
INTRODUCTION
The second leading cause of cancer death worldwide and the sixth most common cancer in the world is hepatocellular carcinoma (HCC). The age-standardized rate per 100,000 ranges from 13.4 (Guinea-Bissau) to 78.1 (Mongolia). Hepatitis B or C virus, alcoholic drinks, and aflatoxins are factors causing HCC; oxidative stress, inflammation, and immune response are important for hepatic tumor growth [1]. In addition, oxidative stress (reactive oxygen species)-induced cellular DNA damage leads to gene mutation and activation of multiple signaling pathways that promote many aspects of hepatocellular-tumor development and progression [2].
In the liver-tumor microenvironment, balancing cellular ROS is important to tumor-cell maintenance. Low doses of ROS upregulate mRNA levels of cyclins to stimulate cell proliferation, promoting tumor growth; however, disproportionately increased cellular ROS induces cancer-cell cycle arrest, senescence, and apoptosis. Therefore, ROS scavengers and antioxidant enzymes are essential to tumorigenesis. Depletion of antioxidant proteins from cells increases cellular ROS causing cell apoptosis, which is one method of cancer therapy [3–5].
Peroxiredoxins (Prxs) are thioredoxin peroxidases that catalyze hydrogen peroxide, organic hydroperoxides, and peroxynitrite [6, 7]. They are important ROS scavengers. Mammalian cells have six isoforms of Prx, divided into three subfamilies based on catalytic cysteine (Cys) residue. Prx I belongs to 2-Cys Prxs. Recent evidence suggests that Prx I intervenes cell signaling by activating several signaling proteins to regulate cell proliferation, differentiation, and apoptosis [8–10]. Moreover, the role of Prx I in human cancer is still controversial and the different role of Prx I in human cancer depends on the specific cellular context. In several malignant cancers like lung cancer, it acts as a tumor suppressor. In K-ras-driven lung tumorigenesis mouse models, Prx I suppresses K-ras-induced ROS/ERK/cyclin D1 signaling pathways to inhibit lung tumorigenesis [11]. In contrast, in breast cancer and prostate cancer, it acts as a pro-oncogenic factor. Expression of Prx I in breast cancer facilitates cancer-cell survival from oxidative stress-induced cell death [12]; in prostate cancer, Prx I binds with TLR4 and thus regulates growth factor VEGF expression to promote prostate carcinogenesis [13].
Ras-family genes (H-ras, K-ras, and N-ras) are the most frequently altered human cancers [14–16]. H-ras mutation was found in HCC patients and contributed to tumor development [17–20]. In normal mice and HCC cell lines, the mutant form of H-rasG12V is not present. Our laboratory generated hepatic carcinoma H-rasG12Vmouse model and stable HCC-H-rasG12V cell lines by transfecting mutant form H-rasG12V into mouse embryo and HCC cell lines. In Tg mice and HCC-H-rasG12V cell lines, mutant oncogenic Ras leads to elevated intracellular ROS levels and activation of the ERK/FoxM1/cyclin D1 pathway to promote hepatic tumorigenesis [21].
Previously reported that Prx I and mutant form of H-ras have well correlated with HCC, respectively [17–20, 27]. To determine the relationship between Prx I and H-ras induced Hepatic tumorigenesis, we generated H-rasG12V/Prx I-/- double mutant mice and analyzed phenotypes of the mice. We investigated molecular events by performing knockdown or overexpression of Prx I in HCC-H-rasG12V cell lines. Our results illustrated that Prx I suppresses ROS-induced apoptosis and positively regulates the Ras/ERK/FoxM1/Nrf2 pathway to promote hepatocacinogenesis. Therefore, Prx I, a novel target ROS, plays a role in hepatic cancer-cell survival, and will be a potential therapeutic target for HCC prevention and treatment.
RESULTS
Prx I was overexpressed in HCC patient tissues, H-rasG12V-HCC cells, and the H-rasG12V Tg mice hepatic-tumor region
Prx I and H-ras were expressed significantly higher in the tumor region of HCC patients than the non-tumor region (Figure 1A). In two distinct Prx I expression pattern HCC cell lines, Huh7 and SK-HEP-1 (Figure 1B), after v-HA-Ras-vector transfection, the Prx I protein level significantly increased in both cells (Figure 1C). At the age of 3 months H-rasG12V Tg mice liver showed dysplasia (Dys) and Prx I expression was no different compared with wild type (WT). However, at 7 months H-rasG12V Tg mouse livers showed carcinoma and Prx I expression increased significantly in the tumor region (Figures 1D and 2E). Consistently, these results indicated that Prx I expression can be regulated by oncogenic Ras, and correlated with tumor progression.
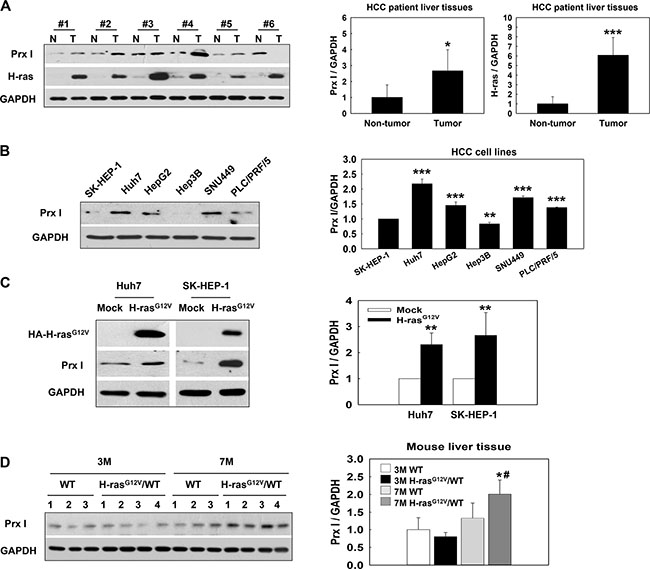
Figure 1: Prx I overexpression linked with oncogenic Ras expression in HCC patient tissues, HCC-H-rasG12V cells, and the H-rasG12V Tg mice hepatic-tumor region. (A) HCC patient-tumor region (T) and non-tumor region (N) were analyzed by Western blotting with Prx I, H-ras, and GAPDH antibodies. GAPDH was used as a loading control. Samples 1, 3, and 6 were from hepatocellular carcinoma patients’ liver tissues. Samples 2, 4, and 5 were from hepatitis B virus patients’ liver tissues. *p < 0.05, ***p < 0.001 compared to non-tumor. (B) Western blotting analysis of Prx I expression in HCC cells. **p < 0.01, ***p < 0.001 compared to SK-HEP-1. (C) The expression level of Prx I in Huh7 and SK-HEP-1 stable cell lines transfected by the pCAG-HA (Mock) or the pCAG-HA-H-rasG12V vector. HA is a tag of H-rasG12V protein. **p < 0.01, compared to Mock cells. (D) Using immunoblotting to detect Prx I expression in C57BL/6 wild type (WT) or H-rasG12V/WT Tg mice liver tissues. *p < 0.05, compared to 3 M H-rasG12V/WT and #p < 0.05 compared to 7 M WT. The data were repeated in at least three separate experiments.
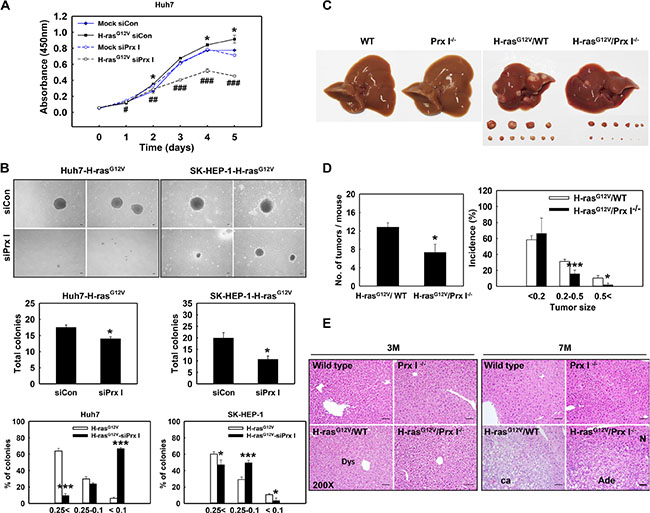
Figure 2: Prx I promoted Ras-induced hepatocarcinogenesis. (A) Huh7-Mock and Huh7-H-rasG12V cells were transiently transfected with scramble siRNA (siCon) or siPrx I. After incubation, cell proliferation was determined by CCK8 assay at the indicated time. *p < 0.05 compared to Huh7-Mock-siCon cells and #p < 0.05, ##p < 0.01, ###p < 0.001 compared to Huh7-H-rasG12V-siCon cells. (B) Anchorage-independent growth in soft agar were performed in Huh7-H-rasG12V and SK-HEP-1-H-rasG12V cells after transfected with siRNA (scramble or Prx I). Cell morphologies were observed under an inverted-phase contrast microscope at × 40 magnification. Scale bars, 100 μm. The number of colonies was determined by counting duplicated plates, *p < 0.05 compared to siCon. The diameters of the colonies were 0.25 mm <, 0.25–0.1 mm, and < 0.1 mm, *p < 0.05, ***p < 0.001 compared to H-rasG12V. (C) The gross appearance of WT, Prx I-/-, H-rasG12V/WT, and H-rasG12V/Prx I-/- mice liver at 7 months. (D) Tumor number and tumor size were measured at the age of 7 months H-rasG12V/WT (n = 6) and H-rasG12V/Prx I-/- (n = 7) mice-liver region. Tumor size; long × short diameter, cm2 (< 0.2 cm2, 0.2–0.5 cm2 and 0.5 < cm2). *p < 0.05, ***p < 0.001. (E) (H&E) staining of livers at 3 months and 7 months of WT, Prx I-/-, H-rasG12V/WT, and H-rasG12V/Prx I-/- mice. Magnification, 200 X. Scale bars, 100 μm. The data were repeated in at least three separate experiments and presented as mean ± SD.
Prx I promoted Ras-induced hepatocarcinogenesis
H-rasG12V transfected HCC cells grew faster than HCC-Mock cells (Figure 2A); H-rasG12V overexpression significantly increased anchorage-independent growth in HCC cells (Figure 2B); H-rasG12V Tg mice at the age of 7 months showed hepatic carcinoma in the liver region (Figure 2C and 2E). To investigate the role of Prx I in H-rasG12V-induced hepatocarcinogenesis, we knocked down Prx I in HCC-H-rasG12V cells by treating with siPrx I, and generated H-rasG12V/Prx I-/- double mutant mice. CCK8 assay data showed that siPrx I significantly decreased the growth speed of HCC-H-rasG12V cells from the 3rd day, dramatically suppressed cell proliferation (Figure 2A). Consistently, soft-agar assay results showed that knockdown of Prx I in HCC-H-rasG12V cells significantly suppressed colony formation (Figure 2B). Tumor numbers of H-rasG12V/Prx I-/- double mutant mice at 7 months decreased significantly; tumor size was markedly smaller than in H-rasG12V/WT mice (Figure 2C and 2D). The histological data showed that deletion of Prx I significantly suppressed H-rasG12V-mediated hepatic tumorigeneisis (Figure 2E). These results suggest that Prx I promotes oncogenic Ras-induced hepatocarcinogenesis.
Prx I modulated tumorigenesis through positive regulation of pERK and cyclin D1 expression
Western blotting data showed that pERK and cyclin D1 were more highly expressed in HCC-H-rasG12V cells and liver tissues of H-rasG12V Tg mice at 3 months than in controls (Figure 3A and 3D). Immunohistochemical data of liver tissues of H-rasG12V Tg mice at 7 months also showed consistent results (Figure 3E). To determine the underlying mechanism of Prx I function in the Ras/ERK/cyclin D1 signaling pathway, we performed knockdown of Prx I in HCC-H-rasG12V cells by siRNA. Western blotting results showed that reduced Prx I protein levels significantly suppressed activation of the pERK/cyclin D1 signaling pathway (Figure 3B). Consistently, null Prx I dramatically down-regulated pERK and cyclin D1 levels in the H-rasG12V Tg mice liver region, and no difference emerged between WT and Prx I-/- mice (Figure 3D and 3E). To confirm regulation of Prx I on pERK and cyclin D1 expression, we overexpressed Prx I in Huh7 and SK-HEP-1 cells. Our results demonstrated that Prx I overexpression can significantly up-regulate pERK and cyclin D1 expression in both cells (Figure 3C). Collectively, these results suggest that Prx I positively affects activation of H-rasG12V-induced pERK and cyclin D1 expression to promote hepatic-tumor growth.
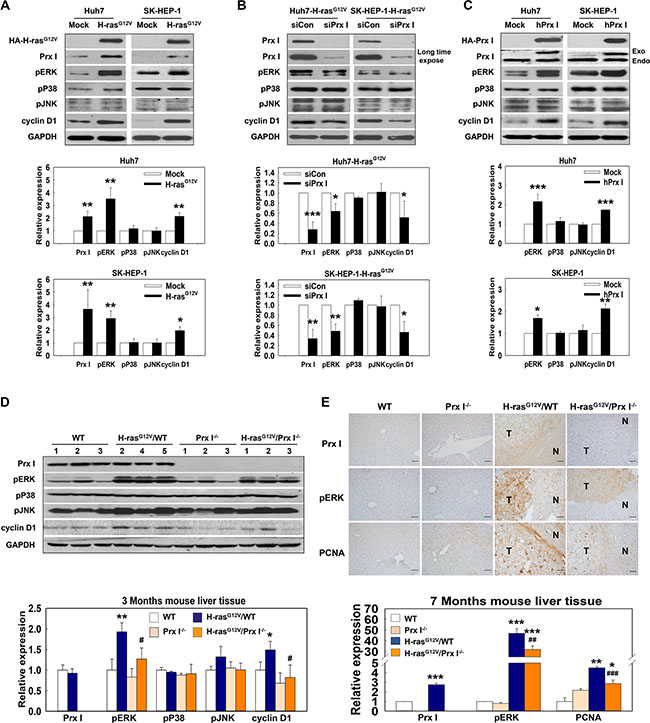
Figure 3: Prx I regulated the Ras-activation pathway. (A–D) Protein expression levels of Prx I, cyclin D1, and GAPDH as well as phosphorylation levels of ERK, P38, and JNK were analyzed in HCC-H-rasG12V cells (A), siPrx I-transfected HCC-H-rasG12V cells (B), pCAGGS-hPrx I-HA-neo-transfected Prx I-HCC cells (C), and 3 months WT, Prx I-/-, H-rasG12V/WT, and H-rasG12V/Prx I-/- mice-liver tissues (D). In (A, B and C), *p < 0.05, **p < 0.01, ***p < 0.001 compared to Mock, in (D), *p < 0.05, **p < 0.01 compared to WT and #p < 0.05 compared to H-rasG12V/WT. (E) Immunohistochemical staining of Prx I, PCNA and pERK at 7 months WT, Prx I-/-, H-rasG12V/WT, and H-rasG12V/Prx I-/- mice-liver tissues. Magnification, 200 X. Scale bars, 100 μm. *p < 0.05, **p < 0.01, ***p < 0.001 compared to WT and ##p < 0.01, ###p < 0.001 compared to H-rasG12V/WT. HCC cells; Huh7 and SK-HEP-1 cells. In C, HA-Prx I and Exo indicate the transfected Prx I expression; Endo indicates the intracellular endogeneous Prx I. The data were repeated in at least three separate experiments.
Prx I is needed for tumor-cell survival by inhibiting ROS-induced cell death
In oncogene-mediated tumorigenicity, ROS are essential to activate critical signaling pathways and promote cellular proliferation [22, 23]. Prx I is a scavenger of ROS that belongs to the peroxidase family of antioxidant enzymes. Knockdown of Prx I in Huh7-H-rasG12V cells and SK-HEP-1-H-rasG12V cells significantly increased the intracellular ROS level (Figure 4A) and cell apoptosis (Figure 4B). Consistently, FACS analysis showed that H-rasG12V/Prx I-/- double mutant mice had high levels of ROS and apoptosis, especially at 7 months (Figure 4C and 4D); Histological analysis showed that ROS-induced DNA damage and cell apoptosis were dramatically higher in liver tissues of H-rasG12V/Prx I-/- double mutant mice at 7 months than H-rasG12V/WT mice (Figure 4E), whereas Prx I overexpression in SK-HEP-1 cells significantly suppressed intracellular ROS and cell apoptosis (Figure 4F). In summary, Prx I expression in the liver is able to protect hepatic carcinoma cells from ROS-induced cell death to promote hepatocarcinogenesis.
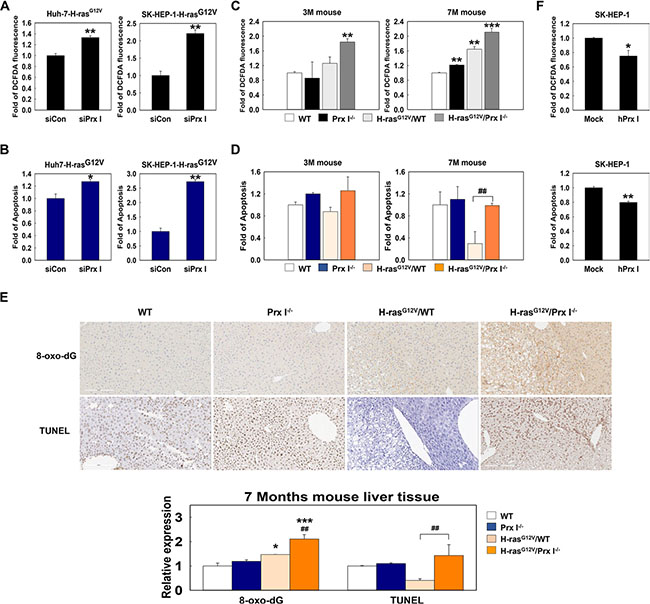
Figure 4: Prx I inhibited ROS induced tumor cell death. (A and B) Using flow cytometry to determine intracellular ROS (A) and cell apoptosis (B) in siRNA (scramble or Prx I)-transfected HCC-H-rasG12V cell lines. *p < 0.05, **p < 0.01 compared to siCon. (C and D) ROS (C) and cell apoptosis (D) level at 3 months (left panels) and 7 months (right panels) WT, Prx I-/-, H-rasG12V/WT, and H-rasG12V/Prx I-/- mice-liver tissues were determined by flow cytometry. **p < 0.01, ***p < 0.001 compared to WT mice and ##p < 0.01 compared to H-rasG12V/WT mice. (E) Paraffin-embedded liver specimens at 7 months WT, Prx I-/-, H-rasG12V/WT, and H-rasG12V/Prx I-/- mice were stained with 8-oxo-dG antibody and the TUNEL assay kit to detect ROS-induced DNA damage and cell death, respectively. Magnification, 200 X. Scale bars, 200 μm. *p < 0.05, ***p < 0.001 compared to WT and ##p < 0.01 compared to H-rasG12V/WT. (F) Intracellular ROS and apoptosis levels in SK-HEP-1-Mock cells and Prx I-SK-HEP-1 cells (SK-HEP-1-hPrx I) transfected by pCAGGS-hPrx I-HA-neo vector were detected by flow cytometry. *p < 0.05, **p < 0.01 compared to Mock. HCC cells; Huh7 and SK-HEP-1 cells. The data were repeated in at least three separate experiments and presented as mean ± SD.
Prx I regulated transcription factor FoxM1 and Nrf2 levels in H-rasG12V induced hepatic tumors
Forkhead box protein M1 (FoxM1) and Nrf2 are both important transcription factors in carcinogenesis [24]. FoxM1 downregulates ROS levels by stimulating expression of ROS scavenger genes, such as MnSOD, catalase and PRDX3 [25]; the Nrf2-Prx I axis is important in human lung-cancer, prostate-cancer, colon-cancer, and breast-cancer development [26]. In our data, FoxM1 and Nrf2 significantly increased in HCC-H-rasG12V cells, accompanied by increased Prx I expression (Figure 5A).
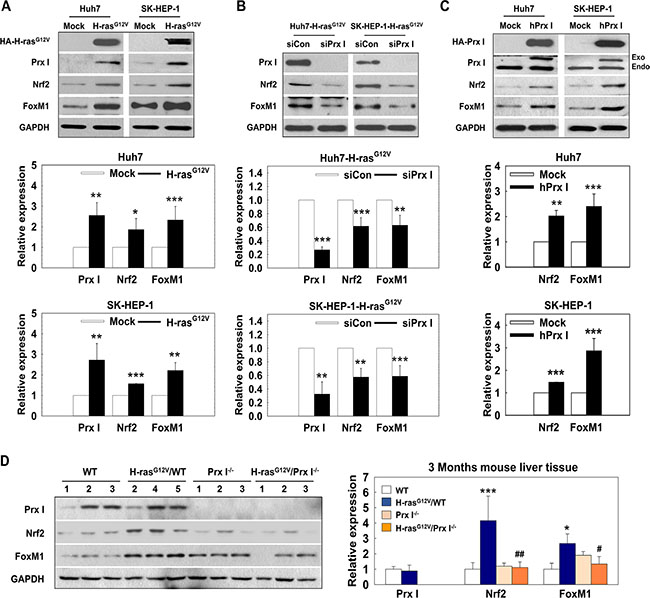
Figure 5: Prx I regulated transcription factor FoxM1 and Nrf2 levels in H-rasG12V-induced hepatic tumors. (A–D). Prx I, Nrf2, FoxM1, and GAPDH expression levels were detected in HCC-Mock cells and HCC-H-rasG12V cells (A); siPrx I-transfection HCC-H-rasG12V cells (B); pCAGGS-hPrx I-HA-neo-transfected Prx I-HCC cells (C); and 3 months WT, Prx I-/-, H-rasG12V/WT, and H-rasG12V/Prx I-/- mice liver tissues (D) by Western blotting. In (A, B and C), *p < 0.05, **p < 0.01, ***p < 0.001 compared to Mock, in D, *p < 0.05, ***p < 0.001 compared to WT and #p < 0.05, ##p < 0.01 compared to H-rasG12V/WT. HCC cells; Huh7 and SK-HEP-1 cells. The data were repeated in at least three separate experiments.
To determine the relationship between Prx I and transcription factors (FoxM1 and Nrf2), we performed Western blotting analysis in HCC-H-rasG12V cells. The data showed that knockdown of Prx I dramatically down-regulated Nrf2 and FoxM1 expression (Figure 5B) and conversely, overexpression of Prx I in HCC cells up-regulated Nrf2 and FoxM1 protein levels (Figure 5C). Mouse-liver tissue samples also showed consistent data (Figure 5D). Taken together, these results suggested that Prx I and transcription factor (FoxM1 and Nrf2) expression closely correlate and Prx I positively upregulates FoxM1 and Nrf2 expression.
Oncogene Ras activates the ERK/FoxM1/Nrf2/Prx I pathway to promote hepatocarcinogenesis
Knockdown of FoxM1 by siRNA caused a reduction of Prx I, Nrf2 and cyclin D1 protein in Huh7-H-rasG12V and SK-HEP-1-HrasG12V cells (Figure 6A). Consistent results are shown in Huh7-H-rasG12V cell treatment with Siomycin A, a potent specific inhibitor of FoxM1 (Figure 6B). However, knockdown of Nrf2 only down-regulated Prx I and cyclin D1 protein levels, and did not affect FoxM1 expression (Figure 6C). CHIP assay showed that in Huh7 cell, Nrf2 binds to the Prx I promoter region (–536 to –528) and (–1429 to –1421) to regulate Prx I gene expression. RT-PCR results, consistent with CHIP assay, showed that Prx I mRNA levels were significantly reduced by knockdown of Nrf2 (Figure 6D and 6E).
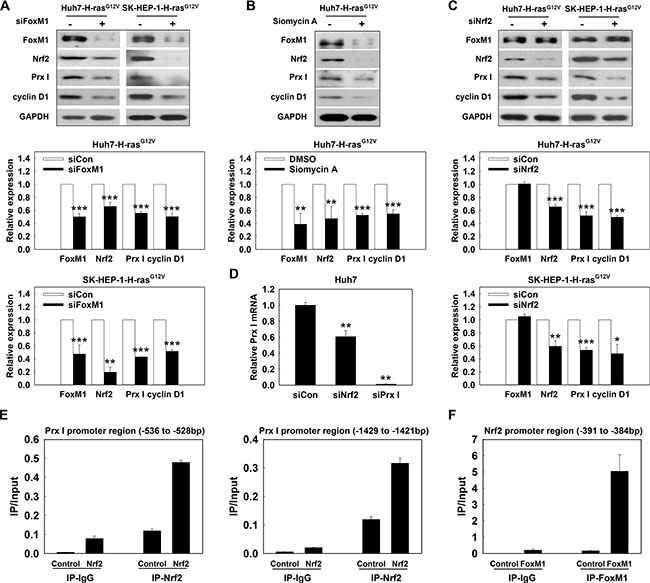
Figure 6: Oncogene Ras activated the ERK/FoxM1/Nrf2/Prx I pathway. (A) Huh7-H-rasG12V cells and SK-HEP-1-H-rasG12V cells were transfected with siFoxM1 for 72 h; (B) Huh7-H-rasG12V cells were treated with Siomycin A (10 μM) for 24 h. After incubation, the cell lysates were used to determine protein expression. (C) After HCC-H-rasG12V cells transfected with siNrf2 for 72 h, the protein expression of FoxM1, Nrf2, Prx I, cyclin D1, and GAPDH were detected in the cell lysates. In A and C, *p < 0.05, **p < 0.01, ***p < 0.001 compared to siCon. In B, **p < 0.01, ***p < 0.001 compared to DMSO. (D) Prx I mRNA level was detected in Huh7 cells transfected with siRNA (scramble, Nrf2 or Prx I) for 48 h, **p < 0.01 compared to siCon. (E and F) CHIP assay was performed to detect the Nrf2 binding sites in the Prx I promoter region (–1429 to –1421), (–536 to –528) (E) or FoxM1 target site in the Nrf2 promoter region (−391 to −384) (F). HCC-H-rasG12V cells; Huh7-H-rasG12V and SK-HEP-1-H-rasG12V cells. The data were repeated in at least three separate experiments.
To investigate whether FoxM1 controls Nrf2 at the transcriptional level, we used silico prediction of transcription factors binding to an Nrf2 promoter, and found that the FoxM1B/FoxA binding site (TTTGTTTGTTTG) was located at the human Nrf2 promoter –391 to –384, upstream of the transcriptional start site. CHIP analysis indicated that FoxM1 binds to the FoxM1 binding site of the Nrf2 promoter region (Figure 6F) and revealed that Nrf2 is a transcription target of FoxM1. Taken together, H-rasG12V modulates hepatic tumorigenesis through regulation of the ERK/FoxM1/Nrf2/Prx I pathway, and Prx I is able to facilitate the ERK/FoxM1/Nrf2 pathway by forming a positive feedback loop between Prx I and the ERK/FoxM1/Nrf2 pathway (Figure 7).
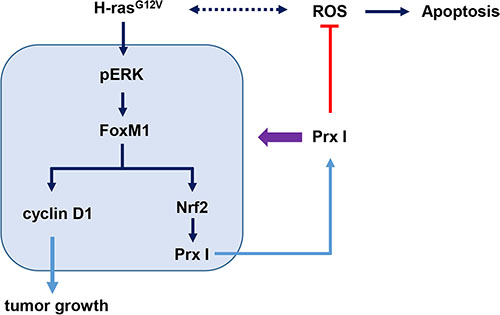
Figure 7: Schematic illustration of Prx I function in oncogenic Ras-induced liver tumorigenesis. H-rasG12V enhanced intracellular ROS levels and promoted downstream pERK/FoxM1 pathway. Transcription factor FoxM1 targeted the cell-cycle-related protein cyclin D1 and Nrf2 expression. Then Nrf2 targeted Prx I promoter to regulate Prx I expression. Overexpressing Prx I positively regulated the ERK/FoxM1/Nrf2 pathway and suppressed ROS-induced cell death to promote hepatic tumorigenesis. However, knockdown Prx I failed to scavenge ROS-induced cell death, which inhibited the tumorigenesis.
DISCUSSION
Prx I is an important antioxidant enzyme in tumor development. In the current study, we demonstrated that Prx I has an essential role in H-ras-induced hepatic cellular carcinoma. Prx I deficiency in HCC-H-rasG12V cells and H-rasG12V/Prx I-/- double mutant mice led to reduced HCC-H-rasG12V cell-colony formation and decreased hepatic-tumor numbers and sizes, respectively. Prx I protected tumor cells from ROS-induced cell death and positively regulated the H-rasG12V-induced ERK/FoxM1/Nrf2 pathway to promote tumorigenesis.
Results from HCC patients’ sample analysis showed that Prx I is a diagnostic and prognostic biomarker of HCC, and high levels of Prx I mRNA in tumor tissue aligned with a shorter overall survival time [27]. Moreover, increased Prx I expression positively correlated with endothelial tumor growth, vessel density, and tumor size [28]. Decreased expression of Prx I in HepG2 cells led to changes in cell morphology and delayed cell growth [29]. These data were consistent with our present data. In the HCC-patient tumor region, Prx I expression was higher than in the non-tumor region. In the H-rasG12V-induced HCC mouse model, hepatic tumor growth was time dependent. Interestingly, Prx I expression also increased in H-rasG12V/WT Tg mice in a time-dependent manner. Knockdown of Prx I expression in HCC-H-rasG12V cells dramatically suppressed cell proliferation and colony formation; Prx I knockout in H-rasG12V Tg mice significantly decreased tumor size and numbers. At 7 months H-rasG12V Tg mice markedly decreased in body weight and increased in liver weight. However, Prx I knockout in HrasG12V Tg mice significantly recovered the body- and liver-weight changes (Supplementary Figure S1). Taken together, Prx I contributes to hepatic-tumor development.
ROS are well known to activate human cancers and their function in cells might be as a double-edged sword. A moderate increase of ROS may promote cell proliferation and survival by triggering a redox-adaptation response, leading to enhanced cellular antioxidant capability to help cells survive under certain levels of oxidative stress [30–33]. However, when ROS are increased to a certain level, they may overwhelm the antioxidant capacity of the cell and trigger cell death [34]. So when K-ras-induced ROS are moderate, they can help in tumorigenicity [23]. In addition, recently it was reported that oncogenic K-ras-activated expression of antioxidant genes via Nrf2 mediated an unusual metabolic pathway of glutamine to generate NADPH, which led to low levels of ROS in pancreatic pre-neoplastic cells. The low level of ROS promotes carcinogenesis in K-ras-mutated pancreatic ductal adenocarcinoma (PDAC) [35]. However, a further increase of ROS may break the antioxidant system and elevating cellular ROS above the threshold level would lead to cell death [22, 34, 36]. Thus, modulating the ROS-induced signaling pathway is important for tumorigenesis.
Prx I is an antioxidant enzyme activated by oxidative stress that regulates multiple signals in different cancers. In breast cancer, Prx I suppressed ROS-induced AKT phosphorylation to inhibit cell proliferation [37]; In lung cancer, Prx I suppressed the ROS-induced ERK/cyclin D1 pathway to inhibit tumor growth [11]; In pancreatic cancer, Prx I modulated P38 activation to promote cancer-cell invasion [38]; In liver cancer, Prx I is an immediate and sensitive marker of oxidative stress. Inhibition of Prx I cysteine activation in hepatocellular carcinoma cells dramatically increased intracellular ROS levels, leading to enhanced cytotoxicity in HCC cells [36, 39]. However, the mechanism of Prx I in HCC was still unclear.
In this study, we used an H-rasG12V-induced HCC model to identify Prx I function. We found that the increased ROS level in H-ras mutant form was under the toxic thresholds that aid in cancer-cell proliferation and survival. When we knocked down or overexpressed the antioxidant Prx I, the redox balance was broken. A small change in the ROS level over the threshold level loses the ability to adapt to oxidative stress, and thus increases cancer cell death. Thus, over time, small changes in ROS and apoptosis between H-rasG12V Tg mice and H-rasG12V/Prx I-/- double mutant mice induced different rates of carcinogenesis.
FoxM1 is a typical proliferation-associated transcription factor overexpressed in numerous cancer-cell lines and human cancers [40–42], implicated in cell migration, invasion, angiogenesis, and metastasis [43]. Nrf2, also an important transcription factor activated by oxidative stress in tumor development [44–46], controls cell fate by transcriptional upregulation of antioxidant enzyme genes to resist oxidative stress in cancer cells. Prx I is an important target gene of Nrf2. The Nrf2–Prx I axis links with tumor biology, enhancing the drug efficacy of cancer therapy and serving as a fruitful target of cancer prognosis [26]. MEK-ERK activation has been shown to be one of the major pathways resulting in the activation of Nrf2 and induction of Nrf2 downstream targets, including phase II detoxifying/antioxidant genes in response to oxidative stress [47]. In this study, ERK phosphorylation was significantly induced in H-RasG12V HCC cells compared with mock cells. In addition, Nrf2 also significantly increased in accordance with increased expression of Prx I. Furthermore, Nrf2 bound to the Prx I promoter region to regulate Prx I gene expression, as reported in lung cancer cells [11, 26]. Like Nrf2, FoxM1 also significantly increased in accordance with increased expression of Prx I in H-RasG12V HCC cells. It is interesting to note that FoxM1 activated Nrf2 via its binding to Nrf2 promoter region, not to Prx I. These data suggest that H-rasG12V induced ERK phosphorylation increased transcriptional expression of Prx I through FoxM1/Nrf2 signaling.
ROS increment by Ras activation was shown to activate expression of pERK, FoxM1 and Nrf2. However, when ROS level was more increased with Prx I knockdown or deletion in H-RasG12V HCC cells, expression levels of FoxM1 and Nrf2 decreased. Why does increased ROS lead to a decrease in FoxM1 and Nrf2 levels? Previously it was reported that FoxM1 and Nrf2 are pERK substrates [41, 47, 48]. In this experiment, the activation of FoxM1 and Nrf2 was depended on the ERK activity. Knockdown or deletion of Prx I down-regulated the ERK activity. Thus the expression of FoxM1 and Nrf2 was reduced even though the ROS level increased. In addition, overexpression of Prx I in HCC cell lines enhanced pERK, then the expression of FoxM1 and Nrf2, although it decreased the ROS level (Figure 3C). However, further studies are needed to understand the reason why increased ROS lead to a decrease in FoxM1 and Nrf2 levels.
The conclusion is that Prx I positively regulates the H-rasG12V-induced ERK/FoxM1/Nrf2 pathway, and suppresses ROS-induced cell death to promote hepatocarcinogenesis. This outcome can be applied when studying a novel mechanism for HCC prognosis and therapy.
MATERIALS AND METHODS
Animal models
H-rasG12V transgenic and Prx I knockout mice were previously described [11, 49]. HrasG12V/Prx I-/- double mutant mice were generated by crossing H-rasG12V Tg mice with Prx I knockout mice. The mouse room was maintained at a constant temperature of 22 ± 1°C, humidity of 55 ± 10%, and 12 hr light/dark cycle. We used genotyping primers, as previously reported [11, 49]. All animal procedures were conducted in accordance with the guidelines of the Insitutional Animal Care and Use Committee, Korea Research Institute of Bioscience and Biotechnology.
Patient tissue sample
Patient HCC tissues were collected from Chonbuk National University hospital. All study patients provided informed consent prior to surgery.
Cell culture, transfection and treatment
Human HCC cell lines—Huh7, SK-HEP-1, HepG2, Hep3B, PLC/PRF/5 and SNU449 (ATCC, Manassas, VA, USA)—were maintained in Dulbecco’s modified Eagle’s medium (DMEM), Roswell Park Memorial Institute 1640 medium (RPMI 1640) containing 10% fetal bovine serum (FBS), penicillin (100 U/ml), and streptomycin (100 mg/ml). Cells were incubated in a humidified atmosphere with 37°C, 5% CO2 and 95% air. The Huh7- and SK-HEP-1-H-rasG12V mutant cell lines were generated by transfection. The transfection vector pCAG-HA-H-rasG12V-neo was constructed as follows: The coding sequences for mutated H-ras (H-rasG12V) were inserted by PCR cloning into the EcoRI site of the pCAG-HA-neo vector and confirmed by restriction enzyme mapping and DNA sequencing. Huh7 and SK-HEP-1 cells were plated in 6-well culture plates for 24 h prior to transfection. Cells were transfected with 3 μg of a pCAG-HA-H-rasG12V-neo construct using a Lipofectamine LTX reagent (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. After 48 h, cells were trypsinized and plated in a medium containing 400 μg/ml neomycin (G418). Following selection for 2 weeks, total populations of neomycin-resistant cells were pooled and single-cells sorted into 96-well plates with a growth medium containing 400 μg/ml of neomycin. Sorted single cells were grown under selection for an additional 2 weeks and expanded into stable cell lines. Candidate clones were analyzed by immunoblotting using a specific H-ras (Santa Cruz Biotechnology, Dallas, TX, USA) or HA (Roche, Basel, Switzerland) antibody. Transfection of pCAGGS-hPrx I-HA-neo vectors or siRNA (Prx I, Nrf2, and FoxM1) to overexpress Prx I or knockdown their proteins level was described in previous papers [11, 49]. Huh7-H-rasG12V cells were treated with 10 uM Siomycin A, FoxM1 inhibitor (Calbiochem, Billerica, MA, USA) for 24 h.
Western blotting
Western blotting was performed as described previously [11]. Liver tissues and cell lysates were put in a lysis buffer (20 mM HEPES, 150 mL NaCl, 2 mM EGTA, 1 mM EDTA, 20 mM glycerol phosphate, 1% Triton X-100 and 10% glycerol) with protease (sigma, St Louise, MO) and a phosphatase-inhibitor cocktail (Roche, Basel, Switzerland). Protein samples were extracted by an SDS-PAGE and transferred to nitrocellulose membranes (Millipore, Bellerica, MA, USA). The membranes were primarily blotted with primary antibodies against Prx I, PCNA, Nrf2 (Abcam, Cambridge, UK); HA (Roche, Basel, Switzerland); GAPDH, FoxM1 (Abfrontier, Seoul, Korea); pERK, pP38, pJNK, cyclin D1 (Cell Signaling Technology, Danvers, MA, USA); and H-ras (Santa Cruz Biotechnology, Dallas, TX, USA).
Cell-proliferation assay
1,000 cells per well were seeded in a 96-well cell-culture plate. After incubating for 12 h, cells were transfected with siCon or siPrx I, then cultured for 5 days. Cell Counting Kit-8 (CCK8, Dojindo Molecular Technologies, Rockville, MD, USA) was added to the plate, incubated at 37°C for 3 h, with absorbance measured by a microplate reader (VERSAmaxTM, Madison, NC, USA) at a wavelength of 450 nm.
Colony-formation assay
5,000 cells were pretreated with siRNA (Scramble or Prx I) for 24 h, suspended in 1 ml DMEM containing 0.3% agar and plated on 0.6% agar in a cell-growth medium, then put in a 37°C and 5% CO2 incubator for 2 weeks. Colonies were observed under an inverted phase-contrast microscope. Data represent at least triplicate independent wells.
Immunohistochemistry
Liver tissues were fixed in 10% paraformaldehyde and embedded in paraffin. Sections (3 μm) were stained with hematoxylin and eosin (H&E) or subjected to immunohistochemistry. Additional details were performed as previously described [50]. The primary antibodies used were Prx I, PCNA (Abcam, Cambridge, UK); pERK (Cell Signaling Technology, Danvers, MA, USA); and 8-oxoguanine (8-oxo-dG; Millipore, Bellerica, MA, USA). The TUNEL assay was performed using the ApopTag Peroxidase Kit (S-7100; Millipore).
Detection of ROS and apoptosis by flow cytometry
To measure the intracellular ROS and cell apoptosis, cells were stained with 10 uM 5, 6-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA; Invitrogen, Carlsbad, CA, USA) and Annexin V (BD Biosciences, Franklin Lakes, NJ, USA) for 15 min at 37°C, and washed with PBS twice by centrifugation at 600 g for 3 min. The cells resuspended with PBS and were analyzed by flow cytometry on a FACSCalibur instrument (BD Biosciences).
CHIP assay
This experiment was performed using the chromatin immunoprecipitation assay (CHIP assay) kit (Cell Signaling Technology, Danvers, MA, USA) according to the supplied protocol. Antibodies Nrf2, FoxM1, and IgG were added to sonicated Huh7 cell lysates for immunoprecipitation at 4°C with rotation overnight; we then added Chip-Grade Protein G Agarose Beads to pull down the protein-DNA complex, and amplified the binding DNA by qPCR, using the Nrf2 binding regions of the human Prx I promoter (–1429 to –1421) or (–536 to -528): 5′-tcactgcaacctctgcctcc-3′ and 5′-gatcgagaccatcctggcca-3′; 5′-ttgccttaccgtgtgggtctg-3′ and 5′-actcctaaacttacgccagagg-3′. FoxM1 binding human Nrf2 promoter regions: 5′-agctcctacaccaacgcctt-3′ and 5′-cggcgttcagtttgccctcc-3′. The nonbinding position (control) of the human Prx I promoter was 5′-gccaacatgatgaaacctcgtc-3′ and 5′-ggtgttggcttactgcaacctcc-3′. A nonimmunoprecipitated chromation fragment was used as an input control.
Statistical analysis
The data were analyzed using SigmaPlot 13 software for Student’s t-test, a value of p less than 0.05 was considered to be statistically significant.
Abbreviations
Ade, adenoma; Ca, carcinoma; Dys, dysplasia; hPrx I, human Prx I; N, non-tumor; Prx I, peroxiredoxin I; siCon, scramble siRNA; T, tumor; 3 M, 3 months; 7 M, 7 months.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest with the contents of this article.
GRANT SUPPORT
This work was supported by the Korea Research Institute of Bioscience and Biotechnology Research Initiative Program (KGM3141312) and partially from the R&D Convergence Program (CAP-15-03-KRIBB) of NST (National Research Council of Science & Technology) of Republic of Korea.
REFERENCES
1. Kumar M, Zhao X, Wang XW. Molecular carcinogenesis of hepatocellular carcinoma and intrahepatic cholangiocarcinoma: one step closer to personalized medicine? Cell Biosci. 2011; 1:5.
2. Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006; 5:14.
3. Liou GY, Storz P. Reactive oxygen species in cancer. Free Radic Res. 2010; 44:479–496.
4. Storz P. Reactive oxygen species in tumor progression. Front Biosci. 2005; 10:1881–1896.
5. Cadenas E. Mitochondrial free radical production and cell signaling. Mol Aspects Med. 2004; 25:17–26.
6. Rhee SG, Kang SW, Netto LE, Seo MS, Stadtman ER. A family of novel peroxidases, peroxiredoxins. Biofactors. 1999; 10:207–209.
7. Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003; 28:32–40.
8. Neumann CA, Cao J, Manevich Y. Peroxiredoxin 1 and its role in cell signaling. Cell Cycle. 2009; 8:4072–4078.
9. Jarvis RM, Hughes SM, Ledgerwood EC. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic Biol Med. 2012; 53:1522–1530.
10. Morinaka A, Funato Y, Uesugi K, Miki H. Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene. 2011; 30:4208–4218.
11. Park YH, Kim SU, Lee BK, Kim HS, Song IS, Shin HJ, Han YH, Chang KT, Kim JM, Lee DS, Kim YH, Choi CM, Kim BY, et al. Prx I suppresses K-ras-driven lung tumorigenesis by opposing redox-sensitive ERK/cyclin D1 pathway. Antioxid Redox Signal. 2013; 19:482–496.
12. Bae JY, Ahn SJ, Han W, Noh DY. Peroxiredoxin I and II inhibit H2O2-induced cell death in MCF-7 cell lines. J Cell Biochem. 2007; 101:1038–1045.
13. Riddell JR, Bshara W, Moser MT, Spernyak JA, Foster BA, Gollnick SO. Peroxiredoxin 1 controls prostate cancer growth through Toll-like receptor 4-dependent regulation of tumor vasculature. Cancer Res. 2011; 71:1637–1646.
14. Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011; 2:344–358. doi: 10.1177/1947601911411084.
15. Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001; 93:1062–1074.
16. Kiaris H, Spandidos D. Mutations of ras genes in human tumors (review). Int J Oncol. 1995; 7:413–421.
17. Gotzmann J, Huber H, Thallinger C, Wolschek M, Jansen B, Schulte-Hermann R, Beug H, Mikulits W. Hepatocytes convert to a fibroblastoid phenotype through the cooperation of TGF-beta1 and Ha-Ras: steps towards invasiveness. J Cell Sci. 2002; 115:1189–1202.
18. Staib F, Hussain SP, Hofseth LJ, Wang XW, Harris CC. TP53 and liver carcinogenesis. Hum Mutat. 2003; 21:201–216.
19. Sui G, Ma X, Liu S, Niu H, Dong Q. Study of the correlation between H-ras mutation and primary hepatocellular carcinoma. Oncol Lett. 2012; 4:779–782.
20. Tsuda H, Hirohashi S, Shimosato Y, Ino Y, Yoshida T, Terada M. Low incidence of point mutation of c-Ki-ras and N-ras oncogenes in human hepatocellular carcinoma. Jpn J Cancer Res. 1989; 80:196–199.
21. Park YH, Kim SU, Kwon TH, Kim JM, Song IS, Shin HJ, Lee BK, Bang DH, Lee SJ, Lee DS, Chang KT, Kim BY, Yu DY. Peroxiredoxin II promotes hepatic tumorigenesis through cooperation with Ras/Forkhead box M1 signaling pathway. Oncogene. 2015; 35:3503–3513.
22. Sun YL, Cai JQ, Liu F, Bi XY, Zhou LP, Zhao XH. Aberrant expression of peroxiredoxin 1 and its clinical implications in liver cancer. World J Gastroenterol. 2015; 21:10840–10852.
23. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol. 2002; 192:1–15.
24. Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, Chandel NS. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010; 107:8788–8793.
25. Toyokuni S. Iron and thiols as two major players in carcinogenesis: friends or foes? Front Pharmacol. 2014; 5:200.
26. Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, Lau LF, Costa RH, Raychaudhuri P. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009; 28:2908–2918.
27. Kim YJ, Ahn JY, Liang P, Ip C, Zhang Y, Park YM. Human prx1 gene is a target of Nrf2 and is up-regulated by hypoxia/reoxygenation: implication to tumor biology. Cancer Res. 2007; 67:546–554.
28. Sun QK, Zhu JY, Wang W, Lv Y, Zhou HC, Yu JH, Xu GL, Ma JL, Zhong W, Jia WD. Diagnostic and prognostic significance of peroxiredoxin 1 expression in human hepatocellular carcinoma. Med Oncol. 2014; 31:786.
29. Aguilar-Melero P, Prieto-Alamo MJ, Jurado J, Holmgren A, Pueyo C. Proteomics in HepG2 hepatocarcinoma cells with stably silenced expression of PRDX1. J Proteomics. 2013; 79:161–171.
30. Xu W, Trepel J, Neckers L. Ras, ROS and proteotoxic stress: a delicate balance. Cancer Cell. 2011; 20:281–282.
31. Zamkova M, Khromova N, Kopnin BP, Kopnin P. Ras-induced ROS upregulation affecting cell proliferation is connected with cell type-specific alterations of HSF1/SESN3/p21Cip1/WAF1 pathways. Cell Cycle. 2013; 12:826–836.
32. Buder-Hoffmann S, Palmer C, Vacek P, Taatjes D, Mossman B. Different accumulation of activated extracellular signal-regulated kinases (ERK 1/2) and role in cell-cycle alterations by epidermal growth factor, hydrogen peroxide, or asbestos in pulmonary epithelial cells. Am J Respir Cell Mol Biol. 2001; 24:405–413.
33. Conde de la Rosa L, Schoemaker MH, Vrenken TE, Buist-Homan M, Havinga R, Jansen PL, Moshage H. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006; 44:918–929.
34. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009; 8:579–591.
35. Kong B, Qia C, Erkan M, Kleeff J, Michalski CW. Overview on how oncogenic Kras promotes pancreatic carcinogenesis by inducing low intracellular ROS levels. Front Physiol. 2013; 4:246.
36. Hou JK, Huang Y, He W, Yan ZW, Fan L, Liu MH, Xiao WL, Sun HD, Chen GQ. Adenanthin targets peroxiredoxin I/II to kill hepatocellular carcinoma cells. Cell Death Dis. 2014; 5:e1400.
37. O'Leary PC, Terrile M, Bajor M, Gaj P, Hennessy BT, Mills GB, Zagozdzon A, O'Connor DP, Brennan DJ, Connor K, Li J, Gonzalez-Angulo AM, Sun HD, et al. Peroxiredoxin-1 protects estrogen receptor alpha from oxidative stress-induced suppression and is a protein biomarker of favorable prognosis in breast cancer. Breast Cancer Res. 2014; 16:R79.
38. Taniuchi K, Furihata M, Hanazaki K, Iwasaki S, Tanaka K, Shimizu T, Saito M, Saibara T. Peroxiredoxin 1 promotes pancreatic cancer cell invasion by modulating p38 MAPK activity. Pancreas. 2015; 44:331–340.
39. Cesaratto L, Vascotto C, D'Ambrosio C, Scaloni A, Baccarani U, Paron I, Damante G, Calligaris S, Quadrifoglio F, Tiribelli C, Tell G. Overoxidation of peroxiredoxins as an immediate and sensitive marker of oxidative stress in HepG2 cells and its application to the redox effects induced by ischemia/reperfusion in human liver. Free Radic Res. 2005; 39:255–268.
40. Pilarsky C, Wenzig M, Specht T, Saeger HD, Grutzmann R. Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia. 2004; 6:744–750.
41. Kopanja D, Pandey A, Kiefer M, Wang Z, Chandan N, Carr JR, Franks R, Yu DY, Guzman G, Maker A, Raychaudhuri P. Essential roles of FoxM1 in Ras-induced liver cancer progression and in cancer cells with stem cell features. J Hepatol. 2015; 63:429–436.
42. Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006; 66:9731–9735.
43. Halasi M, Gartel AL. Targeting FOXM1 in cancer. Biochem Pharmacol. 2013; 85:644–652.
44. Ikeda H, Nishi S, Sakai M. Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis. Biochem J. 2004; 380:515–521.
45. Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. Dual roles of Nrf2 in cancer. Pharmacol Res. 2008; 58:262–270.
46. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012; 12:564–571.
47. Cheung KL, Lee JH, Shu L, Kim JH, Sacks DB, Kong AN. The Ras GTPase-activating-like protein IQGAP1 mediates Nrf2 protein activation via the mitogen-activated protein kinase/extracellular signal-regulated kinase (ERK) kinase (MEK)-ERK pathway. J Biol Chem. 2013; 288:22378–22386.
48. Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003; 23:7198–7209.
49. Wang AG, Moon HB, Lee MR, Hwang CY, Kwon KS, Yu SL, Kim YS, Kim M, Kim JM, Kim SK, Lee TH, Moon EY, Lee DS, Yu DY. Gender-dependent hepatic alterations in H-ras12V transgenic mice. J Hepatol. 2005; 43:836–844.
50. Sun HN, Kim SU, Huang SM, Kim JM, Park YH, Kim SH, Yang HY, Chung KJ, Lee TH, Choi HS, Min JS, Park MK, Kim SK, et al. Microglial peroxiredoxin V acts as an inducible anti-inflammatory antioxidant through cooperation with redox signaling cascades. J Neurochem. 2010; 114:39–50.