
INTRODUCTION
Triple negative breast cancer (TNBC), lacking the expression of estrogen receptor, progesterone receptor and human EGF receptor-2, represents an especially aggressive and hard to treat subtype. Chemotherapy is the only standard treatment available for TNBC patients to date [1]. New targets for TNBC have been proposed to focus on oncogenic lesions and metabolic abnormalities that support the aggressive phenotype [2]. These drivers include; oncogene mutation, tumor suppressor loss, mutation or overexpression of DNA repair proteins and enhanced survival via kinase signaling [2].
Gene expression profiling identified that approximately 80% of sporadic TNBC are “basal-like”, with aberrations mimicking defective homologous recombination (HR) DNA repair and displaying BRCA-ness [3]. BRCA-ness creates a shift from HR, which is dysfunctional, towards alternative DNA repair mechanisms, including those regulated by poly(ADP-ribose)polymerases (PARPs). This has provided the rationale for testing PARP inhibitors in TNBC to create synthetic lethality, the mechanism by which the targeting of two genes is lethal, while individually they are not. However, targeting PARP1/2 has had limited clinical success [4]. PARP inhibition on a BRCA1 background is readily overcome by 53BP1 loss, BRCA1 reversion and upregulation of RAD51, all compensatory mechanisms of DNA repair [5].
In search of a novel and rational therapeutic combination for TNBC, several studies identified RAD51 as target in synthetic lethal screens for PARP inhibition in breast cancer [6-10]. RAD51 has been associated with compensating for functional BRCA1 loss [11] and we have previously published that RAD51 can support metastatic progression of TNBC [12]. We suggested that the pressure on the DNA repair pathways induced by the targeting of PARPi on a BRCAness background forces compensation by RAD51, providing a rationale for its targeting [13]. However, targeting of RAD51 has been shown to increase kinase signaling, suggesting additional mechanisms contributing to loss of synthetic lethality [14, 15]. Importantly, kinase signaling in TNBC is complex and characterized by several rewiring events as compensatory mechanisms for treatment resistance [16, 17]. We recently carried out antibody-based microarray analysis to study protein and phosphoprotein expression in breast cancer, including TNBC. We described that 50% of TNBC lines or cases have very complex kinome profiles, which may explain their resistance to chemotherapy as well as targeted therapies [18].
Following our previous studies on RAD51 [12, 13] and kinome profiling in TNBC [18], in this study we now investigate whether RAD51 inhibition synergizes with inhibitors of kinases that are activated in TNBC. We find that RAD51 inhibition synergizes with several inhibitors, most notably PARP and p38 inhibitors. This synergy is characterized by rewiring of phosphorylation patterns after RAD51 inhibition, which are attenuated by combining with both PARP and p38 inhibition in vitro and in vivo. We propose that kinome rewiring limits the efficacy of targeting RAD51 and that rational targeted combination therapy is required to achieve effective therapy against TNBC.
RESULTS
The effect of RAD51 depletion on the sensitivity of TNBC cells to inhibitors of the DNA-damage response and kinase signaling pathways
Our previous studies showed that depletion of RAD51 limits metastatic progression of TNBC [12], and we suggested that the pressure on the DNA repair pathways induced by the targeting of PARP may force compensation through RAD51 [13], thus rationalizing targeting RAD51 in this setting. Since the targeting of RAD51 has been previously shown to increase kinase signaling [14, 15], we asked whether the combined inhibition of RAD51 and kinase signaling could be at least additive. To this end, we utilized our previous kinome profiling data to identify several activated kinases and overexpressed proteins in TNBC primary tumors and cell lines [18]. These include HSP90, PLCγ, PDK, EGFR, MEK, DNA-PK, GSK3β, PKC, PYK2, PAK, IGF-1R, p38, ERK5, S6K and LIG4. In addition, we also included PARP1, ATM and CHK1 inhibition, based on our hypothesis that targeting of complementary DNA-damage response (DDR) pathways would be most effective in combination with RAD51 depletion.
Using two different siRNA targeting RAD51 in three TNBC cell lines (MDA-MB-231, MDA-MB-436 and PMC42-ET), in the presence of drugs targeting the selected 18 proteins, the effectiveness of the combination was examined using a ratio of IC50 values comparing RAD51 knockdown to scrambled control lines. In line with our hypothesis that targeting complementary DDR pathways should be effective with RAD51 depletion, we found that PARP1 inhibition was most effective in RAD51 knockdown cell lines (average of knockdown over scrambled ratio IC50’s is 0.39 across all cell lines, Table 1). This finding is consistent with several reported synthetic lethality screens involving PARP inhibitors [8-10]. Inhibition of the DNA-dependent protein kinase (DNA-PK) and RAD51 depletion was also a potent combination (average 0.67, Table 1). Of the remaining drugs, 4 were additive in two of the 3 lines (inhibitors of LIG4, HSP90, MEK and EGFR), while 4 were additive across all 3 cell lines (p38, S6K, PAK, PYK2). Interestingly, the inhibition of p38 was more potent than DNA-PK inhibition (p=0.041, Table 1). p38 signaling by constitutively active MKK6 (MKK6E) has been shown to increase level of RAD51 protein, improving its stability without altering the level of RAD51 mRNA [19]. This may explain the effect of combining RAD51 depletion with p38 inhibition.
Table 1: Ratio of IC50s for RAD51 knockdown and drug targets in three TNBC cell lines
IC50 Target |
MDA-MB-231 |
MDA-MB-436 |
PMC42-ET |
Ave |
p Value |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
SCR |
RAD51 |
Ratio |
SCR |
RAD51 |
Ratio |
SCR |
RAD51 |
Ratio |
|||
PARP1 |
32.26 |
9.86 |
0.3056 |
6.92 |
3.22 |
0.4653 |
35.01 |
13.73 |
0.3922 |
0.3877 |
0.028 |
P38 |
16.13 |
12.7 |
0.7874 |
12.92 |
7.877 |
0.6097 |
19.69 |
10.43 |
0.5297 |
0.6422 |
0.041 |
DNA-PK |
1.02 |
0.69 |
0.6330 |
2.02 |
1.6 |
0.7921 |
1.89 |
1.1 |
0.5820 |
0.6690 |
0.051 |
S6K |
9.38 |
3.965 |
0.4227 |
4.838 |
2.944 |
0.6085 |
1.587 |
1.623 |
1.0227 |
0.6846 |
0.268 |
ATM |
20.25 |
9.54 |
0.4711 |
33.94 |
25.07 |
0.7386 |
21.94 |
21.94 |
1.010403R3 |
0.7366 |
0.187 |
PAK |
20.39 |
15.08 |
0.7396 |
13.1 |
11.85 |
0.9046 |
16.95 |
11.9 |
0.7021 |
0.7821 |
0.098 |
LIG4 |
19.01 |
19.4 |
1.0205 |
20.07 |
15.51 |
0.7728 |
12.94 |
7.716 |
0.5963 |
0.7965 |
0.219 |
CHK1 |
0.09 |
0.08 |
0.8889 |
0.03 |
0.03 |
1.010403R3 |
0.15 |
0.1 |
0.6667 |
0.8519 |
0.312 |
PYK2 |
0.098 |
0.088 |
0.9025 |
20.96 |
16 |
0.9025 |
13.17 |
10.55 |
0.9025 |
0.9025 |
0.219 |
HSP90 |
0.14 |
0.12 |
0.8330 |
0.1698 |
0.1816 |
1.0695 |
0.21 |
0.20 |
0.9393 |
0.9472 |
0.52 |
GSK3 |
12.13 |
13.12 |
1.0816 |
20.55 |
17.22 |
0.8380 |
ND |
ND |
ND |
0.9598 |
0.684 |
DNA-PK(2) |
0.097 |
0.094 |
0.9678 |
0.096 |
0.093 |
0.9676 |
0.094 |
0.089 |
0.9501 |
0.9619 |
0.032 |
MEK (2) |
13.23 |
16.89 |
1.2766 |
17.49 |
9.122 |
0.5216 |
14.88 |
17.69 |
1.1888 |
0.9957 |
0.885 |
EGFR |
0.21 |
0.19 |
0.8984 |
0.1705 |
0.2248 |
1.3185 |
0.20 |
0.17 |
0.8485 |
1.0218 |
0.986 |
PDK |
12.58 |
18.38 |
1.4610 |
14.58 |
15.04 |
1.0316 |
18.08 |
15.05 |
0.8324 |
1.1083 |
0.716 |
PKC |
15.41 |
13.75 |
0.8923 |
17.26 |
23.82 |
1.3801 |
11.67 |
12.48 |
1.0694 |
1.1139 |
0.516 |
MEK |
0.5591 |
0.7839 |
1.4021 |
0.097 |
0.091 |
0.9986 |
0.098 |
0.093 |
0.9464 |
1.1157 |
0.451 |
PLC |
16.13 |
22.70 |
1.4073 |
16.13 |
18.27 |
1.1327 |
20.55 |
16.72 |
0.8136 |
1.1179 |
0.643 |
IGF-1R |
9.54 |
17.97 |
1.8836 |
14.51 |
10.34 |
0.7126 |
ND |
ND |
ND |
1.2981 |
0.792 |
ERK5 |
4.151 |
8.943 |
2.1544 |
5.018 |
6.049 |
1.2055 |
3.041 |
2.25 |
0.7399 |
1.3666 |
0.415 |
The IC50 values in μM were established for individual drugs in three TNBC cell lines that express high levels of RAD51. They are ranked based on the difference in IC50 values across the three cell lines for RAD51 depletion with a drug versus no RAD51 depletion. A score below 1.0 was deemed to be additive while above 1.0 was antagonistic.
RAD51 inhibition in combination with p38 and PARP inhibition
Next, we validated our findings using the RAD51 inhibitor B02 [20]. We confirmed the activity of B02 as a potent inhibition of RAD51 foci formation after irradiation (Supplementary Figure S1B). We also confirmed the activity of the PARPi ABT-888, which showed concentration-dependent PARP inhibition of protein PARylation (Supplementary Figure S1A). The activity of the p38 inhibitor LY2228820 (p38i) demonstrated restricted phosphorylation of MK2 (Supplementary Figure S1C), a p38 downstream target [21]. In line with the combination of RAD51 depletion and PARP inhibition, each of the three TNBC cell lines displayed a decrease in IC50 when targeted with the RAD51 inhibitor B02 (RAD51i) in combination with PARPi (2.5 μM ABT-888) compared to PARPi alone (Figures 1A-1C). We also confirmed that the combination of RAD51i and p38i is more potent than each drug individually (Figures 1D-1F). The specific effects of combining RAD51i/PARPi or RAD51i/p38i were supported by the lack of benefit from combining PARPi and p38i (Figure 1G-1I). Given the role of p38 signaling in the stabilization of RAD51 protein (16), we questioned whether p38 inhibition could further enhance the effect of the combination of RAD51 and PARP inhibition. Indeed, p38i significantly potentiated RAD51i/PARPi combination in the three TNBC cells lines (Figures 1J-1L), suggesting that p38 signaling may be an escape mechanism from the combined inhibition of RAD51 and PARP.
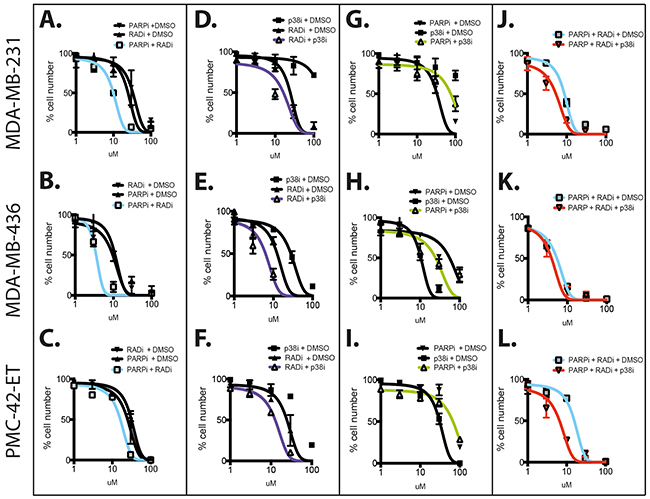
Figure 1: Combination of RAD51, PARP and p38 inhibitors against TNBC cell lines. The three TNBC cell lines MDA-MB-231 (top row), MDA-MB-436 (middle row) and PMC42-ET (bottom row) were used for dose response studies using three molecular inhibitors targeting RAD51 (B02, RAD51i), PARP (ABT-888, PARPi) and p38 (LY2228820, p38i). Dose curves for the single drugs were performed using 0-100 μM escalating doses. The combinations were carried out as follows: A–C. RAD51i escalating doses 0-100 μM with 2.5 μM of PARPi; D–F. RAD51i escalating doses 0-100 μM with 10 μM of p38i; G–I. p38i escalating doses 0-100 μM with 2.5 μM of PARPi J–L. RAD51i escalating doses 0-100 μM with 2.5 μM PARPi alone or with 10 μM of p38i. All experiments were performed over 72 hours in triplicate and graphs represent line of best-fit non-linear regression +/-SEM.
Targeting of RAD51 activates p38 signaling via ERK1/2
To investigate the mechanism of how p38 affects RAD51 inhibition, we examined the kinase signaling network in MDA-MB-231 cells in response to RAD51 inhibition (RAD51i), dual PARP and p38 inhibition (PARPi/p38i), and the triple combination (RAD51i/PARPi/p38i). We hypothesized that key kinases might be induced by RAD51 inhibition and/or dual PARP and p38 inhibition, but are significantly reduced by triple combination. We found significantly increased signaling in 14 of 44 kinases under these treatment conditions compared with DMSO controls (Figure 2A). RAD51i and the dual combination of PARPi/p38i significantly induced the MAPK signaling axis, particularly ERK1/2 and p38 (Figure 2B). Although the triple combination, RAD51i/PARPi/p38i, attenuated this induction, the phosphorylation of ERK1/2 and p38 remained higher than in the vehicle control (Figure 2B and 2C). It is vital to note that the p38 inhibitor LY2228820 does not affect the phosphorylation of p38 but inhibits its activity [21, 22], which is determined by the phosphorylation of MK2. The RAD51 inhibitor significantly increased signaling via p38 with increased phosphorylation of MK2 (Figure 2C). The use of the p38 inhibitor in the triple combination did not significantly attenuate the phosphorylation of p38, but inhibited its activity as expected as judged by reduced MK2 phosphorylation (Figure 2C). Moreover, while the AKT-RSK-p70S6K and AKT-eNOS signaling axes were induced by RAD51i, these axes were significantly reduced by the RAD51i/PARPi/p38i triple combination (Figure 2B). This triple combination reduced pSTAT3 (Y705) under the baseline level of vehicle control, and reverted p53, S15 and S392 phosphorylations to the baseline levels. Collectively, our results show that the triple combination led to the inhibition of the induced p38 activity, AKT signaling pathways, STAT3 phosphorylation and p53 phosphorylation, which were induced by either RAD51 inhibition and/or the dual PARP/p38 inhibition. This significant reduction of survival signaling pathways explains the effectiveness of this combination in vitro.
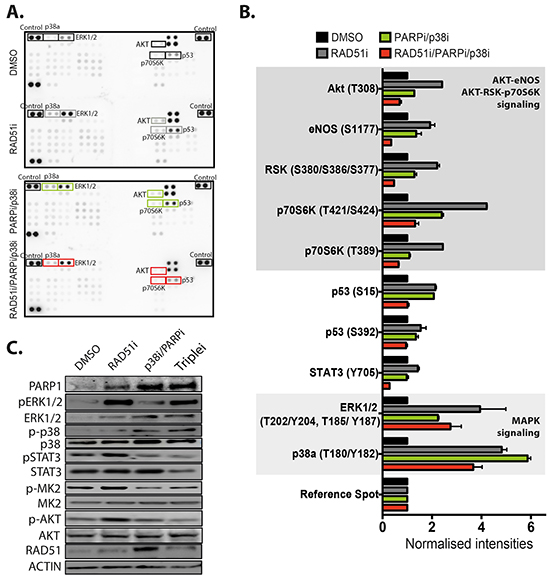
Figure 2: Targeting of RAD51 activates several signaling pathways but attenuated by combining with PARP and p38 inhibition. A. The Human Phospho-Kinase arrays (R&D Systems) were probed with MDA-MB-231 lysate samples that had been treated for 72 hours; samples used are labeled (from top to bottom): (1) DMSO treatment, (2) 10 μM RAD51i, (3) 2.5 μM PARPi/10 μM p38i, and (4) 10 μM RAD51i /2.5 μM PARPi/10 μM p38i triple combination. Highlighted dots represent a significant change in signal over DMSO treated controls. The corners are positive control blots for quantification. B. Quantitation of spot intensity was standardized for cells treated with DMSO and plotted as normalized intensity. Several kinases displayed greater than 2 fold increase in phosphorylation compared to references. Shading represents grouping based on pathway signaling. C. Protein expression and changes in phosphorylation of ERK1/2, p38, STAT3, MK-2 (p38 target) and AKT were confirmed by western blotting.
Effects of targeting RAD51, PARP and p38 in vitro and in vivo
To gain understanding of the consequences of the RAD51i/PARPi/p38i triple combination, we carried out in vitro and in vivo studies using the MDA-MB-231 TNBC cells. The triple combination significantly retarded short-term proliferation of MDA-MB-231 cells compared to vehicle and single drug controls (p<0.001), as well as the two-agent combinations (p<0.008, Figure 3A). Next, we examined the cell cycle profile in response to individual drugs and the triple combination at 72 hours post-treatment. There was no change in the cell cycle profiles of MDA-MB-231 cells in response to any of the individual inhibitors at the selected time point (Figure 3B). In contrast, the triple combination induced significant cell death compared to untreated cells with enhanced sub-G1 population (p=0.05, Figure 3B). The increased apoptosis in the triple combination explains the reduced short-term proliferation (Figure 3A) and the significant reduction of long-term colony formation (p=0.024, Figure 3C). Protein analysis revealed that drug treatment increased p38 and ERK1/2 phosphorylation, except for PARPi treated cells, which retained low ERK1/2 signaling, while RAD51i treatment induced pSTAT3 signaling, in agreement with our earlier observations (Figure 2A). p38 signaling to its downstream target MK2 was significantly reduced in the p38 targeting and triple treatment groups, while pAKT signaling was only enhanced in the RAD51i treated group (Figure 3D). In MDA-MB-436 cells that harbor a pathogenic BRCA1 5396+1G>A mutation, we observed an expected G2 arrest in response to PARPi and the triple combination, with a corresponding increase in the polyploidy population (Supplementary Figure S2A). The treatments also correlated with potent inhibition of colony formation (p=0.027, Supplementary Figure S2B) with the remaining resistant cells retaining a normal cell cycle profile comparable to control cells (Supplementary Figure S2C). This suggests that the G2 arrest and polyploid population (presumably due to mitotic slippage) is another mechanism of response to the triple combination. Protein analysis of MDA-MB-436 cells also showed an induction of the phosphorylated form of p38, but its activity was reduced by the p38 inhibitor significantly in the triple combination, as judged by MK2 phosphorylation (Supplementary Figure S2D). Interestingly, total and phosphorylated ERK1/2 protein was significantly reduced under all conditions in the MDA-MB-436 (Supplementary Figure S2D), in contrast to its increased phosphorylation in the MDA-MB-231 cells.
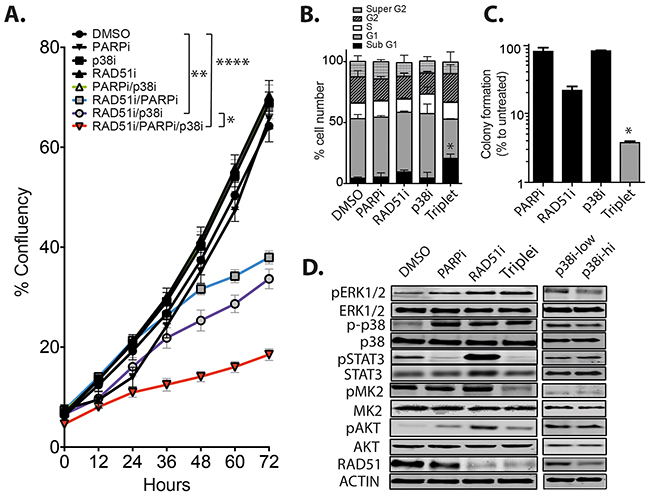
Figure 3: The combination of RAD51/PARP/p38 inhibition retards TNBC growth in vitro. A. MDA-MB-231 cells were incubated in the presence of single, dual or triple drug combinations using 10 μM RAD51i, 10 μM p38i and 2.5 μM PARPi and cell growth followed over 72 hours. Results are average percentage confluency of the well (*p<0.05, **p<0.01, ****p<0.001). B. Cell cycle analysis of MDA-MB-231 cells treated with each inhibitor individually and in combination *p<0.05. C. Long term 14-day colony assays were performed on treated MDA-MB-231 and expressed as percentage of DMSO treated cells *p<0.05. D. Expression of key signaling proteins were examined after single drug treatment or the triple combination, p38i was utilized at low concentration of 10 μM as used in the combination therapies and at 100 μM high dose to observe any changes in expression.
To illustrate the effect of targeting RAD51, PARPi and p38, and the resulting signaling beyond in vitro studies, we examined the triple combination against MDA-MB-231 tumors in vivo (Figure 4A). The use of inhibitors against the individual targets did not significantly reduce primary tumor growth of orthotopic MDA-MB-231 mammary fat pad xenografts (Figure 4B). Of the dual combination therapies, RAD51i and p38i was the most effective compared to control cohort, with significant inhibition of tumor growth after 10 days of treatment (Figure 4B, p=0.019), in agreement with our in vitro results (Figure 3A). The triple combination inhibited tumor growth most significantly (Figure 4B and S3A, p=0.002). Reduced tumor burden was confirmed by assessing ex vivo mammary tumors (Figure 4C). At the ethically allowed maximal tumor size for the control cohort, there was no difference between treated and untreated samples in cancer cell proliferation (Figure 4D). Of note, DMSO control tumor Ki67+ number may have been limited by lack of blood supply to the inner mass. We further examined whether the tumors in the triple combination had sustained alterations in signaling that corroborate our in vitro findings. Indeed, ex vivo immunoblotting displayed induction of ERK1/2 and p38 and a decrease in STAT3 and MK2 phosphorylation in the triple treatment group (Figure 4E), associated with increased apoptosis judged by PARP cleavage (Figure 4E). As RAD51 is a key protein involved in the recombination of both B and T cell receptors, there is a general suggestion that therapeutically targeting of RAD51 in a combination therapy has the potential to induce myelosuppression [23]. Although we utilized immune-compromised nu/nu mice, they still retain functional immune cells except for T-cells, and we were able to detect no significant reduction in whole white blood cell count and lymphocytes in response to the triple combination, which all stayed within normal parameters (Supplementary Figure S3) [24-26]. Taken together, our in vivo studies confirmed our findings that p38 inhibition potentiates the cytotoxic effect of RAD51 inhibition alone and more so when combined with PARP inhibition in triple combination. Similar to our in vitro studies, the strong effect of the triple combination can be explained by the inhibition of survival signaling pathways, which are induced by the single or dual treatments.
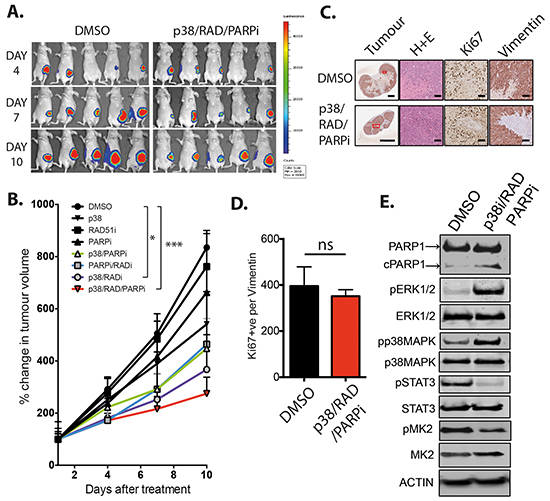
Figure 4: RAD51, PARP and p38 combined inhibition in vivo. A. MDA-MB-231 xenografts were monitored by bioluminescence (luciferin) imaging, DMSO and triple therapy cohorts are represented. B. Tumor burden plotted as percentage change in volume for all single arm controls, double combinations and triple therapy combination compared to days after treatment with all three drugs. *p<0.05, ***p<0.005. C. Histological analysis for overall size, cellularity, proliferation and vimentin expression of DMSO and triple treated tumors (Scale bar =100 μm). D. Quantification of human proliferating cells (Ki67+, Vimentin+). E. Protein expression and signaling analysis of day 10 ex vivo DMSO and triple combination treated tumors.
The rationale for targeting p38 with RAD51 is driven by signaling rather than changes in transcription of key proteins
The rationale behind combining RAD51 depletion and PARP1 inhibition is a well-defined set of mechanisms that coordinate inhibition of concordant DNA repair pathways [6-10]. The mechanisms by which RAD51 depletion combines with p38 inhibition are speculative. Previously, we found that RAD51 can bind c/EBPβ and influence gene expression in MDA-MB-231 cells [12]. Our data here suggest that RAD51 depletion or inhibition induces signaling in the p38 pathway. The significant induction of the transcriptional factor STAT3 suggested a possible transduction of signal from DNA damage to modulate transcription of specific genes [27]. To examine whether this signaling was related to changes in expression, we used the transcriptional repressor actinomycin D. Upon depletion of RAD51, compared to non-targeted controls (Figure 5A-insert), there was an associated depletion of c/EBPβ, p38-alpha and p38-beta transcript (Figure 5A, p<0.01, p<0.05), but not the p38-delta transcript (Supplementary Figure S4A). p38-alpha and p38-beta transcripts were also reduced in response to c/EBPβ depletion (Supplementary Figure S4B). This suggests that in the absence of the RAD51-c/EBPβ complex and active transcription, there is reduced expression or stability of these transcripts. Despite this interesting transcriptional regulation of p38-alpha and p38-beta, it is noteworthy that p38 protein levels after RAD51 targeting in vitro/in vivo or depletion with siRNA, were never down regulated (shown in Figure 5B).
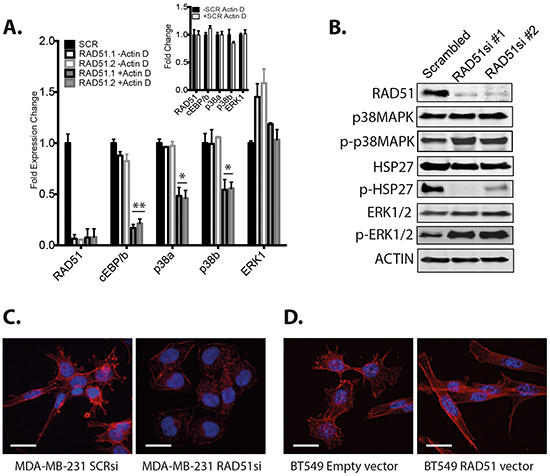
Figure 5: Analysis of p38 pathway signaling. A. MDA-MB-231 cells were depleted of RAD51 and analyzed for expression of the specified genes in the presence or absence of the transcriptional inhibitor actinomycin D. Plots represent the fold expression change over GAPDH and are the average of triplicate experiments +/-SEM. (A-Insert) Controls for non-targeting siRNA +/- actinomycin D. B. Expression of the specified proteins and phosphoproteins examined after RAD51 depletion with two different siRNA. C. Immunofluorescence of MDA-MB-231 cells depleted of RAD51 and stained for morphology. D. Immunofluorescence of BT549 cells engineered for RAD51 overexpression and stained for morphology (Scale bar =100 μm).
Altogether, our transcriptional studies here support the notion that the increased phosphorylation of p38 is a bona fide signaling effect after RAD51 depletion, since p38 mRNA (Figure 5A) and protein (Figure 5B) did not increase even while transcription was active, or reduced when transcription was blocked with actinomycin D. Our initial drug screen supports this conclusion where the p38 inhibitor, but not MEK1 inhibitors, had an additive effect with RAD51 depletion, as p38 phosphorylation was increased as a possible compensatory mechanism due to the stress DNA repair inhibition.
The targeting of p38 and RAD51 mediates additional anti-tumor effects via HSP27
The loss of HSP27 phosphorylation after RAD51 depletion (Figure 5B) provides further support to our conclusion. p38-mediated phosphorylation of HSP27 via MK2 has been demonstrated to regulate alpha-actin turnover [28]. The dephosphorylated form of HSP27 caps the barbed ends of actin filaments, while the phosphorylated form stabilizes long unbranched actin filaments [28]. This phenomenon was observed in morphological cell changes in response to RAD51 expression levels that correlate with HSP27 phosphorylation status, with contracted morphology after RAD51 depletion (Figure 5C) and elongated morphology with RAD51 overexpression (Figure 5D). This observation supports that p38 signaling plays a strong role during RAD51 modulation and further explains the benefit from combining p38 inhibition with RAD51 inhibition alone or when combined with PARP inhibition. The role for HSP27 phosphorylation in association with RAD51 in tumor progression is part of ongoing investigations.
DISCUSSION
Therapies utilizing synthetic lethality with PARPi have been plagued by mechanisms of resistance. We show that by inducing synthetic lethality with PARPi and RAD51i, the addition of targeting p38 induces TNBC cell death in vitro and in vivo. p38 signaling effects associated with RAD51-mediated DNA repair is highly context-dependent, with both DNA-repair dependent and independent mechanisms reported [19, 29]. In contrast to our findings, several studies identified a role for p38 in the activation of Chk1 supporting G2 cell cycle arrest checkpoint assisting ATR signaling and DNA repair [30, 31]. We did not observe a G2 arrest under any single drug conditions except for the well-documented synthetic lethality of targeting PARP in MDA-MB-436. Previous studies also showed the inhibition of p38 activity had no effect on Chk1 activation and DNA repair [30], however, p38 was essential for survival through the expression of prosurvival apoptotic proteins [30]. This implies DNA repair-independent p38 survival activity, via changes in transcription. However, in the cancer setting, p38 can be essential for survival as a sensor for replicative stress, with loss of its downstream target MK2 protecting cells through a DNA-repair dependent activity [32]. Loss of MK2 signaling was observed in MDA-MB-231 cells upon treatment with the p38i/PARPi combination with concordant induction of RAD51 protein expression levels. This mechanism is contradicted by the observation that inhibition of p38 activation enhanced RAD51 protein instability, sensitizing cells to DNA damage [19]. We confirm reduced RAD51 expression observed with high dose p38 inhibitor, however, this was ten times the concentration required for in vivo therapy response. The targeting of p38 did result in loss of MK2 signaling, and the additional targeting RAD51 and PARP1 was able to overcome potentially enhanced DNA repair signaling. Therefore the targeting of p38 may protect cells from DNA damage, but is dependent upon a functional DNA repair pathway.
Several key proteins identified here likely mediate the molecular mechanisms of signaling that underlies our combination therapy. We found that RAD51 depletion and DNA damage activated the AKT pathway with enhanced phosphorylation of key proteins including STAT3 and AKT (Figure 6). The induction of signaling is likely enhanced by the KRAS mutation in MDA-MB-231 cells that results in constitutively activated PI3K signaling via mTOR to STAT3 [33]. As a mechanism of cell survival, STAT3 can induce transcription of specific target genes. We have previously established that RAD51 also contributes to changes in transcription via binding of the transcription factor c/EBPβ [12]. Interestingly STAT3 and c/EBPβ have overlapping consensus sequences in breast cancer promoters, suggesting competitive regulation [34]. Analysis of transcription after RAD51 depletion revealed loss of c/EBPβ transcript stability and p38 (a target gene of c/EBPβ). This scenario could result in the loss of c/EBPβ occupying promoters allowing STAT3 transcription and upregulation of kinase expression that result in the observed enhanced phosphorylation of STAT3.
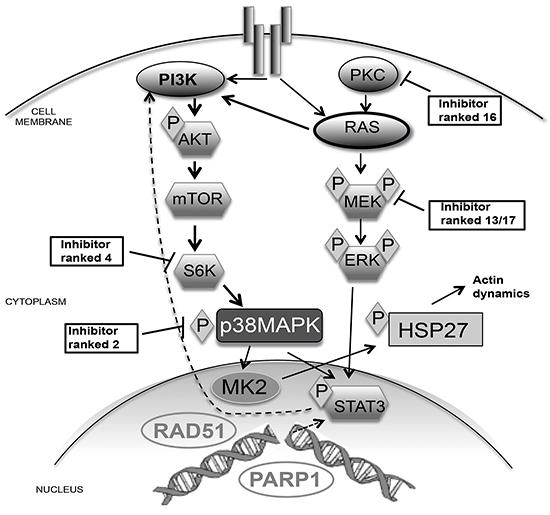
Figure 6: Schematic of p38 pathway signaling. The schematic shows the signaling cascade from a receptor tyrosine kinase to the nucleus. We have highlighted some of the key proteins that were directly inhibited by small molecules and where they ranked in order of efficacy with RAD51 depletion. Due to a K-RAS mutation in MDA-MB-231 cells PI3K is highly active, however RAD51 depletion resulted in ERK1/2 activation. This is why the PKC and MEK were lowly ranked targets compared to S6K and p38 (Ranking obtained from Table 1). We suggest that the ability of RAD51 to bind c/EBPβ can increase expression of key components of the signaling pathway. This is supported by the observation of RAD51-mediated changes in actin dynamics via p38 signaling to HSP27.
Activation of DNA repair increases ERK1/2 phosphorylation [35] and p38 phosphorylation [36]. We observed significant ERK1 signaling in particularly when targeting RAD51 in vitro and both in vitro and in vivo under triple therapy. This might explain the ability of p38 inhibition to potentiate the effect of RAD51 and PARP inhibition. In the case of ERK1/2 phosphorylation after the triple combination, the difference in response observed between MDA-MB-231 cells (p53-mutant/BRCA1-wild type) and MDA-MB-436 cells (p53-low/BRCA1-mutant) may relate to BRCA status. A relation between BRCA1 and ERK1/2 during DNA damage response and cell cycle arrest has been previously described [35]. The different genetic context, particularly p53 status, of MDA-MB-436 compared to that of MDA-MB-231 could also attribute to the cell G2 arrest/mitotic catastrophe [36] in the p53-low MDA-MB-436 compared to apoptosis in the p53-mutant MDA-MB-231 cells. A function independent of signaling, ERK1 is capable of blocking the translocation of c/EBPβ localization to the nucleus via p38 in astrocytes [37]. In the absence of p38 signaling we suggest this function does not occur. Therefore, there may be no c/EBPβ transcriptional activity due to cytoplasmic retention via ERK1 and down regulation of transcript. This could explain the reduced transcript stability of c/EBPβ and p38 observed after RAD51 depletion and previous published observations for RAD51 depletion inhibiting c/EBPβ transcriptional activity [12].
In addition to RAD51 depletion enhancing p38 signaling, we observed depletion of phosphorylated-HSP27. p38 forms a complex with MK2 which can phosphorylate HSP27 in the presence of DNA damage [38]. Under the control of p38/MK2, phosphorylated-HSP27 releases its inhibitory activity on filament capping of actin polymers. This promotes polymerization and cell migration [39]. Thus, loss of HSP27 signaling promotes filament capping. We observed associated changes in cell morphology and migration after RAD51 depletion, which could be accounted for by loss of phosphorylated-HSP27 [12]. Depletion of HSP27 in human breast cancer cell line MDA-MB-231 reduces cell migration and invasion, and this attenuation of invasion correlates in vivo with a decreased ability of breast cancer cells to metastasize and grow in the skeleton [40]. In a comprehensive study it was shown that down-regulation of HSP27 in MDA-MB-436 breast cancer cells induced long-term dormancy in vivo [41] . Thus, the targeting of RAD51, PARPi and p38 also may have the ability to prevent metastatic dissemination of breast cancer cells and this forms part of our ongoing investigations.
The targeting of RAD51, PARP1 and p38 represents a potential effective therapy in TNBC that is not circumvented by “synthetic lethal resistance” observed via pressure on the DNA repair pathway. The unique combination utilizes a “pressure release valve” by targeting p38, which can drive survival by several described signaling mechanisms.
MATERIALS AND METHODS
Animal ethics
All animal procedures were conducted in accordance with Australian National Health and Medical Research regulations on the use and care of experimental animals, and approved by the QIMR Berghofer Medical Research Institute Animal Ethics Committee (approval P1301).
Cell lines
TNBC MDA-MB-231, MDA-MB-436, and BT549 lines were acquired from ATCC, PMC42ET was a gift from Robin Anderson (Peter MacCallum Cancer Centre, Melbourne, Australia). BT549 was engineered for overexpression of RAD51 using Origene letinviral construct and empty vector as control (SKU: RG218333).
Sensitivity to single DNA damaging and signaling inhibitor agents
MDA-MB-231, PMC42-ET and MDA-MB-436 breast cancer cell lines were analyzed for sensitivity to; PARPi (ABT-888-Veliparib), p38i (LY222882), DNA-PKi (KU-57788), S6K (PF 4708671), ATMi (KU-55933), PAKi (IPA3), Ligase IVi (SCR7), CHK1i (AZD7762), PYK2i (PF-562271), HSP90 (AUY922), GSK3i (CHIR-98014), DNA-PKi(2) (NU7026), MEKi(2) (AS703026), EGFRi (W2992-Afatinib), PDKi (GSK2334470), PKCi (LY317615), MEKi (AZD6244), PLCi (U73122), IGF-1R (GSK1904529A) and ERK5i (BIX 02189). All drugs were purchased from Selleck Chem. Each cell line was targeted with two different RAD51 siRNA or scrambled control siRNA and 1x104 cells/well plated in the presence of 0-100 μM inhibitor and incubated for 120 hours. Growth inhibition was measured by MTS assay (Cell titer 96 AQueous Promega) measured at 500 nm. Data was analyzed using GraphPad Prism and plotted as nonlinear regression line of best fit for IC50. IC50 was then compared between non-targeted and RAD51 depleted cells and expressed as a ratio difference. The average between the three different cell lines provided the order of efficacy against TNBC.
Combination therapy dose curves
Each of MDA-MB-231, MDAM-MB-436 or PMC42ET cell lines were incubated in the presence of each drug individually (0-100 μM) plus DMSO controls, as well as in combination with PARPi (2.5 μM ABT-888) or p38i (10 μM LY222882) and measured by MTS assay. The plots were standardized to DMSO untreated cells and represent line of best-fit non-linear regression +/-SEM.
Kinome array
We performed the Proteome ProfilerTM arrays as per our previous studies [18] and manufacturers’ instructions. In summary, cells were washed with cold PBS and collected by trypsinization, lysates from breast cancer cell lines were prepared using the lysis buffer provided in the kit. Lysates were then hybridized to the arrays as per manufacturer instructions (R&D Systems, MN, USA). Phosphorylation levels were detected with the FujiFilm LAS-3000 Imaging System (Fuji Photo Film, Tokyo, Japan) using Millipore Immobilon Western chemiluminescent HRP substrate. Protein band intensity was determined using the “Protein array analyser” quantitation function of Image J v 1.43.
Flow cytometry analysis
Cultures were trypsinized to single cell suspensions, washed with phosphate-buffered saline (PBS)/10% FBS, fixed with 70% cold ethanol and stained with propidium iodide containing RNAse. Flow cytometry analysis was performed on 10,000 gated events per sample. Cells were analysed and gated per subG1, G1, S, G2 and >G2 (Super G2-polyploid) populations.
Long-term colony assays
After incubation with appropriate drug combinations, 1x103 cells were plated per well in a 6-well plate and incubated with DMEM supplemented with 10% FBS and antibiotics. After 14 days incubation the colonies were washed with PBS, fixed with 70% ethanol and stained with 0.5% crystal violet in PBS for 5 minutes. Excess stain was removed with running water and plates scanned and quantitated using Image J software (NIH). Plots represent the mean and standard error of the mean (SEM) of 4 independent experiments.
Immunoblotting
Protein lysates were prepared via whole cell lysis in ice-cold lysis buffer (150 mM NaCl, 10 mM Tris-Cl pH 7.4, 5 mM EDTA, 1% Triton X-100) supplemented with protease inhibitors (Leupeptin, Pepstatin and PMSF, Sigma Aldridge) as previously described [42]. Immunoblots were probed with anti-RAD51 (Santa Cruz Biotech), anti-p38, anti-phospho-p38 (Cell Signaling), anti-PARP (BD Biosciences), anti-MK2 (Cell Signaling), anti-HSP27, anti-phospho-HSP27 (Cell Signaling), anti-ERK1/2, anti-phospho-ERK1/2 (Cell Signaling) and anti-ß-Actin, (Sigma) as a loading control. Membranes were developed using fluorescent-labeled secondary antibodies and visualized using the Odyssey system. Protein expression levels were determined by optical density versus actin loading controls using Image J software (NIH).
Xenograft therapy
Cohorts of 5 week-old female nude NU-Foxn1nu or Balb/c mice with established 40 mm3 luciferase positive inguinal mammary fat pad tumors from 5x106 cells of MDA-MB-231 were used. Mice were treated with 50 mg/kg B02 (Sigma), 25 mg/kg LY228820 (Selleck Chem) and 25mg/kg ABT-888 (Selleck Chem). Mice were treated via intraperitoneal injection. LY228820 and B02 were administered daily while ABT-888 was administered every second day. Tumor growth was monitored by caliper measurement and visualized using luciferase-mediated live animal imaging with the IVIS100 Xenogen system (Caliper Life Sciences).
Expression analysis
RNA was isolated from RAD51 or c/EBPβ depleted MDA-MB-231 cells incubated in the presence or absence of 0.05mg/ml actinomycin D, using QIAGEN RNAEasy Mini Kit. cDNA was synthesized using RT2 First Strand Kit (QIAGEN). cDNA was analyzed on Roche Lightcycler 480 with validated primer sets (Supplementary Table S1). Fold expression change was calculated against actin and expressed as base-two exponential increase in RNA levels (2ΔΔCt)+/- SEM.
Immunofluorescence
Approximately 5x104 cells were seeded onto 18 mm glass coverslips. At 48 hours later, cells were gamma irradiated at 6 Gy. Coverslips were washed in (PBS), the cells were fixed in cold 70% ethanol and permeabilized in 0.5% Triton X-100 solution for 15 min at room temperature. Cells were blocked with 10% FBS in PBS and incubated with primary antibody (or alexa phalloidin) for 1 h and with secondary antibody for 30 min at room temperature. All antibodies were diluted in 5% FBS–PBS. Cells were then washed, counterstained with 4′,6′-diamidino-2-phenylindole (DAPI), and mounted. Primary antibody dilutions were as follows: Rad51, γH2Ax 1:1000 and 1:500 respectively. All secondary antibodies were used at 1:200.
Statistical analysis
Results are presented as mean +/- SEM of replicate analyses and are either representative of or inclusive of at least three independent experiments. All statistical analyses were performed using two-tailed Student’s t-tests in GraphPad PRISM 6 software. In all figures, significant differences between specified pair of conditions, as judged by t-test, are indicated using asterisks (*p<0.05; **p<0.01; ***p<0.005, ****p<0.001). IC50 doses for were calculated by interpolation of the sigmoidal dose response curves (Graphpad Prism 6.0 software).
Abbreviations
TNBC triple negative breast cancer, PARP1 poly(ADP-ribose)polymerase 1, p38 p38 Mitogen-Activated Protein Kinase.
ACKNOWLEDGMENT
APW would like to acknowledge Luke Ishak for performing the drug screen. APW and AM would like to thank the National Breast Cancer Foundation Australia for fellowship and grant support, and all the members of the Tumor Microenvironment lab for critical reading of the manuscript.
CONFLICTS OF INTEREST
All authors declare that they have no competing interests.
FINANCIAL SUPPORT
This work was supported by the National Breast Cancer Foundation Australia (to APW:PF-13-12, to AM: ECF-11-09, NC-13-26), Cancer Council Queensland (to AM: APP1045620), the National Health and Medical Research Council (to AM: APP106851) and ARC Future Fellowship (to FA: FT130101417).
Authors contributions
APW conceived the study, performed most of the experimentation and drafted the manuscript, FA and MM led the experimental design and analysis of the phospho-kinase array and associated Western blotting, SWW carried out the in vivo xenograft therapy experiment, AM supervised experimental design, drafted the manuscript and coordinated the work. All authors read and approved the final version of the manuscript.
REFERENCES
1. Stover DG, Winer EP. Tailoring adjuvant chemotherapy regimens for patients with triple negative breast cancer. Breast. 2015; 24:S132-5.
2. Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016.
3. Anders CK, Winer EP, Ford JM, Dent R, Silver DP, Sledge GW, Carey LA. Poly(ADP-Ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clin Cancer Res.16:4702-10.
4. O’Shaughnessy J, Schwartzberg L, Danso MA, Miller KD, Rugo HS, Neubauer M, Robert N, Hellerstedt B, Saleh M, Richards P, Specht JM, Yardley DA, Carlson RW, Finn RS, Charpentier E, Garcia-Ribas I, Winer EP. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2014;32:3840-7.
5. Dhillon KK, Swisher EM, Taniguchi T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011;102:663-9.
6. Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368-77.
7. Michels J, Vitale I, Saparbaev M, Castedo M, Kroemer G. Predictive biomarkers for cancer therapy with PARP inhibitors. Oncogene. 2014;33:3894-907.
8. Neijenhuis S, Bajrami I, Miller R, Lord CJ, Ashworth A. Identification of miRNA modulators to PARP inhibitor response. DNA Repair (Amst).12:394-402.
9. Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst). 2008;7:2010-9.
10. Daemen A, Wolf DM, Korkola JE, Griffith OL, Frankum JR, Brough R, Jakkula LR, Wang NJ, Natrajan R, Reis-Filho JS, Lord CJ, Ashworth A, Spellman PT, Gray JW, van’t Veer LJ. Cross-platform pathway-based analysis identifies markers of response to the PARP inhibitor olaparib. Breast Cancer Res Treat. 2012;135:505-17.
11. Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, Bishop DK. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res. 2007;67:9658-65.
12. Wiegmans AP, Al-Ejeh F, Chee N, Yap PY, Gorski JJ, Da Silva L, Bolderson E, Chenevix-Trench G, Anderson R, Simpson PT, Lakhani SR, Khanna KK. Rad51 supports triple negative breast cancer metastasis. Oncotarget. 2014;5:3261-72. doi: 10.18632/oncotarget.1923.
13. Wiegmans AP, Yap PY, Ward A, Lim YC, Khanna KK. Differences in expression of key DNA damage repair genes after epigenetic-induced BRCAness dictate synthetic lethality with PARP1 inhibition. Mol Cancer Ther. 2015.
14. Ko JC, Ciou SC, Cheng CM, Wang LH, Hong JH, Jheng MY, Ling ST, Lin YW. Involvement of Rad51 in cytotoxicity induced by epidermal growth factor receptor inhibitor (gefitinib, IressaR) and chemotherapeutic agents in human lung cancer cells. Carcinogenesis. 2008;29:1448-58.
15. Su YJ, Tsai MS, Kuo YH, Chiu YF, Cheng CM, Lin ST, Lin YW. Role of Rad51 down-regulation and extracellular signal-regulated kinases 1 and 2 inactivation in emodin and mitomycin C-induced synergistic cytotoxicity in human non-small-cell lung cancer cells. Mol Pharmacol. 2010;77:633-43.
16. Graves LM, Duncan JS, Whittle MC, Johnson GL. The dynamic nature of the kinome. Biochem J. 2013;450:1-8.
17. Fleuren ED, Zhang L, Wu J, Daly RJ. The kinome ’at large’ in cancer. Nat Rev Cancer. 2016;16:83-98.
18. Al-Ejeh F, Miranda M, Shi W, Simpson PT, Song S, Vargas AC, Saunus JM, Smart CE, Mariasegaram M, Wiegmans AP, Chenevix-Trench G, Lakhani SR, Khanna KK. Kinome profiling reveals breast cancer heterogeneity and identifies targeted therapeutic opportunities for triple negative breast cancer. Oncotarget. 2014;5:3145-58. doi: 10.18632/oncotarget.1865.
19. Chuang SM, Wang LH, Hong JH, Lin YW. Induction of Rad51 protein levels by p38 MAPK decreases cytotoxicity and mutagenicity in benzo[a]pyrene-exposed human lung cancer cells. Toxicology and applied pharmacology. 2008;230:290-7.
20. Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem Biol. 2011;6:628-35.
21. Ishitsuka K, Hideshima T, Neri P, Vallet S, Shiraishi N, Okawa Y, Shen Z, Raje N, Kiziltepe T, Ocio EM, Chauhan D, Tassone P, Munshi N, Campbell RM, Dios AD, Shih C, Starling JJ, Tamura K, Anderson KC. p38 mitogen-activated protein kinase inhibitor LY2228820 enhances bortezomib-induced cytotoxicity and inhibits osteoclastogenesis in multiple myeloma; therapeutic implications. Br J Haematol. 2008;141:598-606.
22. Mader M, de Dios A, Shih C, Bonjouklian R, Li T, White W, Lopez de Uralde B, Sanchez-Martinez C, del Prado M, Jaramillo C, de Diego E, Martin Cabrejas LM, Dominguez C, Montero C, Shepherd T, Dally R, Toth JE, Chatterjee A, Pleite S, Blanco-Urgoiti J, Perez L, Barberis M, Lorite MJ, Jambrina E, Nevill CR, Jr., Lee PA, Schultz RC, Wolos JA, Li LC, Campbell RM, Anderson BD. Imidazolyl benzimidazoles and imidazo[4,5-b]pyridines as potent p38alpha MAP kinase inhibitors with excellent in vivo antiinflammatory properties. Bioorganic & medicinal chemistry letters. 2008;18:179-83.
23. Jekimovs C, Bolderson E, Suraweera A, Adams M, O’Byrne KJ, Richard DJ. Chemotherapeutic compounds targeting the DNA double-strand break repair pathways: the good, the bad, and the promising. Frontiers in oncology. 2014;4:86.
24. Monteiro JP, Benjamin A, Costa ES, Barcinski MA, Bonomo A. Normal hematopoiesis is maintained by activated bone marrow CD4+ T cells. Blood. 2005;105:1484-91.
25. Matsumoto K, Inukai S, Isaka T, Aruga N, Nakamura S, Nagasawa T, Naito J. Cell counts in peripheral blood and bone marrow of male C.B-17 scid/scid mice. Laboratory animals. 1995;29:218-22.
26. Dadachova E, Revskaya E, Sesay MA, Damania H, Boucher R, Sellers RS, Howell RC, Burns L, Thornton GB, Natarajan A, Mirick GR, DeNardo SJ, DeNardo GL, Casadevall A. Pre-clinical evaluation and efficacy studies of a melanin-binding IgM antibody labeled with 188Re against experimental human metastatic melanoma in nude mice. Cancer Biol Ther. 2008;7:1116-27.
27. Barry SP, Townsend PA, Knight RA, Scarabelli TM, Latchman DS, Stephanou A. STAT3 modulates the DNA damage response pathway. International journal of experimental pathology. 2010;91:506-14.
28. Pichon S, Bryckaert M, Berrou E. Control of actin dynamics by p38 MAP kinase - Hsp27 distribution in the lamellipodium of smooth muscle cells. Journal of cell science. 2004;117:2569-77.
29. Lu CH, Lin SC, Yang SY, Pan MY, Lin YW, Hsu CY, Wei YH, Chang JS, Chang CC. Prodigiosin-induced cytotoxicity involves RAD51 down-regulation through the JNK and p38 MAPK pathways in human breast carcinoma cell lines. Toxicology letters. 2012;212:83-9.
30. Phong MS, Van Horn RD, Li S, Tucker-Kellogg G, Surana U, Ye XS. p38 mitogen-activated protein kinase promotes cell survival in response to DNA damage but is not required for the G(2) DNA damage checkpoint in human cancer cells. Mol Cell Biol. 2010;30:3816-26.
31. Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E, Fornace AJ, Jr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001;411:102-7.
32. Kopper F, Bierwirth C, Schon M, Kunze M, Elvers I, Kranz D, Saini P, Menon MB, Walter D, Sorensen CS, Gaestel M, Helleday T, Schon MP, Dobbelstein M. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proc Natl Acad Sci U S A. 2013;110:16856-61.
33. Lee HW, Kyung T, Yoo J, Kim T, Chung C, Ryu JY, Lee H, Park K, Lee S, Jones WD, Lim DS, Hyeon C, Heo WD, Yoon TY. Real-time single-molecule co-immunoprecipitation analyses reveal cancer-specific Ras signalling dynamics. Nature communications. 2013;4:1505.
34. Shackleford TJ, Zhang Q, Tian L, Vu TT, Korapati AL, Baumgartner AM, Le XF, Liao WS, Claret FX. Stat3 and CCAAT/enhancer binding protein beta (C/EBP-beta) regulate Jab1/CSN5 expression in mammary carcinoma cells. Breast Cancer Res. 2011;13:R65.
35. Yan Y, Spieker RS, Kim M, Stoeger SM, Cowan KH. BRCA1-mediated G2/M cell cycle arrest requires ERK1/2 kinase activation. Oncogene. 2005;24:3285-96.
36. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175-89.
37. Fields J, Cisneros IE, Borgmann K, Ghorpade A. Extracellular regulated kinase 1/2 signaling is a critical regulator of interleukin-1beta-mediated astrocyte tissue inhibitor of metalloproteinase-1 expression. PLoS One. 2013;8:e56891.
38. Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cellular and molecular life sciences : CMLS. 2009;66:3289-307.
39. Kotlyarov A, Yannoni Y, Fritz S, Laass K, Telliez JB, Pitman D, Lin LL, Gaestel M. Distinct cellular functions of MK2. Molecular and cellular biology. 2002;22:4827-35.
40. Gibert B, Eckel B, Gonin V, Goldschneider D, Fombonne J, Deux B, Mehlen P, Arrigo AP, Clezardin P, Diaz-Latoud C. Targeting heat shock protein 27 (HspB1) interferes with bone metastasis and tumour formation in vivo. Br J Cancer. 2012;107:63-70.
41. Straume O, Shimamura T, Lampa MJ, Carretero J, Oyan AM, Jia D, Borgman CL, Soucheray M, Downing SR, Short SM, Kang SY, Wang S, Chen L, Collett K, Bachmann I, Wong KK, Shapiro GI, Kalland KH, Folkman J, Watnick RS, Akslen LA, Naumov GN. Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:8699-704.
42. Wiegmans AP, Alsop AE, Bots M, Cluse LA, Williams SP, Banks KM, Ralli R, Scott CL, Frenzel A, Villunger A, Johnstone RW. Deciphering the molecular events necessary for synergistic tumor cell apoptosis mediated by the histone deacetylase inhibitor vorinostat and the BH3 mimetic ABT-737. Cancer research. 2011;71:3603-15.