
INTRODUCTION
A hallmark of cancer cells is their ability to prolong cell survival by avoiding apoptosis. The family of B-cell lymphoma-2 (Bcl-2) proteins is a critical regulator of this process [1–5]. It consists of anti-apoptotic members, including Bcl-2 [6] and Bcl-Xl [7] and pro-apoptotic members like Bax [8]. All the members of the family share at least one of the four conserved α-helical motifs, known as Bcl-2 homology (BH1-4) domains [1, 9]. Many of these proteins exist in more than one isoforms [7, 10–13] and Bcl-2 is not an exception. Two isoforms of Bcl-2, resulting from alternative splicing, were described: Bcl-2α and Bcl-2β [14]. Most of the work until now has been done with Bcl-2α, which is the long isoform and which in addition to the four BH domains contains a C-terminal extension with a putative trans-membrane domain (TMD) (Figure 1A). In contrast, Bcl-2β has a much shorter C-terminus and lacks a TMD [14, 15]. While Bcl-2β is mostly detected in cytosolic fractions, the TMD and a short preceding sequence target Bcl-2α to a variety of intracellular membranes including mitochondrial, endoplasmic reticulum (ER) and nuclear membranes [16–18]. Bcl-2α is the more abundant isoform in both healthy and cancer cells and it remains dominant in cancer cells up-regulating Bcl-2 protein [14, 19]. In virtually all studies published to this date, Bcl-2 refers to Bcl-2α. The anti-apoptotic function of Bcl-2 oncogene was first characterized at the level of the mitochondria, particularly at the outer mitochondrial membrane, where it inhibits Bax/Bak–mediated apoptosis. The mechanism involves a BH3-dependent interaction, where the hydrophobic cleft of Bcl-2 formed by the BH3-BH1-BH2 domains sequesters the BH3 domain of the pro-apoptotic members. This prevents Bax/Bak activation and oligomerization and inhibits the consequent mitochondrial permeabilization and cell death [2, 3, 20, 21].
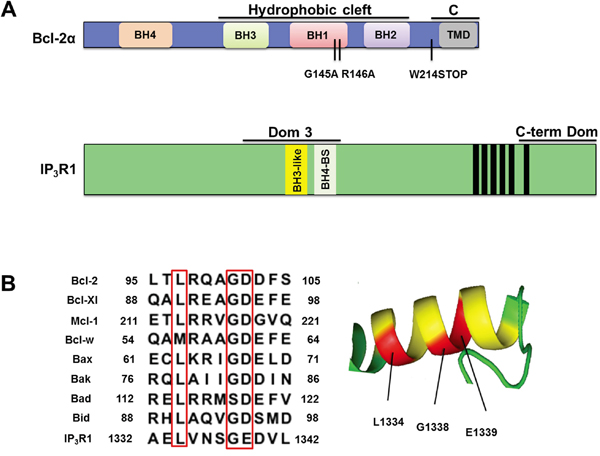
Figure 1: The Dom 3 of IP3R1 contains a BH3 motif. A. Linear representation of Bcl-2 and IP3R1. Bcl-2α is depicted in blue with its BH domains and the trans-membrane domain (TMD). The two functional domains of interest, the hydrophobic cleft formed by BH3-BH1-BH2 domains and the C-terminus (C), are indicated with black lines. The C-terminal region, containing the TMD, is present in Bcl-2α, but not in Bcl-2β. The G145 and R146 residues, located in the BH1 domain were mutated to yield Bcl-2GR/AA. Bcl-2 was truncated at W214 residue to yield Bcl-2ΔC, which correlates with Bcl-2β. A schematic representation of IP3R1 is depicted in green. The Bcl-2-binding fragments of IP3R used in this study, the domain 3 (Dom 3) and the C-terminal domain containing the last TMD (C-term Dom), are indicated with black lines and the six TMDs are shown as black bars. The exact BH4-binding site in the Dom 3 is represented in light grey (BH4-BS). The BH3-like motif in the Dom 3 is represented in yellow. B. The Dom 3 of IP3R1 contains a BH3 motif. Left: Sequence alignment between the BH3 domains of Bcl-2 family members and the Dom 3 of IP3R1 reveals the presence of the conserved residues (LxxxGD/E, pointed in red), required for a typical BH3 motif (46). Right: A secondary structure prediction of the putative BH3 motif of IP3R1 present in the Dom 3 sequence, predicted by the I-TASSER web server and drawn using PyMol. The predicted BH3 motif within the Dom 3 is depicted in yellow and the conserved residues in red.
Ca2+ signaling is another important modulator in cell-fate decisions, which can serve as a survivor factor promoting cell proliferation, but also as a cell-death inducer [22–25]. Bcl-2 was shown to execute its pro-survival function not only via direct inhibition of pro-apoptotic proteins but also via suppression of pro-apoptotic Ca2+ signals. This occurs by direct interaction with inositol 1,4,5-trisphosphate (IP3) receptors (IP3Rs) [26–29], the main intracellular Ca2+ release channels, located at the ER [30–34]. IP3R inhibition by Bcl-2 appears to be an important mechanism that contributes to the oncogenic properties of Bcl-2. Many cancer cells, including leukemia, lymphoma and lung cancer cells, are addicted to IP3R/Bcl-2-complex formation for their survival, since tools that disrupt this complex trigger cell death [35–37]. Over the last years, important insights in the regulation of IP3Rs by Bcl-2 (i.e. Bcl-2α) at the molecular level have been obtained. The suppression of IP3R-mediated Ca2+ release by Bcl-2 was attributed to the interaction of the BH4 domain of Bcl-2 with a 20 amino acid region (a.a. 1389-1408) located in the central modulatory domain, more particularly in the Domain 3 (Dom 3) (a.a. 923-1581) of IP3R [38, 39]. Previous studies, which exploited synthetic peptides covering the BH4 domain of Bcl-2 (BH4-Bcl-2), revealed that this domain is necessary and sufficient to bind to IP3R and to suppress its activity [26, 27, 39, 40]. Nevertheless, the relatively low affinity of inhibition by the BH4 domain (measured in vitro IC50=30μM) [27, 39] cannot explain the potent inhibitory effect of Bcl-2 full-length protein in physiological conditions. This Achilles’ heel of the model suggests that additional domains in Bcl-2 could be responsible for an efficient in cellulo inhibition of IP3R. Interestingly, the C-terminal domain, containing the last 6th TMD of the IP3R (C-term Dom, a.a. 2512-2749), which is in close proximity of the channel pore is also targeted by Bcl-2 [41, 42], but the mechanism and significance of this interaction are not completely solved. The same C-term Dom of IP3R also appeared to be responsible for interaction with other members of the family: Bcl-Xl and Mcl-1 [41].
Here, we aimed to identify the molecular determinants in Bcl-2α responsible for its interaction with the C-term Dom of IP3R and to assess their functional impact on Bcl-2α-mediated inhibition of the channel. We especially focused on two important functional domains in Bcl-2α, i.e. the hydrophobic cleft, involved in BH3-dependent interactions and the C-terminal region, containing the TMD, involved in hydrophobic interactions within the membrane environment (Figure 1A). Using genetic and pharmacological approaches, we could however exclude the hydrophobic cleft as a major player in the formation of the Bcl-2α/IP3R complex. In contrast, we found that Bcl-2α binding to the C-term Dom of IP3R1 depends on the presence of the C-terminus of Bcl-2α. This region of Bcl-2α is required for efficient inhibition of IP3Rs in a cellular context and for inhibition of staurosporine (STS) – induced apoptosis. Furthermore, we demonstrated a direct interaction between a peptide corresponding to the TMD of Bcl-2α (TMD-Bcl-2) and the purified C-terminal fragment of IP3R1. The TMD-Bcl-2 was able to suppress IP3-induced Ca2+ release (IICR) when applied at high concentrations. These results suggest that the C-terminal region, and particularly the TMD, of Bcl-2α not only serves as an anchor for tethering Bcl-2α in the membranes, but is also an important functional regulator of IP3R activity. Since the TMD is only present in Bcl-2α, but not in Bcl-2β, this study is the first one hinting towards important functional difference between the two isoforms with respect to Ca2+-signaling regulation.
RESULTS
Despite the presence of BH3-domain features in the IP3R sequence, the hydrophobic cleft of Bcl-2α is dispensable for interaction with the receptor
We performed a sequence alignment of the BH3 domains of different Bcl-2 proteins with the fragment of the central modulatory domain of IP3R1 (Dom 3), shown in previous studies to bind Bcl-2 [27, 38, 42]. This analysis revealed the presence of a BH3 motif (a.a. 1332-1342) upstream of the previously described region in Dom 3 of IP3R targeted by the BH4 domain of Bcl-2 (a.a. 1389-1408) (Figure 1A) [43]. Figure 1B depicts the presence of the conserved LxxxGD/E motif [44] in the Dom 3 of IP3R and the α-helical secondary structure of this motif as predicted by I-TASSER web server. To determine whether a BH3-dependent mechanism plays a direct role in the interaction between Bcl-2α and IP3R we used two different approaches to antagonize the hydrophobic cleft of Bcl-2α, genetic manipulation and pharmacological inhibition. The genetic approach is based on mutations in the BH1 domain (replacement of G145R146 by AA yielding Bcl-2GR/AA) (Figure 1A), which lead to disruption of the binding between Bcl-2α and Bax [45–47]. The second approach is based on the use of pharmacological inhibitors like the BH3-mimetic compounds [48, 49], designed to occupy the hydrophobic cleft, thereby disrupting interactions between BH3 domain-containing proteins and anti-apoptotic Bcl-2 proteins [48, 49]. Here, we applied ABT-199, a selective Bcl-2 inhibitor which does not target Bcl-Xl [50].
First, we validated that both, the GR/AA mutation or the incubation with ABT-199 (3 μM), prevent Bcl-2α binding to Bax in co-immunoprecipitation experiments. The concentration of ABT-199 that we used in the experiments is well above the documented subnanomolar affinity of this compound for Bcl-2 (Ki < 0.01 nM) [50], thus maximizing the potential effect of ABT-199 on Bcl-2/IP3R interaction. 3xFLAG-tagged proteins (3xFLAG-Bcl-2wt in presence and absence of ABT-199 or 3xFLAG-Bcl-2GR/AA) were overexpressed in COS-1 cells and immunoprecipitated from the cell lysates using anti-FLAG-loaded agarose beads. Immunoblots were stained for FLAG and Bax (Figure 2A).
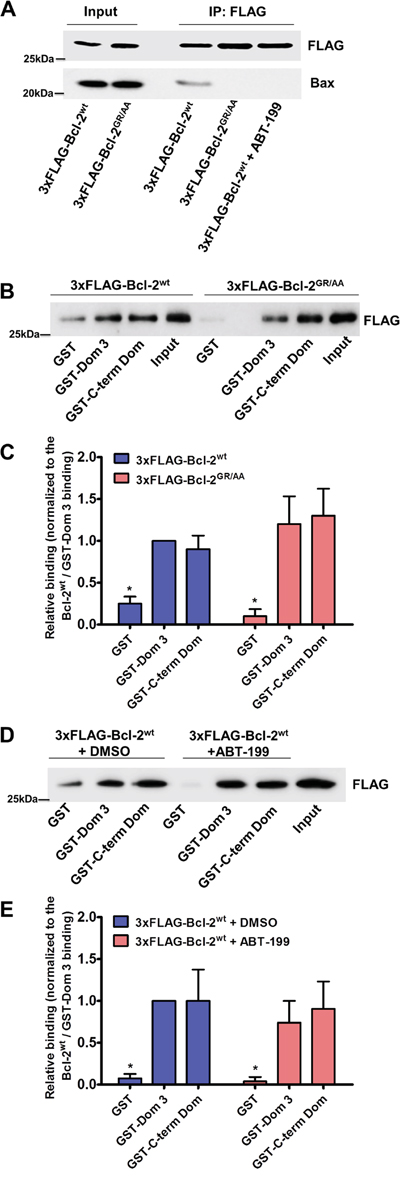
Figure 2: Bcl-2GR/AA and Bcl-2wt exposed to ABT-199 fail to bind pro-apoptotic Bax, but remain capable of binding the Dom 3 and the C-term Dom of IP3R. A. Representative FLAG-co-immunoprecipitation experiment for detection of the 3xFLAG-Bcl-2/Bax interaction is shown. Overexpressed 3xFLAG-Bcl-2wt (in absence and presence of 3 μM ABT-199) or 3xFLAG-Bcl-2GR/AA was immunoprecipitated from COS-1 cell lysates by anti-FLAG-loaded agarose beads. The immunoreactive blots were stained with antibody against FLAG and Bax. 0.1 μg and 0.5 μg of total COS-1 lysates were used as input for the 3xFLAG-proteins and Bax respectively. The experiments were performed 3 times utilizing each time independently transfected cells and freshly prepared lysates. B. Representative GST-pull down experiments with COS-1 cell lysates for comparing the binding of overexpressed 3xFLAG-Bcl-2wt and 3xFLAG-Bcl-2GR/AA to the GST-Dom 3 and GST-C-term Dom are shown. The samples were analyzed via Western blot and stained with anti-FLAG antibody. The binding to the GST was used as a negative control. 0.1 μg of total COS-1 lysates was used as input. C. The immunoreactive bands from 3 independent experiments, utilizing each time independently transfected cells and freshly prepared lysates, were quantified and normalized to the binding of 3xFLAG-Bcl-2wt to GST-Dom 3, which was set as 1. The data are plotted as mean ± S.E.M. D. Representative GST-pull down experiments with COS-1 cell lysates for comparing the binding of overexpressed 3xFLAG-Bcl-2wt in absence and presence of 3 μM ABT-199 to the GST-Dom 3 and GST-C-term Dom are shown. The samples were analyzed via Western blot and stained with anti-FLAG antibody. The binding to the GST was used as a negative control. 0.1 μg of total COS-1 lysates was used as input. E. The immunoreactive bands from 3 independent experiments, utilizing each time independently transfected cells and freshly prepared lysates, were quantified and normalized to the binding of 3xFLAG-Bcl-2wt to GST-Dom 3, which was set as 1. The data are plotted as mean ± S.E.M.
Next we performed two different sets of GST pull-down experiments, using the two purified IP3R domains targeted by Bcl-2, GST-Dom 3 (a.a. 923-1581) and GST-C-term Dom (a.a. 2512-2749). To compare the binding properties of the wild-type Bcl-2 protein versus the mutant for these IP3R fragments we overexpressed 3xFLAG-Bcl-2wt or 3xFLAG-Bcl-2GR/AA in COS-1 cells. The binding of 3xFLAG-Bcl-2wt to GST-Dom 3 was used as reference and all binding values were normalized to this control. Our results show that 3xFLAG-Bcl-2GR/AA remained fully capable of binding to both GST-Dom 3 and GST-C-term Dom to a similar extent as 3xFLAG-Bcl-2wt (Figure 2B).
As a second approach, we examined the interaction between Bcl-2wt and the two GST-fused domains of IP3R in presence or absence of the BH3-mimetic compound ABT-199 (3 μM). Incubation with ABT-199 did not significantly affect the binding of 3xFLAG-Bcl-2wt to the GST-Dom 3, nor to the GST-C-term Dom (Figure 2C).
Taken together, these results suggest that the hydrophobic cleft of Bcl-2 is dispensable for its interaction with IP3R.
The hydrophobic cleft of Bcl-2α does not contribute to the inhibitory effect on IP3Rs
Bcl-2 overexpression results in dampened IP3R-mediated Ca2+ release in intact cells [27, 29, 39, 51], but whether this effect is mediated through the hydrophobic cleft of Bcl-2 is not known. To address this question, we monitored the change in cytosolic Ca2+ levels in response to an IP3R agonist, ATP, using the ratiometric fluorescent Ca2+ dye Fura-2-AM. Similarly to the GST-pull down experiments, we used the mutation (Bcl-2GR/AA) or ABT-199 to antagonize the hydrophobic cleft of Bcl-2α. Intact COS-1 cells overexpressing 1) 3xFLAG-empty vector, 3xFLAG-Bcl-2wt or 3xFLAG-Bcl-2GR/AA and 2) 3xFLAG-empty vector or 3xFLAG-Bcl-2wt in presence or absence of ABT-199 (3 μM), and co-transfected with mCherry plasmid were exposed to ATP (0.5 μM). The proper expression of the 3xFLAG-proteins in the COS-1 cells was assessed via Western blotting using anti-FLAG antibody (Supplementary Figure S1A and S1B). Importantly, the expression levels of 3xFLAG-Bcl-2wt and 3xFLAG-Bcl-2GR/AA proteins were similar, although 3xFLAG-Bcl-2GR/AA tended to be expressed at slightly higher levels. In addition, only cells with similar intensity of mCherry, thus similar levels of 3xFLAG-proteins were subjected to measurement. To chelate the free extracellular Ca2+, the experiments were performed in the presence of BAPTA (3 mM), an extracellular Ca2+ buffer, ensuring that the ATP-induced [Ca2+] rise is only due to Ca2+ release from intracellular stores. The ER Ca2+-store content was also assessed by applying thapsigargin (Tg, 1 μM), an irreversible SERCA inhibitor, in the presence of BAPTA (Supplementary Figure S1A and Supplementary Figure S1B). Consistent with our previous studies, overexpression of 3xFLAG-Bcl-2wt inhibited ATP-induced Ca2+ release without affecting the ER Ca2+-stores content [27]. In line with our GST-pull down experiments, neither the overexpression of 3xFLAG-Bcl-2GR/AA (Figure 3A-3C), nor the presence of ABT-199 (Figure 4A-4D) prevented this effect. The quantitative analysis indicated that 3xFLAG-Bcl-2wt, 3xFLAG-Bcl-2GR/AA (Figure 3D) and 3xFLAG-Bcl-2wt in presence of ABT-199 (Figure 4E) were equally potent in inhibiting IP3R-mediated Ca2+ release.
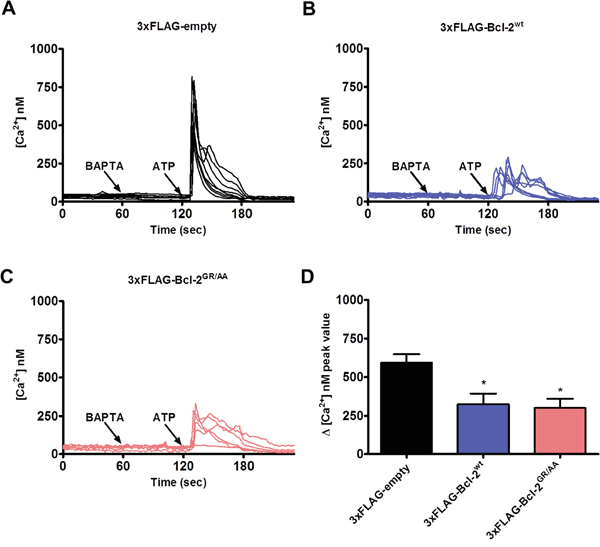
Figure 3: Bcl-2GR/AA remains capable of inhibiting agonist-induced Ca2+ release. A-C. Intracellular Ca2+ release in response to 0.5 μM ATP was followed in the mCherry-positive Fura-2-AM loaded COS-1 cells overexpressing 3xFLAG-empty vector (A), 3xFLAG-Bcl-2wt (B) or 3xFLAG-Bcl-2GR/AA (C). The free extracellular Ca2+ was buffered by addition of 3 mM BAPTA. The obtained Fura-2 fluorescence signals (F340/F380) were calibrated and representative traces are plotted as [Ca2+]. D. Quantitative analysis of the amplitude of the ATP-induced Ca2+ signals from at least 3 independent experiments (n > 80 cells) is plotted as mean ± S.E.M.
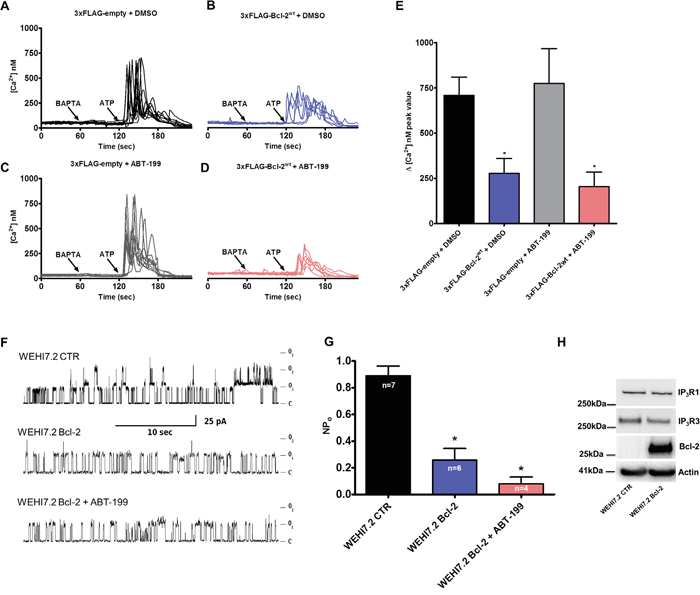
Figure 4: ABT-199 does not impact Bcl-2’s ability to suppress IP3R activity in single-cell measurements and in patch-clamp single-channel recordings. A-D. Intracellular Ca2+ release in response to 0.5 μM ATP was followed in the mCherry-positive Fura-2-AM loaded COS-1 cells overexpressing 3xFLAG-empty vector (A, C) or 3xFLAG-Bcl-2wt (B, D) in absence (A, B) or presence of 3 μM ABT-199 (C, D). The free extracellular Ca2+ was buffered by addition of 3 mM BAPTA. The obtained Fura-2 fluorescence signals (F340/F380) were calibrated and representative traces are plotted as [Ca2+]. E. Quantitative analysis of the amplitude of ATP-induced Ca2+ signals from at least 3 independent experiments (n > 80 cells) is plotted as mean ± S.E.M. F. Representative IP3R currents in ER-containing membrane fractions from control (WEHI7.2 CTR) (top) and Bcl-2-expressing WEHI7.2 cells without (middle) or with (bottom) application of 1 μM ABT-199. The IP3R activity was triggered by 5 μM IP3 and 1 μM Ca2+ G. The mean levels of IP3R activity (NPo) under these conditions are summarized and the data are plotted as mean ± S.E.M. The total number of recordings for each condition is indicated within every bar. H. Western blot analysis of the expression levels of Bcl-2, IP3R1 and IP3R3 in WEHI7.2 CTR and WEHI7.2 Bcl-2 cells. 5 μg of total lysate was loaded and the immunoreactive bands were stained against Bcl-2, IP3R1, IP3R3 and actin.
Finally, to underpin that IP3R inhibition by Bcl-2α is not affected by ABT-199, we performed direct IP3R single-channel measurements by using patch-clamp recordings on giant unilamellar vesicles (GUVs) prepared from the ER membrane fractions of native WEHI7.2 cells, which do not express any of the Bcl-2 isoforms (WEHI7.2 control) or Bcl-2α-overexpressing WEHI7.2 cells (WEHI7.2 Bcl-2). Figure 4F, 4G presents a comparison of the measured IP3R-mediated channel current after application of IP3 (5 μM) and Ca2+ (1 μM). The results demonstrate a significant inhibition of IP3R activity in the presence of Bcl-2α, measured as the open probability (NPo). The NPo value of 0.89 ± 0.07 for the WEHI7.2-control cells decreased to 0.26 ± 0.09 for WEHI7.2 Bcl-2 cells. Application of ABT-199 (1 μM) could not alleviate the inhibitory effect of Bcl-2α on IP3R single-channel opening (NPo 0.08 ± 0.05).
Collectively, these functional experiments based on independent approaches exclude a major contribution of the hydrophobic cleft of Bcl-2α for inhibiting IP3R-mediated Ca2+ flux.
The C-terminal region of Bcl-2α is critical for its interaction with the C-term Dom, but not with the Dom 3 of IP3R1
After demonstrating that the hydrophobic cleft of Bcl-2α is not involved in the binding to and inhibition of IP3R, we investigated whether the C-terminal region containing the TMD of Bcl-2 could serve as an IP3R-interaction domain. We studied the binding of 3xFLAG-Bcl-2 lacking its C-terminal region (3xFLAG-Bcl-2ΔC) to purified GST-Dom 3 and GST-C-term Dom using GST-pull-down assays. In these experiments, consistent with our previous results, 3xFLAG-Bcl-2wt bound with equal efficiency both IP3R GST-domains [42]. In line with previous data, showing that the BH4 domain of Bcl-2 is sufficient to bind to the Dom 3 [27, 39], 3xFLAG-Bcl-2ΔC remained capable to bind to this domain. Yet, the interaction with GST-C-term Dom was severely impaired (Figure 5A, 5B). These results suggest that while the C-terminal region of Bcl-2α is not crucial for interaction with the Dom 3, it is essential for binding to the C-term Dom of IP3R.
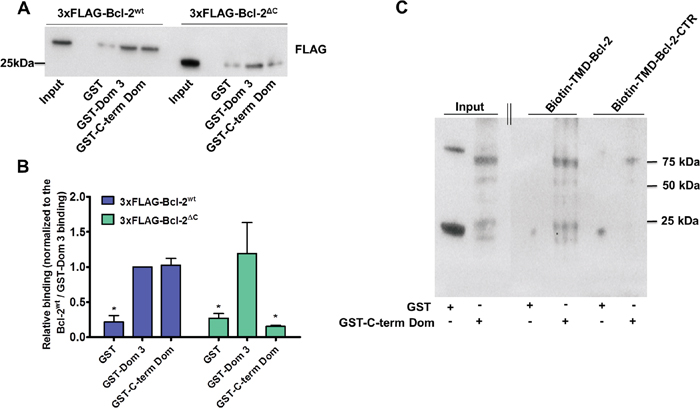
Figure 5: Bcl-2 requires its TMD for binding to the C-term Dom, but not to the Dom 3 of IP3R1. A. Representative GST-pull down experiments to compare the binding properties of 3xFLAG-Bcl-2wt versus 3xFLAG-Bcl-2ΔC overexpressed in COS-1 cells for GST-Dom 3 and GST-C-term Dom are shown. The binding to GST is used as a negative control. 0.1 μg of total COS-1 lysates was used as input. B. The immunoreactive bands from 4 independent experiments, utilizing each time independently transfected cells and freshly prepared lysates, were quantified and normalized to the binding of 3xFLAG-Bcl-2wt to GST-Dom 3, which was set as 1. The data are plotted as mean ± S.E.M. C. Representative biotin-pull down experiment to study the binding of biotin-TMD-Bcl-2 or biotin-TMD-Bcl-2-CTR peptide to the purified GST or GST-C-term Dom is shown. The immunoblots were stained for GST. The experiment was performed 3 times. 0.2 μg of purified GST and GST-C-term Dom was loaded as input. The double line indicates that two parts of the same immunoblot and exposure time were merged together.
3xFLAG-Bcl-2wt, 3xFLAG-Bcl-2GR/AA and 3xFLAG-Bcl-2ΔC bind to the full-size IP3R
3xFLAG-Bcl-2 mutants seem to have differential binding properties for the different IP3R domains. However, the performed FLAG-co-immunoprecipitation experiments with lysates from COS-1 cells overexpressing 3xFLAG-empty vector, 3xFLAG-Bcl-2wt, 3xFLAG-Bcl-2GR/AA or 3xFLAG-Bcl-2ΔC revealed that the wild type and both mutated proteins are able to interact with the endogenous IP3R1 (Supplementary Figure S2). These data are consistent with previous studies showing that the BH4 domain of Bcl-2 is the major determinant for binding to IP3Rs [27].
We also compared the binding properties of 3xFLAG-Bcl-2wt, 3xFLAG-Bcl-2GR/AA and 3xFLAG-Bcl-2ΔC for endogenous pro-apoptotic Bax. As expected, 3xFLAG-Bcl-2GR/AA failed to interact with Bax. The truncated Bcl-2 displayed equal efficiency for binding Bax as the wild type Bcl-2, confirming that the hydrophobic cleft is the major binding determinant in Bcl-2 interactions with pro-apoptotic proteins (Supplementary Figure S2).
The TMD of Bcl-2α directly interacts with the C-term Dom of IP3R1
As a next step we performed pull-down experiments using neutravidin-coated beads that captured the biotinylated peptides corresponding either to the TMD of Bcl-2 (biotin-TMD-Bcl-2) or to a control version in which several hydrophobic residues were substituted by charged amino acids (biotin-TMD-Bcl-2-CTR) in the presence of either purified parental GST or purified GST-C-term Dom of IP3R1. After incubation and washing steps, the resulting pull-down samples were analysed via immunoblotting using anti-GST antibody (Figure 5C). This analysis revealed a direct interaction between the GST-C-term Dom of IP3R1 and biotin-TMD-Bcl-2.
The lack of the C-terminus leads to loss of Bcl-2α's ability to suppress IP3R-mediated Ca2+ release
Next, we studied the role of the C-terminus in Bcl-2α’s inhibitory function on IP3R-mediated Ca2+ signaling. Similar experiments were performed as described in Figure 4, comparing the effect of 3xFLAG-Bcl-2wt versus 3xFLAG-Bcl-2ΔC overexpression on ATP-induced IP3R-mediated Ca2+ release. In contrast to 3xFLAG-Bcl-2wt, which reduced Ca2+ release in response to ATP (0.5 μM), 3xFLAG-Bcl-2ΔC was not able to suppress IP3R-mediated Ca2+ release (Figure 6). The 3xFLAG-proteins displayed similar expression levels (Supplementary Figure S1C), indicating that the failure of 3xFLAG-Bcl-2ΔC to inhibit IP3Rs is not due to a lower expression level compared to the 3xFLAG-Bcl-2wt protein. The ER Ca2+-store content was not changed in either of the conditions, pointing that the difference in ATP-induced Ca2+ rise is not due to a decreased ER Ca2+-store content (Supplementary Figure S1C).
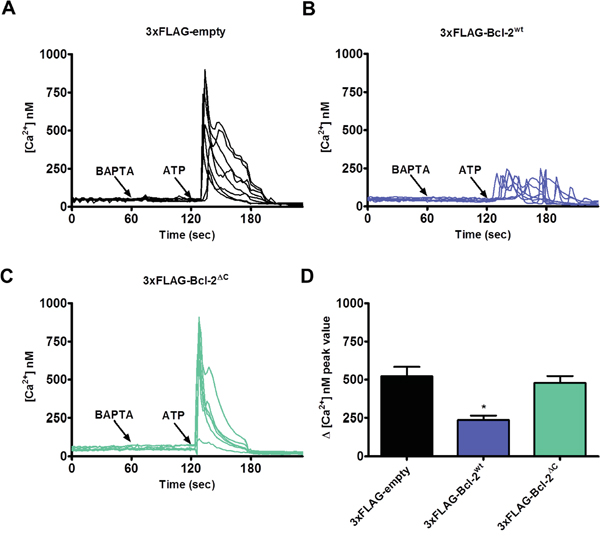
Figure 6: Bcl-2ΔC fails to inhibit IP3R-mediated Ca2+ release. A-C. Intracellular Ca2+ release in response to 0.5 μM ATP was followed in the mCherry-positive Fura-2-AM loaded COS-1 cells overexpressing 3xFLAG-empty vector (A), 3xFLAG-Bcl-2wt (B) or 3xFLAG-Bcl-2ΔC (C). The free extracellular Ca2+ was buffered by addition of 3 mM BAPTA. The obtained Fura-2 signals (F340/F380) were calibrated and representative traces are plotted as [Ca2+]. D. Quantitative analysis of the amplitude of the ATP-induced Ca2+ signals from 5 independent experiments (n > 110 cells) is plotted as mean ± S.E.M.
The TMD of Bcl-2α suppresses IICR in permeabilized cells and in single-channel recordings
We demonstrated that the TMD of Bcl-2α directly binds to the C-term Dom of IP3R and that Bcl-2ΔC does not inhibit IP3R-mediated Ca2+ release. Next, we assessed whether the TMD of Bcl-2α by itself could affect the Ca2+-flux properties of the IP3R. Therefore, we performed unidirectional 45Ca2+ flux assays in saponin-permeabilized mouse embryonic fibroblasts (MEFs), in which non-mitochondrial Ca2+ stores were loaded with 45Ca2+. After loading, the unidirectional Ca2+ flux was measured in the presence of EGTA (1 mM) and in presence of Tg (4 μM). We quantified 45Ca2+ release triggered by IP3 (3 μM) in presence or absence of different concentrations of the synthetic peptides corresponding to the TMD of Bcl-2 or its mutated version. Peptides were applied 2 min before till 2 min after IP3 application. All conditions were matched to the vehicle control (DMSO). Data are plotted as the fractional loss (%/2 min) over time. These experiments indicated that high concentrations of TMD-Bcl-2 (30 μM and higher), but not of TMD-Bcl-2-CTR, suppress IICR without affecting ER Ca2+ level (Figure 7A, 7B). To further underpin these observations, IP3R1 single-channel recordings were performed using the nuclear-membrane patch-clamp technique on isolated nuclei obtained of triple-IP3R-knockout DT40 cells ectopically expressing IP3R1 [52]. This approach allows a direct measurement of the activity of the IP3R1 channel. IP3R1 single-channel activity was recorded in response to submaximal concentrations of IP3 (1 μM) in the presence of ATP (5 mM) and Ca2+ (200 nM). Figure 7C shows different representative traces of IP3R1 single-channel openings at a pipette holding potential of -100 mV in control conditions or in the presence of TMD-Bcl-2 or TMD-Bcl-2-CTR peptides, both at 60 μM final concentrations. TMD-Bcl-2 decreased the Po of the IP3R1 channel from about 0.25 in the control conditions to about 0.15, whereas the TMD-Bcl-2-CTR peptide did not have any significant impact.
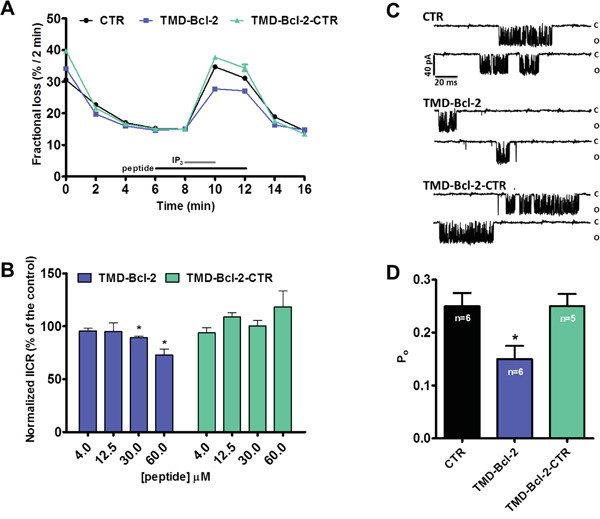
Figure 7: The TMD of Bcl-2 is sufficient to inhibit IP3Rs in permeabilized cell systems and single-channel recordings. A. Typical experiment of unidirectional 45Ca2+ fluxes in permeabilized MEFs. Ca2+ release was induced by 3 μM IP3 (grey bar) in control condition or in presence of 60 μM peptides, TMD-Bcl-2 or TMD-Bcl-2-CTR (black bar). The results are plotted as fractional loss after 2 min of incubation with IP3 minus the fractional loss before the addition of IP3 (%/2 min) as a function of time. B. Quantification of the IICR from 5 independent experiments. The values of IICR measured as fractional loss were calculated as percentage of the IICR in control condition, which was set as 100%. C. Representative IP3R1 single-channel recordings from DT40 cells ectopically expressing IP3R1 evoked by 1 μM IP3 at 200 nM Ca2+ and 5 mM ATP, in control condition or in presence of the TMD-Bcl-2 or TMD-Bcl-2-CTR peptides. D. Histogram depicting the open probability (Po) ± SD for the IP3R1 under the previously described conditions. The total number of recordings for each condition is indicated within every bar.
Bcl-2α requires its TMD to suppress STS-induced apoptosis
Finally, we studied the potency of overexpressed 3xFLAG-Bcl-2wt, 3xFLAG-Bcl-2GR/AA and 3xFLAG-Bcl-2ΔC to protect against STS, an apoptotic trigger that acts in part through Ca2+ signalling [53]. As a marker of apoptosis, we monitored the cleavage of poly-(ADP-ribose)-polymerase 1 (PARP1), which is a downstream target of activated Caspase-3. Compared to the control cells (transfected with an empty vector), the overexpression of 3xFLAG-Bcl-2wt significantly reduced the levels of cleaved PARP1 upon STS treatment (1 μM, 6h). 3xFLAG-Bcl-2GR/AA failed to prevent PARP1 cleavage, in line with its failure to bind Bax (Supplementary Figure S2). Despite the fact that 3xFLAG-Bcl-2ΔC was equally efficient as the 3xFLAG-Bcl-2wt to bind endogenous Bax (Supplementary Figure S2), it was much less efficient in preventing STS-induced apoptosis in COS-1 cells (Figure 8).
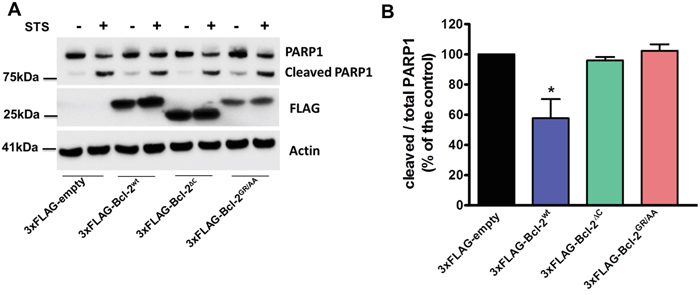
Figure 8: The TMD of Bcl-2 is required for STS-induced apoptosis. A. Western-blot analysis for monitoring PARP1 cleavage upon staurosporine (STS) treatment (1 μM for 6 h) in COS-1 cells overexpressing 3xFLAG-empty, 3xFLAG-Bcl-2wt, 3xFLAG-Bcl-2GR/AA or 3xFLAG-Bcl-2ΔC. 7 μg of total cell lysate were loaded and the immunoblots were stained for PARP1, FLAG and Actin. B. Quantification of the ratio of the immunoreactive bands of cleaved over full-length PARP1 from 4 independent experiments, utilizing each time independently transfected COS-1 cells and freshly prepared lysates. The ratio of cleaved over full-length PARP1 obtained for control cells was set as 100% and the other ratios were normalized to this value. The data are plotted as ± S.E.M.
DISCUSSION
Here, we demonstrate that the efficient in cellulo suppression of IP3R activity by Bcl-2α protein requires the C-terminal region, containing the TMD, but not the hydrophobic cleft of Bcl-2α. Consistent with this finding, Bcl-2α lacking the TMD is less effective to protect cells against Ca2+-dependent pro-apoptotic stimuli like staurosporine. Since the TMD is present only in Bcl-2α, but not in Bcl-2β, our study is the first one that indicates a possible difference between the functional effects of the Bcl-2 isoforms on IP3R activity (Figure 9) and thus on Ca2+-dependent apoptosis. Furthermore, our data indicate that BH3-mimetic compounds like ABT-199, which selectively antagonizes Bcl-2, do not interfere with the functional regulation of IP3Rs by Bcl-2.
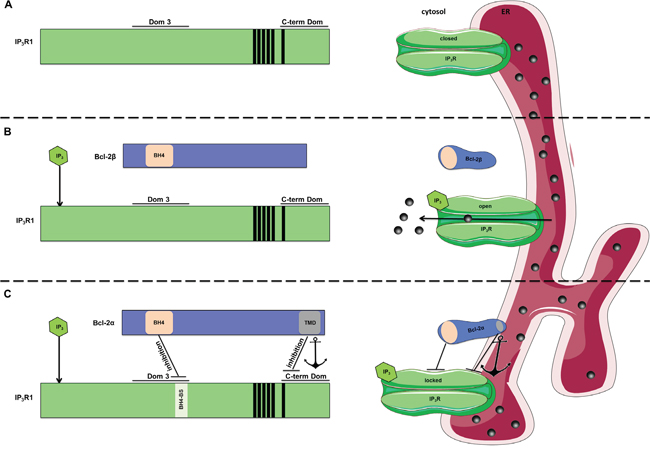
Figure 9: Model for inhibition of IP3Rs by Bcl-2 proteins. The left side of the picture shows linear representation of the multi-domain interaction between Bcl-2 proteins and IP3R. On the right side these interactions are depicted within the ER membrane environment. A. Without IP3 present, IP3R is in closed conformation and no Ca2+ release occurs. B. Upon stimulation, IP3 binds to the N-terminal ligand-binding domain of IP3R and leads to change in the conformation of the channel from closed to open state. This results in IP3R-mediated Ca2+ release. Bcl-2β, which similarly to our Bcl-2ΔC, contains the BH4 domain, but lacks the TMD, might result in ineffective binding and regulation of the channel in cellulo. C. Efficient IP3R inhibition by Bcl-2α in cellulo requires multi-domain interaction between the two proteins, which involves binding of the BH4 domain of Bcl-2α to the Dom 3 of IP3R and binding between their C-termini. Here, we hypothesize that due to this multi-domain interaction, the IP3R is “locked” in a rigid conformation, leading to decreased Ca2+ release through the channel even in presence of IP3. We propose a model, in which the interaction between the TMD of Bcl-2α and the C-term Dom of IP3R can “concentrate” the BH4 domain in the proximity of the Dom 3 by serving as an anchoring mechanism (indicated with an anchor). This “concentration effect” could overcome the inherent low affinity of inhibition by the BH4 domain. In addition to its anchoring role, the TMD of Bcl-2α has an inhibitory effect by itself.
Using genetic and pharmacological approaches, we firmly ruled out a major role for the hydrophobic cleft of Bcl-2 in inhibiting IP3R function, despite the presence of previously suggested [54] or identified throughout this study putative BH3 motifs within the IP3R sequence (Figure 1B). This is in striking contrast to the regulation of IP3Rs by Bcl-Xl, very recently described to occur via a BH3-dependent mechanism, involving an interaction between the hydrophobic cleft of Bcl-Xl and two BH3 motifs in the C-term Dom of IP3R [54, 55]. Disruption of these interactions resulted in diminished cell viability. The authors speculated that similar BH3-dependent interactions might underlie the Bcl-2/IP3R complex [55]. Therefore, our work suggests that despite the similarities in their structure and function as inhibitors of the canonical Bax/Bak-dependent apoptosis, Bcl-2 and Bcl-Xl target and regulate IP3Rs by different mechanisms. The data reported here might concede another striking difference in addition to the documented selective function of Bcl-2 versus Bcl-Xl in regulating IP3Rs at the level of their BH4 domains [27]. Of note, selective BH3-mimetic molecules that could occupy the hydrophobic cleft of Bcl-2, but not that of Bcl-Xl, have been developed, indicating important differences in the molecular determinants contributing to the hydrophobic cleft of Bcl-2 and Bcl-Xl [50]. Hence, the BH3 motifs present in the IP3R might be suited for binding the hydrophobic cleft of Bcl-Xl, but not the one of Bcl-2. In addition, the hydrophobic cleft of Bcl-2 was recently excluded as a major contributor in the inhibition of another family of intracellular Ca2+-release channels, namely ryanodine receptors (RyRs) [45].
Previously we reported that the absence of the 6th TMD of IP3R results in impaired Bcl-2 binding to the C-terminus of the channel [42]. Here, we demonstrate that the TMD of Bcl-2α is also required for this interaction, which likely occurs within the ER membrane. We propose that the TMD of Bcl-2α provides a concentration effect of Bcl-2 and its BH4 domain. This indicates that the membrane-dependent interaction between Bcl-2α and IP3R is critical for effective in cellulo inhibition of IP3R-mediated Ca2+ signaling and subsequent protection against Ca2+-dependent apoptosis. Our results also hint towards an unappreciated function of the TMD of Bcl-2α beyond its anchoring role for protein insertion into the membranes. Indeed, the TMD by itself is sufficient to inhibit IP3Rs as shown in unidirectional 45Ca2+ fluxes and single IP3R-channel recordings, correlating with previous findings that TMDs of other Bcl-2 family members play an important role in the protein functioning [56, 57]. However, it should be noted that even 60 μM TMD-Bcl-2 only partially inhibited IP3R-mediated Ca2+ release in permeabilized cells, indicating that also other Bcl-2 domains, particularly the BH4 domain, are required for efficient IP3R inhibition by Bcl-2α. Of note, the binding of Bcl-2α lacking its C-terminal region to the purified Dom 3 or to the full-length IP3Rs was not significantly disturbed, supporting the idea that the BH4 domain of Bcl-2 is sufficient for binding to the Dom 3 of IP3R and that this binding indeed occurs with relatively high affinity as documented via previous surface plasmon resonance analysis. We propose a model according to which the efficient IP3R inhibition relies on a complex multi-domain binding between Bcl-2α and IP3R, involving interactions between the BH4 domain of the former and the Dom 3 of the channel, and between the C-terminal regions of both proteins. We propose that the C-terminus of Bcl-2α (Figure 9C), but not the one of the Bcl-2β (Figure 9B), mediates the inhibitory effect of the BH4 domain by increasing its local concentration in the proximity of the Dom 3 of IP3R. In addition, the dual targeting of IP3Rs by Bcl-2α via its C-terminus and its BH4 domain might affect the conformational flexibility of the IP3R, by locking it in a rigid conformation and limiting the opening of the Ca2+-channel pore in response to IP3 (Figure 9). A particularly challenging aspect of our model is that, based on the most recent cryo-electron microscopy high-resolution structure of the IP3R1 [58], the 6th TMD of the IP3R may not be readily available for interaction with other proteins. Yet, the published structure is in the absence of IP3 and thus likely represents the closed state. Hence, changes in the IP3R structure might arise in different IP3/Ca2+ conditions impacting the accessibility of the 6th TMD of the IP3R to proteins like Bcl-2. Vice versa, it is also possible that the structure of IP3Rs loaded with Bcl-2 is different from the structure of IP3Rs in the absence of Bcl-2, thereby impacting the structural environment of the 6th TMD of IP3R. Finally, we also would like to note that the molecular foundation for this model is mainly based on binding studies, using IP3R1-expression constructs and the electrophysiological analysis of IP3R1 channels. However, some of the cell models used for the functional analysis mainly express IP3R3 and IP3R1 isoforms [59]. As such, we anticipate that the important role of Bcl-2’s TMD for efficient IP3R inhibition is not limited to IP3R1 channels, but further detailed molecular and functional work would be needed to firmly proof this. Of note, the BH4-domain-binding site present in IP3R1 is completely conserved in IP3R2 and IP3R3 [60], consistent with Bcl-2’s ability to bind to the central domain of all three IP3R isoforms [27].
The importance of the multi-domain interaction between IP3Rs and Bcl-2 is underpinned by the fact that peptides antagonizing Bcl-2 at its BH4 domain (like Bcl-2/IP3 Receptor Disrupter-2; BIRD-2) are able to trigger pro-apoptotic Ca2+ signaling in a variety of cancer-cell models, including lymphoma, leukemia and lung cancer cells [35, 37, 61]. Thus, development of inhibitors targeting Bcl-2’s TMD and interfering with the IP3R/Bcl-2 complex at the level of the TMD/C-term Dom interaction might further potentiate BH4-domain-antagonizing tools by helping to destabilize the Bcl-2/IP3R complex. Yet, given the hydrophobic nature of TMD/C-term Dom interactions, such small-molecule developments may prove to be very challenging.
We conclude that efficient IP3R regulation by Bcl-2α requires the TMD, a unique feature that discriminates Bcl-2α from Bcl-2β. Bcl-2α, via its TMD, likely “concentrates” its BH4 domain in the proximity of the central, modulatory domain of the IP3R, thereby facilitating its ability to efficiently suppress IP3R-mediated Ca2+ signaling and subsequent apoptosis.
MATERIALS AND METHODS
Peptides
The following peptides, obtained from Life Tein (Hillsborough, NJ, USA) with purity ≥ 85% were used: the peptide corresponding to the TMD of Bcl-2, Bcl-2-TMD: KTLLSLALVGACITLGAYLGHK (also used with biotin tag); the control peptide containing several mutations of hydrophobic residues, Bcl-2-TMD-CTR: KTRRSLADRGACRTRGAYDGHK (also used with biotin tag) and the peptide used to compete with the 3xFLAG tag, Anti-DYKDDDDK-tag peptide: MDYKDHDGDYKDHDIDYKDDDDK.
Antibodies
The following antibodies were used: mouse monoclonal HRP-conjugated anti-FLAG M2 (1:1000; Sigma-Aldrich, Munich, Germany); mouse anti-FLAG M2 (1:1000, Sigma-Aldrich); mouse monoclonal HRP-conjugated anti-Bcl-2 (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-GST (1:5000; Cell Signaling Technology, Danvers, Massachusetts, USA); mouse monoclonal anti-βActin (1:20 000, Sigma-Aldrich); rabbit anti-BAX (1:1000; Santa Cruz), rabbit anti-IP3R1 (1:1000; Rbt03 [62]); rabbit polyclonal anti-PARP-1 (1:1000, Alexis-Enzo Life Sciences, Farmingdale, NY, USA) as primary antibodies and secondary mouse and rabbit anti-IgG HRP conjugated antibodies (1:2500, Cell Signaling Technology).
Plasmids, constructs and protein purification
pCMV24-3xFLAG-Myc constructs for expression of 3xFLAG–Bcl-2 and 3xFLAG-Bcl-2GR/AA were obtained as previously described [45]. The 3xFLAG-Bcl-2ΔC mutant, in which a stop codon was introduced at amino acid W214, was developed via PCR site-directed mutagenesis utilizing the following primers: Forward: 5' GTTTGATTTCTCCTGACTGTCTCTGAAGACTC 3' and Reverse: 5' GAGTCTTCAGAGACAGTCAGGAGAAATCAAAC 3'.
BL21(DE3) Escherichia coli cells were transformed with pGEX-6p2 constructs containing cDNAs of parental GST, GST-Dom 3 of IP3R1 (a.a. 923-1581) or GST-C-term Dom of IP3R1 (a.a. 2512–2749), which were obtained as previously described [42]. The expressed parental GST or GST-fusion proteins were purified as previously described [42] and dialysed against standard phosphate-buffered saline (PBS) without Ca2+ and Mg2+ (Invitrogen, Merelbeke, Belgium) using Slide-A-Lyzer cassettes with a cut-off of 10 kDa (Thermo Fisher Scientific, Pittsburg, PA, USA). The concentration of the purified and dialysed proteins was determined using the Bradford assay (Sigma–Aldrich). Purity and quality were assessed after SDS–PAGE via total protein staining using the GelCode Blue Stain Reagent (Thermo Scientific, Rockford, IL, USA).
Cell culture and transfections
All media and supplements used in this paper were purchased from Life Technologies (Ghent, Belgium) unless stated otherwise. COS-1 cells were cultured at 37°C, 10% CO2 in Dulbecco's Modified Eagle's medium (DMEM), containing 10% fetal calf serum (Sigma-Aldrich), 100 IU/ml penicillin, 100 μg/ml streptomycin, 2.5 μg/ml fungizone and 2 mM glutamax. MEF cells were cultured at 37°C in a 10% CO2 incubator in DMEM/Ham's F12 medium supplemented with 10% fetal calf serum, 3.8 mM L-glutamine, 85 IU/ml penicillin and 85 μg/ml streptomycin.
24 hours after seeding COS-1 cells were transiently transfected with empty p3xFLAG-Myc-CMV-24 (3xFLAG-empty) or with the same vector containing either Bcl-2wt or the mutants Bcl-2GR/AAor Bcl-2ΔC. For co-IP and pull-down experiments JETPrime transfection reagent (Polyplus Transfections, Illkirch, France) was used according to the manufacturer’s instructions. For single-cell cytosolic [Ca2+] measurements COS-1 cells were seeded in two-chamber slides. The same construct, in combination with a pcDNA 3.1(-) mCherry-encoding vector at a 3:1 ratio as selection marker, were introduced 24 hours after seeding using X-tremeGene HP DNA (Roche, Basel, Switzerland) as a transfection reagent according to the manufacturer's instructions.
GST-pull down assays
48 hours after transfection COS-1 cells overexpressing 3xFLAG-Bcl-2wt, 3xFLAG-Bcl-2GR/AA or 3xFLAG-Bcl-2ΔC were harvested and lysed in a buffer containing 25 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 0.5 mM DTT, 1% Triton X-100 and protease inhibitor cocktail tablets (Roche). After 30 min of incubation at 4°C the clear lysates were collected via centrifugation for 2 min at 10 000 rpm at 4°C. Parental GST, GST-Dom 3 or GST-C-term Dom (0.5 μM) were incubated together with 100 μg lysate in the lysing buffer (final volume 500 μl) at 4°C. After 1 hour the GST-proteins, used as bait, were immobilized on glutathione-Sepharose 4B beads (GE Healthcare, Diegem, Belgium) for 1.5 hour at 4°C. In order to study the effect of the BH3-mimetic compound, 3 μM ABT-199 (Active Biochem, Germany) or the vehicle control DMSO (Sigma-Aldrich, St Louis, MO) was added during the last hour of incubation. The beads were washed 5 times with the Triton X-100 buffer. The GST-complexes were eluted in 40 μl 2×LDS (Life Technologies) supplemented with 1:200 β-mercaptoethanol by boiling for 5 min at 95°C. Samples (10 μl) were analyzed via SDS-PAGE and the quantification was performed as previously described [63].
Biotin-pull down assays
Equal amounts of the peptides (30 μg), biotin-TMD-Bcl-2 or biotin-TMD-Bcl-2-CTR, dissolved in 100% DMSO were incubated with 0.35 μM purified GST-C-term Dom of IP3R1 or parental GST (control) in interaction buffer (50 mM Tris-HCl, 200 mM NaCl, 0.1% NP-40, 1% BSA and protease inhibitor cocktail, pH 7.0) in a final volume of 400 μl. The incubation was performed over night at 4°C in a head-over-head rotator. The biotinylated peptides were immobilized on neutravidin agarose beads (Thermo Fisher Scientific, Pierce, Erembodegem, Belgium) and placed in a head-over-head rotator for 2 hours at 4°C. The beads were washed 7 times with the interaction buffer and the peptide-protein complexes were eluted by incubating the beads with 35 μl LDS supplemented with 1:200 β-mercaptoethanol for 3 min at 95°C. The eluates were collected after centrifuging at 2000 g for 1 min, using spin columns (Pierce) and 10 μl was analysed on NuPAGE 4–12% Bis/Tris SDS–polyacrylamide gels using MES/SDS-running buffer (Invitrogen).
FLAG-co-immunoprecipitation assay
48 hours after transfection COS-1 cells overexpressing the control vector 3xFLAG-empty, 3xFLAG-Bcl-2wt, 3xFLAG-Bcl-Bcl-2GR/AA or 3xFLAG-Bcl-2ΔC were harvested and lysed in buffer containing 10 mM Hepes (pH 7.5), 0.25% NP-40, 142 mM KCl, 5 mM MgCl2, 2 mM EDTA, 2 mM EGTA and protease inhibitor cocktail tablets (Roche) as described for the GST-pull down assay. 100 μg of lysate was mixed with 30 μl anti-DYKDDDDK-tag conjugated resin (Biolegend, San Diego, CA) in the lysis buffer in total volume of 400 μl. The samples were incubated for 2.5 hours using a head-over-head rotor at 4°C. The beads were washed 2 times with washing buffer (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS) via centrifugation for 1 min at 3000g using spin columns. The FLAG-complexes were eluted by competitive incubation with the Anti-DYKDDDDK-tag peptide (10 μg, dissolved in 50mM Tris-HCl and 150mM NaCl) for 30 min at 15°C. To the resulting eluates, collected via centrifugation for 1 min at 500g, 25 μl LDS supplemented with 1:200 β-mercaptoethanol was added. 15 μl of each sample was analysed on NuPAGE 4–12% Bis/Tris SDS–polyacrylamide gels using MES/SDS-running buffer.
Single-cell cytosolic Ca2+ imaging
Fura-2-AM [Ca2+] measurements in COS-1 cells were performed as previously described [27]. The effect of ABT-199 was studied by incubating the cells with 3 μM of the compound or DMSO for 1 hour (during the incubation procedure with Fura-2 AM). BAPTA (3 mM) was added for 1 minute prior to the stimulation with ATP or Tg to chelate all free extracellular Ca2+. Cytosolic Ca2+ rises in response to 0.5 μM ATP or 2.5 μM Tg were measured in mCherry-positive (excitation 546 nm, emission 610 nm) and Fura-2-loaded cells. Intracellular cytoplasmic Ca2+ concentrations were calculated as previously described [27].
Unidirectional 45Ca2+-flux assay
The unidirectional 45Ca2+-flux experiments were performed in permeabilized MEFs as previously described [27]. IICR was triggered during the unidirectional 45Ca2+-efflux phase by the addition of 3 μM IP3 for 2 min. Peptides were added 2 min before IP3 till 2 min after IP3. IICR was plotted as fractional loss, representing the amount of Ca2+ leaving the store in a 2-min time period divided by the total store Ca2+ content at that time point as a function of time [64].
Preparation of GUVs and electrophysiological analysis
Isolation of the ER-containing membrane fractions from control and Bcl-2-expressing WEHI7.2 cells and preparation of the GUVs were carried out as described previously [65]. GUVs were prepared from the 1:5 mixtures of the ER-containing fraction with 10:1 diphytanoylphosphatidylcholine/cholesterol lipid combination (5 mM). The Patch-clamp experiments were carried out using Axopatch 200B amplifier and pClamp 10.0 software (Molecular Devices, Union City, CA) for data acquisition and analysis. Patch pipettes were fabricated from borosilicate glass capillaries (World Precision Instr., Inc., Sarasota, FL) on a horizontal puller (Sutter Instruments Co., Novato, CA) and had a resistance in the range of 7-10 MΩ. Prepared vesicles were immersed in a bath solution containing 150 mM KCl, 10 mM Hepes, 5 mM glucose, pH 7.2. Patch pipettes were filled with the same solution.
Isolation of nuclei and electrophysiological analysis
Isolated DT40 nuclei were prepared by homogenization as previously described [52]. A 3 μl aliquot of nuclear suspension was placed in 3 ml of bath solution which contained 140 mM KCl, 10 mM Hepes, 500 μM BAPTA and 246 nM free Ca2+, pH 7.1. Nuclei were allowed to adhere to a plastic culture dish for 10 min prior to patching. Single IP3R channel potassium currents (ik) were measured in the on-nucleus patch clamp configuration using pCLAMP 9 and an Axopatch 200B amplifier (Molecular Devices, Sunnydale, CA, USA) as previously described [66]. Pipette solution contained 140 mM KCl, 10 mM Hepes, 1 μM IP3, 5 mM ATP, and 200 nM free Ca2+ as well as 60 μM TMD-Bcl-2 or TMD-Bcl-2-CTR peptides. Traces were consecutive 3 s sweeps recorded at −100 mV, sampled at 20 kHz and filtered at 5 kHz. A minimum of 15 s of recordings were considered for data analyses. Pipette resistances were typically 20 MΩ and seal resistances were >5 GΩ. Single channel openings were detected by half-threshold crossing criteria using the event detection protocol in Clampfit 9. We assumed that the number of channels in any particular nuclear patch is represented by the maximum number of discrete stacked events observed during the experiment. Only patches with one apparent channel were considered for analyses.
Apoptosis induction and analysis
COS-1 cells were transiently transfected with 3xFLAG-vectors and treated with 1 μM STS (Sigma-Aldrich). After 6h the cells were harvested and lysed in a buffer containing 25 mM Hepes (pH 7.5), 1% Triton X-100, 10% glycerol, 0.3 M NaCl, 1.5 mM MgCl2, 1 mM DTT, 2 mM EDTA, 2 mM EGTA and protease inhibitor cocktail tablets (Roche). Apoptosis progression was monitored via Western-blotting analysis of PARP1 cleavage in 10 μg total lysate.
Sequence alignment and secondary-structure predictions
The amino acid sequences of the BH3 domains of Bcl-2 proteins and the Dom 3 of IP3R were taken from the National Center for Biotechnological Information’s nonredundant database. The I-TASSER v 2.1 webserver [67, 68] was used to predict the secondary structure of the BH3-like motif identified in the Dom 3 of IP3R1. I-TASSER builds protein models using iterative assembling procedures and multiple threading alignments from template structures libraries. An estimate of accuracy of the predictions is given by the confidence score. The most accurate I-TASSER model was downloaded as PDB file and imported in PyMOL, a molecular graphic software (http://www.pymol.org).
Statistical analysis
Two-tailed unpaired Student's t-tests were performed when two conditions were compared. When comparing three or more conditions a repeated measure ANOVA with Bonferroni post test was performed. * indicates significantly different results with p < 0.05.
ACKNOWLEDGMENTS
The authors thank Anja Florizoone and Marina Crabbe for the excellent technical help. The authors thank Dr. Clark Distelhorst (Case Western Reserve University, Cleveland, OH) for providing WEHI7.2 cells overexpressing Bcl-2. The authors thank all lab members for the fruitful discussions.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
FUNDING
Work performed in the authors’ laboratory was supported by grants from the Research Foundation-Flanders (FWO grants 6.057.12, G.0819.13, G.0C91.14 and G.0A34.16), by the Research Council of the KU Leuven (OT grant 14/101) and by the Interuniversity Attraction Poles Program (Belgian Science Policy; IAP-P7/13). HI is a recipient of a doctoral fellowship of the FWO. GM is a recipient of a post-doctoral fellowship of the FWO.
REFERENCES
1. Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998; 281:1322-1326.
2. Gross A, McDonnell JM, Korsmeyer SJ. BCL-2 family members and the mitochondria in apoptosis. Genes & Development. 1999; 13:1899-1911.
3. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014; 15:49-63.
4. Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010; 37:299-310.
5. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004; 116:205-219.
6. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988; 335:440-442.
7. Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G, Thompson CB. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993; 74:597-608.
8. Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993; 74:609-619.
9. Aouacheria A, Rech de Laval V, Combet C, Hardwick JM. Evolution of Bcl-2 homology motifs: homology versus homoplasy. Trends Cell Biol. 2013; 23:103-111.
10. O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S, Huang DC. Bim: a novel member of the Bcl-2 family that promotes apoptosis. Embo J. 1998; 17:384-395.
11. Laetsch TW, Liu X, Vu A, Sliozberg M, Vido M, Elci OU, Goldsmith KC, Hogarty MD. Multiple components of the spliceosome regulate Mcl1 activity in neuroblastoma. Cell Death Dis. 2014; 5:e1072.
12. U M, Miyashita T, Shikama Y, Tadokoro K, Yamada M. Molecular cloning and characterization of six novel isoforms of human Bim, a member of the proapoptotic Bcl-2 family. FEBS Lett. 2001; 509:135-141.
13. Fu NY, Sukumaran SK, Kerk SY, Yu VC. Baxβ: a constitutively active human Bax isoform that is under tight regulatory control by the proteasomal degradation mechanism. Mol Cell. 2009; 33:15-29.
14. Tsujimoto Y, Croce CM. Analysis of the structure, transcripts, and protein products of bcl-2, the gene involved in human follicular lymphoma. Proc Natl Acad Sci U S A. 1986; 83:5214-5218.
15. Chen-Levy Z, Nourse J, Cleary ML. The bcl-2 candidate proto-oncogene product is a 24-kilodalton integral-membrane protein highly expressed in lymphoid cell lines and lymphomas carrying the t(14;18) translocation. Mol Cell Biol. 1989; 9:701-710.
16. de Jong D, Prins F, van Krieken HH, Mason DY, van Ommen GB, Kluin PM. Subcellular localization of bcl-2 protein. Curr Top Microbiol Immunol. 1992; 182:287-292.
17. Krajewski S, Tanaka S, Takayama S, Schibler MJ, Fenton W, Reed JC. Investigation of the subcellular distribution of the bcl-2 oncoprotein: residence in the nuclear envelope, endoplasmic reticulum, and outer mitochondrial membranes. Cancer Res. 1993; 53:4701-4714.
18. Kaufmann T, Schlipf S, Sanz J, Neubert K, Stein R, Borner C. Characterization of the signal that directs Bcl-xL, but not Bcl-2, to the mitochondrial outer membrane. J Cell Biol. 2003; 160:53-64.
19. Ghassemifar R, Forster L, Finlayson J, Calogero T, Augustson B, Joske D, Cull G. Differential expression of the Bcl-2 and Bax isoforms in CD19 positive B-lymphocytes isolated from patients diagnosed with chronic lymphocytic leukaemia. Pathology. 2012; 44:632-637.
20. Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC. Inhibition of Bax channel-forming activity by Bcl-2. Science. 1997; 277:370-372.
21. Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998; 3:697-707.
22. Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003; 4:552-565.
23. Mattson MP, Chan SL. Calcium orchestrates apoptosis. Nat Cell Biol. 2003; 5:1041-1043.
24. Fleckenstein A, Janke J, Doring HJ, Leder O. Myocardial fiber necrosis due to intracellular Ca overload-a new principle in cardiac pathophysiology. Recent Adv Stud Cardiac Struct Metab. 1974; 4:563-580.
25. Berridge M, Lipp P, Bootman M. Calcium signalling. Curr Biol. 1999; 9:R157-159.
26. Rong YP, Barr P, Yee VC, Distelhorst CW. Targeting Bcl-2 based on the interaction of its BH4 domain with the inositol 1,4,5-trisphosphate receptor. Biochim Biophys Acta. 2009; 1793:971-978.
27. Monaco G, Decrock E, Akl H, Ponsaerts R, Vervliet T, Luyten T, De Maeyer M, Missiaen L, Distelhorst CW, De Smedt H, Parys JB, Leybaert L and Bultynck G. Selective regulation of IP3-receptor-mediated Ca2+ signaling and apoptosis by the BH4 domain of Bcl-2 versus Bcl-Xl. Cell Death Differ. 2012; 19:295-309.
28. Chang MJ, Zhong F, Lavik AR, Parys JB, Berridge MJ, Distelhorst CW. Feedback regulation mediated by Bcl-2 and DARPP-32 regulates inositol 1,4,5-trisphosphate receptor phosphorylation and promotes cell survival. Proc Natl Acad Sci U S A. 2014; 111:1186-1191.
29. Zhong F, Davis MC, McColl KS, Distelhorst CW. Bcl-2 differentially regulates Ca2+ signals according to the strength of T cell receptor activation. J Cell Biol. 2006; 172:127-137.
30. Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007; 87:593-658.
31. Parys JB, De Smedt H. Inositol 1,4,5-trisphosphate and its receptors. Adv Exp Med Biol. 2012; 740:255-279.
32. Mikoshiba K. The IP3 receptor/Ca2+ channel and its cellular function. Biochem Soc Symp. 2007:9-22.
33. Bezprozvanny I. The inositol 1,4,5-trisphosphate receptors. Cell Calcium. 2005; 38:261-272.
34. Berridge MJ. Inositol trisphosphate and calcium signalling mechanisms. Biochim Biophys Acta. 2009; 1793:933-940.
35. Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, Rong YP, Molitoris JK, Lam M, Ryder C, Matsuyama S, Distelhorst CW. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood. 2011; 117:2924-2934.
36. Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, Molgo J, Distelhorst CW, Missiaen L, Mikoshiba K, Parys JB, De Smedt H, Bultynck G. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013; 4:e632.
37. Greenberg EF, McColl KS, Zhong F, Wildey G, Dowlati A, Distelhorst CW. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 2015; 6:e2034.
38. Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, Matsuyama S, Herlitze S, Roderick HL, Bootman MD, Mignery GA, Parys JB, De Smedt H, Distelhorst CW. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Mol Cell. 2008; 31:255-265.
39. Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc Natl Acad Sci U S A. 2009; 106:14397-14402.
40. Monaco G, Decrock E, Nuyts K, Wagner LE 2nd, Luyten T, Strelkov SV, Missiaen L, De Borggraeve WM, Leybaert L, Yule DI, De Smedt H, Parys JB, Bultynck G. Alpha-helical destabilization of the Bcl-2-BH4-domain peptide abolishes its ability to inhibit the IP3 receptor. PLoS One. 2013; 8:e73386.
41. Eckenrode EF, Yang J, Velmurugan GV, Foskett JK, White C. Apoptosis protection by Mcl-1 and Bcl-2 modulation of inositol 1,4,5-trisphosphate receptor-dependent Ca2+ signaling. J Biol Chem. 2010; 285:13678-13684.
42. Monaco G, Beckers M, Ivanova H, Missiaen L, Parys JB, De Smedt H, Bultynck G. Profiling of the Bcl-2/Bcl-XL-binding sites on type 1 IP3 receptor. Biochem Biophys Res Commun. 2012; 428:31-35.
43. Aouacheria A, Combet C, Tompa P, Hardwick JM. Redefining the BH3 Death Domain as a 'Short Linear Motif'. Trends Biochem Sci. 2015; 40:736-748.
44. Galindo-Moreno J, Iurlaro R, El Mjiyad N, Diez-Perez J, Gabaldon T, Munoz-Pinedo C. Apolipoprotein L2 contains a BH3-like domain but it does not behave as a BH3-only protein. Cell Death Dis. 2014; 5:e1275.
45. Vervliet T, Lemmens I, Welkenhuyzen K, Tavernier J, Parys JB, Bultynck G. Regulation of the ryanodine receptor by anti-apoptotic Bcl-2 is independent of its BH3-domain-binding properties. Biochem Biophys Res Commun. 2015; 463:174-179.
46. Petros AM, Olejniczak ET, Fesik SW. Structural biology of the Bcl-2 family of proteins. Biochim Biophys Acta. 2004; 1644:83-94.
47. Yin XM, Oltvai ZN, Korsmeyer SJ. BH1 and BH2 domains of Bcl-2 are required for inhibition of apoptosis and heterodimerization with Bax. Nature. 1994; 369:321-323.
48. Ni Chonghaile T, Letai A. Mimicking the BH3 domain to kill cancer cells. Oncogene. 2008; 27:S149-157.
49. Billard C. BH3 mimetics: status of the field and new developments. Mol Cancer Ther. 2013; 12:1691-1700.
50. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19:202-208.
51. Rong Y, Distelhorst CW. Bcl-2 protein family members: versatile regulators of calcium signaling in cell survival and apoptosis. Annu Rev Physiol. 2008; 70:73-91.
52. Wagner LE 2nd, Yule DI. Differential regulation of the InsP3 receptor type-1 and -2 single channel properties by InsP3, Ca2+ and ATP. J Physiol. 2012; 590:3245-3259.
53. Akimzhanov AM, Barral JM, Boehning D. Caspase 3 cleavage of the inositol 1,4,5-trisphosphate receptor does not contribute to apoptotic calcium release. Cell Calcium. 2013; 53:152-158.
54. Foskett JK, Yang J, Cheung K-H, Vais H. Bcl-xL Regulation of InsP3 Receptor Gating Mediated by Dual Ca2+ Release Channel BH3 Domains. Biophysical Journal. 2009; 96:391a.
55. Yang J, Vais H, Gu W, Foskett JK. Biphasic regulation of InsP3 receptor gating by dual Ca2+ release channel BH3-like domains mediates Bcl-xL control of cell viability. Proc Natl Acad Sci U S A. 2016; 113:E1953-1962.
56. Todt F, Cakir Z, Reichenbach F, Youle RJ, Edlich F. The C-terminal helix of Bcl-xL mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. 2013; 20:333-342.
57. Wilfling F, Weber A, Potthoff S, Vogtle FN, Meisinger C, Paschen SA, Hacker G. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ. 2012; 19:1328-1336.
58. Fan G, Baker ML, Wang Z, Baker MR, Sinyagovskiy PA, Chiu W, Ludtke SJ, Serysheva, II. Gating machinery of InsP3R channels revealed by electron cryomicroscopy. Nature. 2015; 527:336-341.
59. Ivanova H, Vervliet T, Missiaen L, Parys JB, De Smedt H, Bultynck G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim Biophys Acta. 2014; 1843:2164-2183.
60. Monaco G, Vervliet T, Akl H, Bultynck G. The selective BH4-domain biology of Bcl-2-family members: IP3Rs and beyond. Cell Mol Life Sci. 2013; 70:1171-1183.
61. Akl H, Bultynck G. Altered Ca2+ signaling in cancer cells: proto-oncogenes and tumor suppressors targeting IP3 receptors. Biochim Biophys Acta. 2013; 1835:180-193.
62. Parys JB, De Smedt H, Missiaen L, Bootman MD, Sienaert I, Casteels R. Rat basophilic leukemia cells as model system for inositol 1,4,5-trisphosphate receptor IV, a receptor of the type II family: functional comparison and immunological detection. Cell Calcium. 1995; 17:239-249.
63. Vervliet T, Decrock E, Molgo J, Sorrentino V, Missiaen L, Leybaert L, De Smedt H, Kasri NN, Parys JB, Bultynck G. Bcl-2 binds to and inhibits ryanodine receptors. J Cell Sci. 2014; 127:2782-2792.
64. Decuypere JP, Monaco G, Kiviluoto S, Oh-hora M, Luyten T, De Smedt H, Parys JB, Missiaen L, Bultynck G. STIM1, but not STIM2, is required for proper agonist-induced Ca2+ signaling. Cell Calcium. 2010; 48:161-167.
65. Bidaux G, Borowiec AS, Gordienko D, Beck B, Shapovalov GG, Lemonnier L, Flourakis M, Vandenberghe M, Slomianny C, Dewailly E, Delcourt P, Desruelles E, Ritaine A, et al. Epidermal TRPM8 channel isoform controls the balance between keratinocyte proliferation and differentiation in a cold-dependent manner. Proc Natl Acad Sci U S A. 2015; 112:E3345-3354.
66. Betzenhauser MJ, Wagner LE 2nd, Park HS, Yule DI. ATP regulation of type-1 inositol 1,4,5-trisphosphate receptor activity does not require walker A-type ATP-binding motifs. J Biol Chem. 2009; 284:16156-16163.
67. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015; 12:7-8.
68. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010; 5:725-738.