGF-2 and IGF-binding protein (IGFBP)-3. European journal of cancer. 2007; 43:1611–1616.
53. Ramljak D, Jones AB, Diwan BA, Perantoni AO, Hochadel JF, Anderson LM. Epidermal growth factor and transforming growth factor-alpha-associated overexpression of cyclin D1, Cdk4, and c-Myc during hepatocarcinogenesis in Helicobacter hepaticus-infected A/JCr mice. Cancer research. 1998; 58: 3590–3597.
54. MacLean CH, Newberry SJ, Mojica WA, Khanna P, Issa AM, Suttorp MJ, Lim YW, Traina SB, Hilton L, Garland R, Morton SC. Effects of omega-3 fatty acids on cancer risk: a systematic review. Jama. 2006; 295:403–415.
55. Higurashi T, Takahashi H, Endo H, Hosono K, Yamada E, Ohkubo H, Sakai E, Uchiyama T, Hata Y, Fujisawa N, Uchiyama S, Ezuka A, Nagase H, et al. Metformin efficacy and safety for colorectal polyps: a double-blind randomized controlled trial. BMC cancer. 2012; 12:118.
56. Hull MA, Sandell AC, Montgomery AA, Logan RF, Clifford GM, Rees CJ, Loadman PM, Whitham D. A randomized controlled trial of eicosapentaenoic acid and/or aspirin for colorectal adenoma prevention during colonoscopic surveillance in the NHS Bowel Cancer Screening Programme (The seAFOod Polyp Prevention Trial): study protocol for a randomized controlled trial. Trials. 2013; 14:237.
57. Gaddy JA, Radin JN, Loh JT, Zhang F, Washington MK, Peek RM, Jr., Algood HM, Cover TL. High dietary salt intake exacerbates Helicobacter pylori-induced gastric carcinogenesis. Infection and immunity. 2013; 81:2258–2267.
58. Nam SY, Kim N, Lee CS, Choi KD, Lee HS, Jung HC, Song IS. Gastric mucosal protection via enhancement of MUC5AC, MUC6 by geranylgeranylacetone. Digestive diseases and sciences. 2005; 50:2110–2120.
59. Goo MJ, Ki MR, Lee HR, Yang HJ, Yuan DW, Hong IH, Park JK, Hong KS, Han JY, Hwang OK, Kim DH, Do SH, Cohn RD, et al. Helicobacter pylori promotes hepatic fibrosis in the animal model. Laboratory investigation. 2009; 89:1291–1303.
60. Lacy ER, Ito S. Microscopic analysis of ethanol damage to rat gastric mucosa after treatment with a prostaglandin. Gastroenterology. 1982; 83:619–625.
61. Nowak J, Weylandt KH, Habbel P, Wang J, Dignass A, Glickman JN, Kang JX. Colitis-associated colon tumorigenesis is suppressed in transgenic mice rich in endogenous n-3 fatty acids. Carcinogenesis. 2007; 28:1991–1995.