
INTRODUCTION
MiRNAs are one category of short non-coding RNAs (~22nt) which could inhibit the protein production and gene expression through binding to the 3’-UTRs of the target mRNAs at the post-transcriptional and translational level [1–4]. However, miRNAs could also serve as positive regulators according to some studies [5, 6]. In the recent several years, thousands of miRNAs have been detected based on various experimental methods and computational models since the first two miRNAs (Caenorhabditis elegans lin-4 and let-7) were discovered more than twenty years ago [7–10]. There are 26845 entries in the latest version of miRBase, including more than 1000 human miRNAs [11]. Furthermore, accumulating evidences indicated that miRNAs are important components in cells, which could play critical roles in multiple important biological processes, including cell proliferation [12], development [13], differentiation [14], and apoptosis [15], metabolism [16, 17], aging [16, 17], signal transduction [18], and viral infection [14]. Therefore, it is no surprise that miRNAs have close associations with the development, progression, and prognosis of many human diseases [19–24]. For example, the miRNA deregulation is closely related to the development of various cancers [25–28]. Calin et al. firstly clarified that miR-15 and miR-16 are deleted in more than half cases of B-cell chronic lymphocytic leukemia (B-CLL), and this discovery also become the first evidence for the fact that miRNAs are involved in cancer formation [29]. He et al. firstly reported that there are links between the enhanced expression of miR-17 cluster in B-cell lymphomas and the development of c-Myc-induced tumorigenesis [30]. Besides, miR-122 suppresses cell proliferation and tumorigenesis by targeting IGF1R in some breast cancer cases [31]. Experiments further showed that the regulation of Ad6 by miR-122 could significantly improves the safety profile of the whole body after systemic administration, which allows increasing therapeutic doses and therefore improves anticancer efficacy of prostate cancer [32]. Therefore, identifying disease-related miRNAs could effectively promote disease biomarker detection for the treatment, diagnosis and prevention of human complex diseases [33]. Considering vast amount of miRNA-related biological datasets has been generated, it is urgent to develop powerful computational models to predict novel human disease-miRNA associations [34–46].
Many computational methods have been proposed to predict potential miRNA-disease associations based on the assumption that miRNAs with similar functions tend to be related to phenotypically similar diseases [24, 47–51]. Jiang et al. [52] presented a hypergeometric distribution-based computational model to predict novel miRNA-disease associations. This model is mainly based on the integration of disease phenotype similarity network, miRNA functional similarity network, and the known human disease-miRNA association network. Only adopting miRNA neighbor information seriously influences the prediction performance of this model. Shi et al. [53] further proposed a computational model to exploit the functional associations between miRNA and disease by implementing the algorithm of random walk on protein-protein interaction (PPI) network. Considering the assumption that disease tends to be associated with miRNAs whose target genes also have associations with this disease, they paid attentions to the functional links between disease genes and miRNA targets in PPI network by integrating the information of miRNA–target interactions, disease–gene associations, and PPIs. In addition, Mork et al. [54] proposed the computational model of miRPD by integrating protein–disease associations and miRNA–protein interactions to further predict novel miRNA-disease associations. Xu et al. [55] presented an integrated disease-specific miRNA prioritization approach without the rely on known disease-miRNA associations. This method integrates known disease–gene associations and context-dependent miRNA-target interactions. They converted the association probability of a miRNA-disease pair into the functional similarity calculation between the targets of this miRNA and known associated genes of this diseases. However, the predict performances of above several methods were seriously limited by miRNA-target interactions with high false-positive and false-negative results or the incomplete disease-gene association network.
Under the basic assumption that functionally similar miRNAs are regarded to be involved in similar diseases and vice versa, Xuan et al. [56] proposed reliable computational model of HDMP by combining the distribution of miRNAs related with the disease in the k neighbors and miRNA functional similarity to predict the potential disease-related miRNAs. The miRNA functional similarity used in HDMP was integrated by disease phenotype similarity, disease semantic similarity based on the disease terms information content, and known miRNA-disease associations. The important improvement of HDMP over previous studies lies in that it assigned higher weights to members in the same miRNA cluster or family when miRNA functional similarity was calculated. However, HDMP cannot be applied to the new diseases which do not have any known related miRNAs. In addition, HDMP is local network similarity-based computational model, which does not make full use of global network similarity information, which could effectively benefit the prediction performance improvement as demonstrated by many previous studies. Chen et al. [57] proposed the first global network similarity-based computational model, RWRMDA, to predict novel human miRNA–disease associations by considering the information of human miRNA–miRNA functional similarity and known human miRNA–disease associations. The new associations were predicted by adopting the method of random walk on miRNA functional similarity network. RWRMDA has obtained excellent prediction performance based on cross validation and case studies of several important human cancers. However, it also has the important limitation that it could not work for new diseases which do not have any known related miRNAs. Recently, Chen et al. [40] developed a novel computational method of WBSMDA by integrating known miRNA-disease associations, miRNA functional similarity, disease semantic similarity, and Gaussian interaction profile kernel similarity for diseases and miRNAs. WBSMDA could be implemented for the prediction of potential related miRNAs for the diseases which do not have any known related miRNAs and new miRNAs which do not have any known associated diseases. However, the performance of WBSMDA is still not very satisfactory.
Some studies developed machine learning-based computational models to predict novel miRNA-disease associations. For example, Xu et al. [58] constructed a heterogeneous miRNA-target dysregulated network (MTDN) which combines miRNA-target interactions and the expression profiles of miRNAs and mRNAs in tumor and non-tumor tissues. In addition, they performed feature extraction based network topology information and constructed support vector machine (SVM) classifier to identify positive miRNA–disease associations from negative associations. It is well-known that collecting known negative associations is a very difficult and even impossible task. Therefore, inaccurate selection of negative samples would seriously decrease the prediction performance of supervised classifier such as SVM. By integrating disease semantic similarity, miRNA functional similarity, and known miRNA-disease associations, Chenet al. [59] proposed a novel computational model of RLSMDA in the framework of semi-supervised learning to predict potential disease-related miRNAs. RLSMDA could be applied to the diseases without any known related miRNAs. Furthermore, RLSMDA did not need the information of negative miRNA-disease associations. The limitation of RLSMDA lies in the selection of parameter values and the combination of two classifiers in the different spaces.
In this study, we developed a novel computational model of HGIMDA for potential miRNA-disease association prediction. HGIMDA showed superior performance to four classical miRNA-disease association prediction methods (WBSMDA [40], RLSMDA [59], RWRMDA [57], and HDMP [56]). In the case studies of several important human cancers, 45, 44, and 44 out top 50 predicted miRNAs for Colon Neoplasms, Esophageal Neoplasms, and Kidney Neoplasms were verified by recent experimental reports.
RESULTS
Performance evaluation
We implemented Local and global LOOCV based on the recorded miRNA-disease associations in the HMDD database [60] to evaluate the prediction accuracy of HGIMDA (See Figure 1) and four state-of-the-art computational models for miRNA-disease association prediction: WBSMDA [40], RLSMDA [59], RWRMDA [57], and HDMP [56]. In the validation framework of LOOCV, each known association was treated as test sample in turn and other known associations were used for model training. The difference between local and global LOOCV lies in whether we simultaneously investigated all the diseases. In the local LOOCV, test sample was ranked with the candidate samples composed of all the miRNAs without any known associations with the investigated disease. However, in the global LOOCV, test sample was ranked with all the miRNA-disease pairs without any known confirmed associations. The test samples which obtained ranks higher than the given threshold were considered as successful predictions. Furthermore, we drew Receiver operating characteristics (ROC) curve by plotting the true positive rate (TPR, sensitivity) against the false positive rate (FPR, 1-specificity) at different thresholds. Sensitivity denotes the percentage of the test samples which obtained ranks higher than the given threshold. Meanwhile, specificity denotes the percentage of negative miRNA-disease pairs with ranks lower than the threshold. Area under the ROC curve (AUC) is calculated to demonstrate the prediction ability of HGIMDA. AUC=1 indicates the model has perfect prediction performance; AUC=0.5 indicates the model only has random prediction performance.
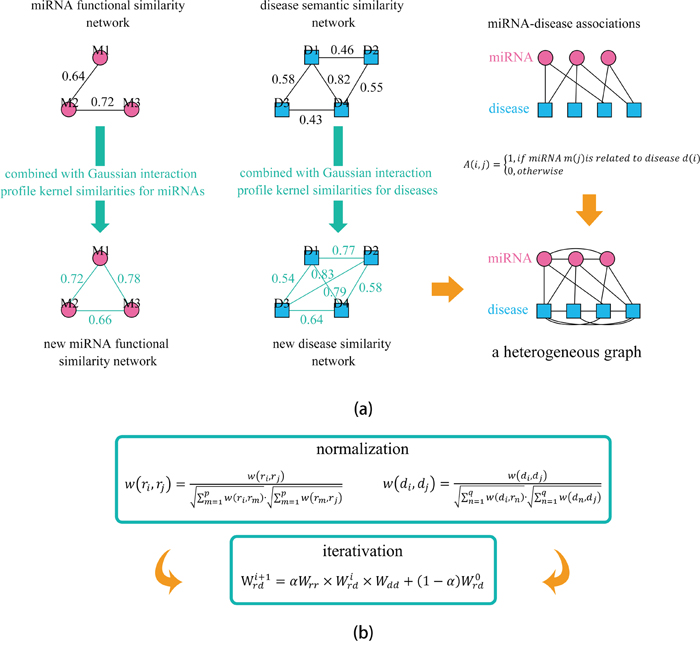
Figure 1: Flowchart of potential disease-miRNA association prediction based on the computational model of HGIMDA. a. Constructing the heterogeneous graph by integrating miRNA functional similarity, disease semantic similarity, Gaussian interaction profile kernel similarity, and experimentally verified miRNA-disease associations; b. Predicting potential miRNA-disease associations based on an iterative equation and obtaining the stable association probability.
The performance comparisons in the framework of local and global LOOCV have been shown in Figure 2. As a result, HGIMDA, WBSMDA, RLSMDA, HDMP obtained AUCs of 0.8781, 0.8030, 0.8426, and 0.8366 in the global LOOCV, respectively. For local LOOCV, HGIMDA, WBSMDA, RLSMDA, HDMP, RWRMDA obtained AUCs of 0.8077, 0.8031, 0.6953, 0.7702, and 0.7891, respectively. Global LOOCV cannot be implemented for RWRMDA model, for the reason that this model cannot uncover the missing associations for all the diseases simultaneously. In conclusion, HGIMDA has shown reliable and effective prediction performance and potential application value for potential miRNA–disease association prediction.
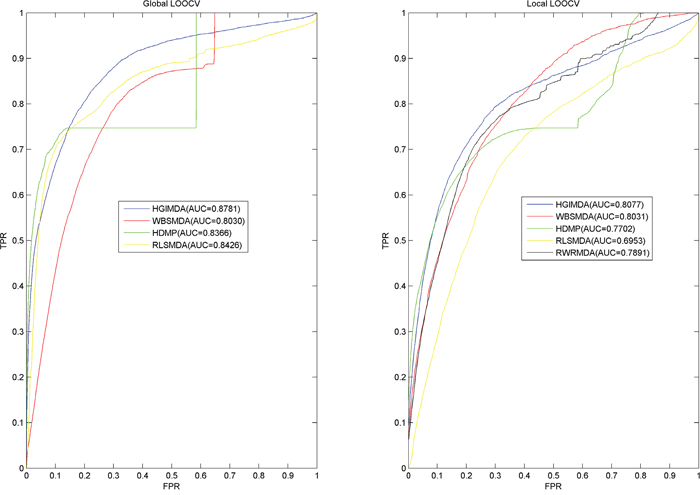
Figure 2: Performance comparisons between HGIMDA and four state-of-the-art disease-miRNA association prediction models (BSMDA, RLSMDA, HDMP, and RWRMDA) in terms of ROC curve and AUC based on local and global LOOCV, respectively. As a result, HGIMDA achieved AUCs of 0.8781 and 0.8031 in the global and local LOOCV, significantly outperforming all the previous classical models.
Case studies
Here, we further implement case studies of several important human complex diseases to further validate the prediction ability of HGIMDA. Predictive results were verified by checking recent experimental reports from another two databases about miRNA-disease associations, miR2Disease [61] and dbDEMC [62].
Colon Neoplasms is one of the biggest threatens to human life worldwide [63, 64]. Studies show that about half of the Colon Neoplasms patients die of metastatic disease within 5 years from diagnosis [65, 66]. With the rapid development of high-throughput sequencing technologies, researchers have identified several miRNAs associated with Colon Neoplasms. For example, miR-126, which is usually lost in Colon Neoplasms, takes phosphatidylinositol 3-kinase signaling as a target and suppresses neoplastic cells growth [67]. It is also found that miR-145 could inhibit Colon Neoplasms cells growth by targeting the insulin receptor substrate-1 [68]. By implementing HGIMDA to identify potential miRNAs associated with Colon Neoplasms, 10 out of the top 10 and 45 out of the top 50 predicted Colon Neoplasms related miRNAs were confirmed based on miR2Disease and dbDEMC (See Table 1). For example, miR-20a and miR-155 were confirmed to be up-regulated in Colon Neoplasms [69]. MiR-20a and miR-19b shown differential expression between neoplastic conditions and non-tumoral colon tissues [70]. MiR-18a was confirmed to be upregulated in colon cancer tissues which suggested that miR-18a is correlated with Colon Neoplasms [71]. An inverse correlation of miR-21 was found in 10 colorectal cell lines which suggested it is a useful diagnostic biomarker for Colon Neoplasms prognosis [72, 73].
Table 1: Here, we implemented HGIMDA to predict potential Colon Neoplasms-related miRNAs
miRNA |
Evidence |
miRNA |
Evidence |
|---|---|---|---|
hsa-mir-20a |
dbDEMC |
hsa-mir-106b |
dbDEMC |
hsa-mir-155 |
dbDEMC |
hsa-mir-143 |
dbDEMC |
hsa-mir-18a |
dbDEMC |
hsa-mir-200a |
unconfirmed |
hsa-mir-21 |
dbDEMC |
hsa-mir-9 |
dbDEMC |
hsa-mir-19b |
dbDEMC |
hsa-mir-1 |
dbDEMC |
hsa-mir-34a |
dbDEMC |
hsa-mir-15a |
dbDEMC |
hsa-mir-19a |
dbDEMC |
hsa-mir-34c |
miR2Disease |
hsa-let-7a |
dbDEMC |
hsa-let-7g |
dbDEMC |
hsa-mir-125b |
dbDEMC |
hsa-mir-146b |
unconfirmed |
hsa-mir-221 |
dbDEMC |
hsa-mir-141 |
dbDEMC |
hsa-mir-92a |
dbDEMC |
hsa-mir-125a |
dbDEMC |
hsa-let-7b |
dbDEMC |
hsa-mir-200c |
dbDEMC |
hsa-mir-146a |
dbDEMC |
hsa-mir-214 |
dbDEMC |
hsa-mir-29b |
dbDEMC |
hsa-mir-34b |
dbDEMC |
hsa-let-7c |
dbDEMC |
hsa-mir-29c |
dbDEMC |
hsa-mir-200b |
dbDEMC |
hsa-mir-101 |
unconfirmed |
hsa-mir-16 |
dbDEMC |
hsa-mir-181b |
dbDEMC |
hsa-let-7d |
dbDEMC |
hsa-mir-210 |
dbDEMC |
hsa-mir-199a |
unconfirmed |
hsa-mir-205 |
dbDEMC |
hsa-mir-29a |
dbDEMC |
hsa-mir-24 |
miR2Disease |
hsa-let-7e |
dbDEMC |
hsa-mir-133a |
dbDEMC |
hsa-mir-223 |
dbDEMC |
hsa-mir-25 |
dbDEMC |
hsa-let-7f |
dbDEMC |
hsa-mir-132 |
miR2Disease |
hsa-mir-222 |
dbDEMC |
hsa-mir-181a |
dbDEMC |
hsa-let-7i |
dbDEMC |
hsa-mir-429 |
unconfirmed |
As a result, 10 out of the top 10 and 45 out of the top 50 predicted Colon Neoplasms related miRNAs were confirmed based on miR2Disease and dbDEMC (1st column: top 1–25; 2nd column: top 26–50).
Esophageal Neoplasms is reported as the sixth-leading cause of deaths related with cancers and the eighth most common cancer worldwide based on the pathological characteristics [74]. The number of male patients is three to four times higher than the number of the female patients [75]. The overall 5-year survival ranges from 15% to 25% [76]. It is suggested that the survival rate could increase to 90% if the tumors could be diagnosed at an early stage [77]. Therefore, the early detection of Esophageal Neoplasms is vital to cancer treatment [78, 79]. There are a lot of miRNAs which have been confirmed to be connected with Esophageal Neoplasms. For example, miR-98 and miR-214 could suppress migration and invasion in human esophageal squamous cell carcinoma by post-transcriptionally regulating enhancer of zeste homolog 2 [80]. HGIMDA was implemented to identify potential related miRNAs for Esophageal Neoplasms based on known associations in the HMDD database. As a result, 9 out of the top 10 and 44 out of the top 50 predicted Esophageal Neoplasms related miRNAs were experimentally confirmed by reports from dbDEMC (See Table 2).
Table 2: We implemented HGIMDA to prioritize candidate miRNAs for Esophageal Neoplasms based on known associations in the HMDD database
miRNA |
Evidence |
miRNA |
Evidence |
|---|---|---|---|
hsa-mir-17 |
dbDEMC |
hsa-mir-30c |
dbDEMC |
hsa-mir-18a |
dbDEMC |
hsa-mir-127 |
dbDEMC |
hsa-mir-19b |
dbDEMC |
hsa-mir-24 |
dbDEMC |
hsa-mir-200b |
dbDEMC |
hsa-mir-10b |
dbDEMC |
hsa-mir-125b |
dbDEMC |
hsa-mir-181a |
dbDEMC |
hsa-let-7d |
dbDEMC |
hsa-mir-106a |
dbDEMC |
hsa-mir-221 |
dbDEMC |
hsa-mir-7 |
dbDEMC |
hsa-let-7e |
dbDEMC |
hsa-mir-191 |
dbDEMC |
hsa-mir-29b |
dbDEMC |
hsa-mir-142 |
dbDEMC |
hsa-let-7f |
unconfirmed |
hsa-mir-20b |
unconfirmed |
hsa-let-7i |
dbDEMC |
hsa-mir-18b |
dbDEMC |
hsa-mir-16 |
dbDEMC |
hsa-mir-195 |
dbDEMC |
hsa-mir-29a |
dbDEMC |
hsa-mir-30d |
dbDEMC |
hsa-mir-222 |
dbDEMC |
hsa-mir-182 |
dbDEMC |
hsa-mir-106b |
dbDEMC |
hsa-mir-199b |
dbDEMC |
hsa-mir-9 |
dbDEMC |
hsa-mir-30a |
dbDEMC |
hsa-mir-1 |
dbDEMC |
hsa-mir-194 |
dbDEMC |
hsa-let-7g |
dbDEMC |
hsa-mir-302b |
dbDEMC |
hsa-mir-125a |
dbDEMC |
hsa-mir-15b |
unconfirmed |
hsa-mir-146b |
dbDEMC |
hsa-mir-92b |
dbDEMC |
hsa-mir-218 |
unconfirmed |
hsa-mir-302c |
dbDEMC |
hsa-mir-429 |
dbDEMC |
hsa-mir-107 |
dbDEMC |
hsa-mir-181b |
dbDEMC |
hsa-mir-30e |
unconfirmed |
hsa-mir-132 |
dbDEMC |
hsa-mir-373 |
dbDEMC |
hsa-mir-93 |
dbDEMC |
hsa-mir-219 |
unconfirmed |
As a result, 9 out of the top 10 and 44 out of the top 50 predicted Esophageal Neoplasms related miRNAs were confirmed by experimental reports from dbDEMC (1st column: top 1–25; 2nd column: top 26–50)
Kidney Neoplasm is a nonhomogeneous cancer which accounts for 3% of adult malignancies [81]. There has been an increasing trend for the incidence and mortality rates of Kidney Neoplasm over the past few years. Specifically, more than 250,000 new cases of kidney cancer are diagnosed every year [82]. As the most common form of adult Kidney Neoplasm [83], renal cell carcinoma (RCC) is comprised of several different types of cancer [84–86], including chromophobe RCC (chRCC), collecting duct carcinoma (CDC), clear cell RCC (ccRCC), and papillary RCC (PRCC) [87–89]. Experiments indicated that the histopathology of Kidney Neoplasm has been connected with different genetic changes [90, 91]. Recently, accumulating studies have shown that many miRNAs are associated with Kidney Neoplasms. For example, miR-215, miR-200c, miR-192, miR-194 and miR-141 were downregulated in Kidney Neoplasms [92]. What’s more, their common target ACVR2B was found to have strong expression in renal childhood neoplasms [92]. Furthermore, miR-21 was up-regulated in Kidney Neoplasms which corresponds to lower Kidney Neoplasms survival [93]. Finally, we implemented HGIMDA on Kidney Neoplasms for potential disease-related miRNA prediction. As a result, 9 out of the top-10 candidates and 44 out of the top-50 candidates of Kidney Neoplasm related miRNAs were verified by dbDEMC (See Table 3). As for the top 5 confirmed Kidney Neoplasms related miRNAs, miR-17 was found differentially expressed in Kidney Neoplasms compared to normal cell tissues [94]. MiR-20a, miR-155, and miR-18a were found up-regulated in Kidney Neoplasms while miR-145 was found down-regulated.
Table 3: We implemented HGIMDA on Kidney Neoplasms for potential disease-related miRNA prediction
miRNA |
Evidence |
miRNA |
Evidence |
|---|---|---|---|
hsa-mir-17 |
dbDEMC |
hsa-mir-222 |
dbDEMC |
hsa-mir-20a |
dbDEMC |
hsa-let-7i |
dbDEMC |
hsa-mir-155 |
dbDEMC |
hsa-mir-200a |
dbDEMC |
hsa-mir-18a |
dbDEMC |
hsa-mir-106b |
dbDEMC |
hsa-mir-145 |
dbDEMC |
hsa-mir-143 |
dbDEMC |
hsa-mir-19b |
dbDEMC |
hsa-mir-9 |
dbDEMC |
hsa-mir-34a |
dbDEMC |
hsa-mir-1 |
dbDEMC |
hsa-mir-19a |
dbDEMC |
hsa-mir-34c |
dbDEMC |
hsa-let-7a |
dbDEMC |
hsa-mir-146b |
dbDEMC |
hsa-mir-125b |
unconfirmed |
hsa-let-7g |
dbDEMC |
hsa-mir-126 |
dbDEMC |
hsa-mir-125a |
dbDEMC |
hsa-mir-221 |
unconfirmed |
hsa-mir-34b |
dbDEMC |
hsa-mir-92a |
unconfirmed |
hsa-mir-214 |
dbDEMC |
hsa-mir-146a |
dbDEMC |
hsa-mir-29c |
dbDEMC |
hsa-mir-200b |
dbDEMC |
hsa-mir-101 |
dbDEMC |
hsa-let-7b |
unconfirmed |
hsa-mir-181b |
dbDEMC |
hsa-mir-29b |
dbDEMC |
hsa-mir-205 |
unconfirmed |
hsa-mir-199a |
dbDEMC |
hsa-mir-210 |
dbDEMC |
hsa-let-7c |
dbDEMC |
hsa-mir-133a |
unconfirmed |
hsa-let-7d |
dbDEMC |
hsa-mir-429 |
dbDEMC |
hsa-mir-16 |
dbDEMC |
hsa-mir-25 |
dbDEMC |
hsa-mir-29a |
dbDEMC |
hsa-mir-93 |
dbDEMC |
hsa-let-7e |
dbDEMC |
hsa-mir-181a |
dbDEMC |
hsa-mir-223 |
dbDEMC |
hsa-mir-24 |
dbDEMC |
hsa-let-7f |
dbDEMC |
hsa-mir-218 |
dbDEMC |
As a result, 9 out of the top 10 and 44 out of the top 50 predicted Kidney Neoplasms related miRNAs were confirmed by dbDEMC
The results in cross validation and independent case studies exploring on three important human complex diseases have fully indicated the outstanding prediction ability of HGIMDA. Therefore, we further used HGIMDA to prioritize candidate miRNAs for all the diseases investigated in HMDD (See Supplementary Table 1). We anticipate that these prediction results could be confirmed by experimental research in the future.
DISCUSSION
Recently, more and more researchers start to propose new computational models to search novel miRNA-disease associations. In this paper, considering the hypothesis that functional similar miRNAs are likely to be involved in similar diseases and vice versa, we presented the computational model of HGIMDA to predict new human complex diseases related miRNAs by integrating Gaussian interaction profile kernel similarity, disease semantic similarity, miRNA functional similarity, and known miRNA-disease associations into a heterogeneous graph. The excellent performance of HGIMDA has been demonstrated by the reliable results from both case studies and cross validation of Colon Neoplasms, Esophageal Neoplasms and Kidney Neoplasms. It could be anticipated that HGIMDA can serve as an effective tool for predicting potential miRNA-disease associations, and will be helpful in human disease prevention, treatment, diagnosis, and prognosis.
The reasons of reliable performance of HGIMDA may come from the following several factors. Firstly, the success of HGIMDA is mainly dependent on the integration of several reliable biological datasets into a heterogeneous graph. Especially, the number of known miRNA-disease associations used in this method significantly increases compared with known associations used for previous methods. Secondly, similar to the process of random work, HGIMDA is an iterative process to find the optimal solutions based on global network similarity information, whose improvement over local network-similarity-based models has been fully indicated by the previous studies. However, there are essential differences between HGIMDA and traditional random walk. Traditional random walk set the initial probability vector only based on known related miRNAs with the investigated disease. Therefore, when this disease has no known related miRNAs, random walk can’t work. Here, various disease similarity measures, various miRNA similarity measures, and known miRNA-disease association were combined to implement prediction, which ensures that HGIMDA could be used to predict related miRNAs for new diseases which have no known related miRNAs and miRNAs without any known associated diseases. Therefore, the application scope of classical random walk has been significantly broadened. This distinct advantage overcomes the important limitations of many previous computational models. Furthermore, HGIMDA could effectively uncover the missing miRNA-disease associations for all the diseases simultaneously. Limitations also exist in this method. Firstly, the known miRNA-disease associations with experimental evidences are still insufficient. By integrating more available biological information in the future, the prediction performance of HGIMDA could be further improved [95–97]. Secondly, HGIMDA may cause bias to miRNAs which have more associated disease records. Finally, the selection of the parameter value in formula (11) is still not well solved.
MATERIALS AND METHODS
Human miRNA-disease associations
Accumulating biological experiments have produced plenty of miRNA–disease associations. The human miRNA-disease association dataset used in this study was downloaded from HMDD database (June, 2013) [60], including 5430 distinct experimentally confirmed human miRNA-diseases associations about 383 diseases and 495 miRNAs. Adjacency matrix A is defined to represent known miRNAs-disease associations. If miRNA m(i) is related to disease d(j), the entity A(m(i), d(j)) is 1, otherwise 0. Furthermore, variables nm and nd are denoted as the number of miRNAs and diseases in the known association dataset, respectively.
MiRNA functional similarity
Based on the assumption that miRNAs with similar functions tend to be associated with similar diseases and vice versa [24, 47–49, 56], Wang et al. [48] proposed the method of miRNA functional similarity calculation. We obtained miRNA functional similarity from http://www.cuilab.cn/files/images/cuilab/misim.zip and established miRNA functional similarity matrix FS to represent the miRNA functional similarity network, in which FS(i,j) is the functional similarity score between miRNA m(i) and m(j).
Disease semantic similarity
The relationships among different diseases can be described as a Directed Acyclic Graph (DAG). Disease D can be represented as DAG(D)=(D,T(D),E(D)), where T(D) represents all ancestor nodes of D and D itself, E(D) represents all direct edges from parent nodes to child nodes. Disease MeSH descriptors were downloaded from the National Library of Medicine (http://www.nlm.nih.gov) [98], including Category A for anatomic terms, Category B for organisms, Category C for diseases, Category D for drugs and chemicals and so on. Here, we selected the MeSH descriptor of Category C to construct disease DAGs. The location of each disease term in the DAG could be decided by the tree number of each MeSH descriptor.
The contribution of disease d in DAG(D) to the semantic value of disease D is defined as follows:
(1)
Here, Δ is the semantic contribution factor. The contribution score for disease d is inversely proportional to the distance between disease d and D. The semantic value of disease D could be defined as follows:
(2)
It is obviously that two diseases with larger shared part of their DAGs may have greater similarity score. Therefore, the semantic similarity score between disease d(i) and d(j) is defined as follows:
(3)
Gaussian interaction profile kernel similarity
Gaussian interaction profile kernel similarity for diseases are constructed based on the assumption that similar diseases tend to be associated with miRNAs with similar functions and vice versa [24, 47–49]. Binary vector IP(d(u)) is defined to represent the interaction profiles of disease d(u) by observing whether there are known associations between disease d(u) and each miRNA or not. Therefore, Gaussian interaction profile kernel similarity of diseases d(u) and d(v) is defined as follows.
(4)
Here, is used for kernel bandwidth control, which is obtained by normalizing a new bandwidth parameter by the average number of associated miRNAs per disease.
(5)
Similarly, Gaussian interaction profile kernel similarity between miRNA m(i) and m(j) is constructed as follows:
(6)
(7)
Integrated similarity for miRNAs and diseases
Considering that miRNA functional similarity scores do not cover all the miRNAs, we integrate miRNA functional similarity scores and Gaussian interaction profile kernel similarity scores for miRNAs to calculate the new integrated similarity scores. That is to say, for the miRNA pair without known functional similarity score, we use Gaussian interaction profile kernel similarity score as integrated similarity; for the miRNA pair with known functional similarity score, we use the average value of Gaussian interaction profile kernel similarity score and functional similarity score as integrated similarity. Therefore, the integrated similarity between miRNA m(i) and m(j) is defined as follows:
(8)
Similarly, the integrated similarity between diseases d(u) and d(v) is defined as follows:
(9)
HGIMDA
We developed the computational model of HGIMDA by integrating miRNA functional similarity, disease semantic similarity, Gaussian interaction profile kernel similarity, and experimentally verified miRNA-disease associations to predict potential miRNA-disease associations. Based on the similar nature of miRNA-disease associations, miRNA similarity, disease similarity, and known miRNA-disease associations could be combined together to predict potential associations. For example, for disease d and miRNA m, we could define their potential association probability as follows if they have no known associations.
(10)
This equation means that we can infer potential association between disease d and miRNA m by summarizing all paths with the length equal to three. We consider the iteration of above procedure and represent the equation as matrix multiplications. Therefore, the iterative equation could be obtained as follows:
(11)
Here, α is a decay factor similar to the restart probability in the random walk with restart. According to previous literature [99], association probability matrix P will converge when SM and SD are properly normalized utilizing equation (12) and (13), respectively.
(12)
(13)
After some steps, the iteration is stable (the change between P(i) and P(i + 1) measured by L1 norm is less than a given cutoff, here we adopt the cutoff as 10−6).
ACKNOWLEDGMENTS
XC was supported by the National Natural Science Foundation of China under Grant No. 11301517. ZHY was supported by the National Natural Science Foundation of China under Grant No. 61572506. GYY was supported by the National Natural Science Foundation of China under Grant No. 11371355 and National Center for Mathematics and Interdisciplinary Sciences, CAS.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Ambros V. The functions of animal microRNAs. Nature. 2004; 431:350-355.
2. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281-297.
3. Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004; 431:343-349.
4. Ambros V. microRNAs: tiny regulators with great potential. Cell. 2001; 107:823-826.
5. Jopling CL, Yi MK, Lancaster AM, Lemon SM, Sarnow P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science. 2005; 309:1577-1581.
6. Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007; 318:1931-1934.
7. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. cell. 1993; 75:843-854.
8. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993; 75:855-862.
9. Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, Enright AJ. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006; 34:D140-D144.
10. Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011; 39:D152-D157.
11. Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ. miRBase: tools for microRNA genomics. Nucleic acids Res. 2008; 36:D154-D158.
12. Cheng AM, Byrom MW, Shelton J, Ford LP. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005; 33:1290-1297.
13. Karp X, Ambros V. Encountering microRNAs in cell fate signaling. Science. 2005; 310:1288-1289.
14. Miska EA. How microRNAs control cell division, differentiation and death. Curr Opin Genet Dev. 2005; 15:563-568.
15. Xu P, Guo M, Hay BA. MicroRNAs and the regulation of cell death. Trends Genet. 2004; 20:617-624.
16. Alshalalfa M, Alhajj R. Using context-specific effect of miRNAs to identify functional associations between miRNAs and gene signatures. BMC bioinformatics. 2013; 14:S1.
17. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009; 136:215-233.
18. Cui Q, Yu Z, Purisima EO, Wang E. Principles of microRNA regulation of a human cellular signaling network. Mol Syst Biol 2006; 2:46.
19. Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005; 132:4653-4662.
20. Meola N, Gennarino VA, Banfi S. microRNAs and genetic diseases. Pathogenetics. 2009; 2:7.
21. Lynam-Lennon N, Maher SG, Reynolds JV. The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc. 2009; 84:55-71.
22. Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006; 6:259-269.
23. Latronico MV, Catalucci D, Condorelli G. Emerging role of microRNAs in cardiovascular biology. Circ Res. 2007; 101:1225-1236.
24. Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, Cui Q. An analysis of human microRNA and disease associations. PloS one. 2008; 3:e3420.
25. Vandenboom Ii TG, Li Y, Philip PA, Sarkar FH. MicroRNA and cancer: tiny molecules with major implications. Curr Genomics. 2008; 9:97-109.
26. Gregory RI, Shiekhattar R. MicroRNA biogenesis and cancer. Cancer Res. 2005; 65:3509-3512.
27. DeSano JT, Xu L. MicroRNA regulation of cancer stem cells and therapeutic implications. AAPS J. 2009; 11:682-692.
28. Sassen S, Miska EA, Caldas C. MicroRNA—implications for cancer. Virchows Archiv. 2008; 452:1-10.
29. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E, Aldler H, Rattan S, Keating M, Rai K. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2002; 99:15524-15529.
30. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ. A microRNA polycistron as a potential human oncogene. Nature. 2005; 435:828-833.
31. Wang B, Wang H, Yang Z. MiR-122 inhibits cell proliferation and tumorigenesis of breast cancer by targeting IGF1R. PLoS One. 2012; 7:e47053.
32. Zhang Z, Zhang X, Newman K, Liu X. MicroRNA regulation of oncolytic adenovirus 6 for selective treatment of castration-resistant prostate cancer. Mol Cancer Ther. 2012; 11:2410-2418.
33. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6:857-866.
34. Chen X. Predicting lncRNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci Rep. 2015; 5:13186.
35. Chen X, Yan CC, Zhang X, Li Z, Deng L, Zhang Y, Dai Q. RBMMMDA: predicting multiple types of disease-microRNA associations. Sci Rep. 2015; 5:13877.
36. Chen X, Yan CC, Luo C, Ji W, Zhang Y, Dai Q. Constructing lncRNA functional similarity network based on lncRNA-disease associations and disease semantic similarity. Sci Rep. 2015; 5:11338.
37. Chen X, Yan GY. Novel human lncRNA–disease association inference based on lncRNA expression profiles. Bioinformatics. 2013; 29:2617-2624.
38. Chen X, Liu MX, Cui QH, Yan GY. Prediction of Disease-Related Interactions between MicroRNAs and Environmental Factors Based on a Semi-Supervised Classifier. PloS One. 2012; 7:e43425.
39. Chen X. KATZLDA: KATZ measure for the lncRNA-disease association prediction. Sci Rep. 2015; 5:16840.
40. Chen X, Yan CC, Zhang X, You Z-H, Deng L, Liu Y, Zhang Y, Dai Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Scientific reports. 2016; 6:21106
41. Huang Y, Chen X, You Z, Huang D, Chan K. ILNCSIM: improved lncRNA functional similarity calculation model. Oncotarget. 2016; 7:25902-25914. doi: 10.18632/oncotarget.8296.
42. Chen X. miREFRWR: a novel disease-related microRNA-environmental factor interactions prediction method. Mol Biosyst. 2016; 12:624-633.
43. Liu MX, Chen X, Chen G, Cui QH, Yan GY. A computational framework to infer human disease-associated long noncoding RNAs. PLoS One. 2014; 9:e84408.
44. Chen X, Huang Y, Wang X, You Z, Chan K. FMLNCSIM: fuzzy measure-based lncRNA functional similarity calculation model. Oncotarget. 2016. doi: 10.18632/oncotarget.10008.
45. Chen X, Yan CC, Zhang X, You ZH. Long non-coding RNAs and complex diseases: from experimental results to computational models. Briefings in Bioinformatics. 2016:bbw060.
46. Huang YA, You ZH, Chen X, Chan K, Luo X. Sequence-based prediction of protein-protein interactions using weighted sparse representation model combined with global encoding. BMC bioinformatics. 2016; 17:184.
47. Bandyopadhyay S, Mitra R, Maulik U, Zhang MQ. Development of the human cancer microRNA network. Silence. 2010; 1:6.
48. Wang D, Wang J, Lu M, Song F, Cui Q. Inferring the human microRNA functional similarity and functional network based on microRNA-associated diseases. Bioinformatics. 2010; 26:1644-1650.
49. Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007; 104:8685-8690.
50. Pasquier C, Gardès J. Prediction of miRNA-disease associations with a vector space model. Scientific Reports. 2016; 6:27036.
51. Le TD, Zhang J, Liu L, Li J. Computational methods for identifying miRNA sponge interactions. Briefings in bioinformatics. 2016:bbw042.
52. Jiang Q, Hao Y, Wang G, Juan L, Zhang T, Teng M, Liu Y, Wang Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst Biol. 2010; 4:S2.
53. Shi H, Xu J, Zhang G, Xu L, Li C, Wang L, Zhao Z, Jiang W, Guo Z, Li X. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst Biol. 2013; 7:101.
54. Mørk S, Pletscher-Frankild S, Caro AP, Gorodkin J, Jensen LJ. Protein-driven inference of miRNA–disease associations. Bioinformatics. 2013; 30:392-397.
55. Xu C, Ping Y, Li X, Zhao H, Wang L, Fan H, Xiao Y, Li X. Prioritizing candidate disease miRNAs by integrating phenotype associations of multiple diseases with matched miRNA and mRNA expression profiles. Mol BioSyst. 2014; 10:2800-2809.
56. Xuan P, Han K, Guo M, Guo Y, Li J, Ding J, Liu Y, Dai Q, Li J, Teng Z. Prediction of microRNAs Associated with Human Diseases Based on Weighted k Most Similar Neighbors. PLoS One. 2013; 8:e70204.
57. Chen X, Liu MX, Yan GY. RWRMDA: predicting novel human microRNA–disease associations. Mol Biosyst. 2012; 8:2792-2798.
58. Xu J, Li CX, Lv JY, Li YS, Xiao Y, Shao TT, Huo X, Li X, Zou Y, Han QL. Prioritizing candidate disease miRNAs by topological features in the miRNA target–dysregulated network: Case study of prostate cancer. Mol Cancer Ther 2011; 10:1857-1866.
59. Chen X, Yan GY. Semi-supervised learning for potential human microRNA-disease associations inference. Sci Rep 2014; 4:5501.
60. Li Y, Qiu C, Tu J, Geng B, Yang J, Jiang T, Cui Q. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014; 42:D1070-D1074.
61. Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009; 37:D98-D104.
62. Yang Z, Ren F, Liu C, He S, Sun G, Gao Q, Yao L, Zhang Y, Miao R, Cao Y. dbDEMC: a database of differentially expressed miRNAs in human cancers. BMC genomics. 2010; 11:S5.
63. Ogata-Kawata H, Izumiya M, Kurioka D, Honma Y, Yamada Y, Furuta K, Gunji T, Ohta H, Okamoto H, Sonoda H. Circulating exosomal microRNAs as biomarkers of colon cancer. PLoS One. 2014; 9:e92921.
64. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69-90.
65. Drusco A, Nuovo GJ, Zanesi N, Di Leva G, Pichiorri F, Volinia S, Fernandez C, Antenucci A, Costinean S, Bottoni A. MicroRNA profiles discriminate among colon cancer metastasis. PLoS One. 2014; 9:e966760.
66. Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005; 55:74-108.
67. Guo C, Sah JF, Beard L, Willson JK, Markowitz SD, Guda K. The noncoding RNA, miR-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer. 2008; 47:939-946.
68. Shi B, Sepp-Lorenzino L, Prisco M, Linsley P, Baserga R. Micro RNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J Biol Chem. 2007; 282:32582-32590.
69. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006; 103:2257-2261.
70. Arndt GM, Dossey L, Cullen LM, Lai A, Druker R, Eisbacher M, Zhang C, Tran N, Fan H, Retzlaff K. Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC cancer. 2009; 9:374.
71. Motoyama K, Inoue H, Takatsuno Y, Tanaka F, Mimori K, Uetake H, Sugihara K, Mori M. Over-and under-expressed microRNAs in human colorectal cancer. Int J Oncol. 2009; 34:1069-1075.
72. Asangani I, Rasheed S, Nikolova D, Leupold J, Colburn N, Post S, Allgayer H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008; 27:2128-2136.
73. Schetter AJ, Leung SY, Sohn JJ, Zanetti KA, Bowman ED, Yanaihara N, Yuen ST, Chan TL, Kwong DL, Au GK. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. JAMA. 2008; 299:425-436.
74. He B, Yin B, Wang B, Xia Z, Chen C, Tang J. microRNAs in esophageal cancer. [Review]. Mol Med Rep. 2012; 6:459-465.
75. Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003; 349:2241-2252.
76. Kim T, Grobmyer SR, Smith R, Ben-David K, Ang D, Vogel SB, Hochwald SN. Esophageal cancer—the five year survivors. J Surg Oncol. 2011; 103:179-183.
77. Daly JM, Fry WA, Little AG, Winchester DP, McKee RF, Stewart AK, Fremgen AM. Esophageal cancer: results of an American College of Surgeons patient care evaluation study. J Am Coll Surg. 2000; 190:562-572.
78. Xie Z, Chen G, Zhang X, Li D, Huang J, Yang C, Zhang P, Qin Y, Duan Y, Gong B. Salivary microRNAs as promising biomarkers for detection of esophageal cancer. PLoS One 2013; 8:e57502.
79. Wan J, Wu W, Che Y, Kang N, Zhang R. Insights into the potential use of microRNAs as a novel class of biomarkers in esophageal cancer. Dis Esophagus. 2015.
80. Huang SD, Yuan Y, Zhuang CW, Li BL, Gong DJ, Wang SG, Zeng ZY, Cheng HZ. MicroRNA-98 and microRNA-214 post-transcriptionally regulate enhancer of zeste homolog 2 and inhibit migration and invasion in human esophageal squamous cell carcinoma. Mol Cancer. 2012; 11:51.
81. Jemal A, Siegel R, Ward E, Murray T, Xu J, Smigal C, Thun MJ. Cancer statistics, 2006. CA Cancer J Clin. 2006; 56:106-130.
82. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012; 62:10-29.
83. Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009; 373:1119-1132.
84. Linehan WM. Genetic basis of kidney cancer: role of genomics for the development of disease-based therapeutics. Genome Res. 2012; 22:2089-2100.
85. Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol. 2003; 170:2163-2172.
86. Linehan WM, Zbar B. Focus on kidney cancer. Cancer cell. 2004; 6:223-228.
87. Zambrano NR, Lubensky IA, Merino MJ, Linehan WM, Walther MM. Histopathology and molecular genetics of renal tumors: toward unification of a classification system. J Urol. 1999; 162:1246-1258.
88. Kovacs G, Akhtar M, Beckwith BJ, Bugert P, Cooper CS, Delahunt B, Eble JN, Fleming S, Ljungberg B, Medeiros LJ. The Heidelberg classification of renal cell tumours. J Pathol. 1997; 183:131-133.
89. Thoenes W, Rumpelt H. Histopathology and classification of renal cell tumors (adenomas, oncocytomas and carcinomas): the basic cytological and histopathological elements and their use for diagnostics. Pathol Res Pract. 1986; 181:125-143.
90. Kovacs G. Molecular differential pathology of renal cell tumours. Histopathology. 1993; 22:1-8.
91. Gottardo F, Liu CG, Ferracin M, Calin GA, Fassan M, Bassi P, Sevignani C, Byrne D, Negrini M, Pagano F. Micro-RNA profiling in kidney and bladder cancers. Urol Oncol. 2007; 25:387-392.
92. Senanayake U, Das S, Vesely P, Alzoughbi W, Fröhlich LF, Chowdhury P, Leuschner I, Hoefler G, Guertl B. miR-192, miR-194, miR-215, miR-200c and miR-141 are downregulated and their common target ACVR2B is strongly expressed in renal childhood neoplasms. Carcinogenesis. 2012; 33:1014-1021.
93. Zaman MS, Shahryari V, Deng G, Thamminana S, Saini S, Majid S, Chang I, Hirata H, Ueno K, Yamamura S. Up-regulation of microRNA-21 correlates with lower kidney cancer survival. PLoS One. 2012; 7:e31060.
94. Tsz-fung FC, Youssef YM, Lianidou E, Romaschin AD, Honey RJ, Stewart R, Pace KT, Yousef GM. Differential expression profiling of microRNAs and their potential involvement in renal cell carcinoma pathogenesis. Clin Biochem. 2010; 43:150-158.
95. Liu Y, Tennant DA, Zhu Z, Heath JK, Yao X, He S. DiME: a scalable disease module identification algorithm with application to glioma progression. PloS one. 2014; 9:e86693.
96. Wong L, You ZH, Ming Z, Li J, Chen X, Huang YA. Detection of interactions between proteins through rotation forest and local phase quantization descriptors. International journal of molecular sciences. 2016; 17:21.
97. You ZH, Zhu L, Zheng CH, Yu HJ, Deng SP, Ji Z. Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC bioinformatics. 2014; 15:S9.
98. Lipscomb CE. Medical subject headings (MeSH). Bull Med Libr Assoc. 2000; 88:265-266.
99. Wang W, Yang S, Li J. Drug target predictions based on heterogeneous graph inference. Pac Symp Biocomput. 2013:53-64.