
INTRODUCTION
High-grade serous ovarian cancer is the most lethal gynecological cancer due to high rates of relapse and acquired platinum resistance after conventional chemotherapy [1]. Since sensitivity to platinum determines the prognosis of patients with ovarian cancer, it is important to investigate the factors associated with this condition. The identification and differentiation of ovarian cancer patients in terms of their response to platinum-based treatment is central to advancing ovarian cancer management and has been the subject of intense research. Germline or somatic mutations in the BRCA1 or BRCA2 gene have prognostic value in ovarian cancer since ovarian cancer patients with BRCA1/2 mutations are reported to have a better response to platinum-based treatment [2] and subsequently a longer survival duration than patients without such mutations [3-5]. Although BRCA1/2 mutations have been found in 11%-20.3% of patients in several studies [6-8], the overall chemosensitivity rates are approximately 70% [9], suggesting that there are other mutations associated with platinum sensitivity [10].
In our previous study, we reported that approximately 10.4% of a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS) family genes were mutated in patients with high-grade serous ovarian carcinoma. Patients harboring these mutations had significantly longer overall survival (OS), progression-free survival and platinum-free survival independent of BRCA1 or BRCA2 mutation, stage, residual tumor, or age, according to the results of a computational analysis of the whole-exome sequencing data from 512 patients in the Cancer Genome Atlas [11]. This finding suggests that ADAMTS mutations partially account for BRCAness [12]. In that study, mutations of ADAMTS 1, 6, 8, 9, 15, 16, 18, and L1 were detected. ADAMTS16 was one of the most commonly mutated genes.
ADAMTS protease is a secreted extracellular peptidase that mainly consists of three basic structures: a pro-domain, catalytic domain, and ancillary domain [13]. The ADAMTS family has 19 members, and the ADAMTS-like glycoproteins (ADAMTSL) family is often considered the same family because they share similar structures [14-16]. Since the first member of the ADAMTS family was identified in 1997 [17], studies have revealed that the family plays important roles in extracellular matrix development, maintenance, degradation, angiogenesis [13], microfibril biogenesis [16], von Willebrand factor maturation [18], and embryogenesis [19]. The ADAMTS family has been linked to various clinical diseases, including arthritis [20, 21] and thrombotic thrombocytopenic purpura [22, 23]. There is also an increasing number of studies demonstrating the important roles the ADAMTS family plays in the pathogenesis of cancer [24-27]. Mutations of ADAMTS family genes have been detected in various types of cancers, including colorectal, breast, esophageal, and pancreatic cancers and glioblastoma (2, 28-36).
However, the functional role of these mutations in ovarian cancer cells is largely unknown. In this study, we hypothesized that ADAMTS16 mutations sensitize ovarian cancer cells to platinum-based chemotherapy. To test this hypothesis, we performed in vitro studies to compare the drug response in ovarian cancer cells with and without one of the six ADAMTS16 missense mutations. We also used a well-characterized in vivo mouse model to evaluate the response of ovarian cancer cells with ADAMTS16 mutations to platinum-based therapy.
RESULTS
Generation of ADAMTS16 mutant ovarian cancer stable cell lines
With no prior knowledge of the biological function of ADAMTS16 mutations in ovarian cancer cells, we first generated six ADAMTS16 mutants on the basis of the missense mutations detected on this gene [11]: C274R, F660I, S787Y, A1155V, S1170L and K1206 (Figure 1A). A1155V and S1170L are in one of the Thrombospondin 1 domains, S787Y is in the ADAM Spacer 1 domain, K1206M is in the protease and lacunin domain (PLAC) and C274R and F660I are in non-functional designated domains. To determine the role of these mutations in ovarian cancer cells, we established ovarian cancer cell lines that stably expressed empty vector (EV) or WT or each of the six ADAMTS16 missense mutants. ADAMTS16 protein level of stable cell lines was detected in both the cell lysate and conditioned medium (Figure 1B) as previously reported in esophageal squamous cell carcinoma [33].
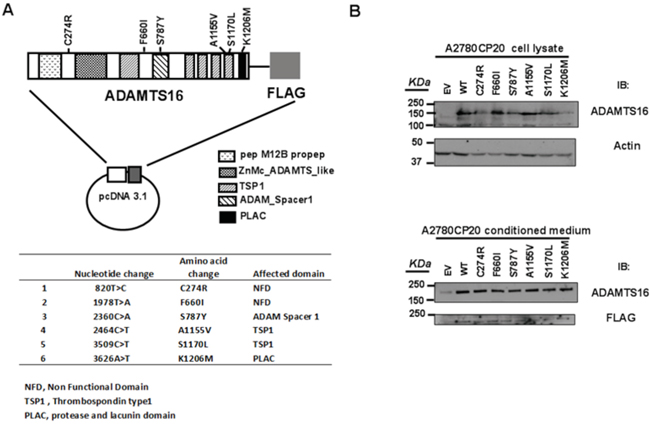
Figure 1: ADAMTS16 mutations and the creation of stable cell lines. Panel A. shows the vector map, and panel B shows the immunoblotting of transiently transfected A2780CP20 cells. Each vector had wild-type (WT) ADAMTS16 or a mutant ADAMTS16 construct (C274R, F660I, S787Y, A1155V, S1170L, or K1206M). After cells had been transfected with vectors, including empty vector, they were selected with neomycin for 2 weeks. Panel B. shows ADAMTS16 protein expression in both the cell lysate and conditioned medium, confirmed via immunoblotting using anti-ADAMTS16 antibody.
ADAMTS16 missense mutations inhibit cell growth
To determine whether ADAMTS16 mutations affect cell viability, we first analyzed ovarian cancer cells that stably expressed EV, or one of six ADAMTS16 missense mutants (C274R, F660I, S787Y, A1155V, S1170L, or K1206M) using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay over a period of 6 days (Figure 2A solid lines). All six stable ADAMTS16 mutant cell lines showed significantly decreased viability compared to cells transfected with EV (p=0.0002, p<0.0001, p=0.0017, p<0.0001, p=0.0011, and p<0.0001, respectively, for C274R, F660I, S787Y, A1155V, S1170L, and K1206M).
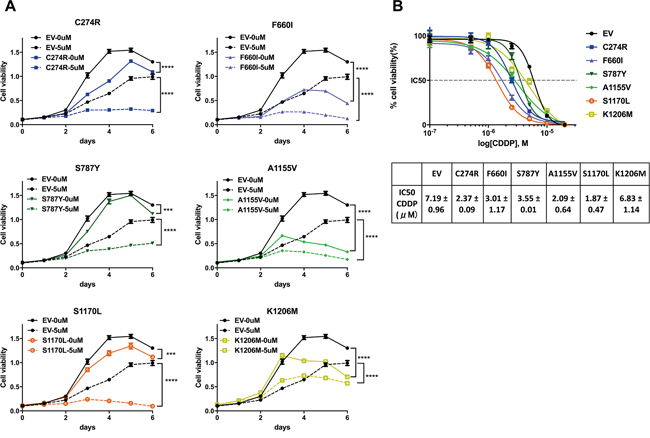
Figure 2: ADAMTS16 mutation improved the response to cisplatin. Panel A. shows the viability of A2780CP20 cells expressing empty vector (EV) or one of six ADAMTS16 missense mutations (C274R, F660I, S787Y, A1155V, S1170L, or K1206M) with no treatment or 5 μM cisplatin. Panel B. shows the dose-response curves obtained by plotting the viability of EV and all six mutations in seven different cisplatin concentrations (0.1 μM, 0.5 μM, 1.0 μM, 2.5 μM, 5.0 μM, 10 μM, and 20 μM), normalized to the control in a semi-log scale. The cellular 50% inhibitory concentrations (IC50s) of cisplatin for each cell line 5 days after treatment were 7.19±0.48, 2.37±0.09, 3.01±1.17, 3.55±0.01, 2.09±0.64, 1.87±0.47, and 6.83±1.14, respectively (average and SEM), for C274R, F660I, S787Y, A1155V, S1170L, and K1206M. Each average and SEM was obtained from two independent experiments. The IC50s of C274R, F660I, S787Y, A1155V, and S1170L were significantly reduced compared to that of EV (p=0.026, p=0.0144, p=0.0071, p=0.0034, and p=0.0023, respectively; two-tailed unpaired t test).
ADAMTS16 missense mutation sensitizes ovarian cancer cells to cisplatin
Next, we determined whether ADAMTS16 mutation increases sensitivity to cisplatin. After treatment with 5.0 μM cisplatin, all six stable ADAMTS16 mutant cell lines showed significantly decreased viability compared to those treated with EV control (p<0.0001) (Figure 2A, dotted lines). Next we calculated the 50% inhibitory concentration (IC50) of cisplatin in the EV control and all six stable ADAMTS16 mutant cell lines by plotting their normalized viability at seven different concentrations (0.1 μM, 0.5 μM, 1.0 μM, 2.5 μM, 5.0 μM, 10 μM or 20 μM) in a semi-log scale. The IC50s were 7.19±0.48, 2.37±0.09, 3.01±1.17, 3.55±0.01, 2.09±0.64, 1.87±0.47, and 6.83±1.14, respectively (Figure 2B, mean and SEM). The IC50s of C274R, F660I, S787Y, A1155V, and S1170L were significantly lower than that of EV (p=0.026, p=0.0144, p=0.0071, p=0.0034, and p=0.0023, respectively). To further assess the effect of a longer incubation period of the mutations on cells with and without cisplatin treatment, we performed a colony formation assay. Without cisplatin treatment, C274R, F660I, A1155V, and K1206M cells formed significantly fewer colonies than did EV control after 14 days incubation (p=0.0151, p=0.0160, p=0.0237, and p=0.0191, respectively) (Figures 3A and 3B). Fourteen days after cisplatin treatment, all six stable ADAMTS16 mutant cells formed significantly fewer colonies than did the EV control (p=0.0023, p=0.0003, p=0.0423, p=0.0003, p=0.0162, and p=0.0177) (Figure 3B).
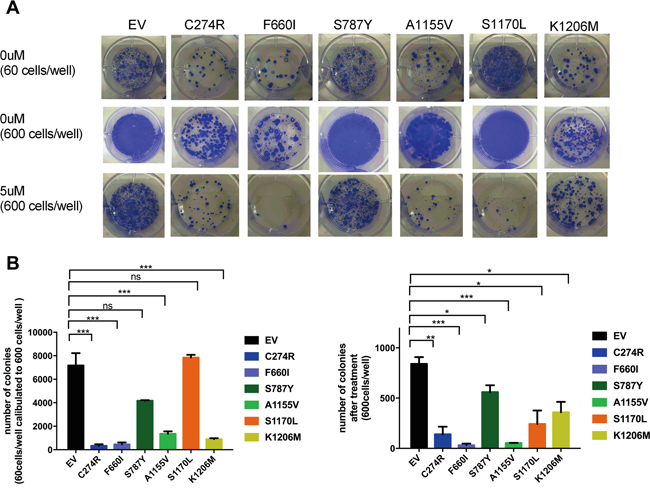
Figure 3: The effect of ADAMTS16 mutations on cancer cells was also observed over time. Panel A. shows images from the colony formation assay. The first row includes the empty vector (EV) and six mutant cells, seeded at 60 cells/well and incubated for 2 weeks without cisplatin treatment. The second and third rows are the EV and six mutant cells, seeded at 600 cells/well and incubated for 2 weeks without cisplatin treatment and with 5 μM cisplatin. In panel B. the bar graph shows the colony number of each cell type after 14 days of incubation without cisplatin treatment (left) and with 5 μM cisplatin (right). As shown in the left graph, we calculated the number obtained from cells seeded at 60 cells/well to 10 times because some cells that were seeded at 600 cells/well showed overgrowth without cisplatin treatment. Panel B shows the average number of colonies without treatment (left) and after 5 μM cisplatin treatment (right). Each bar represents EV, C274R, F660I, S787Y, A1155V, S1170L, and K1206M, respectively. C274R, F660I, S787Y, A1155V, S1170L, and K1206M are respectively compared with EV by the two-tailed unpaired t test (p=0.0151, p=0.0160, p>0.05, p=0.0237, p>0.05, and p=0.0191, left : p=0.0023, p=0.0003, p=0.0423, p=0.0003, p=0.0162, and p=0.0177, right).
Ovarian cancer cells overexpressing WT ADAMTS16 did not affect cell growth but made cells resistant to cisplatin
To determine whether cells that over-express WT ADAMTS16 have a proliferative phenotype or are resistant to cisplatin, we used stable WT ADAMTS16 cells. These cells showed slightly less viability than did the EV control without treatment (p=0.0248), but there was no significant difference between the EV control and WT cells in the cell growth and IC50 (Figures 4A and 4B). In the colony formation assay, there was no significance difference in the number of colonies between the EV control and WT cells after 14 days of incubation, and WT cells formed significantly more colonies than did the EV control at 14 days after treatment with 10μM cisplatin (p=0.0040) (Figures 4C and 4D).
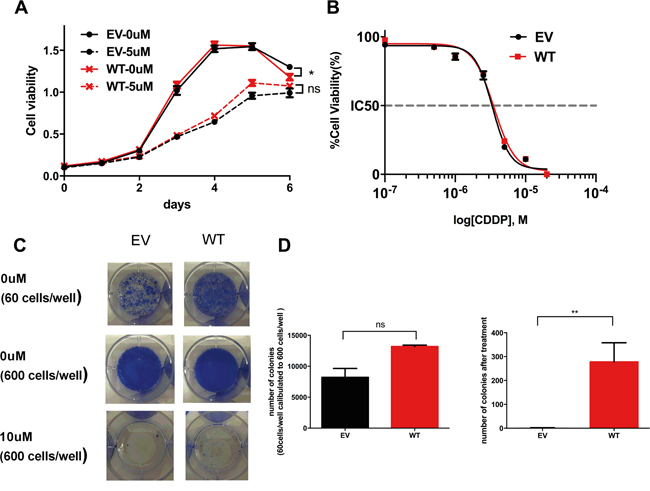
Figure 4: The effect of wild-type (WT) ADAMTS16 on cancer cells. Panel A. shows the viability of A2780CP20 cells expressing empty vector (EV) and WT ADAMTS16 with no treatment or 5 μM cisplatin treatment. Panel B. shows the dose-response curves obtained by plotting the viability of EV and WT in seven different cisplatin concentrations (0.1 μM, 0.5 μM, 1.0 μM, 2.5 μM, 5.0 μM, 10 μM, and 20 μM), normalized to the control in a semi-log scale. The cellular 50% inhibitory concentrations (IC50s) of cisplatin for each cell line 5 days after treatment were 7.19±0.48 and 8.92±1.88 (average and SEM). Each average and SEM was obtained from two independent experiments. There were no significant differences (p=0.2738; two-tailed unpaired t test). Panel C. shows images of the colony formation assay. The first row shows EV and WT cells, seeded at 60 cells/well and incubated for 2 weeks without cisplatin treatment. The second and third rows show the EV and six mutant cells, seeded at 600 cells/well and incubated for 2 weeks without cisplatin or with 10 μM cisplatin. In panel D. the bar graph shows the quantification of the colony number of each cell type after 14 days of incubation without cisplatin treatment (left) or with 10 μM cisplatin treatment (right p=0.0040; two-tailed unpaired t test). As shown in the left graph, we calculated the number obtained from cells seeded at 60 cells/well to 10 times because some cells seeded at 600 cells/well showed overgrowth without cisplatin treatment.
ADAMTS16 mutations significantly inhibit cell migration and invasion compared to WT
A previous study reported that deletion of ADAMTS16 in esophageal cancer cells inhibited invasion [33]. To determine whether the presence of ADAMTS16 mutation in ovarian cancer cells also changes the phenotype, we performed a migration and invasion assay. Compared to WT ADAMTS16 cells, all six stable ADAMTS16 mutant cell lines showed significantly less migration (p=0.0047, p=0.0360, p=0.0395, p=0.0036, p=0.0401, p=0.0112, respectively, for C274R, F660I, S787Y, A1155V, S1170L, and K1206M) and invasion (p<0.0001, p=0.0064, p=0.0107, p=0.0003, p=0.0036, p=0.0002, respectively, for C274R, F660I, S787Y, A1155V, S1170L, and K1206M)(Figure 5).
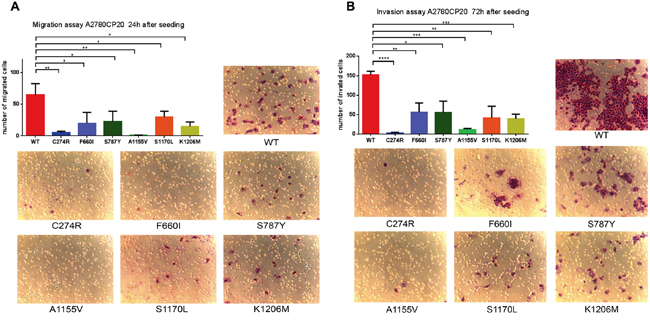
Figure 5: Functional analysis of ADAMTS16 mutations by migration assay and invasion assay. Panel A. shows the number of migrated cells. Data are presented as the means (±SEM) from triplicate wells. The number of migrated cells in C274R (p=0.0047; two-tailed unpaired t test), F660I (p=0.0360), S787Y (p=0.0395), A1155V (p=0.0036), S1170L (p=0.0401), and K1206M (p=0.0112) cells was significantly lower than that in cells expressing wild-type (WT). Panel B. shows representative images from the migration assay. Panel C quantifies the number of invaded cells. Data are presented as the mean (±SEM) from triplicate wells. The number of invaded cells in C274R (p<0.0001; two-tailed unpaired t-test), F660I (p=0.0064), S787Y (p=0.0107), A1155V (p=0.0003), S1170L (p=0.0036), and K1206M (p=0.0002) cells was significantly lower than that in cells expressing WT. Panel D shows representative images of the invasion assay.
Mutated ADAMTS16 cells had a better response to cisplatin in the mouse model
Next, we determined the effect of ADAMTS16 mutations on response to cisplatin in vivo using an orthotopic mouse model. The experiment was performed with four groups (7 mice/group), WT A2780-CP20, A2780-CP20 transfected with EV, and A2780-CP20 with ADAMTS16 mutation at S787Y or S1170L. All the mice received intraperitoneal cisplatin (160μg in 200μl/mouse) once per week, starting 1 week after cell injection. They were all killed when mice from any group became moribund (Figure 6A). Compared to the WT controls, the EV group had no effect on response to cisplatin, however, both of the mutated ADAMTS16 (S787Y and S1170L) cell lines had a significantly better response to cisplatin (p<0.05 and p<0.01, respectively), as indicated by a reduction in tumor weight or the number of tumor nodules (Figure 6B).
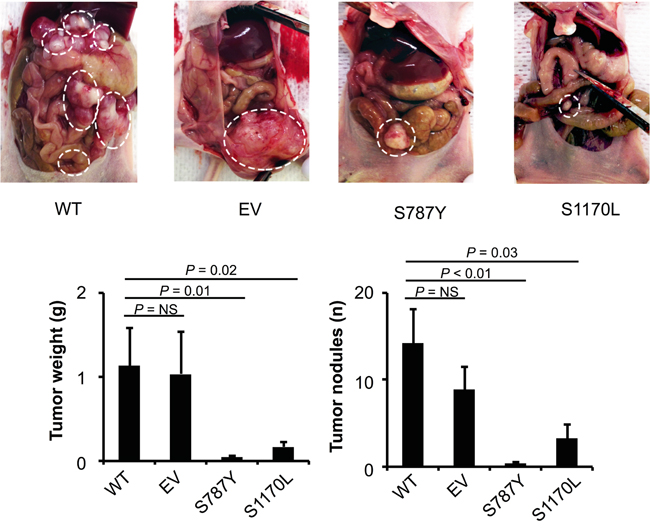
Figure 6: The effect of ADAMTS16 mutation in a mouse model. Panel A. show the extent of metastatic spread in treated mice; metastatic areas are outlined with dotted white lines. Nude mice were injected with either A2780-CP wild-type (WT), empty vector (EV), or mutated ADAMTS16 (S787Y or S1170L) cells. All mice received intraperitoneal cisplatin (160 μg once per week). The tumor weight and number of tumor nodules were compared in the four treated groups (n=7 per group). The results represent the average of the seven treated mice in each group, with error bars representing the SEM. *p<0.05, **p<0.01, comparison with WT by the Mann-Whitney U test (Panel B).
DISCUSSION
In our previous study, we demonstrated that mutations of eight members of the ADAMTS family (including ADAMTS16 gene) were significantly associated with chemotherapy sensitivity and longer survival in patients with ovarian cancer, independent of BRCA1 or BRCA2 mutations [11]. The identification of the effect of ADAMTS mutations has important implications for clinical prediction. However, unlike that of BRCA1/2 mutations, the functional role of these ADAMTS mutations in ovarian cancer cells is largely unknown.
To fill in this knowledge gap, we systematically carried out multiple in vitro and in vivo experimental assays and demonstrated that the introduction of ADAMTS16 mutants into ovarian cancer cells resulted in both improved sensitivity to cisplatin treatment and reduced cell migration and invasion, providing experimental evidence to support the genomic observation in a large population of patient cohort. The findings of this study, together with those from bioinformatics analyses, offer a cohesive view of the relationship between ADAMTS mutations and chemotherapy response in ovarian cancer patients, and may lead to the identification of novel targets of therapeutic intervention in patients with ovarian cancer. To the best of our knowledge, this is the first report revealing the functional effect of ADAMTS16 mutations in ovarian cancer cells.
In general, ADAMTS16 mRNA is highly expressed in the adult brain and ovaries [37]. Although studies have demonstrated an association between ADAMTS16 and disease such as hypertension [38, 39], osteoarthritis [40], premature ovarian failure [41] and Dupuytren’s disease [42], less is known about the function of the gene than about other members of the ADAMTS family [30]. Few studies have examined the role of ADAMTS16 in the pathogenesis of cancer. Sakamoto et al showed that ADAMTS16 mRNA was upregulated in esophageal squamous cell carcinoma, and cell growth and invasion were inhibited upon depletion of ADAMTS16 [33]. Castellana et al reported that mRNA expression of ADAMTS16 was upregulated in invasive ductal carcinoma compared to in ductal carcinoma in situ [30]. On the other hand, a study reported that ADAMTS16 overexpression resulted in significantly reduced proliferation in chondrosarcoma cells [40]. It is possible that ADAMTS16 has a different effect on malignancies of epithelial origin and mesenchymal origin, like matrix metalloproteinase 1 [43].
In our in vitro study, we observed that cells that expressed mutant ADAMTS16 C274R, F660I, A1155V, and K1206M had significantly suppressed proliferation without treatment. On the other hand, cells that expressed mutant ADAMTS16 S787Y and S1170Y had almost no proliferation effects without treatment but showed substantial inhibition after treatment. The latter finding was supported by the result of our mouse model. The results of our study suggests that each missense mutation leads to different cellular changes that affect the response to cisplatin. All six stable ADAMTS16 mutant cells also showed significantly less migration and invasion than those of WT. This finding supports those of prior studies investigating the role of ADAMTS16 in tumor invasion [30, 33]. ADAMTS16 is known to degenerate extracellular protein [37]. Therefore, we think that decrease in cell invasion and migration likely results from the decreased ability of ADAMTS16 to degenerate extracellular proteins without alternating their gene expression.
Our study has limitations. We evaluated phenotypes of ovarian cancer cells with ADAMTS16 mutations. However, the exact molecular mechanisms of how these mutations sensitize tumor cells to platinum remain unclear because the original function of ADAMTS16 has not been revealed yet. We assume that once ADAMTS16 gene is mutated, the secreted protein reduces its activity to denature extracellular protein. It is possible that this altered enzymatic function plays an important role in impairing tumor microenvironment favorable for cancer cells, thus platinum-sensitivity and migration/invasion are both altered in these cancer cells. Our future investigation to reveal this mechanism will surely bring new insight of chemotherapy in recurrent ovarian cancer patients because therapy approaching to tumor microenvironment would be crucial to cancer cells and administering drug against ADAMTS16 is safer than drugs targeting whole matrix metalloproteinases.
Second, we only used A2780-CP20 in our functional study, because most available ovarian cancer cell lines are relatively sensitive to cisplatin. Therefore, we are limited to cell line selection since we are investigating a molecular event that make resistant cells to sensitivity to chemotherapy. We believe that this weakness was compensated by the fact that we have evaluated six different mutations therefore greatly reducing the possibility that the observation is accidental.
In summary, our investigations revealed that exogenously expressed ADAMTS16 missense mutations lead to cellular changes that enhance cisplatin sensitivity or inhibit cell growth and suppress tumor invasion and migration in platinum-resistant ovarian cancer. ADAMTS16 is a potential therapeutic target in patients with platinum-resistant ovarian cancer. Further evaluations are needed to reveal the detailed effects of ADAMTS16 mutation in this disease.
MATERIALS AND METHODS
Cell culture
A2780CP20 cells, platinum resistant human epithelial ovarian cancer cells that are derived from A2780 platinum-sensitive cells, are obtained from Dr. Anil K. Sood [44]. Cells were cultured at 37°C in 5% CO2 in RPIM1640 (Cellgro) supplemented with 10% fetal bovine serum (Hyclone) [45, 46].
ADAMTS16 expression plasmid
Full-length wild-type (WT) human ADAMTS16 cDNA, kindly provided by Dr. Ian M. Clark (University of East Anglia) [40], was cloned into pcDNA3.1 vector (Invitrogen) with a c-terminal Flag tag (Figure 1A). The six missense mutations of ADAMTS16 were introduced using the QuikChange MultiSite-Directed mutagenesis kit (Agilent Technologies) according to the manufacturer’s protocols. All mutations were confirmed by DNA sequencing, as previously described [47].
Transfection and generation of stable cell lines
A2780CP20 cells were transfected in six-well plates with WT or one of six different mutant ADAMTS16 expression plasmids using FuGENE HD transfection reagent (Roche Applied Science) according to the manufacturer’s instructions. After 24 hours, cells were tripsinized and seeded in 10-cm dishes, and the remaining cells were used to confirm transfection by Western blot analysis. Cells were cultured in G418 (Invitrogen)- containing media for 2 weeks. After the selection, the mixtures of stable cells were confirmed by checking the cell lysate and the conditioned media by Western blot analysis [47].
Validation of stable cells by western blot analysis
Cells were seeded in six-well plates in serum-containing media for 24 hours. After 24 hours of culture in serum-free media, conditioned media were harvested, centrifuged, and stored at -20°C. Whole Cell lysates were harvested and lysed using RIPA buffer with a protease and phosphatase inhibitors cocktail (Thermo Scientific). Samples (conditioned media and whole cell lysate) were separated on 7% polyacrylamide gels and transferred to nitrocellulose membranes. After blocking in PBS containing 5% nonfat-milk, membranes were incubated overnight at 4°C with anti-ADAMTS16 and Anti-Actin (Santa Cruz) antibodies and for 1 hour at ambient temperature with secondary antibodies (Santa Cruz). The proteins were visualized using the chemiluminescent substrate (Rockford, IL).
Cell viability assay
Cells were seeded at 3000 cells/well in a 96-well plate in quadruplicate. After 12 hours, they were treated with 0 μM, 5 μM, or 10 μM cisplatin. Cell viability was measured at 0 h as day 0 and every day for 6 days after treatment with 0.5 mg/ml MTT reagent in PBS (Sigma-Aldrich) at 590 nm using the Tecan SpectraFluor microplate Reader and Magellan 6 software (Tecan Group, Ltd.). For the dose dependent curve, cells were treated with 0 μM, 0.5 μM,1 μM, 2.5 μM, 5 μM, 10 μM, or 20 μM cisplatin. Cell viability was measured 5 days after drug treatment [45, 46].
Colony formation assay
Cells were seeded in six-well plates at 60 and 600 cells/well. After 12 hours, they were treated with 0 μM, 5 μM, or 10 μM cisplatin. The plates were incubated for 2 weeks and the medium was changed twice per week. Colonies consisting of more than 50 cells were counted under a microscope after staining.
Cell migration and invasion assays
The cell migration and invasion assay was performed in duplicate using Matrigel-coated transwell chambers. The cells were plated in 500 μl of serum-free medium and allowed to migrate or invade towards a 10% FBS medium for 24 h or 72 h. Cells that invaded into the underside of the filter were fixed and stained with Hema-Diff solution (Fisher). The numbers of invaded cells from 5 randomly chosen fields were counted for each membrane, as previously described [48].
Mouse model
Female athymic nude mice were purchased from Taconic Farms and maintained in pathogen-free conditions. They were cared for according to the guidelines of the American Association for Accreditation of Laboratory Animal Care and the U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals. All in vivo experiments and protocols were approved by MD Anderson’s Institutional Animal Care and Use Committee. The development and characterization of the orthotopic mouse models of epithelial ovarian cancer have been described previously [49, 50] The experiment was performed with four groups (seven mice/group): A) WT A2780-CP20 (WT), B) A2780-CP20 transfected with empty vector (EV), and C and D) A2780-CP20 with ADAMTS mutations (S787Y and S1170L). These two mutations were selected for in vivo model experiments because they did not exhibit significant inhibition on cell growth compared to the empty vector (Figure 2A and 4A). For the cell injections, cells were trypsinized at 60%-80% confluence and centrifuged at 1200 RPM for 6 min at 4°C. They were then washed twice with phosphate-buffered saline (PBS) and reconstituted in Hanks balanced salt solution (HBSS) to a desired concentration (5.0 X 106 cells/mL). Two hundred microliters of the cell suspension containing 1.0 X 106 cells were injected into the peritoneal cavity of each mouse. The treatment was started 1 week after injecting the cells. All the mice in all four groups received intraperitoneal cisplatin treatment (160μg in 200μl/mouse) once per week. All the mice were killed when mice from any group became moribund. Tumor weight and the number of tumor nodules were recorded.
Statistical analysis
Each experiment was repeated two times. All data are represented as mean ± SEM. Statistical analyses were performed using GraphPad Prism6 Software (Graph Pad).
ACKNOWLEDGMENTS
Dr. Zhang is supported by the Hanes and Willis Family Endowed Professorship in Cancer.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This study was supported by grants from National Cancer Institute (CA 143835, CA 151668).
REFERENCES
1. Cancer Genome Atlas Research N. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474: 609-15.
2. Acar M, Ocak Z, Erdogan K, Cetin EN, Hatipoglu OF, Uyeturk U, Gunduz E, Gunduz M. The effects of hypericin on ADAMTS and p53 gene expression in MCF-7 breast cancer cells. J BUON. 2014; 19: 627-32.
3. Chetrit A, Hirsh-Yechezkel G, Ben-David Y. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J Clin Oncol. 2008; 26: 20-5.
4. Yang D, Khan S, Sun Y, Hess K, Shmulevich I, Sood AK, Zhang W. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. 2011; 306: 1557-65.
5. Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Pharoah PD. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA. 2012; 307: 382-90.
6. TCGA. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474: 609-15.
7. Hennessy BT, Timms KM, Carey MS, Lu KH, Mills GB. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly(ADP ribose) polymerase inhibitors in ovarian cancer. J Clin Oncol. 2010; 28: 3570-6. doi: 10.1200/JCO.2009.27.2997. Epub 2010 Jul 6.
8. Pal T, Permuth-Wey J, Betts JA, Krischer UP, Sutphen R. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005; 104: 2807-16.
9. Dressman HK, Berchuck A, Chan G, Zhai J, Bild A, Sayer R, Cragun J, Lancaster JM. An integrated genomic-based approach to individualized treatment of patients with advanced-stage ovarian cancer. J Clin Oncol. 2007; 25: 517-25.
10. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A. Signatures of mutational processes in human cancer. Nature. 2013; 500: 415-21.
11. Liu Y, Yasukawa M, Chen K, Hu L, Broaddus RR, Ding L, Mardis ER, Spellman P, Levine DA, Mills GB, Shmulevich I, Sood AK, Zhang W. Association of Somatic Mutations of ADAMTS Genes With Chemotherapy Sensitivity and Survival in High-Grade Serous Ovarian Carcinoma. JAMA Oncol. 2015; 1: 486-94.
12. Zhang M, Liu G, Xue F, Edwards R, Sood AK, Zhang W, Yang D. Copy number deletion of RAD50 as predictive marker of BRCAness and PARP inhibitor response in BRCA wild type ovarian cancer. Gynecol Oncol. 2016; 141: 57-64.
13. Kelwick R, Desanlis I, Wheeler GN, Edwards DR. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biol. 2015; 16: 113.
14. Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005; 386: 15-27.
15. Dubail J, Apte SS. Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics. Matrix Biol. 2015; 44-46: 24-37.
16. Hubmacher D, Apte SS. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015; 47: 34-43.
17. Kuno K, Kanada N, Nakashima E, Fujiki F, Ichimura F, Matsushima K. Molecular cloning of a gene encoding a new type of metalloproteinase-disintegrin family protein with thrombospondin motifs as an inflammation associated gene. J Biol Chem. 1997; 272: 556-62.
18. Budde U, Schneppenheim R. Interactions of von Willebrand factor and ADAMTS13 in von Willebrand disease and thrombotic thrombocytopenic purpura. Hamostaseologie. 2014; 34: 215-25.
19. Nandadasa S, Foulcer S, Apte SS. The multiple, complex roles of versican and its proteolytic turnover by ADAMTS proteases during embryogenesis. Matrix Biol. 2014; 35: 34-41.
20. Lin EA, Liu CJ. The role of ADAMTSs in arthritis. Protein Cell. 2010; 1: 33-47.
21. Dancevic CM, McCulloch DR. Current and emerging therapeutic strategies for preventing inflammation and aggrecanase-mediated cartilage destruction in arthritis. Arthritis Res Ther. 2014; 16: 429.
22. Chapman K, Seldon M, Richards R. Thrombotic microangiopathies, thrombotic thrombocytopenic purpura, and ADAMTS-13. Semin Thromb Hemost. 2012; 38: 47-54.
23. Adler M, Kremer Hovinga JA, Lammle B. [Thrombotic thrombocytopenic purpura—an often missed diagnosis]. Rev Med Suisse. 2014; 10: 2280-4.
24. Rocks N, Paulissen G, El Hour M, Quesada F, Crahay C, Gueders M, Foidart JM, Noel A, Cataldo D. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie. 2008; 90: 369-79.
25. Turner SL, Blair-Zajdel ME, Bunning RA. ADAMs and ADAMTSs in cancer. Br J Biomed Sci. 2009; 66: 117-28.
26. Cal S, Lopez-Otin C. ADAMTS proteases and cancer. Matrix Biol. 2015.
27. Sun Y, Huang J, Yang Z. The roles of ADAMTS in angiogenesis and cancer. Tumour Biol. 2015; 36: 4039-51.
28. Przemyslaw L, Boguslaw HA, Elzbieta S, Malgorzata SM. ADAM and ADAMTS family proteins and their role in the colorectal cancer etiopathogenesis. BMB Rep. 2013; 46: 139-50.
29. Alonso S, Gonzalez B, Ruiz-Larroya T, Duran Dominguez M, Kato T, Matsunaga A, Suzuki K, Strongin AY, Gimenez-Bonafe P, Perucho M. Epigenetic inactivation of the extracellular matrix metallopeptidase ADAMTS19 gene and the metastatic spread in colorectal cancer. Clin Epigenetics. 2015; 7: 124.
30. Castellana B, Escuin D, Peiro G, Garcia-Valdecasas B, Vazquez T, Pons C, Perez-Olabarria M, Barnadas A, Lerma E. ASPN and GJB2 Are Implicated in the Mechanisms of Invasion of Ductal Breast Carcinomas. J Cancer. 2012; 3: 175-83.
31. Ocak Z, Acar M, Gunduz E, Gunduz M, Demircan K, Uyeturk U, Ozlu T. Effect of hypericin on the ADAMTS-9 and ADAMTS-8 gene expression in MCF7 breast cancer cells. Eur Rev Med Pharmacol Sci. 2013; 17: 1185-90.
32. Fontanil T, Rua S, Llamazares M, Moncada-Pazos A, Quiros PM, Garcia-Suarez O, Vega JA, Sasaki T, Mohamedi Y, Esteban MM, Obaya AJ, Cal S. Interaction between the ADAMTS-12 metalloprotease and fibulin-2 induces tumor-suppressive effects in breast cancer cells. Oncotarget. 2014; 5: 1253-64. doi: 10.18632/oncotarget.1690.
33. Sakamoto N, Oue N, Noguchi T, Sentani K, Anami K, Sanada Y, Yoshida K, Yasui W. Serial analysis of gene expression of esophageal squamous cell carcinoma: ADAMTS16 is upregulated in esophageal squamous cell carcinoma. Cancer Sci. 2010; 101: 1038-44.
34. Bohm M, Gerlach R, Beecken WD, Scheuer T, Stier-Bruck I, Scharrer I. ADAMTS-13 activity in patients with brain and prostate tumors is mildly reduced, but not correlated to stage of malignancy and metastasis. Thromb Res. 2003; 111: 33-7.
35. Nakada M, Miyamori H, Kita D, Takahashi T, Yamashita J, Sato H, Miura R, Yamaguchi Y, Okada Y. Human glioblastomas overexpress ADAMTS-5 that degrades brevican. Acta Neuropathol. 2005; 110: 239-46.
36. Held-Feindt J, Paredes EB, Blomer U, Seidenbecher C, Stark AM, Mehdorn HM, Mentlein R. Matrix-degrading proteases ADAMTS4 and ADAMTS5 (disintegrins and metalloproteinases with thrombospondin motifs 4 and 5) are expressed in human glioblastomas. Int J Cancer. 2006; 118: 55-61.
37. Gao S, De Geyter C, Kossowska K, Zhang H. FSH stimulates the expression of the ADAMTS-16 protease in mature human ovarian follicles. Mol Hum Reprod. 2007; 13: 465-71.
38. Joe B, Saad Y, Dhindaw S, Lee NH, Frank BC, Achinike OH, Luu TV, Gopalakrishnan K, Toland EJ, Farms P, Yerga-Woolwine S, Manickavasagam E, Rapp JP, et al. Positional identification of variants of Adamts16 linked to inherited hypertension. Hum Mol Genet. 2009; 18: 2825-38.
39. Gopalakrishnan K, Kumarasamy S, Abdul-Majeed S, Kalinoski AL, Morgan EE, Gohara AF, Nauli SM, Filipiak WE, Saunders TL, Joe B. Targeted disruption of Adamts16 gene in a rat genetic model of hypertension. Proc Natl Acad Sci U S A. 2012; 109: 20555-9.
40. Surridge AK, Rodgers UR, Swingler TE, Davidson RK, Kevorkian L, Norton R, Waters JG, Goldring MB, Parker AE, Clark IM. Characterization and regulation of ADAMTS-16. Matrix Biol. 2009; 28: 416-24.
41. Pyun JA, Kim S, Cha DH, Kwack K. Epistasis between polymorphisms in TSHB and ADAMTS16 is associated with premature ovarian failure. Menopause. 2013.
42. Johnston P, Larson D, Clark IM, Chojnowski AJ. Metalloproteinase gene expression correlates with clinical outcome in Dupuytren's disease. J Hand Surg Am. 2008; 33: 1160-7.
43. Jawad MU, Garamszegi N, Garamszegi SP, Correa-Medina M, Diez JA, Wen R, Scully SP. Matrix metalloproteinase 1: role in sarcoma biology. PLoS One. 2010; 5: e14250.
44. Moreno-Smith M, Halder JB, Meltzer PS, Gonda TA, Mangala LS, Rupaimoole R, Lu C, Nagaraja AS, Gharpure KM, Kang Y, Rodriguez-Aguayo C, Vivas-Mejia PE, Zand B, et al. ATP11B mediates platinum resistance in ovarian cancer. J Clin Invest. 2013; 123: 2119-30.
45. Liu G, Sun Y, Ji P, Li X, Cogdell D, Yang D, Parker Kerrigan BC, Shmulevich I, Chen K, Sood AK, Xue F, Zhang W. MiR-506 suppresses proliferation and induces senescence by directly targeting the CDK4/6-FOXM1 axis in ovarian cancer. J Pathol. 2014; 233: 308-18.
46. Liu G, Yang D, Rupaimoole R, Pecot CV, Sun Y, Mangala LS, Li X, Ji P, Cogdell D, Hu L, Wang Y, Rodriguez-Aguayo C, Lopez-Berestein G, et al. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J Natl Cancer Inst. 2015; 107.
47. Parker BC, Annala MJ, Cogdell DE, Granberg KJ, Sun Y, Ji P, Li X, Gumin J, Zheng H, Hu L, Yli-Harja O, Haapasalo H, Visakorpi T, et al. The tumorigenic FGFR3-TACC3 gene fusion escapes miR-99a regulation in glioblastoma. J Clin Invest. 2013; 123: 855-65.
48. Sun Y, Hu L, Zheng H, Bagnoli M, Guo Y, Rupaimoole R, Rodriguez-Aguayo C, Lopez-Berestein G, Ji P, Chen K, Sood AK, Mezzanzanica D, Liu J, et al. MiR-506 inhibits multiple targets in the epithelial-to-mesenchymal transition network and is associated with good prognosis in epithelial ovarian cancer. J Pathol. 2015; 235: 25-36.
49. Lu C, Han HD, Mangala LS, ali-Fehmi R, Newton C, Ozbun L, Armaiz-Pena G, Hu W, Stone RL, Munkarah A, Ravoori MK, Shahzad MM, Lee JW, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 2010; 18: 185-97.
50. Stone RL, Nick AM, McNeish IA, Balkwill F, Han HD, Bottsford-Miller J, Rupaimoole R, Armaiz-Pena GN, Pecot CV, Coward J, Deavers MT, Vasquez HG, Urbauer D, et al. Paraneoplastic thrombocytosis in ovarian cancer. N Engl J Med. 2012; 366: 610-8.