
INTRODUCTION
Lung cancer remains the leading cause of cancer-related mortality in the United States and worldwide with an overall 5 year survival rate of 19.3% [1]. Adenocarcinoma is the most common form of lung cancer and accounts for 38% of all newly diagnosed lung cancers [2]. Recent advancements in targeted therapies such as EGFR tyrosine kinase inhibitors and ALK inhibitors have led to modest improvements in survival times for certain subgroups of patients with lung adenocarcinoma, however, additional therapeutic strategies are desperately needed [3, 4]. Once patients have developed metastatic disease only 15% will be alive after one year and there are virtually no longer term survivors [5], underlying the importance in developing treatment strategies which target the mechanisms leading to tumor invasion and metastasis.
Epithelial-mesenchymal transition (EMT) is a process in which cells lose their cell-cell adhesive properties and gain migratory and invasive potential. EMT is essential for events in embryonic development, wound healing, fibrosis, and in cancer progression and metastasis. It has been well established that epithelial cancer cells undergo EMT for tumor progression and in order to invade local tissue and metastasize [6, 7]. EMT process is regulated by various signaling pathways including Sonic Hedgehog (SHh) which lead to many complex changes in the expression and function of proteins. The loss of the cell-to-cell adhesive protein E-Cadherin, which is inactivated in many cancers, is a fundamental event in EMT. During EMT, E-Cadherin is replaced with N-Cadherin, which enhances the motility of tumor cells [8]. EMT confers on cells the critical traits required for seeding metastasis and for developing the stem cell properties that allow for launching of new cancer cell colonies [9]. Acquisition of EMT features has been associated with poor prognosis as well as resistance to chemotherapy [10–12]. Further knowledge of the process of EMT can improve our understanding of tumor recurrence and metastasis and identify potential therapeutic targets.
The role of SHh pathway in cancer development is suspected to be via the transformation of adult stem cells into cancer stem cells. Activated SHh has been implicated in tumorigenesis and metastasis in multiple types of cancers including lung, brain, breast, prostate, and skin. In the canonical SHh pathway, the absence of the SHh ligand leads the transmembrane receptor Patched (Ptch) to inhibit the transmembrane receptor Smoothened (Smo). Inhibited Smo causes cleavage of Gli to the N-terminal repressor form. Therefore when SHh binds to Ptch, the inhibitory effect on Smo is released and active full length Gli is transported into the nucleus and activates transcription of Gli-dependent target genes such as Gli1, Ptch1, CyclinD1 and Wnt [13–16]. However, non-canonical Gli activation independent of SHh, has been shown in many cancer cells types [17, 18], and there is evidence for mechanisms of Gli activation independent of SHh, stimulated by other oncogenic signaling pathways such as transforming growth factor β (TGFβ), epidermal growth factor receptor (EGFR), RAS and AKT/PI3K pathways [19–23]. As Gli transcription factors constitute the final effectors of the SHh pathway, and are implicated in multiple other oncogenic signaling pathways, they represent an important downstream target for potential cancer therapeutics [17].
The relationship of SHh pathway to EMT has not been previously studied in lung adenocarcinomas and the existing data from other solid tumors is controversial. There is a growing body of literature that shows that SHh/Gli inhibition blocks EMT, however the exact mechanisms remain to be elucidated. Some studies in melanoma and pancreatic cancers have suggested that Gli facilitates cancer cell migration and invasion via E-Cadherin [24, 25]. In lung squamous cell cancer (SCC) and in hepatocellular carcinoma, Gli expression has been shown to be inversely correlated with E-Cadherin expression and in lung SCC inhibition of the SHh pathway increases E-Cadherin expression [26, 27]. In hepatocellular cancer, Gli1 over-expression is correlated with capsular invasion, advanced tumor stage, vascular invasion and intrahepatic metastasis and interfering with Gli transcription suppresses cell migration by down-regulating matrix metalloprotease (MMP)-2 and MMP-9 [28]. In vitro down-regulation of Gli1 with siRNA reduced hepatoceullular cancer cell invasion and increased E-Cadherin expression [27]. However there is some conflicting data that showed inhibition of Gli promoted EMT in pancreatic cancer [29]. We have recently demonstrated increased SHh signaling in lung SCC and that Gli1 expression is inversely correlated with the EMT marker E-Cadherin. Inhibition of the SHh pathway up-regulates E-Cadherin expression and suppresses lung SCC cell migration [26].
Here, we report the Gli activation in two cohorts of patients with lung adenocarcinomas and show that Gli1 and EMT markers are inversely correlated in lung adenocarcinoma. Inhibition of Gli suppresses migration of lung adenocarcinoma cells and up-regulates E-Cadherin expression in vitro. We also demonstrate a reduction in lung adenocarcinoma growth in vivo by a small molecule Gli inhibitor.
RESULTS
Gli expression inversely correlates with E-Cadherin expression in lung adenocarcinoma
We investigated the expression of Gli proteins and E-Cadherin in lung adenocarcinoma patient tissues from the Lung Cancer Center at Tianjin Medical University Cancer Institute and Hospital, Tianjin and the Thoracic Oncology Program at University of California, San Francisco. The expression of Gli1, Gli2 and E-Cadherin was evaluated by immunohistochemistry (IHC) with 68 formalin-fixed, paraffin-embedded tissue specimens from the Tianjin cohort. Clinical and demographic information from the Tianjin cohort is summarized in Table 1. Tumor samples with high Gli1 or Gli2 expression showed lower E-Cadherin expression while low Gli expression showed high expression with an epithelial growth pattern (Figure 1A). The protein expressions of Gli1, Gli2, and E-Cadherin were scored a “high” or “low” expression based on IHC as previously described [26]. Statistical analysis with Kendall’s tau-b correlation tests revealed that both Gli1 and Gli2 were significantly inversely correlated with E-Cadherin expression (p-value 0.01 and 0.03 respectively) (Figure 1B). In addition, we found E-Cadherin exhibited a heterogeneous pattern in some tissue samples, and Gli expression seemed inversely correlated with E-Cadherin within the same specimen. To confirm the inverse correlation between Gli and E-Cadherin, we conducted co-immunofluorescence of Gli2 and E-Cad on 17 tissue samples from UCSF, and 13 of these 17 (76%) samples exhibited a clear inverse correlation between Gli2 and E-Cadherin. Four of 17 (24%) samples exhibited heterogeneous expression patterns of Gli2 and E-Cadherin. Within one representative sample (Figure 1C), high Gli2 areas showed low E-Cadherin expression whereas low Gli2 areas showed high E-Cadherin expression with a clear epithelial pattern, further verifying the inverse correlation of E-Cadherin and Gli. No available combination of antibodies allowed us to perform co-staining of Gli1 and E-Cadherin; however, we demonstrated that Gli1 and Gli2 expression correlated significantly in lung adenocarcinoma by IHC on FFPE samples of the Tianjin cohort (Figure 1B) and by RT-PCR on 63 freshly frozen samples of the UCSF cohort (Figure 1D). Consistent with the tissue expression analysis, Gli expression negatively correlated with E-Cadherin expression in nine human lung adenocarcinoma cell lines assessed by Western blotting (Figure 1E). Taken together, our results demonstrated Gli expression inversely correlates with E-Cadherin expression in lung adenocarcinoma.
Table 1: Patient Information
Characteristics |
Number |
Percent |
|---|---|---|
Age (Years) |
||
<60 |
44 |
64.70% |
≥60 |
24 |
35.30% |
Gender |
||
Male |
40 |
85.30% |
Female |
28 |
14.70% |
Smoking history |
||
Never |
32 |
47.10% |
Smoker |
36 |
52.90% |
Surgical Procedure |
||
Lobectomy |
60 |
88.20% |
Pneumonectomy |
8 |
11.80% |
T stage |
||
T1 |
23 |
7.90% |
T2 |
39 |
57.40% |
T3 |
6 |
8.80% |
Postoperative chemotherapy |
||
Yes |
44 |
64.70% |
No |
24 |
35.30% |
Postoperative radiotherapy |
||
Yes |
10 |
14.70% |
No |
58 |
85.30% |
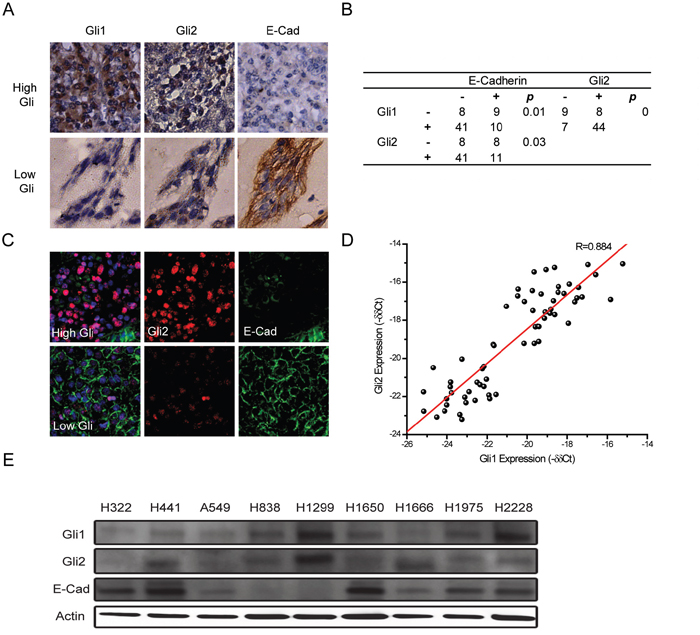
Figure 1: Gli expression inversely correlates with E-Cadherin expression in lung adenocarcinoma. A. Expression of Gli1, Gli2 and E-Cadherin (E-Cad) in two representative tissue specimens assessed by immunohistochemistry in the Tianjin cohort showing high Gli expression (upper panels) and low Gli expression (lower panels). B. Correlation between Gli1, Gli2 and E-Cadherin in the Tianjin cohort. Statistical analyses were performed between Gli1 and E-Cad, and also Gli2 and E-Cad. C. Expression of Gli2 (red) and E-Cadherin (E-Cad) (green) in two representative tissue specimens was analyzed by immunofluorescence in the UCSF cohort with high Gli expression (upper panels) and low Gli expression (lower panels). DAPI (blue) was used to stain the nuclei of cells. D. Correlation between Gli1 and Gli2 mRNA levels by quantitative RT-PCR in the UCSF cohort. E. Gli1, Gli2 and E-Cad protein expression in human lung adenocarcinoma cell lines assessed by Western blotting. Actin was used as a loading control.
SHh/Gli signaling promotes cell migration and invasion
Reduced levels of E-Cadherin have been shown to promote cancer cell mobility and invasive capability. To elucidate whether SHh/Gli signaling enhances cell migration, we carried out a wound-healing assay in two lung adenocarcinoma cell lines A549 and H1666. To inhibit SHh/Gli signaling, we applied Gli-I and Vismodegib (also known as GDC-0449). Our lab has previously developed Gli-I, a small molecule inhibitor targeting Gli, and showed it effectively inhibited cell proliferation both in vitro and in vivo by interfering Gli transcriptional activity [30, 31]. Vismodegib is a Smo inhibitor approved by the U.S. Food and Drug Administration to treat adult patients with basal cell carcinoma [32–35]. It is currently being investigated in clinical trials to treat other types of cancer due to its ability to selectively target SHh signaling [32, 36]. To stimulate the pathway, we treated cells with a recombinant SHh protein.
Our results illustrated that down-regulation of SHh/Gli at different points in the signaling pathway with either Gli-I or Vismodegib reduced cell mobility significantly in both cell lines, while up-regulation of the pathway enhanced cell migration. Addition of Gli-I significantly reduced cell migration in A549 (p<0.01) and in H1666 (p<0.001). Vismodegib similarly reduced cell migration in A549 (p<0.01) and in H1666 (p<0.05) (Figure 2). To evaluate if SHh/Gli signaling promotes cell invasiveness, we performed in vitro Matrigel 3D invasion assays on A549 with Gli-I, Vismodegib and SHh treatment, and observed cell invasion on days 1, 3 and 6. The inhibition of SHh/Gli signaling significantly suppressed the invasive capability of cells, while SHh stimuli induced dramatic cell invasion. Quantification was carried out by measuring the distance between the invasive cell frontier and spheroid edge. The addition of SHh recombinant proteins significantly increased cell invasion at Day 3 (p<0.001) and Day 6 (p<.001). In contrast, use of the Smo inhibitor, Vismodegib significantly reduced cell invasion on Day 3 (p<0.01) and Day 6 (p<0.001) and similarly use of a novel Gli inhibitor significantly reduced cell invasion on Day 3 (p<0.05) and Day 6 (p<0.001) (Figure 3). Our results strongly suggest that SHh/Gli signaling promotes cell migration and invasion in lung adenocarcinoma and that inhibitors of this pathway that target Gli or Smo significantly suppress this invasion in vitro.
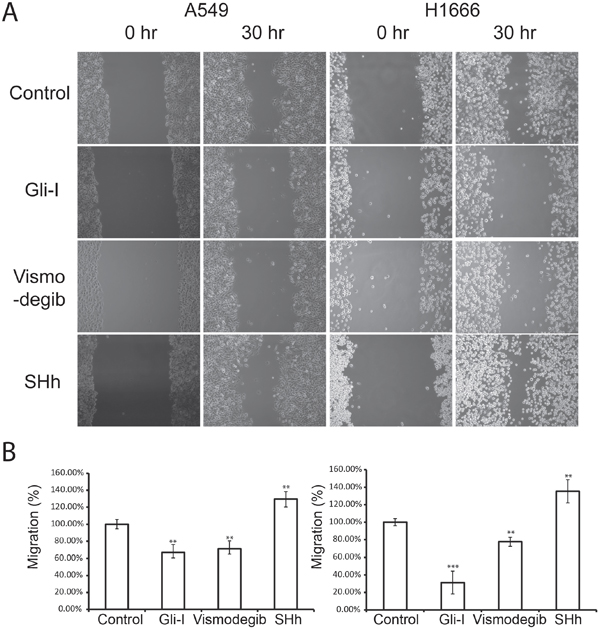
Figure 2: SHh/Gli signaling promotes cell migration in lung adenocarcinoma. A. Wound healing assays of lung adenocarcinoma A549 cells (left) and H1666 (right) treated with Gli-I, Vismodegib, and recombinant SHh proteins. Representative images shown at 0 hr and 30 hr were captured using a light microscope (100X). B. Quantification of wound healing assays. The migration distance of cells at 0 hr was set as 100%. Statistical analysis was performed between control and treated groups. p values of <0.05 <0.01 or <0.001 are indicated as *, ** or *** respectively.
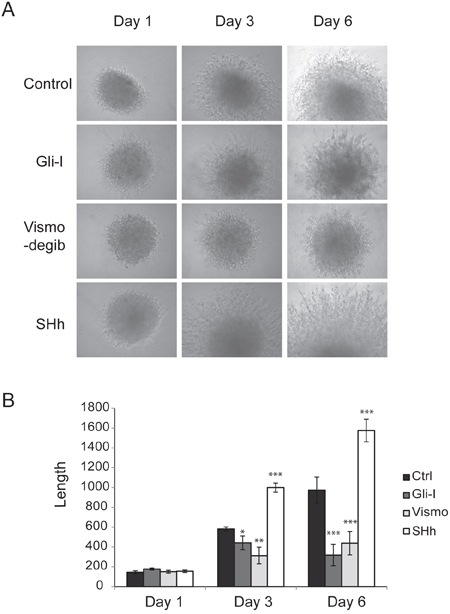
Figure 3: SHh/Gli signaling enhances cell invasion. A. 3D spheroid cell invasion assay of lung adenocarcinoma A549 cells treated with Gli-I, Vismodegib, and recombinant SHh proteins. Representative images shown at day 1, day 3 and day 6 were captured using a light microscope (100X). B. Quantification of 3D spheroid cell invasion assays. Quantification was carried out by measuring the distance between the invasive cell frontier and spheroid edge. Statistical analysis was performed between control and treated groups. p-values of <0.05 <0.01 or <0.001 were indicated as *, ** or *** respectively.
Inhibition of SHh/Gli signaling restores E-Cadherin expression in vitro
To determine whether E-Cadherin expression is regulated by the SHh/Gli pathway, we examined E-Cadherin expression upon Gli-I, Vismodegib and SHh treatment in cell lines A549 and H1666. E-Cadherin expression was restored and enhanced upon SHh inhibition with Vismodegib or the Gli inhibitor, while E-Cadherin expression was decreased upon SHh pathway stimulation with SHh recombinant proteins (Figure 4).
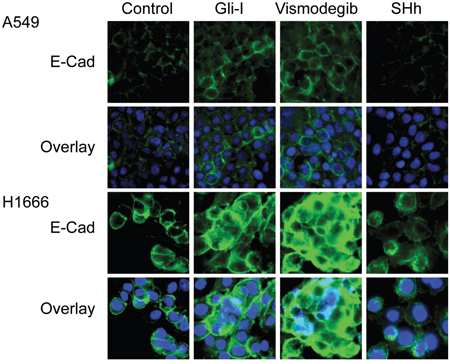
Figure 4: SHh/Gli signaling reduces E-Cadherin expression. Immunofluorescent staining of E-Cadherin (green) in lung adenocarcinoma A549 and H1666 cells treated with Gli-I, Vismodegib, and recombinant SHh proteins. DAPI (blue) was used to stain the nuclei cells.
In vivo inhibition of SHh/Gli reduces tumor growth and restores E-Cadherin expression
We further examined whether inhibition of SHh/Gli signaling could restore E-Cadherin expression, by conducting in vivo analyses in a xenograft lung tumor model of A549 cells. Ten days after implantation of A549 cells, mice received daily intraperitoneal injections of Gli-I at 25 mg/kg for 14 days, and another round of treatment for 10 days after an interval of 10 days. Gli-I treatment significantly inhibited tumor growth by 69% (p<0.001) at the end of the second round of treatment compared with the control group (Figure 5A). Neither an obvious change of body weight nor any noticeable toxicity in major organs form the treated mice were observed during the course of the treatments (data not shown). We examined the resected xenograft tumor specimens after the completion of the in vivo experiment by immunohistochemistry, and two representative samples from each group are shown in Figure 5B. Gli1 and Gli2 proteins were down-regulated in Gli-I-treated tumors compared with the control group. A decrease in Ki-67 levels confirmed the reduction of proliferation in Gli-I-treated tumors. Consistent with our in vitro data in cultured cells, E-Cadherin expression was induced, and then restored with a typical cell-cell junction pattern (Figure 5B).
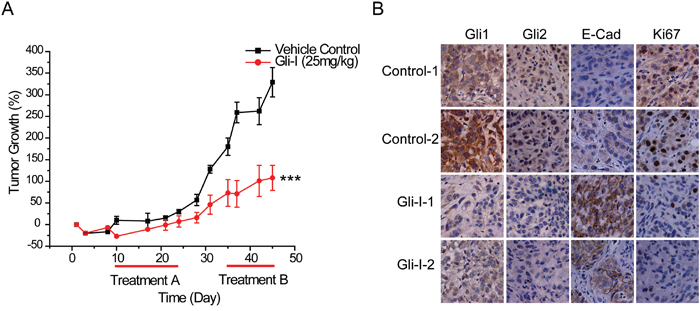
Figure 5: Inhibition of SHh/Gli signaling restores E-Cadherin expression in vivo. A. A549 xenograft tumor growth post Gli-I treatment. Treatment was performed from day 10 for 14 days (Treatment A), followed by a 10-day interval, and a second 10-day treatment (Treatment B). Tumor size was measured every 3-4 days, and was calculated by using the equation x2y (where x<y), and presented as percentage of its initial volume on day 1. Statistical analysis was performed between control and the treated group at the end of the treatment. A p value of <0.001 was indicated as ***. B. Gli1, Gli2, E-Cad and Ki67 expression in the resected xenograft tumor assessed by immunohistochemistry in two representative samples in control and treated groups respectively. Mice treated with Gli-I show decreased Gli expression, increased E-Cadherin expression, and decreased Ki67 expression.
Our results demonstrate that inhibition of SHh/Gli signaling have the capability to restore E-Cadherin expression both in vitro and in vivo, indicating that SHh/Gli signaling regulates EMT in lung adenocarcinoma through suppression of E-Cadherin.
DISCUSSION
Our study is the first to demonstrate that activation of the SHh/Gli pathway is associated with changes in expression of EMT markers in lung adenocarcinoma. We are also the first to show the effect of SHh/Gli pathway on inhibition of lung adenocarcinoma cell migration in vitro and reduction of tumor growth using a Gli inhibitor in vitro.
The SHh/Gli pathway is known to play an important role in many solid malignancies. The downstream effects of the SHh pathway are involved with EMT and the cellular players that orchestrate the process are potential drug targets to prevent cell invasion and metastasis. Vismodegib is a Smo inhibitor that prevents the Smo transmembrane protein from transducing the SHh signal to the nucleus, thereby preventing the activation of SHh target genes. Vismodegib is approved for clinical use in patients with basal cell carcinoma and is in trials to expand its use to other cancers [32, 36]. Inhibition of Gli may be more effective than inhibition of Smo in preventing downstream SHh gene activation given evidence for non-canonical Gli activation independent of SHh in cancer cells and evidence that Gli is a target of other oncogenic signaling pathways [17–22]. Here we provide strong preliminary evidence that in vitro and in vivo Gli inhibition impairs the migratory and invasive properties of lung adenocarcinoma cells by inhibition of EMT.
In this study we show that lung adenocarcinoma specimens from two cohorts of patients, Gli1 and Gli2 are inversely correlated with the cell adhesions protein E-Cadherin, suggesting that high levels of Gli are associated with markers of EMT. We further demonstrate that the addition of either a Smo inhibitor or a Gli inhibitor significantly reduced migration of lung adenocarcinoma cells in vitro, whereas the addition of SHh ligand significantly increased their migration. These findings are reproduced in a 3D cell invasion simulation assay. To elucidate potential mechanisms responsible for these declines in cell migration and invasion with inhibition of the SHh pathway, we examined expression levels of the EMT marker E-Cadherin. When the SHh pathway was inhibited with Smo or Gli inhibitors E-Cadherin expression was enhanced and when the SHh pathway was stimulated with SHh recombinant proteins, E-Cadherin expression declined.
The regulation of EMT is a sophisticated process. We speculate that SHh signaling might regulate EMT at different levels. It has been suggested that the transcriptional targets of SHh signaling includes key genes that are involved in the EMT process, such as snail1 [37, 38]. The cross-talks between SHh and TGF-beta signaling may play an important role in the regulation [37, 38]. Besides, the possible protein-level interaction between Gli proteins and beta-catenin at cell-cell junctions might also contribute to the regulation [39]. The molecular mechanism how the SHh pathway regulates EMT in lung adenocarcinoma remains to be further explored.
Most importantly, we demonstrated the successful use of a Gli inhibitor in vivo to suppress the growth of lung adenocarcinoma in a xenograft model without any toxicity and in these tumors showed histologically that Gli inhibition led to decreased Gli expression, increased E-Cadherin, and decreased markers of cellular proliferation.
While Vismodegib has shown promise at reducing tumor size and lengthening progression free survival times [34], resistance mechanisms have already been identified. A Smo mutation, D477G, that prevents Vismodegib from binding to Smo was identified in a medulloblastoma patient who had relapsed after an initial response [40], and another Smo mutation, E518K, has also been identified in humans [41]. Research is underway into overcoming these resistance mechanisms with second generation Smo inhibitors such as Itraconazole, which interacts with Smo at a different binding site, and arsenic trioxide which promotes the degradation of Gli2 transcription factor before it can translocation to the nucleus and affect gene expression. Itraconazole and arsenic trioxide both inhibit the growth of medulloblastoma and basal cell carcinoma in vitro and in vivo, and prolong survival of mice with intracranial drug-resistant Smo [42]. Inhibiting the SHh pathway with a Gli inhibitor is favored due to avoidance of potential upstream resistance mechanisms that have posed a challenge to the currently approved Smo inhibitor. In addition, direct Gli inhibition can block both canonical and non-canonical activation of Gli, thereby preventing gene transcription that is a target of multiple oncogenic pathways. While further research is still required, Gli inhibition is a promising area for further investigation and potential drug development.
MATERIALS AND METHODS
Tissue specimens
Tissue specimens of the UCSF cohort were collected from 17 patients who underwent surgical resection for lung adenocarcinoma at the Thoracic Oncology Program at UCSF. Tissue specimens of the Tianjin cohort were collected from 68 patients who underwent surgical resection for lung adenocarcinoma at the Tianjin Medical University Cancer Institute and Hospital. Samples were fixed in formalin and embedded in paraffin to make tissue slides. The study with UCSF patient tissues was approved by the Committee on Human Research (CHR approval number: H8714-11647-10) at the University of California, San Francisco (UCSF), and that with Tianjin cohort was approved by the Tianjin Medical University Cancer Institute and Hospital. Written, informed consent was obtained from each patient before specimen collection.
Immunohistochemistry, immunofluorescence and western blot
Immunohistochemistry (IHC), immunofluorescence (IF) and western blot were performed following standard procedures. Antibodies applied to detect protein expressions in IHC and IF were Gli1 1:100 (sc-20687 Santa Cruz Biotechnology, Santa Cruz, CA), Gli2 1:150 (Abcam, Cambridge, MA), E-Cadherin 1:100 (EMD Millipore, Billerica, MA), and the cell proliferation marker Ki67 1:200 (Abcam, Cambridge, MA). Antibodies used in Western blot were Gli1 1:100 (sc-20687 Santa Cruz Biotechnology), Gli2 1:150 (Abcam, Cambridge, MA), E-Cadherin 1:100 (EMD Millipore), and Actin 1:500 (Sigma-Aldrich, St. Louis, MO). Total protein extraction was performed with M-PER Mammalian Protein Extraction Solution (Thermo Scientific, Waltham, MA), and 40 μg of protein extracts were analyzed by Western blotting.
RNA extraction and RT-PCR
Total RNA was isolated from tissue or cultured cells using an RNeasy kit (Qiagen, Germany). Genomic DNA contamination was eliminated by DNase I treatment. Reverse transcription was conducted with 250 ng RNA using the iScript cDNA synthesis kit (Bio-Rad, Herculas, CA). The resulting cDNAs were analyzed with real-time RT-PCR using Gene Expression Assays in a 7900 Real-Time PCR System (Applied Biosystems, Foster City, CA) for 40 cycles (96°C for 15 seconds and 60°C for 1 minute). Gene expression was normalized to 18S expression. Experiments were performed in triplicate and three times.
Cell culture, drug treatment, migration assay and 3D culture
Human lung adenocarcinoma cell lines H322, H441, H838, H1299, H1650, H1666, H1975, and H2228 were purchased from American Type Culture Collection (Manassas, VA). The cell lines were cultured in RPMI 1640 (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and antibiotics. Cells were seeded one day before treatment with Gli-I and Vismodegib (Selleck) at different concentrations and SHh recombinant proteins (eBioscience) were incubated for 30 hours, with vehicle (DMSO) as controls. Cells were then subjected to the following analyses of immunofluorescence, migration assay and Cultrex® 3D Spheroid Cell Invasion Assay (Trevigen, Gaithersburg, MD). In migration assays, four wounds were made in each condition, and cell migration was presented by the average of distance differences between 30 hours and 0 hours. In 3D Spheroid Cell Invasion Assay, distances between the invasive cell frontier and spheroid edge were measured on day 1, day 3 and day 6. In order to observe the restoration of E-Cadherin, the cells were incubated at different conditions for 24 hours before immunofluorescence. All experiments have been conducted more than three times, and representative results were included in the text.
Animal studies
Nude mice were subcutaneously injected with 10 million A549 cells with BD Matrigel Matrix (BD Biosciences, San Jose, CA). Ten days after inoculation, mice were randomized and intraperitoneally injected with either Gli-I at 25 mg/kg (n=10) or vehicle alone (n=10) for 14 consecutive days, followed by a 10-day observation and a 10-day Gli-I treatment regimen. Tumor volumes were measured every 3-4 days from day 1 to day 44 post-inoculation. Growth was calculated using the equation x2y (where x and y were the largest width and length dimensions and x<y), and presented as a percentage of the initial volume on day 1. Tumors and organs were dissected on day 44 to make paraffin blocks for follow-up IHC analyses. All experiments were performed at the University of California, San Francisco Preclinical Core. The experimental protocol was approved by and performed in accordance with guidelines from the Institutional Animal Care and Use Committee at University of California, San Francisco.
Statistical analysis
A Kappa test was used for correlation analysis between the expressions of SHh pathway components Gli1 and Gli2 and EMT maker E-Cadherin. IHC scores of 1-3 were grouped as positive “+”, and 0 was grouped as “-“for dichotomous analysis. Two-sided student’s t-test was performed for proliferation assays, migration assays, and 3D spheroid cell invasion assays analyses. A p value <0.05 was indicated as *, 0.01 as **, and 0.001 as *** in corresponding figures. Data analysis was performed using SPSS 17.0 software.
ACKNOWLEDGMENTS
The authors acknowledge the important contributions of the other members of the University of California San Francisco Thoracic Oncology Program and Biao He’s research team including Fleur Leguay, BS; Ngoc Hoang, BS; Luis Acevedo, BS.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by NIH/NCI R01CA125030, and the Eileen D. Ludwig Endowed for Thoracic Oncology Research (to B. He); The Bonnie J. Addario Lung Cancer Foundation, the Kazan, McClain, Abrams, Fernandez, Lyons, Greenwood, Harley & Oberman Foundation, the Ziegelmam Family Foundation, and the Barbara Isackson Lung Cancer Research Fund (to D.M. Jablons); The Key Program for Anti-cancer Research of Tianjin Municipal Science and Technology Commission (12ZCDZSY15400) (to C.L. Wang); the Science & Technology Support Project of Hebei Province (132077127D) (to L. Wang); and the National Natural Science Foundation of China (No. 81501983) (to D.S. Yue).
REFERENCES
1. Howlader N NA, Krapcho M, Garshell J, Neyman N, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Cho H, Mariotto A, Lewis DR, Chen HS, et al. SEER Cancer Statistics Review, 1975-2010, National Cancer Institute. Bethesda, MD, http://seer.cancer.gov/csr/1975_2010/, based on November 2012 SEER data submission, posted to the SEER web site. 2013.
2. Travis WD. Pathology of lung cancer. Clin Chest Med. 2011; 32:669-692.
3. Yang X, Yang K, Kuang K. The efficacy and safety of EGFR inhibitor monotherapy in non-small cell lung cancer: a systematic review. Curr Oncol Rep. 2014; 16:390.
4. Rossi A, Maione P, Sacco PC, Sgambato A, Casaluce F, Ferrara ML, Palazzolo G, Ciardiello F, Gridelli C. ALK inhibitors and advanced non-small cell lung cancer (review). Int J Oncol. 2014; 45:499-508.
5. Groome PA, Bolejack V, Crowley JJ, Kennedy C, Krasnik M, Sobin LH, Goldstraw P. The IASLC Lung Cancer Staging Project: validation of the proposals for revision of the T, N, and M descriptors and consequent stage groupings in the forthcoming (seventh) edition of the TNM classification of malignant tumours. J Thorac Oncol. 2007; 2:694-705.
6. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer. 2013; 13:97-110.
7. Hong KO, Kim JH, Hong JS, Yoon HJ, Lee JI, Hong SP, Hong SD. Inhibition of Akt activity induces the mesenchymal-to-epithelial reverting transition with restoring E-cadherin expression in KB and KOSCC-25B oral squamous cell carcinoma cells. J Exp Clin Cancer Res. 2009; 28:28.
8. Voulgari A, Pintzas A. Epithelial-mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim Biophys Acta. 2009; 1796:75-90.
9. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011; 331:1559-1564.
10. Soltermann A, Tischler V, Arbogast S, Braun J, Probst-Hensch N, Weder W, Moch H, Kristiansen G. Prognostic Significance of Epithelial-Mesenchymal and Mesenchymal-Epithelial Transition Protein Expression in Non-Small Cell Lung Cancer. Clin Cancer Res. 2008; 14:7430-7437.
11. Vendrell JA, Thollet A, Nguyen NT, Ghayad SE, Vinot S, Bieche I, Grisard E, Josserand V, Coll JL, Roux P, Corbo L, Treilleux I, Rimokh R et al. ZNF217 Is a Marker of Poor Prognosis in Breast Cancer That Drives Epithelial-Mesenchymal Transition and Invasion. Cancer Res. 2012; 72:3593-3606.
12. Van den Broeck A, Vankelecom H, Van Eijsden R, Govaere O, Topal B. Molecular markers associated with outcome and metastasis in human pancreatic cancer. J Exp Clin Cancer Res. 2012; 31.
13. Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005; 6:306-317.
14. Huangfu D, Anderson KV. Signaling from Smo to Ci/Gli: Hedgehog pathways from Drosophila to vertebrates. Development. 2006; 133:3-14.
15. Ruiz i Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007; 17:438-447.
16. Mullor JL, Dahmane N, Sun T, Ruiz i Altaba A. Wnt signals are targets and mediators of Gli function. Curr Biol. 2001; 1:769-773.
17. Lauth M, Toftgard R. Non-canonical activation of GLI transcription factors. Cell Cycle. 2007; 6:2458-2463.
18. Mimeault M, Batra SK. Frequent Deregulations in the Hedgehog Signaling Network and Cross-Talks with the Epidermal Growth Factor Receptor Pathway Involved in Cancer Progression and Targeted Therapies. Pharmacol Rev. 2010; 62:497-524.
19. Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009; 19:71-88.
20. Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009; 69:1284-1292.
21. Pasca di Magliano M, Sekine S, Ermilov A, Ferris J, Dlugosz AA, Hebrok M. Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 2006; 20:3161-3173.
22. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V. Melanomas require HEDGEHOG-GLI signaling reaulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci USA. 2007; 104:5895-5900.
23. Sharma N, Nanta R, Sharma J, Gunewardena S, Singh KP, Shankar S, Srivastava RK. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget. 2015; 6:32039-32060. doi: 10.18632/oncotarget.5055.
24. Alexaki VI, Javelaud D, Van Kempen LCL, Mohammad KS, Dennler S, Luciani F, Hoek KS, Juarez P, Goydos JS, Fournier PJ, Sibon C, Bertolotto C, Verrecchia F, et al. GLI2-Mediated Melanoma Invasion and Metastasis. J Natl Cancer Inst. 2010; 102:1148-1159.
25. Inaguma S, Kasai K, Ikeda H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene. 2012; 30:714-723.
26. Yue D, Li H, Che J, Zhang Y, Tseng HH, Jin JQ, Luh TM, Giroux-Leprieur E, Mo M, Zheng Q, Shi H, Zhang H, Hao X, et al. Hedgehog/Gli promotes epithelial-mesenchymal transition in lung squamous cell carcinomas. J Exp Clin Cancer Res. 2014; 33:34.
27. Chen JS, Li HS, Huang JQ, Zhang LJ, Chen XL, Wang Q, Lei J, Feng JT, Liu Q, Huang XH. Down-regulation of Gli-1 inhibits hepatocellular carcinoma cell migration and invasion. Mol Cell Biochem. 2014; 393:283-291.
28. Chen JS, Huang XH, Wang Q, Huang JQ, Zhang LJ, Chen XL, Lei J, Cheng ZX. Sonic hedgehog signaling pathway induces cell migration and invasion through focal adhesion kinase/AKT signaling-mediated activation of matrix metalloproteinase (MMP)-2 and MMP-9 in liver cancer. Carcinogenesis. 2013; 34:10-19.
29. Joost S, Almada LL, Rohnalter V, Holz PS, Vrabel AM, Fernandez-Barrena MG, McWilliams RR, Krause M, Fernandez-Zapico ME, Lauth M. GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells. Cancer Res. 2012; 72:88-99.
30. Bosco-Clement G, Zhang F, Chen Z, Zhou HM, Li H, Mikami I, Hirata T, Yagui-Beltran A, Lui N, Do HT, Cheng T, Tseng HH, Choi H, et al. Targeting Gli transcription activation by small molecule suppresses tumor growth. Oncogene. 2013; 33:2087-2097.
31. Li H, Lui N, Cheng T, Tseng HH, Yue D, Giroux-Leprieur E, Do HT, Sheng Q, Jin JQ, Luh TW, Jablons DM, He B. Gli as a novel therapeutic target in malignant pleural mesothelioma. PLoS One. 2013; 8:e57346.
32. Ng JM, Curran T. The Hedgehog's tale: developing strategies for targeting cancer. Nat Rev Cancer. 2011; 11:493-501.
33. LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin Cancer Res. 2011; 17:2502-2511.
34. Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, Marmur E, Rudin CM, Chang AL, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N Engl J Med. 2012; 366:2171-2179.
35. Dlugosz A, Agrawal S, Kirkpatrick P. Vismodegib. Nat Rev Drug Discov. 2012; 11:437-438.
36. Agarwal V, Lind MJ, Cawkwell L. Targeted epidermal growth factor receptor therapy in malignant pleural mesothelioma: where do we stand? Cancer Treat Rev. 2011; 37:533-542.
37. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014; 7:re8.
38. Zhang J, Tian XJ, Xing J. Signal Transduction Pathways of EMT Induced by TGF-beta, SHH, and WNT and Their Crosstalks. J Clin Med. 2016; 5.
39. Li X, Deng W, Lobo-Ruppert SM, Ruppert JM. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by beta-catenin. Oncogene. 2007; 26:4489-4498.
40. Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, Bazan JF, Kan Z, Seshagiri S, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009; 326:572-574.
41. Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, Scales SJ, Gould SE, Yauch RL, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011; 71:435-444.
42. Kim J, Aftab BT, Tang JY, Kim D, Lee AH, Rezaee M, Chen B, King EM, Borodovsky A, Riggins GJ, Epstein EH, Jr, Beachy PA, Rudin CM. Itraconazole and arsenic trioxide inhibit Hedgehog pathway activation and tumor growth associated with acquired resistance to smoothened antagonists. Cancer Cell. 2013; 23:23-34.