
INTRODUCTION
Acute myeloid leukemia (AML) is a complex neoplastic disease of the hematopoietic system resulting in maturation arrest and aberrant proliferation of leukemic cells. Despite the use of cytogenetic and molecular risk stratification for treatment guidance, the majority of AML patients still do not achieve long-term survival. A better knowledge of the disease biology and novel targeted therapeutic approaches may improve cure rates.
Recently, we and others reported that the deregulated expression of microRNAs (miRs) – small non-coding RNA molecules regulating post-transcription protein expression – is associated with AML [1, 2]. Assessing the expression levels of some miRs refines patients’ molecular risk classification and helps selecting treatment regimens [1–11]. These results are being translated into the clinic, and early clinical trials targeting miRs have been initiated [12–16].
The miR-181 family comprises four mature miRs (miR-181a, miR-181b, miR-181c, miR-181d) and has been associated with the regulation of inflammatory mechanisms [17, 18]. Physiologically, miR-181 may accelerate the megakaryocyte differentiation of CD34-positive hematopoietic cells [19]. Furthermore, these miRs have been found to be deregulated in several types of human cancers, including leukemias [2, 9, 20–26]. In solid tumors the role of miR-181 seems to be organ-specific. High expression of miR-181 has been associated with poor clinical outcomes in patients with colorectal cancer [20] and lymph node metastasis in oral squamous cell carcinoma [21]. However, in glioma high expression of miR-181 seems to have tumor suppressor activity [22]. In hematologic malignancies higher expression of miR-181 is associated with better outcomes [2, 9, 26–28]. Indeed, we recently reported the favorable impact of higher miR-181a expression in both AML cytogenetically normal (CN) or abnormal (CA) patients [2, 9, 28]. To date, however the molecular basis for the attenuation of disease aggressiveness by miR-181a remains to be fully elucidated.
RAS proto-oncogenes encode small GTPase proteins, that is, KRAS, NRAS and HRAS, that are involved in homeostatic mechanisms of proliferation, differentiation and apoptosis of normal cells [29]. Whereas KRAS and NRAS are frequently mutated and activated in AML, HRAS mutations are rare, and HRAS wild-type expression is the lowest with respect to the other RAS isoforms in the hematopoietic system [29]. Aberrant activation of RAS signal transduction is often found in human neoplasia [30–43]. In hematopoietic malignancies, including AML, activating oncogenic RAS mutations contribute to malignant phenotypes by phosphorylating and activating downstream effectors such as the mitogen-activated protein kinase kinase (MAPKK, also known as MEK), mitogen-activated protein kinase (MAPK), and the PI3K-AKT downstream effectors, thereby promoting aberrant cell proliferation and survival [29]. However, to date, an effective therapeutic approach targeting RAS directly remains to be developed.
Recently, KRAS was shown to be a direct miR-181a target in oral squamous cell carcinoma [44]. Additionally, NRAS and the RAS-downstream effector MAPK1 are in silico predicted to be putative miR-181a targets. We hypothesized that higher miR-181a levels attenuate AML aggressiveness by targeting RAS and/or its downstream effectors in myeloid blasts, thereby reducing proliferation and decreasing the apoptotic threshold. Therefore, we reasoned that the delivery of synthetic miR-181a mimics may increase the low endogenous levels of miR-181a in AML blasts and lead to anti-leukemic activity.
RESULTS
Anti-leukemic activity of miR-181a
We previously reported that chemotherapy-treated patients with AML with higher miR-181a expression achieved complete remission (CR) more frequently and had longer survival compared to lower miR-181a expressing patients [2, 9]. In line with these clinical observations, we and others showed that miR-181a expression is associated with a higher sensitivity to cytarabine in AML cell lines [45, 46].
These findings led us to postulate a tumor suppressor activity of miR-181a that we first tested by overexpressing or knocking-down miR-181a in the FLT3-ITD positive MV4-11 AML cell line by lentiviral infection (Figure 1A). Overexpression of miR-181a (lenti-181a) inhibited cell growth (Figure 1B; lenti-181a vs. lenti-sc: P = 0.009), whereas downregulation of miR-181a (lenti-anti-181a) enhanced cell proliferation compared to cells transfected with a vector carrying a scramble sequence (lenti-sc) (Figure 1B; lenti-sc vs. lenti-anti-181a: P = 0.028). We next engrafted 5 × 106 virally transduced MV4-11 cells into NOD/SCID mice subcutaneously (n = 3 in each group). On day 11, the average tumor weights for animals engrafted with the lenti-anti-181a or lenti-sc transduced cells were 1.642 ± 0.65 g and 0.076 ± 0.022 g, respectively (Figure 1C). No tumor growth was evident in animals engrafted with lenti-181a transduced cells. On day 23, the average tumor weights for the lenti-sc and the lenti-181a transduced cell-engrafted groups were 0.65 ± 0.49 g and 0.037 ± 0.025 g, respectively (Figure 1C).
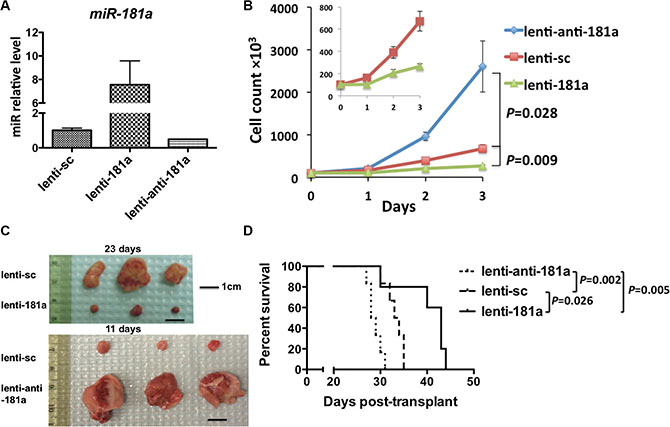
Figure 1: Higher levels of miR-181a are associated with a less aggressive phenotype in AML cells and longer survival in a murine AML model. (A) miR-181a expression in MV4-11 cells after lentiviral infection. (B) Growth curve of MV4-11 cells transduced with lentiviral constructs either overexpressing miR-181a (lenti-181a), expressing a scramble sequence (lenti-sc; = control) or a knock-down construct of miR-181a (lenti-anti-181a). (C) Five million lentiviral transduced cells were engrafted subcutaneously in NOD/SCID mice. At day 11, tumors from lenti-anti-181a and lenti-sc group (n = 3 in each group) were isolated and weighed (no tumor in lenti-181a group). At day 23, tumors from lenti-181a and lenti-sc group (n = 3 in each group) were isolated and weighed. (D) 1.5 million lentiviral transduced MV4-11 cells were engrafted into NSG mice. Survival curves of the mice in the three groups.
To further support the putative tumor suppressor activity of miR-181a, we engrafted NSG mice with virally transduced MV4-11 cells through a tail vein. The median survival for the animals engrafted with the lenti-miR-181a, lenti-sc and lenti-anti-181a transduced cells were 43, 33.5 and 28.5 days, respectively (Figure 1D). Compared to the control group (lenti-sc), the lenti-anti-181a mice lived significantly shorter (P = 0.002, log-rank test) and lenti-miR-181a mice significantly longer (P = 0.02). Though the mice in three groups showed survival time differences, they all died from AML-like disease (Supplementary Figure S1).
We concluded that higher miR-181a expression leads to a less aggressive AML phenotype, thereby functionally validating the previously reported prognostic results [2, 9, 28].
KRAS, NRAS and MAPK1 are direct targets of miR-181a
The RAS-MAPK1 and RAS-AKT-pathways are often aberrantly activated in AML and are known to contribute to myeloid leukemogenesis [29–43]. KRAS has been shown to be a direct miR-181a target in oral squamous cell carcinoma [44]. Here, we first tested whether KRAS and other genes involved in these pathways, including NRAS and its downstream effectors (i.e., MAPK1), were miR-181a targets in AML. Utilizing in silico tools (targetscan.org, http://diana.imis.athena-innovation.gr/ and microrna.org) we first identified putative miR-181a-binding sites in the 3′-untranslated regions (3′-UTRs) of KRAS, NRAS and MAPK1. In contrast, we could not identify putative miR-181a binding sites in the 3′-UTR of HRAS, which is rarely mutated in AML. We then tested whether miR-181a was able to reduce the expression of these genes in AML cells. miR181a overexpression by a lenti-181a vector reduced KRAS, NRAS, and MAPK1 protein levels 5.2, 2.1, and 6.5-fold, respectively, compared to scramble expressing controls in MV4-11 cells (Figure 2A). Consistent with these results, knock-down of miR-181a by a lenti-anti-181a increased KRAS, NRAS and MAPK1 1.5, 1.5 and 1.8-fold compared to scramble controls (Figure 2A).
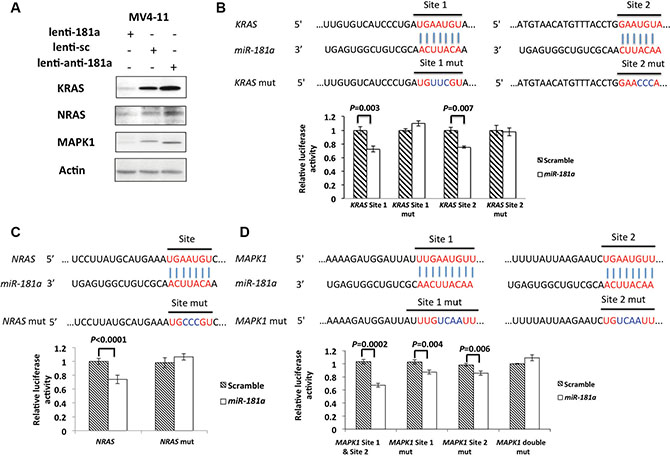
Figure 2: NRAS, KRAS and MAPK1 are direct targets of miR-181a. (A) KRAS, NRAS and MAPK1 protein expression in infected MV4-11 and OCI-AML3 cells with lenti-181a, lenti-sc or lenti-anti-181a. Dual luciferase assays of HEK293T cells co-transfected with firefly luciferase constructs containing the KRAS (B), NRAS (C) and MAPK1 (D) wild-type or mutated 3′-UTRs and miR-181a mimics or scramble mimics (as controls). The firefly luciferase activity was normalized to Renilla luciferase activity. The data are shown as relative luciferase activity of miR-181a mimic transfected cells with respect to the scramble control of nine data points from three independent transfections. Error bars represent the standard deviation (SD).
Next we showed that the modulation of KRAS, NRAS and MAPK1 expression by miR-181a was caused by direct binding to the respective 3′-UTRs. We first validated KRAS as a direct miR-181a target. We identified two miR-181a-binding sites in the KRAS 3′-UTR and observed a 28 ± 4% (P = 0.003) and a 25 ± 1% (P = 0.007) downregulation of luciferase activity on site 1 and site 2 after co-transfecting 293T cells with miR-181a compared with scramble expressing controls. Mutations in the seed sequences of the KRAS 3′-UTRs rescued the miR-181a-induced downregulation (Figure 2B). Next, to demonstrate that NRAS is also a direct miR-181a target, we cloned the predicted miR-181a-binding-site in the NRAS 3′-UTR into a luciferase reporter, and we observed a 26 ± 6% (P < 0.0001) downregulation of luciferase activity. An introduced mutation in the seed sequence rescued the miR-181a-induced downregulation (Figure 2C). We also identified two putative miR-181a binding sites in the MAPK1 3′-UTR. Because of the short distance between the two binding sites (149 base pairs), we cloned the two binding sites into the same luciferase reporter construct. We observed a 33 ± 2% (P = 0.0002) downregulation of luciferase activity with miR-181a treatment compared to cells with scramble control treatment. When we mutated the two sites separately, we observed a 13 ± 3% (site 1; P = 0.004) and a 15 ± 3% (site 2; P = 0.006) miR-181a- induced downregulation of the luciferase activity. However, mutations on both sites of MAPK1 could completely rescue the miR-181a-induced downregulation (Figure 2D). Collectively, these results support that KRAS, NRAS and MAPK1 are direct miR-181a targets.
Delivery of synthetic miR-181a mimic by transferrin (Tf)-conjugated nanoparticles (NP) enhanced miR-181a levels and inhibited RAS-dependent signaling pathways in AML
Because higher miR-181a levels are associated with improved outcomes in AML [2, 9, 26–28], and because miR-181a downregulation contributed to leukemia growth (Figure 1) and directly targeted KRAS, NRAS and MAPK1, we reasoned that increasing miR-181a may have therapeutic value in AML. We have previously demonstrated the successful delivery of miR mimics to AML blasts via transferrin (Tf)-targeted anionic lipid-based lipopolyplex nanoparticles (NP) [47]. Here, we used a similar approach to deliver synthetic miR-181a mimics. We chose KG1a, MV4-11 and OCI-AML cells as models because of the relatively low miR-181a levels and activated RAS pathways (Supplementary Figure S2). Following treatment with Tf-NPs encapsulating miR-181a double-stranded mimic molecules (Tf-NP-miR-181a; 10 nM) or scramble control molecules (Tf-NP-sc; 10 nM), levels of mature miR-181a were measured by qRT-PCR. After 24 hours exposure, mature miR-181a levels increased 211 ± 31, 880 ± 10 and 142 ± 10-fold in KG1a, OCI-AML3 and MV4-11 cells, respectively, whereas levels of miR-181b and unrelated miR-140 remained unchanged (Figure 3A).
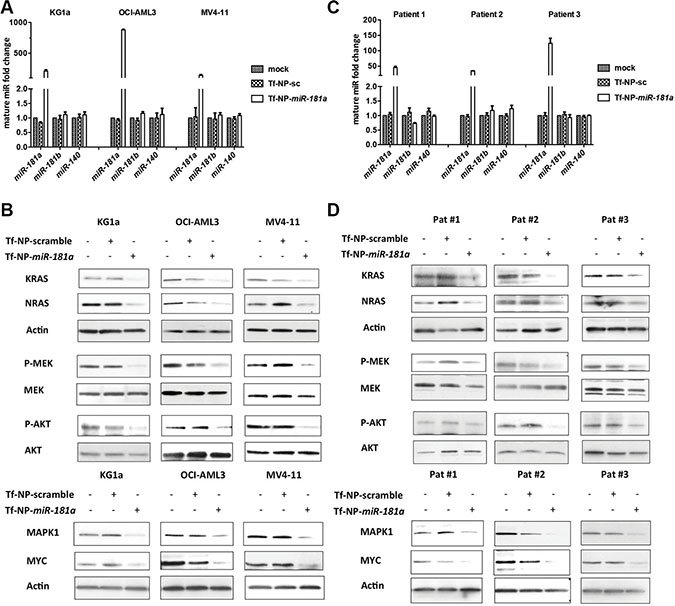
Figure 3: Treatment with Tf-NP-miR-181a increased mature miR-181a levels; downregulated KRAS, NRAS, and MAPK1; and inhibited the RAS-MAPK1 signaling pathway. Mature miR-181a, miR-181b and miR-140 expression levels in KG1a, OCI-AML3 and MV4-11 cells (A) and primary patient blasts (n = 3) (C). NRAS, KRAS, p-MEK, MEK, p-AKT, AKT, MAPK1, and MYC expression in KG1a, OCI-AML3 and MV4-11 cells (B) and primary patient blasts (n = 3) (D) treated with mock, Tf-NP-sc and Tf-NP-miR-181a.
Having shown that the Tf-NP-delivery-system was able to deliver miR-181a to AML blasts, we next tested the impact of Tf-NP-miR-181a on RAS activity. First, we found that the delivered synthetic miR-181a was functional, as it downregulated KRAS, NRAS and MAPK1 proteins (KG1a: 4.3, 4.4 and 5.5-fold; OCI-AML3: 3.2, 3.9 and 2.2-fold; MV4-11: 1.5, 4.4 and 4.6-fold, respectively) compared to Tf-NP-sc treatment (Figure 3B). Compared to Tf-NP-sc, Tf-NP-miR-181a decreased p-MEK protein by 6.8, 2.2 and 4.5-fold and p-AKT protein by 2.0, 2.5 and 5.7-fold in KG1a, OCI-AML3 and MV4-11 cells, respectively (Figure 3B). Finally, we assessed the expression of the oncogenic transcription factor MYC, whose protein stability is enhanced by the RAS-MAPK1 phosphorylation pathway [48]. There was a 4.8, 4.3 and 7.8-fold reduction of MYC protein in KG1a, OCI-AML3 and MV4-11 treated with Tf-NP-miR-181a compared to those treated with Tf-NP-sc control (Figure 3B). Consistent with these results, anti-miR-181a treatment resulted in upregulation of the KRAS, NRAS and MAPK1 proteins in HL60 cells that present with higher levels of endogenous miR-181a (Supplementary Figures S2A and S3).
To validate these results, we treated primary AML blasts having activated RAS from three AML patients (Patient No 1-3; Supplementary Table S2) (Supplementary Figure S2) with Tf-NP-miR-181a and again observed an increase in miR-181a (Figure 3C). After 24 hours, mature miR-181a levels increased 45 ± 4, 35 ± 0.1 and 125 ± 16-fold, respectively, in the three patient blasts samples treated with Tf-NP-miR-181a compared to the Tf-NP-sc treated controls, whereas levels of miR-181b and miR-140 remained unchanged (Figure 3C). Increased levels of miR-181a resulted in decreased protein levels of KRAS, NRAS and MAPK1 by 6.3, 6.8 and 5.6-fold in patient 1; 6.4, 1.6 and 19.7-fold in patient 2; and 2.3, 2.4 and 3.4-fold in patient 3, respectively (Figure 3D). Downregulation of RAS and MAPK1 resulted in RAS-MAPK1 inhibition, decreased MEK and AKT phosphorylation and decreased MYC levels. We observed a 1.4, 3.5 and 2.0-fold decrease of p-MEK, 1.8, 9.3 and 2.0-fold decrease of p-AKT, as well as a 5.3, 7.6 and 2.8-fold decrease of MYC normalized in the patient blasts treated with Tf-NP-miR-181a compared to Tf-NP-sc treatment (Figure 3D).
In summary, we showed the effective delivery of miR-181a via Tf-conjugated nanoparticles and in turn downregulation of KRAS, NRAS and MAPK1 and inhibition of the RAS-MAPK1 and RAS-AKT-kinase signaling cascade.
Tf-NP-miR-181a treatment in AML cells
Next, we demonstrated the anti-leukemic activity of the Tf-NP-miR-181a, which led to reduced proliferation of KG1a cells by 40% (P = 0.015), OCI-AML3 cells by 25% (P = 0.023) and MV4-11 cells by 32% (P < 0.0001) after 72 hours compared to Tf-NP-sc control (Figure 4A). To validate the RAS-MAPK1 and RAS-AKT-kinase-pathways as relevant anti-leukemic miR-181a targets, we treated KG1a and MV4-11 cells with Tf-NP loaded with siRNAs for KRAS, NRAS and MAPK1 (Supplementary Figure S4A). Following this treatment, we observed a similar anti-leukemic effect. The combined siRNA treatment reduced proliferation of KG1a cells by 32% and MV4-11 cells by 30% compared with scramble siRNA treatment (Supplementary Figure S4B). The reduced proliferation induced by Tf-NP-miR-181a treatment was reversed by lentiviral expression of KRAS, NRAS and MAPK1 in OCI-AML3 cells (Supplementary Figure S5A–S5C; Supplementary Table S3) attenuating the anti-leukemic activity of Tf-NP-miR-181a and thereby supporting the relevance of these targets to leukemogenesis. We also observed a more than 50% reduction of colony formation following Tf-NP-miR-181a treatment after 2 weeks (Figure 4B). The average number of colonies formed with mock treatment (buffer only), Tf-NP-sc control and Tf-NP-miR-181a treatment were, respectively, 145 ± 7, 145 ± 11 and 44 ± 3 (P = 0.0002 compared to Tf-NP-sc) for KG1a, 176 ± 11, 172 ± 8 and 80 ± 6 (P < 0.0001 compared to Tf-NP-sc) for OCI-AML3 and 217 ± 42, 180 ± 17 and 82 ± 15 (P = 0.0001 compared to Tf-NP-sc) for MV4-11.
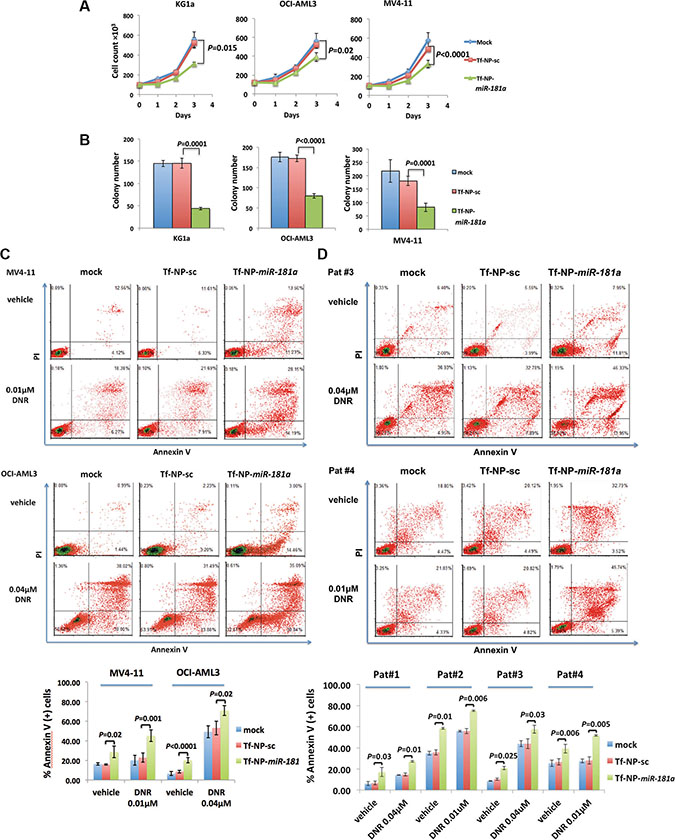
Figure 4: Treatment with Tf-NP-miR-181a had anti-leukemic activity in AML cells. Cell growth curve (A) and colony numbers (B) of KG1a, OCI-AML3 and MV4-11 cells treated with Tf-NP-miR-181a, Tf-NP-sc or mock. Error bars represent SD. Annexin V assays in MV4-11 and OCI-AML3 cells (C) as well as patient blast cells (D) treated with Tf-NP-miR-181a, Tf-NP-sc or mock in the presence or absence of daunorubicin (DNR, 0.01 μM for MV4-11, 0.04 μM for OCI-AML3, 0.01 μM for patient 1 [Pat #1] and patient 3 [Pat #3], 0.04 μM for patient 2 [Pat #2] and patient 4 [Pat #4] blasts). DNR was added 24 hours after priming cells with nanoparticle-miR treatment for another 72 hours.
Treatment with Tf-NP-miR-181a induced apoptosis in both MV4-11 (28.69 ± 5.88% vs. 15.92 ± 0.7% annexinV+, P = 0.02) and OCI-AML3 cells (20.15 ± 2.58% vs. 8.54 ± 1.42% annexinV+, P < 0.0001) compared to Tf-NP-sc treatment at 96 hours (Figure 4C). Following a combined siRNA treatment with Tf-NP loaded with siRNAs for KRAS, NRAS and MAPK1, we observed similar effects in MV4-11 and OCI-AML3 cells (Supplementary Figure S4C). In addition, after 24 hours of priming cells with miR-181a, daunorubicin (DNR) was added to treat the cells for another 72 hours. We observed that miR-181a treatment enhanced the apoptotic effect of DNR in MV4-11 (miR-181a - > 0.01 μM DNR: 45.27 ± 5.99% vs. scramble - > 0.01 μM DNR: 22.88 ± 4.61% annexinV+, P = 0.001) and OCI-AML3 (miR-181a - > 0.04 μM DNR: 70.92 ± 5.01% vs. scramble - > 0.04 μM DNR: 53.25 ± 7.06% annexinV+, P = 0.02; Figure 4C). We also observed similar effects priming MV4-11 and OCI-AML3 cells with siRNAs for KRAS, NRAS and MAPK1 (Supplementary Figure S4C). We then validated our observation in primary patient blasts. Tf-NP-miR-181a induced apoptosis in all four patient blast samples compared to Tf-NP-sc controls (patient 1: 17.04 ± 4.22% vs. 6.66 ± 1.73% annexinV+, P = 0.03; patient 2: 58.53 ± 0.81% vs. 35.73 ± 2.41% annexinV+, P = 0.01; patient 3: 20.86 ± 1.55% vs. 10.32 ± 1.1% annexinV+, P = 0.025; patient 4: 39.28 ± 4.19% vs. 26.70 ± 2.95% annexinV+, P = 0.006; Figure 4D). When exposed to DNR for 72 hours, the Tf-NP-miR-181a treated cells exhibited increased apoptosis compared with control cells (patient 1 exposed to 0.04 μM DNR: 27.28 ± 0.87% vs. 14.75 ± 1.36% annexinV+, P = 0.01; patient 2 exposed to 0.01 μM DNR: 75.16 ± 0.71 vs. 55.91 ± 2.42% annexinV+, P = 0.006; patient 3 exposed to 0.04 μM DNR: 57.61 ± 3.77% vs. 43.99 ± 4.7% annexinV+, P = 0.03; patient 4 exposed to 0.01 μM DNR: 51.61 ± 0.68% vs. 28.06 ± 3.42% annexinV+, P = 0.005; Figure 4D).
Systemic delivery of Tf-NP-miR-181a in an AML mouse model
Next, we examined the anti-leukemic activity of Tf-NP-miR-181a in vivo. Saline (control), Tf-NP-sc or Tf-NP-miR-181a were administrated (1.5 mg/kg/d miR three times/week) through a tail vein 10 days after the engraftment of MV4-11 cells in NSG mice (each group n = 11). Randomly, three mice from each group (i.e. saline, Tf-NP-sc or Tf-NP-miR-181a treated group) were sacrificed after eight treatment doses. The spleen weights were measured and resulted in 187.3 ± 25.93 mg, 174.3 ± 13.65 mg and 77 ± 50 mg (vs. Tf-NP-sc; P = 0.03) in the saline, Tf-NP-sc and Tf-NP-miR-181a groups, respectively (Figure 5A). The spleen weight was 58.3 ± 10.5 mg for age-matched blank control mice (Supplementary Figure S6). Cytospins of bone marrow cells and histopathology of sternum, spleen and liver sections from MV4-11 cell engrafted mice treated with either saline or Tf-NP-sc showed infiltration of blast cells. In contrast, cytospins of bone marrow cells and histopathology of sternum, spleen and liver from Tf-NP-miR-181a treated leukemic mice were similar to that of the age-matched blank control groups (Figure 5B). Furthermore, the population of leukemic cells in spleen samples, measured by flow cytometry, was significantly reduced in mice treated with Tf-NP-miR-181a compared to mice treated with Tf-NP-sc or saline (Figure 5C). We observed a 2.6-fold and a 35-fold increase of miR-181a levels in MV4-11 cells harvested and sorted from bone marrow and spleens, respectively, in the Tf-NP-miR-181a treated mice compared to Tf-NP-sc (Figure 5D and 5E). In these cells, RAS and MAPK1 proteins were downregulated in the Tf-NP-miR-181a treated mice (Figure 5D and 5F).
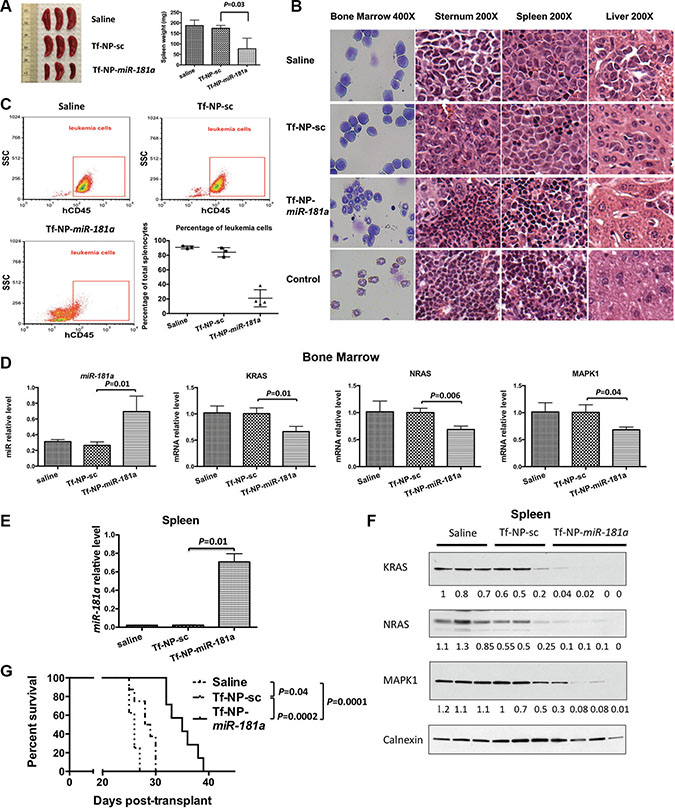
Figure 5: In vivo evaluation of Tf-NP-miR-181a treatment. (A) Spleens and spleen weights from mice sacrificed after 8 doses of treatment from each group: saline, Tf-NP-sc and Tf-NP-miR-181a (n = 3). (B) May-Grünwald/Giemsa staining of bone marrow cells and H&E staining of sections from sternum, spleen and liver of MV4-11 engrafted mice treated with saline, Tf-NP-sc and Tf-NP-miR-181a. NSG mice without MV4-11 engraftment were also used as controls. (C) Leukemic cell population from the spleens harvested from differently treated mice and assessed by flow cytometry. (D) Mature miR-181a levels and KRAS, NRAS and MAPK1 RNA levels in sorted MV4-11 cells from bone marrow samples harvested from differently treated mice. Error bars represent SD. (E) Mature miR-181a levels in sorted MV4-11 cells from spleens harvested from differently treated mice. Error bars represent SD. (F) KRAS, NRAS and MAPK1 protein expression in sorted MV4-11 cells from spleens harvested from differently treated mice. (G) Survival curves of the mice according to the indicated treatment.
The median survival time of the remaining mice was 26, 28.5 and 35 days for the animal groups treated with saline, Tf-NP-sc and Tf-NP-miR-181a, respectively. Tf-NP-miR-181a treatment significantly reduced the disease burden and prolonged survival compared to Tf-NP-sc (P = 0.0002) or saline (P = 0.0001) treatment (Figure 5G). Interestingly, Tf-NP-sc treatment also had some minor anti-leukemic effects compared to the saline treated control group (P = 0.04).
DISCUSSION
MiRs have been implicated in leukemogenesis, and the expression levels of several miRs have been shown to impact the prognosis of AML patients [1–9, 12–14]. Relatively low expression of miR-181a is associated with worse outcomes in AML patients [2, 9, 28]. Here, we provided evidence that AML cells with reduced levels of miR-181a had a more aggressive AML phenotype, and we validated this clinical observation functionally.
In other types of cancers miR-181a has been associated with both tumor suppressor and oncogene functions [20–28], implying context-specific effects. Whereas in colorectal cancer [20] and lymph node metastasis in oral squamous cell carcinoma [21] a high miR-181 level seems to be associated with worse clinical outcomes, in glioma this miR has tumor suppressor function [22]. In these brain tumors miR-181a was shown to target the anti-apoptotic genes BCL2 and MCL1, and downregulated miR-181a reduced glucose deprivation-induced apoptosis and caused mitochondrial dysfunction in astrocytes [22, 49, 50]. The miR-181-family has been reported to be an effector in inflammatory response by TNF-α, IL-6, IL-1β, IL-8 and IL-10 [17, 18, 51–53]. With regard to AML, we previously provided preliminary evidence that miR-181 may target elements of the “inflammasome” that ultimately lead to NF-κB activation and leukemia growth, while Li et al. showed that miR-181 promoted apoptosis, reduced viability and delayed leukemogenesis in MLL-rearranged AML by downregulating the homeobox gene PBX3 [28]. Bai et al. also demonstrated that miR-181a may reduce BCL2 and thus enhance chemosensitivity of AML cells [46]. However, the mechanisms through which miR-181a attenuates disease aggressiveness and the full spectrum of its targets still remain to be fully understood in AML.
Here, we first demonstrated that miR-181a targets the RAS-MAPK1 and RAS-AKT pathways, which have been found to be activated and support AML leukemogenesis [54–58]. Despite extensive efforts, the direct therapeutic targeting of these pathways with small molecule inhibitors remains challenging [59]. Our results show that KRAS, NRAS and MAPK1 proteins may be effectively reduced by utilizing RNA compounds mimicking miR-181a. The efficient delivery of miR-181a mimics by Tf-NPs decreased the targets and their downstream effectors (AKT, MEK, MYC). Altogether, our results support miR-181a replacement as a potential anti-leukemic, RAS targeting strategy in AML. The therapeutic advantage of using miR mimics is in the simultaneous targeting of cross-talking signal transduction pathways (STPs) [38]. Although a use of synthetic mimics may be of relatively difficult in therapeutic application especially compared to the use of anti-miR oligonucleotide, it has been postulated for several types of cancers and is currently being tested in clinical trials (e.g. for miR-34 in NCT01829971). One of the limitations of miR-based therapies is in the optimal delivery of these oligonucleotides as they are subject to rapid hepatic uptake and metabolism and are easily degraded by endonucleases in biological matrices. Nevertheless, we recently reported a novel anionic lipopolyplex nanocarrier system that was designed for the purpose of allowing for efficient miR delivery to AML cells [47]. Here we show that this system could be adapted to the delivery of miR-181a mimics and exert an efficient inhibitory effect on the RAS-MAPK1 and RAS-AKT kinase pathways, thereby resulting in a significant anti-leukemic activity. Interestingly, a very mild anti-leukemic effect and a slight downregulating effect of Tf-NP-scramble treatment on NRAS, KRAS, and MAPK1, as well as on MEK phosphorylation and MYC expression in OCI-AML3 cells was observed. This effect was likely mediated by one of the components of our nanoparticle system, for example, linoleic acid. It has been reported that some fatty acids have anti-tumor activity [60–62].
Other strategies to increase miR-181a have also been tested by our group with significant results. In a previous study, we demonstrated that lenalidomide increases endogenous miR-181a [45], by enhancing the expression of C/EBPα isoforms, which bind to the miR-181a promoter and induce the transcription of miR-181a. However, lenalidomide has several unwanted side-effects at the doses necessary to achieve plasma concentrations at which miR-181a was increased. Thus, the targeting NPs that we reported here may present the advantage to be more specifically directed to AML blasts, thereby sparing normal tissues and perhaps reducing unwanted toxicity. Our preclinical studies showed encouraging results with no toxicity in NP-treated mice at doses inducing anti-leukemic effects [47]. It should also be underscored that we and others have reported that increased levels of miR-181 lead to enhancement of sensitivity to chemotherapy in AML models [45, 46, 63]. Furthermore, patients with higher levels of miR-181a have a better complete remission rate and longer survival compared with those with lower levels, further supporting a role of this miR as a modifier of the response to chemotherapy [9]. Thus, we envision that potential clinical benefit of miR-181a replacement will be more likely if applied in combination with chemotherapy.
In summary, we unveil here a previously unreported activity of miR-181a that directly downregulates NRAS, KRAS and MAPK1 and RAS-dependent downstream signals supporting leukemogenesis. We showed that a nanoparticle-based delivery system could be used to efficiently increase otherwise low levels of miR-181a and achieve anti-leukemic activity in AML models with no evident toxicity. On the basis of our results, miR-181a-NP may warrant further evaluation for potential clinical applications in AML and other RAS-dependent malignancies.
MATERIALS AND METHODS
Cell lines and patient samples
KG1a, MV4-11, HL60, HEK 293T and HEK 293TN cells were obtained from ATCC (Manassas, VA); OCI-AML3 cells were obtained from DSMZ (Braunschweig, Germany). Primary, unselected AML blasts from apheresis samples collected from nine patients were obtained from The Ohio State University (OSU) Leukemia Tissue Bank. Patients signed an informed consent to store and use their tissue for discovery studies according to OSU institutional guidelines.
Lentiviral infections
The lentiviral infections were performed as previously described [7]. The stemloop of miR-181a with 200 bp flanking sequence was cloned into the HIV based lentiviral dual promoter vector (pCDH-CMV-MCS-EF1-copGFP+Puro cDNA; System Biosciences, Mountain View, CA). The miRZip anti-miR-181a (lenti-anti-181a) and scramble vectors were purchased from System Biosciences.
Luciferase assays
Luciferase assays were carried out as previously described [7]. 293T cells were co-transfected with luciferase vector (pGL4.24), Renilla control vector and miR-181a mimic or scramble control. Luciferase activity was normalized to Renilla activity. See supplementary material for more detailed information.
Nanoparticle preparation and treatment
The synthetic double-stranded miR-181a, miR-scramble (sc), and KRAS, NRAS and MAPK1 siRNAs were purchased from Ambion. Nanoparticle preparation was performed as previously described [47, 64, 65]. Briefly, polyethylenimine was used to capture miRs/siRNAs, and the complex was loaded to pre-made anionic liposomal nanoparticles which consists of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dimyristoyl-sn-glycerol, methoxypolyethylene glycol (DMG-PEG) and linoleic acid. Transferrin was first conjugated with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000 maleimide) and then post-inserted to the miR loaded nanoparticle to form the final product. The final concentration of the miRs/siRNAs was 10 nM and was used for all in vitro studies. Protein was collected at 24 and 48 hours for western blot analysis.
Quantitative RT-PCR (qRT-PCR)
Total RNA was extracted with TRIzol reagent (Invitrogen). cDNA was synthesized using Superscript III (Invitrogen) or the Taqman miR Reverse Transcription kit (Applied Biosystems, Foster City, CA) for miR-181a, miR-181b, miR-140 and U44. qRT-PCR was performed with Taqman gene expression assays (Applied Biosystems) following the manufacturer’s protocols. miR-181a, miR-181b and miR-140 expression were normalized to U44. KRAS, NRAS and MAPK1 expression were normalized to GAPDH. The comparative cycle threshold (CT) method as previously described was used for relative quantification of gene expression [47].
Western blot analysis
Anti-KRAS (ab55391) antibodies were purchased from Abcam (Cambridge, MA). Anti-NRAS (C-20, sc-519) and Anti-MYC (N-262, sc-764) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-MAPK1, –MEK1/2 (L38C12), –p-MEK1/2 (S217/221,41G9), AKT and p-AKT (S473, D9E) antibodies were purchased from Cell Signaling Technology (Beverly, MA). Equivalent gel loading was confirmed by probing with antibodies against actin (sc-1616; Santa Cruz) or calnexin (C5C9; Cell Signaling). The intensity of the resulting bands was measured by ImageJ 1.48 s (http://imagej.nih.gov/ij). The intensity ratio of each band respective to the corresponding actin intensity was used for relative quantification.
Growth curves
Lentivirally transduced MV4-11 cells (1 × 105/mL) were plated in 12-well plates. KG1a, OCI-AML3 and MV4-11 cells (1 × 105/mL) were plated in 12-well plates and treated with nanoparticles (Tf-NP-sc or Tf-NP-miR-181a at a final concentration of 10 nM) or were mock treated (buffer only). Cells were harvested and counted at 24-hour intervals using a Bio-Rad TC20 Automated Cell Counter (Bio-Rad, Berkeley, CA). Each sample was run in triplicate.
Colony assays
Methylcellulose colony formation assays were carried out as previously described [66] and counted after 15 days.
Apoptosis assays
MV4-11 and OCI-AML3 cells and four AML patient blast samples cells were treated with Tf-NP-miR-181a, siRNAs, Tf-NP-sc and mock for 24 hours. The cells were then subsequently treated with daunorubicin (DNR; 0.01 μM for MV4-11, 0.04 μM for OCI-AML3, 0.04 μM for patient #1 and #3, 0.01 μM for patient #2 and #4 blasts; Sigma-Aldrich, St Louis, MO) or vehicle control (phosphate-buffered saline; Sigma-Aldrich) for another 72 hours. Annexin V/propidium iodide (PI) stain (BD Biosciences, San Jose, CA) was performed.
In vivo studies
Animal studies were performed according to the Ohio State University institutional guidelines. A total of 5 million lentiviral transduced MV4-11 cells were injected subcutaneously into eight-week female NOD/SCID gamma mice (NSG; The Jackson Laboratory, Bar Harbor, ME). At day 11, 3 mice from each lenti-anti-181a and lenti-sc group were sacrificed, and tumors were weighed. At day 23, 3 mice from each lenti-sc and lenti-181a group were sacrificed, and tumors were weighed.
For the functional study, six-week-old NSG mice were injected with 0.15 million lentivirally transduced MV4-11 cells intravenously through a tail vein (n = 6 in each group: lenti-anti-181a, lenti-sc and lenti-181a).
For the therapeutic study, six-week-old NSG mice were injected with 0.3 million MV4-11 cells intravenously through a tail vein. The treatment started 10 days after the engraftment. Mice were treated with saline, Tf-NP-sc or Tf-NP-miR-181a (1.5 mg/kg/d three times/week). Randomly, 3 mice of each group were sacrificed after 8 doses of treatment for pathology analysis. Age-matched NGS mice without MV4-11 cell engraftment were used as blank control. The treatment was continued for the remaining mice. Eight mice from each group were monitored for survival. The experiment was repeated for biomarker analysis. Bone marrow and spleen cells were isolated from scarified mice and sorted for human CD45-positive cells for further analysis.
Statistical analysis
Data are presented as mean ± SD of at least 3 independent experiments and analyzed by the two-tailed Student’s t-test. The mean and SD were calculated and displayed in bar graphs as the height and the corresponding error bar, respectively. Mouse survival was calculated using the Kaplan–Meier method, and survival curves were compared by the log-rank test.
ACKNOWLEDGMENTS
We thank Donna Bucci and the OSU Leukemia Tissue Bank for assistance with the AML patient samples. We thank Bin Zhang and James Sanchez from The Gehr Family Center for Leukemia Research, City of Hope for their assistance in reviewing the manuscript.
CONFLICTS OF INTEREST
The authors declare no competing financial interests.
GRANT SUPPORT
Supported by CA135243, CA102031, CA140158, EEC-0914790, and the Pelotonia Fellowship Program (A.K.E).
REFERENCES
1. Marcucci G, Mrozek K, Radmacher MD, Garzon R, Bloomfield CD. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood. 2011; 117:1121–1129.
2. Marcucci G, Radmacher MD, Maharry K, Mrozek K, Ruppert AS, Paschka P, Vukosavljevic T, Whitman SP, Baldus CD, Langer C, Liu CG, Carroll AJ, Powell BL, et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008; 358:1919–1928.
3. Jiang X, Huang H, Li Z, He C, Li Y, Chen P, Gurbuxani S, Arnovitz S, Hong GM, Price C, Ren H, Kunjamma RB, Neilly MB, et al. MiR-495 is a tumor-suppressor microRNA down-regulated in MLL-rearranged leukemia. Proc Natl Acad Sci USA. 2012; 109:19397–19402.
4. Diaz-Beya M, Brunet S, Nomdedeu J, Tejero R, Diaz T, Pratcorona M, Tormo M, Ribera JM, Escoda L, Duarte R, Gallardo D, Heras I, Queipo de Llano MP, et al. MicroRNA expression at diagnosis adds relevant prognostic information to molecular categorization in patients with intermediate-risk cytogenetic acute myeloid leukemia. Leukemia. 2014; 28:804–812.
5. Rucker FG, Russ AC, Cocciardi S, Kett H, Schlenk RF, Botzenhardt U, Langer C, Krauter J, Frohling S, Schlegelberger B, Ganser A, Lichter P, Zenz T, et al. Altered miRNA and gene expression in acute myeloid leukemia with complex karyotype identify networks of prognostic relevance. Leukemia. 2013; 27:353–361.
6. Sun SM, Rockova V, Bullinger L, Dijkstra MK, Dohner H, Lowenberg B, Jongen-Lavrencic M. The prognostic relevance of miR-212 expression with survival in cytogenetically and molecularly heterogeneous AML. Leukemia. 2013; 27:100–106.
7. Eisfeld AK, Marcucci G, Maharry K, Schwind S, Radmacher MD, Nicolet D, Becker H, Mrozek K, Whitman SP, Metzeler KH, Mendler JH, Wu YZ, Liyanarachchi S, et al. miR-3151 interplays with its host gene BAALC and independently affects outcome of patients with cytogenetically normal acute myeloid leukemia. Blood. 2012; 120:249–258.
8. Marcucci G, Maharry KS, Metzeler KH, Volinia S, Wu YZ, Mrozek K, Nicolet D, Kohlschmidt J, Whitman SP, Mendler JH, Schwind S, Becker H, Eisfeld AK, et al. Clinical role of microRNAs in cytogenetically normal acute myeloid leukemia: miR-155 upregulation independently identifies high-risk patients. J Clin Oncol. 2013; 31:2086–2093.
9. Schwind S, Maharry K, Radmacher MD, Mrozek K, Holland KB, Margeson D, Whitman SP, Hickey C, Becker H, Metzeler KH, Paschka P, Baldus CD, Liu S, et al. Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. 2010; 28:5257–5264.
10. Eisfeld AK, Schwind S, Patel R, Huang X, Santhanam R, Walker CJ, Markowitz J, Hoag KW, Jarvinen TM, Leffel B, Perrotti D, Carson WE 3rd, Marcucci G, et al. Intronic miR-3151 within BAALC drives leukemogenesis by deregulating the TP53 pathway. Sci Signal. 2014; 7:ra36.
11. Havelange V, Ranganathan P, Geyer S, Nicolet D, Huang X, Yu X, Volinia S, Kornblau SM, Andreeff M, Croce CM, Marcucci G, Bloomfield CD, Garzon R. Implications of the miR-10 family in chemotherapy response of NPM1-mutated AML. Blood. 2014; 123:2412–2415.
12. Blum W, Garzon R, Klisovic RB, Schwind S, Walker A, Geyer S, Liu S, Havelange V, Becker H, Schaaf L, Mickle J, Devine H, Kefauver C, et al. Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc Natl Acad Sci USA. 2010; 107:7473–7478.
13. Blum W, Schwind S, Tarighat SS, Geyer S, Eisfeld AK, Whitman S, Walker A, Klisovic R, Byrd JC, Santhanam R, Wang H, Curfman JP, Devine SM, et al. Clinical and pharmacodynamic activity of bortezomib and decitabine in acute myeloid leukemia. Blood. 2012; 119:6025–6031.
14. Mims A, Walker AR, Huang X, Sun J, Wang H, Santhanam R, Dorrance AM, Walker C, Hoellerbauer P, Tarighat SS, Chan KK, Klisovic RB, Perrotti D, et al. Increased anti-leukemic activity of decitabine via AR-42-induced upregulation of miR-29b: a novel epigenetic-targeting approach in acute myeloid leukemia. Leukemia. 2013; 27:871–878.
15. Blum W, Klisovic RB, Walker A, Schwind S, Jiang Y, Wang J, Phelps M, Geyer S, Devine SM, Walsh K. Priming of Mir-181a in Acute Myeloid Leukemia (AML) to Increase Chemosensitivity: A Phase I Trial of Lenalidomide (LEN) Followed by Idarubicin and Cytarabine. Blood (ASH Annual Meeting Abstracts) 2012; 120:2619.
16. Khalife J, Radomska HS, Santhanam R, Huang X, Neviani P, Saultz J, Wang H, Wu YZ, Alachkar H, Anghelina M, Dorrance A, Curfman J, Bloomfield CD, et al. Pharmacological targeting of miR-155 via the NEDD8-activating enzyme inhibitor MLN4924 (Pevonedistat) in FLT3-ITD acute myeloid leukemia. Leukemia. 2015.
17. Henao-Mejia J, Williams A, Goff LA, Staron M, Licona-Limon P, Kaech SM, Nakayama M, Rinn JL, Flavell RA. The microRNA miR-181 is a critical cellular metabolic rheostat essential for NKT cell ontogenesis and lymphocyte development and homeostasis. Immunity. 2013; 38:984–997.
18. Zietara N, Lyszkiewicz M, Witzlau K, Naumann R, Hurwitz R, Langemeier J, Bohne J, Sandrock I, Ballmaier M, Weiss S, Prinz I, Krueger A. Critical role for miR-181a/b-1 in agonist selection of invariant natural killer T cells. Proc Natl Acad Sci USA. 2013; 110:7407–7412.
19. Li X, Zhang J, Gao L, McClellan S, Finan MA, Butler TW, Owen LB, Piazza GA, Xi Y. MiR-181 mediates cell differentiation by interrupting the Lin28 and let-7 feedback circuit. Cell Death Differ. 2012; 19:378–386.
20. Pichler M, Winter E, Ress AL, Bauernhofer T, Gerger A, Kiesslich T, Lax S, Samonigg H, Hoefler G. miR-181a is associated with poor clinical outcome in patients with colorectal cancer treated with EGFR inhibitor. J Clin Pathol. 2014; 67:198–203.
21. Yang CC, Hung PS, Wang PW, Liu CJ, Chu TH, Cheng HW, Lin SC. miR-181 as a putative biomarker for lymph-node metastasis of oral squamous cell carcinoma. J Oral Pathol Med. 2011; 40:397–404.
22. Shi L, Cheng Z, Zhang J, Li R, Zhao P, Fu Z, You Y. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008; 1236:185–193.
23. Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013; 123:150–163.
24. Zhu Y, Wu J, Li S, Ma R, Cao H, Ji M, Jing C, Tang J. The function role of miR-181a in chemosensitivity to adriamycin by targeting Bcl-2 in low-invasive breast cancer cells. Cell Physiol Biochem. 2013; 32:1225–1237.
25. Jiao X, Zhao L, Ma M, Bai X, He M, Yan Y, Wang Y, Chen Q, Zhao X, Zhou M, Cui Z, Zheng Z, Wang E, et al. MiR-181a enhances drug sensitivity in mitoxantone-resistant breast cancer cells by targeting breast cancer resistance protein (BCRP/ABCG2). Breast Cancer Res Treat. 2013; 139:717–730.
26. Zhu DX, Zhu W, Fang C, Fan L, Zou ZJ, Wang YH, Liu P, Hong M, Miao KR, Liu P, Xu W, Li JY. miR-181a/b significantly enhances drug sensitivity in chronic lymphocytic leukemia cells via targeting multiple anti-apoptosis genes. Carcinogenesis. 2012; 33:1294–1301.
27. Lin S, Pan L, Guo S, Wu J, Jin L, Wang JC, Wang S. Prognostic role of microRNA-181a/b in hematological malignancies: a meta-analysis. PLoS One. 2013; 8:e59532.
28. Li Z, Huang H, Li Y, Jiang X, Chen P, Arnovitz S, Radmacher MD, Maharry K, Elkahloun A, Yang X, He C, He M, Zhang Z, et al. Up-regulation of a HOXA-PBX3 homeobox-gene signature following down-regulation of miR-181 is associated with adverse prognosis in patients with cytogenetically abnormal AML. Blood. 2012; 119:2314–2324.
29. Chung E, Kondo M. Role of Ras/Raf/MEK/ERK signaling in physiological hematopoiesis and leukemia development. Immunol Res. 2011; 49:248–268.
30. Tyner JW, Erickson H, Deininger MW, Willis SG, Eide CA, Levine RL, Heinrich MC, Gattermann N, Gilliland DG, Druker BJ, Loriaux MM. High-throughput sequencing screen reveals novel, transforming RAS mutations in myeloid leukemia patients. Blood. 2009; 113:1749–1755.
31. Illmer T, Thiede C, Fredersdorf A, Stadler S, Neubauer A, Ehninger G, Schaich M. Activation of the RAS pathway is predictive for a chemosensitive phenotype of acute myelogenous leukemia blasts. Clin Cancer Res. 2005; 11:3217–3224.
32. Kim WI, Matise I, Diers MD, Largaespada DA. RAS oncogene suppression induces apoptosis followed by more differentiated and less myelosuppressive disease upon relapse of acute myeloid leukemia. Blood. 2009; 113:1086–1096.
33. Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood. 2006; 108:2349–2357.
34. Towatari M, Iida H, Tanimoto M, Iwata H, Hamaguchi M, Saito H. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia. 1997; 11:479–484.
35. Wu J, Wong WW, Khosravi F, Minden MD, Penn LZ. Blocking the Raf/MEK/ERK pathway sensitizes acute myelogenous leukemia cells to lovastatin-induced apoptosis. Cancer Res. 2004; 64:6461–6468.
36. Grandage VL, Gale RE, Linch DC, Khwaja A. PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappaB, Mapkinase and p53 pathways. Leukemia. 2005; 19:586–594.
37. Min YH, Eom JI, Cheong JW, Maeng HO, Kim JY, Jeung HK, Lee ST, Lee MH, Hahn JS, Ko YW. Constitutive phosphorylation of Akt/PKB protein in acute myeloid leukemia: its significance as a prognostic variable. Leukemia. 2003; 17:995–997.
38. Xu Q, Simpson SE, Scialla TJ, Bagg A, Carroll M. Survival of acute myeloid leukemia cells requires PI3 kinase activation. Blood. 2003; 102:972–980.
39. Kornblau SM, Womble M, Qiu YH, Jackson CE, Chen W, Konopleva M, Estey EH, Andreeff M. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006; 108:2358–2365.
40. Min YH, Cheong JW, Kim JY, Eom JI, Lee ST, Hahn JS, Ko YW, Lee MH. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res. 2004; 64:5225–5231.
41. Brandwein JM, Hedley DW, Chow S, Schimmer AD, Yee KW, Schuh AC, Gupta V, Xu W, Kamel-Reid S, Minden MD. A phase I/II study of imatinib plus reinduction therapy for c-kit-positive relapsed/refractory acute myeloid leukemia: inhibition of Akt activation correlates with complete response. Leukemia. 2011; 25:945–952.
42. Martelli AM, Nyakern M, Tabellini G, Bortul R, Tazzari PL, Evangelisti C, Cocco L. Phosphoinositide 3-kinase/Akt signaling pathway and its therapeutical implications for human acute myeloid leukemia. Leukemia. 2006; 20:911–928.
43. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007; 7:295–308.
44. Shin KH, Bae SD, Hong HS, Kim RH, Kang MK, Park NH. miR-181a shows tumor suppressive effect against oral squamous cell carcinoma cells by downregulating K-ras. Biochem Biophys Res Commun. 2011; 404:896–902.
45. Hickey CJ, Schwind S, Radomska HS, Dorrance AM, Santhanam R, Mishra A, Wu YZ, Alachkar H, Maharry K, Nicolet D, Mrozek K, Walker A, Eiring AM, et al. Lenalidomide-mediated enhanced translation of C/EBPalpha-p30 protein up-regulates expression of the antileukemic microRNA-181a in acute myeloid leukemia. Blood. 2013; 121:159–169.
46. Bai H, Cao Z, Deng C, Zhou L, Wang C. miR-181a sensitizes resistant leukaemia HL-60/Ara-C cells to Ara-C by inducing apoptosis. J Cancer Res Clin Oncol. 2012; 138:595–602.
47. Huang X, Schwind S, Yu B, Santhanam R, Wang H, Hoellerbauer P, Mims A, Klisovic R, Walker AR, Chan KK, Blum W, Perrotti D, Byrd JC, et al. Targeted delivery of microRNA-29b by transferrin-conjugated anionic lipopolyplex nanoparticles: a novel therapeutic strategy in acute myeloid leukemia. Clin Cancer Res. 2013; 19:2355–2367.
48. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000; 14:2501–2514.
49. Chen G, Zhu W, Shi D, Lv L, Zhang C, Liu P, Hu W. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol Rep. 2010; 23:997–1003.
50. Ouyang YB, Lu Y, Yue S, Giffard RG. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion. 2012; 12:213–219.
51. Hutchison ER, Kawamoto EM, Taub DD, Lal A, Abdelmohsen K, Zhang Y, Wood WH, 3rd, Lehrmann E, Camandola S, Becker KG, Gorospe M, Mattson MP. Evidence for miR-181 involvement in neuroinflammatory responses of astrocytes. Glia. 2013; 61:1018–1028.
52. Cichocki F, Felices M, McCullar V, Presnell SR, Al-Attar A, Lutz CT, Miller JS. Cutting edge: microRNA-181 promotes human NK cell development by regulating Notch signaling. J Immunol. 2011; 187:6171–6175.
53. Chen CZ, Schaffert S, Fragoso R, Loh C. Regulation of immune responses and tolerance: the microRNA perspective. Immunol Rev. 2013; 253:112–128.
54. Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012; 120:3397–3406.
55. Borthakur G, Kantarjian H, Ravandi F, Zhang W, Konopleva M, Wright JJ, Faderl S, Verstovsek S, Mathews S, Andreeff M, Cortes JE. Phase I study of sorafenib in patients with refractory or relapsed acute leukemias. Haematologica. 2011; 96:62–68.
56. Zhao S, Zhang Y, Sha K, Tang Q, Yang X, Yu C, Liu Z, Sun W, Cai L, Xu C, Cui S. KRAS (G12D) Cooperates with AML1/ETO to Initiate a Mouse Model Mimicking Human Acute Myeloid Leukemia. Cell Physiol Biochem. 2014; 33:78–87.
57. Gritsman K, Yuzugullu H, Von T, Yan H, Clayton L, Fritsch C, Maira SM, Hollingworth G, Choi C, Khandan T, Paktinat M, Okabe RO, Roberts TM, et al. Hematopoiesis and RAS-driven myeloid leukemia differentially require PI3K isoform p110alpha. J Clin Invest. 2014; 124:1794–1809.
58. Kothe S, Muller JP, Bohmer SA, Tschongov T, Fricke M, Koch S, Thiede C, Requardt RP, Rubio I, Bohmer FD. Features of Ras activation by a mislocalized oncogenic tyrosine kinase: FLT3 ITD signals through K-Ras at the plasma membrane of acute myeloid leukemia cells. J Cell Sci. 2013; 126:4746–4755.
59. Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014; 25:272–281.
60. Germain E, Chajes V, Cognault S, Lhuillery C, Bougnoux P. Enhancement of doxorubicin cytotoxicity by polyunsaturated fatty acids in the human breast tumor cell line MDA-MB-231: relationship to lipid peroxidation. Int J Cancer. 1998; 75:578–583.
61. Menendez JA, del Mar Barbacid M, Montero S, Sevilla E, Escrich E, Solanas M, Cortes-Funes H, Colomer R. Effects of gamma-linolenic acid and oleic acid on paclitaxel cytotoxicity in human breast cancer cells. Eur J Cancer. 2001; 37:402–413.
62. Menendez JA, Vellon L, Colomer R, Lupu R. Effect of gamma-linolenic acid on the transcriptional activity of the Her-2/neu (erbB-2) oncogene. J Natl Cancer Inst. 2005; 97:1611–1615.
63. She X, Yu Z, Cui Y, Lei Q, Wang Z, Xu G, Luo Z, Li G, Wu M. miR-181 subunits enhance the chemosensitivity of temozolomide by Rap1B-mediated cytoskeleton remodeling in glioblastoma cells. Med Oncol. 2014; 31:892.
64. Dorrance AM, Neviani P, Ferenchak GJ, Huang X, Nicolet D, Maharry KS, Ozer HG, Hoellarbauer P, Khalife J, Hill EB, Yadav M, Bolon BN, Lee RJ, et al. Targeting leukemia stem cells in vivo with ANTAGOMIR-126 nanoparticles in acute myeloid leukemia. Leukemia. 2015; 29:2143–2153.
65. Wang X, Huang X, Yang Z, Gallego-Perez D, Ma J, Zhao X, Xie J, Nakano I, Lee LJ. Targeted delivery of tumor suppressor microRNA-1 by transferrin-conjugated lipopolyplex nanoparticles to patient-derived glioblastoma stem cells. Curr Pharm Biotechnol. 2014; 15:839–846.
66. Neviani P, Santhanam R, Oaks JJ, Eiring AM, Notari M, Blaser BW, Liu S, Trotta R, Muthusamy N, Gambacorti-Passerini C, Druker BJ, Cortes J, Marcucci G, et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia. J Clin Invest. 2007; 117:2408–2421.