
INTRODUCTION
Colorectal cancer (CRC) is one of the most common malignancy with high morbidity and mortality worldwide, accounting for almost 1.4 million new cases and 0.7 million deaths in 2012 [1]. In China, the estimated new cases of CRC were 376,300, with 191,000 deaths occurring in 2015 [2]. Despite of improvements in diagnosis and treatment for CRC, its annual new incidence and mortality is still growing.
CRC is a heterogeneous disease with complex multi-pathways. Most of CRC follows the chromosomal instability (CIN) pathway which is the most well characterized pathway type [3]. The Wnt signaling pathway [4], RAS pathway [5], p53 system [6], and other pathways involved in CIN [7, 8] are frequently present to be dysregulated during the initiation, progression and metastasis of CRC. A number of patients have quite different treatment responses and prognoses although their tumor types are histologically identical. Therefore it is necessary to look deeply into those abnormal molecular pathways and develop individual strategies for CRC diagnosis and therapy. To date, a lot of efforts have been put to identify molecular markers, however, with limited success achieved when focusing on single protein or gene mutation [9]. Gene expression profiling has been verified as a promising tool to classify tumors and predict the prognosis of cancer [10]. Identification of molecular subtypes [11, 12], discovery of progression markers [13, 14], and construction of different prognostic models [15–18] in CRC have been processed and confirmed to potentially improve the diagnosis and therapy of CRC. However, all the developed prognostic signatures are still difficult to apply commonly because of the heterogeneity of CRC.
For most of these studies, disease free survival (DFS) or relapse free survival (RFS) was used as the endpoint to develop molecular markers or to evaluate the validity of prognosis. Considering that overall survival (OS) is traditionally regarded as the ultimate measure of treatment benefits, we would like to investigate whether it is possible to build a robust gene signature to predict the OS in CRC patients. By performing univariate survival analysis, 6487 genes associated with OS were discovered in patients with CRC from a Chinese cohort. Of them, a seven-gene signature was developed using a robust likelihood-based survival modelling approach, and further trained by BRB-array tool to generate a prognostic model. Importantly, the prognostic value of this seven-gene signature was validated in other two independent cohorts, indicating its potential use for identifying CRC patients with high risk of mortality.
RESULTS
The overall flowchart of this work was summarized in Figure 1. We employed Cox proportional hazard regression model and forward selection method to identify a seven-gene signature which can predict OS for colorectal cancer patients based on our gene microarray datasets of Chinese patients. To further evaluate the performance of this gene signature, two cohorts on different platforms from other countries were subsequently validated.
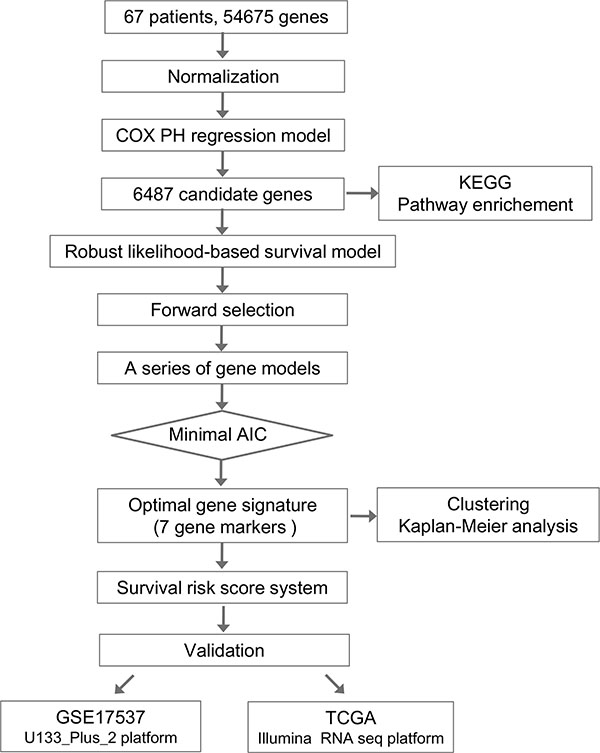
Figure 1: Flow chart of methods for building the seven-gene signature based on 64 Chinese colorectal cancer (CRC) samples. Briefly, 6487 genes associated with the overall survival (OS) in CRC patients were first produced by univariate survival analysis. Next a robust likelihood-based survival modelling approach was used to select the optimal gene signature for prognosis prediction. The survival risk score system was built based on seven-gene signature (NHLRC3, ZDHHC21, PRR14L, CCBL1, PTPRB, PNPO, and PPIP5K2), which divided patients into high- and low-risk groups. Finally, this seven-gene signature was validated in two independent cohorts on different platforms.
Identification of genes associated with OS
Our data included expression values of 54675 genes and 64 samples. Each sample had observed (survival or censoring) time and censoring status. We first selected an initial set of genes by performing univariate survival analysis using Cox proportional hazard regression model, with the threshold of p-value set as 0.05. Total 6487 genes associated with the overall survival were initially identified.
To investigate the key pathways that were associated with patient survival, we next performed KEGG pathway enrichment analysis for the 6487 genes. These genes were found to be enriched in the signalling pathways such as endocytosis, axon guidance, spliceosome, Wnt signalling and ubiquitin mediated proteolysis (Figure 2A).
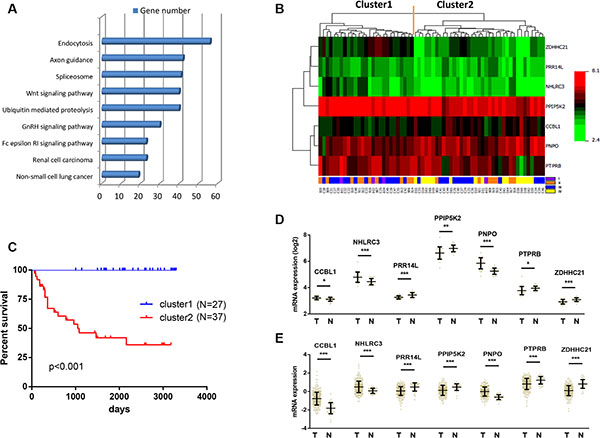
Figure 2: Identification of optimal gene signature for OS prediction. (A) Enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways analysis for 6487 genes associated with the OS. Top 10 pathways were shown. (B) The 6487 genes in 64 samples were shown in a heat map (The green and the red colors represent lower and higher expression value, respectively). Unsupervised hierarchical clustering analysis was applied, which divided patients into two clusters. (C) Kaplan–Meier curves for patients in different clusters. (D) The mRNA expression of CCBL1, NHLRC3, PNPO, PRR14L, PPIP5K2, PTPRB and ZDHHC21 in 26 pairs of CRC tissues and adjacent normal tissues in our cohort. Data is represented by mean ± SD and p values were obtained by Wilcoxon matched-pairs test. (*p < 0.05, **p < 0.01, ***p < 0.001). (E) The mRNA expression of CCBL1, NHLRC3, PNPO, PRR14L, PPIP5K2, PTPRB and ZDHHC21 in 623 CRC tissues and 51 adjacent normal tissues in TCGA cohort. Data is represented by mean ± SD and p values were obtained by Mann–Whitney U test. (*p < 0.05, **p < 0.01, ***p < 0.001).
Screening of seven-gene signature
We next screened the optimal survival-associated signature genes based on a partial likelihood of the Cox proportional hazard regression model [21]. Considering larger variability of the data, a cross-validation technique was applied by separating the samples into training and validation sets (See Materials and Methods). A forward selection was employed to generate a series of gene models and the optimal model was then selected by using the criterion of minimal AIC (Table 1). Seven genes (NHLRC3, ZDHHC21, PRR14L, CCBL1, PTPRB, PNPO and PPIP5K2) were selected as signature genes that can optimally predict the OS of patients with CRC. With the selected gene signature, unsupervised hierarchical clustering analysis was performed, and the patient population was classified into two sub-classes: Cluster 1 and Cluster 2 (Figure 2B). Compared with Cluster 1 patients, all Cluster 2 patients in this study were still alive during follow-up (Figure 2C). Therefore this seven-gene signature may have important application in predicting the OS for patients with CRC.
Table 1: Survival-associated gene signature screening using forward selection
Gene ID |
nloglik |
AIC |
Gene Symbol |
|---|---|---|---|
236953_s_at |
63.51 |
129.01* |
NHLRC3 |
243835_at |
55.24 |
114.47* |
ZDHHC21 |
236941_at |
53.96 |
113.92* |
PRR14L |
206037_at |
50.62 |
109.24* |
CCBL1 |
230250_at |
49.53 |
109.06* |
PTPRB |
222653_at |
46.85 |
105.70* |
PNPO |
203253_s_at |
43.87 |
101.73* |
PPIP5K2 |
203031_s_at |
43.86 |
103.73 |
|
1557118_a_at |
42.42 |
102.84 |
|
242793_at |
42.32 |
104.65 |
|
205462_s_at |
41.73 |
105.45 |
|
1552319_a_at |
40.57 |
105.15 |
Differential expressions of seven-gene signature in colorectal cancer and adjacent normal tissue
We further compared the expressions of these seven genes between CRC tissues and adjacent normal tissues in two independent data sets. In our cohort, the mRNA expression of CCBL1, NHLRC3 and PNPO were significantly down-regulated in 26 CRC as compared with paired adjacent normal tissues (Figure 2D). On the other hand, PRR14L, PPIP5K2, PTPRB and ZDHHC21 were determined to be over-expressed in CRC (Figure 2D). Consistent result was obtained using TCGA cohort that contained 623 CRC and 51 adjacent normal tissues (Figure 2E). Therefore, these seven genes may play important roles as oncogenes or tumor suppressor genes during the development of CRC.
Construction of survival risk score system based on seven-gene signature
We subjected these seven genes to BRB-arrayTools to construct a survival risk score system by using the 64 training samples. The regression coefficient for each gene was generated. The survival risk score was calculated as follows: risk score = (–1.004× expression level of PPIP5K2) + (0.823× expression level of CCBL1) + (–0.715× expression level of PNPO) + (–0.008× expression level of PTPRB) + (–1.077× expression level of PRR14L) + (–0.133× expression level of NHLRC3) + (–0.781× expression level of ZDHHC21). Higher score indicated greater mortality risk for patient with CRC. By performing cross-validated time-dependent ROC curves, the area under the respective ROC Curves (AUC) was 0.814 (Figure 3A), confirming the prediction accuracy of this model. 64 patients were divided into high- and low-risk groups. The OS for patients in high-risk group was significantly poorer compared with patients in low-risk group (hazard ratio (HR) = 25.79, P < 0.001) (Figure 3B).
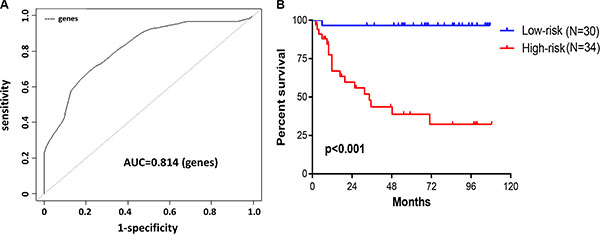
Figure 3: Construction of survival risk score system based on seven-gene signature. (A, B) A prognostic model was developed in BRB-ArrayTools software using penalized Cox regression of the seven-gene signature. Leave-one-out cross-validation (LOOCV) was employed to evaluate the accuracy of predicting OS in 64 CRC patients. (A) The cross-validated time-dependent ROC curve was generated for survival predictions with an AUC of 0.814. (B) Patients were divided into high- and low-risk groups by cross-validated Kaplan–Meier curve.
External validation of seven-gene signature
To evaluate the robustness and effectiveness of the seven-gene signature, we used two independent sets of CRC patients with OS information and gene expressions on different platforms. The first one was from the Vanderbilt Medical Center (GSE17537, N = 55) using the same gene expression array platform as we did. The second one, TCGA dataset (N = 584), used Illumina RNA Sequencing method. The survival risk score of each patient was calculated based on expressions of seven-gene signature. ROC curve analyses demonstrated that this seven-gene signature was capable of predicting OS in patients with CRC (for cohort I, AUC = 0.715, p = 0.011; for cohort II, AUC = 0.585, p = 0.009) (Figure 4A and 4C). We further divided the patients into two risk groups, based on optimal cut-off risk scores (Figure 4A and 4C). For the first validation data set, 38 (69.1%) and 17 (30.9%) patients were distinguished as the low and high risk groups, respectively. For the second validation data set, 418 (71.6%) and 166 (28.4%) patients were classified as low and high risk groups, respectively. Kaplan-Meier plots indicated significant differences between 5-year OS of two groups in both two validation data sets: GSE17537 (p < 0.001, Figure 4B), and TCGA (p = 0.002, Figure 4D). By univariate (Table 2) and multivariate analyses (Table 3), this seven-gene signature significantly predicted the 5-year OS of patients. In multivariate analyses, this seven-gene signature showed prognostic significance for 5-year OS risk in both two validation data sets: GSE17537 (p = 0.002) and TCGA (p = 0.005). Therefore, the seven-gene signature was an independent prognostic factor in predicting the OS of patients with CRC.
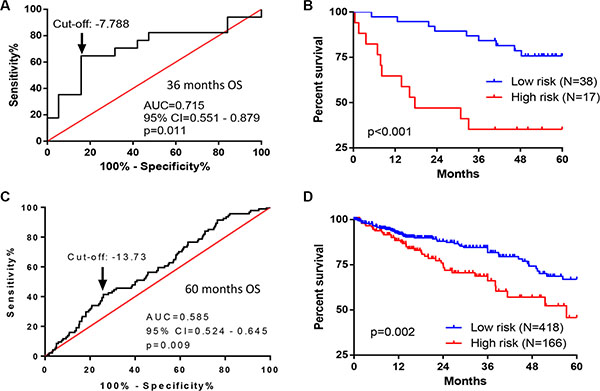
Figure 4: Performance of seven-gene signature in predicting OS in CRC patients from two independent cohorts. (A, B) ROC and Kaplan–Meier curves for the seven-gene signature in dataset from Vanderbilt Medical Center (GSE17537, N = 55). (A) The ROC curve was generated for 3-year overall survival predictions with an AUC of 0.715 (p = 0.011). Optimal cut-off value (–7.788) was obtained to divide the patients into low and high risk groups. (B) Patients in high risk group had poorer OS as compared with patients in low risk group (HR = 4.475, p < 0.001). (C, D) ROC and Kaplan–Meier curves for the seven-gene signature in the TCGA dataset (N = 584). (C) The ROC curve was generated for 5-year overall survival predictions with an AUC of 0.585 (p = 0.009). Optimal cut-off value (–13.73) was obtained. (D) Patients in high risk group had poorer OS as compared with patients in low risk group (HR = 1.915, p = 0.002).
Table 2: Univariate Cox regression analysis of potential prognostic factors for patients with CRC
Cohort I |
Cohort II |
||||||
|---|---|---|---|---|---|---|---|
Characteristics |
No. of Patients |
5 yr SR (%) |
p value |
Characteristics |
No. of Patients |
5 yr SR (%) |
p value |
Age |
0.311 |
Age |
0.042 |
||||
< = 60 |
25 |
72.0% |
< = 60 |
190 |
90.0% |
||
> 60 |
30 |
56.7% |
> 60 |
394 |
81.0% |
||
Gender |
0.395 |
Gender |
0.534 |
||||
Female |
29 |
58.6% |
Female |
267 |
84.6% |
||
Male |
26 |
69.2% |
Male |
317 |
83.3% |
||
Pathologic_stage |
0.100 |
Pathologic_stage |
< 0.001 |
||||
I, II |
19 |
78.9% |
I |
103 |
96.1% |
||
II |
210 |
89.5% |
|||||
III, IV |
36 |
55.6% |
III |
172 |
82.0% |
||
IV |
85 |
62.4% |
|||||
Risk |
0.001 |
Risk |
0.002 |
||||
Low |
38 |
76.3% |
Low |
418 |
86.8% |
||
High |
17 |
35.3% |
High |
166 |
76.5% |
||
Table 3: Multivariate Cox regression analysis of potential prognostic factors for patients with CRC
Cohort I |
Cohort II |
||||||||
|---|---|---|---|---|---|---|---|---|---|
Characteristics |
Exp(B) |
95.0% CI for Exp(B) |
p value |
Characteristics |
Exp(B) |
95.0% CI for Exp (B) |
p value |
||
Lower |
Upper |
Lower |
Upper |
||||||
Age |
Age |
||||||||
< = 60 |
1.000 (ref) |
< = 60 |
1.000 (ref) |
||||||
> 60 |
1.836 |
0.718 |
4.694 |
0.204 |
> 60 |
2.384 |
1.367 |
4.158 |
0.002 |
Gender |
Gender |
||||||||
Female |
1.000 (ref) |
Female |
1.000 (ref) |
||||||
Male |
0.908 |
0.361 |
2.286 |
0.838 |
Male |
0.991 |
0.648 |
1.517 |
0968 |
Pathologic_stage |
Pathologic_stage |
||||||||
I, II |
1.000 (ref) |
I |
1.000 (ref) |
||||||
II |
1.950 |
0.670 |
5.677 |
0.221 |
|||||
III, IV |
2.382 |
0.748 |
7.591 |
0.142 |
III |
4.418 |
1.554 |
12.562 |
0.005 |
IV |
11.795 |
4.166 |
33.396 |
< 0.001 |
|||||
Risk |
Risk |
||||||||
Low |
1.000 (ref) |
Low |
1.000 (ref) |
||||||
High |
4.062 |
1.646 |
10.027 |
0.002 |
High |
1.833 |
1.196 |
2.810 |
0.005 |
To evaluate this seven-gene signature with respect to the prognosis among patients in TNM stage II and III, we further investigated the association between risk scores and OS in 382 CRC patients from TCGA. Stage III tumors were associated with a higher rate of 5-year mortality than stage II tumors (HR = 2.160, p = 0.004) (Figure 5A). Meanwhile, patients in high risk group demonstrated poorer survival as compared with patients in low risk group (HR = 2.918, p < 0.001) (Figure 5B). Moreover, this increased 5-year mortality in high risk group was marked in both stage II (HR = 3.124, p = 0.005) (Figure 5C) and stage III (HR = 2.415, p = 0.012) (Figure 5D). Therefore, this seven-gene signature might be able to help predict prognosis for patients with stage II and stage III CRC.
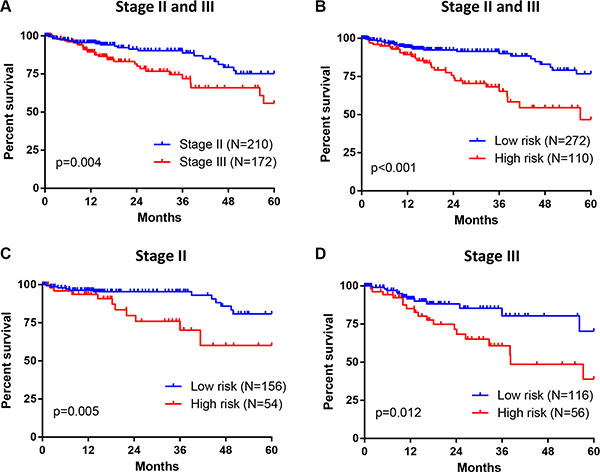
Figure 5: Association between seven-gene signature and OS in patients with stage II and III CRC. (A) For 382 CRC patients from TCGA, Stage III tumors were associated with greater 5-year mortality than stage II tumors. (B) 382 CRC patients were divided into high and low risk group based on their survival risk scores. The association between seven-gene signature (risk scores) and 5-year OS in patients with stage II and III CRC was assessed simultaneously (panel B) or individually (stage II in panel (C); stage III in panel (D) by Kaplan–Meier curves.
DISCUSSION
In this study, we presented the development and validation of a robust seven-gene signature which was able to predict the OS for CRC patients. The signature identified patients with high risk of mortality who may need potential interventions and individual therapies.
A Chinese cohort containing 64 CRC patients was used and total 6487 genes were identified to be associated with the overall survival. Of them, several genes have been reported to present prognostic value for CRC patients, such as CDX2 [19], EPHA2 [20], SLC2A1 [21], CDKN2A [22] and ITGA3 [23]. By performing KEGG analysis, these genes were enriched in the signalling pathways such as endocytosis, axon guidance, spliceosome, Wnt signalling and ubiquitin mediated proteolysis (Figure 2A). The activation of the Wnt signalling pathway is a critical event which frequently occur during CRC development, making it as therapeutic targets for cancer therapy [24]. The ubiquitin-proteasome system is important for cell growth and apoptosis regulation, making it also a potential molecular target for cancer treatment and prevention [25]. We further narrowed down the genes size and selected an optimal seven-gene signature (NHLRC3, ZDHHC21, PRR14L, CCBL1, PTPRB, PNPO and PPIP5K2) for prognosis prediction. By searching NCBI (http://www.ncbi.nlm.nih.gov/gene/), none of these seven genes have been reported previously in cancer study. Of note, NHLRC3 was predicted to play role in protein modification process through ubiquitination; CCBL1, PNPO, PPIP5K2 and ZDHHC21 were possibly involved in cellular metabolism regulation or acting as cell signaling molecules; PTPRB is a member of the protein tyrosine phosphatase (PTP) family which is known to regulate a variety of cellular processes including oncogenic transformation [26]; the function of PRR14L remains unknown. Interestingly, we found that these genes were differentially expressed in CRC tissues compared with adjacent normal tissues in two different cohorts (Figure 2D and 2E). This suggests that these genes might be novel oncogenes or tumor suppressor genes, and that their functions need further investigation.
The prognostic signature was validated in two independent patient sets on different platforms from different countries, with further validation study from China currently underway. Indeed, By ROC curve analyses, this seven-gene signature was capable of predicting OS in patients with CRC from these two datasets with an AUC of 0.715 and 0.585, respectively (Figure 4A and 4C). By multivariate analyses, this seven-gene signature was validated as an independent prognostic factor for predicting 5-year OS of CRC patients (Table 3). Comparing with other studies that constructed different prognostic models [15–18], our study obtained a signature with few genes (only seven) to successfully predict the OS in CRC patients. This is important to develop a RT-PCR assay for clinical practice. In our future study, the high-risk patients identified by seven-gene signature will be further studied for their responses to different therapies in an independent Chinese cohort.
The American Joint Committee on Cancer (AJCC) TNM staging system is a golden standard to determine the treatment and prognosis for patients with CRC. However, the limitation of TNM staging system to accurately predict prognosis has been also demonstrated in clinical practice. For example, the 5-year survival rate of stage IIIA patients (85.4%) was higher than stage II patients (79.2%) [27]. Increasing attention has been focused on patients with stage II and stage III CRC, as the use of adjuvant chemotherapy to a proportion of these patients may cause potential under treatment or overtreatment [28–30]. To date, there are several tests (such as Oncotype Dx [31], a 12-gene RT-PCR assay, and Coloprint, an 18-gene microarray [32]) being developed in clinical laboratories, which are intending to predict the prognosis of patients with stage II CRC and help guide the use of chemotherapy. Still, all of them are under evaluation in multiple validation cohorts. Our seven-gene signature is able to predict the OS for patients in stage II and III in TCGA cohort (Figure 5). However, the sample size is limited. Hence large samples are required to be collected in future to further validate the prognostic value of this seven-gene signature on CRC patients in stage II and III. Moreover, we will study whether the seven-gene signature could benefit selection of adjuvant chemotherapy for individual patient in stage II and III.
In conclusion, our seven-gene signature provides new promising biomarkers for CRC prognosis and potential therapeutic targets for CRC treatment.
MATERIALS AND METHODS
Human CRC Samples
Frozen tumor specimens (n = 64) and paired adjacent non-tumor tissue (n = 26) of 64 patients with colorectal cancer were retrieved from the Second Affiliated Hospital of Zhejiang University, College of Medicine, China. All patients who received surgical operation and were diagnosed as colon or rectal adenocarcinoma were included in this study. All pathologic information and follow-up data were obtained by reviewing the hospital records [33]. Notably, 19 of the 64 patients died of cancer during follow-up. The clinical data of patients was presented in Table 4. This study was approved by the ethics committee of Zhejiang University.
Table 4: Patient characteristics
Variables |
Case number (N = 67) N (%) or mean (range) |
|---|---|
Gender |
|
Male |
41 (61.2%) |
Female |
26 (38.8%) |
Age (years) |
|
Male |
61 (36–92) |
Female |
54.5 (19–83) |
TNM stage (T) |
|
T1 |
1 (1.5%) |
T2 |
6 (9.0%) |
T3 |
35 (52.2%) |
T4 |
25 (37.3%) |
TNM stage (N) |
|
N0 |
25 (37.3%) |
N1 |
25 (37.3%) |
N2 |
17 (25.4%) |
TNM stage (M) |
|
M0 |
49 (73.1%) |
M1 |
18 (26.9%) |
TNM stage |
|
I |
4 (6.0%) |
II |
17 (25.4%) |
III |
28 (41.8%) |
IV |
18 (26.9%) |
Gene expression microarray and data analysis
Fresh CRC samples were immediately snap frozen in liquid nitrogen and stored at –80°C until further use. Total RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, California, USA) and sent out to company (Capitalbio Corporation, Beijing, China) for RNA quality control (QC) performance. Only high quality RNA (OD260/280 = 1.8~2.2; 28S:18S ≥ 1.5; RIN (RNA Integrity Number) ≥ 8) were used for Affymetrix HG- U133plus 2.0 gene array. Statistical analyses of microarray data were performed in the R language environment (http://www.r-project.org) [34]. The data was normalized and transformed to expression values by using justRMA( ) function in “affy” library. Signal values for all genes were transformed to the log base 2. Quantile normalization was applied to obtain equal distributions of the probe signal intensities.
Gene signature identification
A robust likelihood-based survival modeling approach [35] was used to select the gene signature. We implemented our analysis by using the “rbsurv” package in R. The detailed algorithm is summarized as follows:
(i). We randomly divided the samples into the training set with N*(1 − p) samples and the validation set with N*p samples, with p = 1/3. We fitted a gene to the training set of samples and obtained the parameter estimate for this gene. Then we evaluated log likelihood with the parameter estimate and the validation set of samples. This evaluation was repeated for each gene.
(ii). We repeated the above procedure 10 times, thus obtaining 10 log likelihoods for each gene. The best gene, g(1), with the largest mean log likelihood was selected.
(iii). We searched the next best gene by evaluating every two-gene model and selected an optimal one with the largest mean log likelihood.
(iv). We continued this forward gene selection procedure, resulting in a series of models. Akaike information criterions (AICs) for all the candidate models were computed and an optimal model with the smallest AIC was selected finally.
Unsupervised hierarchical clustering and Kaplan-Meier analysis
Unsupervised hierarchical clustering analysis [36] was performed in R using “hclust” function with Euclidean distance. Kaplan-Meier curves for two distinct groups of patients were plotted using “survfit” function in survival package. P value from log rank test was computed using “survdiff” function.
Development and validation of 7-gene survival risk score system
We subjected the seven genes to BRB-array Tools (http://linus.nci.nih.gov/BRB-ArrayTools.html), using survival risk group prediction tool [37], to calculate the regression coefficient for each gene using the 64 training samples from Zhejiang University. The Survival Risk Score is the sum of the product of the expression level of a gene and its corresponding regression coefficient. The patients were divided into two groups at high and low risk using the 50th percentile. Leave-one-out cross-validation method was performed to determine the robustness.
Gene expression data from the Vanderbilt Medical Center (GSE17537, HG-U133_Plus_2 platform; N = 55) with overall survival information was normalized by justRMA( ) and used as the first validation data set [14]. The CRC dataset (N = 623 for primary CRC tumors and N = 51 for adjacent non-tumor tissues) from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/; Illumina GA or Hiseq platform) was analyzed (publicly available TCGA pre-processed data was downloaded from Broad GDAC Firehose (https://gdac.broadinstitute.org/)), and those with overall survival information (N = 584) were used as the second validation set. For each independent cohort, the risk score of patients was calculated using the coefficient derived from the training data set. Receiver operating characteristic (ROC) curve analyses were generated for the 3- and 5-year survival predictions to evaluate the specificity and sensitivity of the gene signature and to estimate discriminatory power of the prognostic gene expression signatures. The area under the curve (AUC) was calculated and a bootstrap method was used to calculate the 95% confident internal (CI) for AUC. Optimal cut- off value based on ROC curve was obtained to divide the patients into low and high risk groups [38]. Kaplan-Meier curves for the two groups of patients were plotted using “survfit” function in survival package.
Enrichment analysis of kyoto encyclopedia of genes and genomes pathways
Genes that were associated with the overall survival of patients evaluated by Cox proportional hazard regression model (P < 0.05) were included in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis using the Gene Set Analysis Toolkit V2 (http://bioinfo.vanderbilt.edu/webgestalt/). The hypergeometric test statistical method and the BH multiple test adjustment method were used. All genes from human beings were used as reference. Top 10 pathways with at least 10 genes involved were considered significantly enriched.
Statistical analysis
Mann–Whitney U test and Wilcoxon matched-pairs test were performed to compare the gene expression between CRC and adjacent normal tissues. All statistical tests were performed using Graphpad Prism 5.0 (GraphPad Software Inc, San Diego, CA, USA), and a 2-tailed P value of less than 0.05 was considered statistically significant
ACKNOWLEDGMENTS
We gratefully acknowledge financial supported by the National High Technology Research and Development Program of China (863 Program) (2012AA02A506), the National High Technology Research and Development Program of China (863 Program) (2012AA02A204), Key Projects in the National Science & Technology Pillar Program during the Twelfth Five-year Plan Period (2014BAI09B07), the Training Program of the Major Research Plan of the National Natural Science Foundation of China(91229104), National Natural Science Foundation of China (61503419), Guangdong Nature Science Foundation (2014A030310355, 2016A030313234) the “985 project” of Sun Yat-sen University (50000-31101302).
CONFLICTS OF INTEREST
The authors declare they have no competing interests.
Authors’ contributions
SZ and HRC conceived and designed the experiments. WG and RB collected the CRC tumor tissues with clinical information. HRC, XQS and YQ analyzed the data. HC and XQS wrote the paper. HRC, XQS and SZ revised the manuscript. All authors read and approved the final manuscript.
REFERENCES
1. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015; 136:E359–386.
2. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016; 66:115–132.
3. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology. 2010; 138:2059–2072.
4. Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998; 58:1130–1134.
5. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003; 3:459–465.
6. Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JKV, Hamilton S, Vogelstein B. P53 Gene-Mutations Occur in Combination with 17p Allelic Deletions as Late Events in Colorectal Tumorigenesis. Cancer Res. 1990; 50:7717–7722.
7. Deming DA, Leystra AA, Nettekoven L, Sievers C, Miller D, Middlebrooks M, Clipson L, Albrecht D, Bacher J, Washington MK, Weichert J, Halberg RB. PIK3CA and APC mutations are synergistic in the development of intestinal cancers. Oncogene. 2014; 33:2245–2254.
8. Kern SE, Fearon ER, Tersmette KW, Enterline JP, Leppert M, Nakamura Y, White R, Vogelstein B, Hamilton SR. Clinical and pathological associations with allelic loss in colorectal carcinoma [corrected]. J Am Med Assoc. 1989; 261:3099–3103.
9. Anwar S, Frayling IM, Scott NA, Carlson GL. Systematic review of genetic influences on the prognosis of colorectal cancer. Br J Surg. 2004; 91:1275–1291.
10. Quackenbush J. Microarray analysis and tumor classification. N Engl J Med. 2006; 354:2463–2472.
11. Sadanandam A, Lyssiotis CA, Homicsko K, Collisson EA, Gibb WJ, Wullschleger S, Ostos LCG, Lannon WA, Grotzinger C, Del Rio M, Lhermitte B, Olshen AB, Wiedenmann B, et al. A colorectal cancer classification system that associates cellular phenotype and responses to therapy. Nat Med. 2013; 19:619–625.
12. Schlicker A, Beran G, Chresta CM, McWalter G, Pritchard A, Weston S, Runswick S, Davenport S, Heathcote K, Castro DA, Orphanides G, French T, Wessels LF. Subtypes of primary colorectal tumors correlate with response to targeted treatment in colorectal cell lines. BMC Med Genomics. 2012; 5:66.
13. Jorissen RN, Gibbs P, Christie M, Prakash S, Lipton L, Desai J, Kerr D, Aaltonen LA, Arango D, Kruhoffer M, Orntoft TF, Andersen CL, Gruidl M, et al. Metastasis-Associated Gene Expression Changes Predict Poor Outcomes in Patients with Dukes Stage B and C Colorectal Cancer. Clin Cancer Res. 2009; 15:7642–7651.
14. Smith JJ, Deane NG, Wu F, Merchant NB, Zhang B, Jiang A, Lu P, Johnson JC, Schmidt C, Bailey CE, Eschrich S, Kis C, Levy S, et al. Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology. 2010; 138:958–968.
15. Salazar R, Roepman P, Capella G, Moreno V, Simon I, Dreezen C, Lopez-Doriga A, Santos C, Marijnen C, Westerga J, Bruin S, Kerr D, Kuppen P, et al. Gene expression signature to improve prognosis prediction of stage II and III colorectal cancer. J Clin Oncol. 2011; 29:17–24.
16. Lin YH, Friederichs J, Black MA, Mages J, Rosenberg R, Guilford PJ, Phillips V, Thompson-Fawcett M, Kasabov N, Toro T, Merrie AE, van Rij A, Yoon HS, et al. Multiple gene expression classifiers from different array platforms predict poor prognosis of colorectal cancer. Clin Cancer Res. 2007; 13:498–507.
17. Eschrich S, Yang I, Bloom G, Kwong KY, Boulware D, Cantor A, Coppola D, Kruhoffer M, Aaltonen L, Orntoft TF, Quackenbush J, Yeatman TJ. Molecular staging for survival prediction of colorectal cancer patients. J Clin Oncol 2005; 23:3526–3535.
18. Nguyen MN, Choi TG, Nguyen DT, Kim JH, Jo YH, Shahid M, Akter S, Aryal SN, Yoo JY, Ahn YJ, Cho KM, Lee JS, Choe W, et al. CRC-113 gene expression signature for predicting prognosis in patients with colorectal cancer. Oncotarget. 2015; 6:31674–31692. doi: 10.18632/oncotarget.5183.
19. Dalerba P, Sahoo D, Paik S, Guo XQ, Yothers G, Song N, Wilcox-Fogel N, Forgo E, Rajendran PS, Miranda SP, Hisamori S, Hutchison J, Kalisky T, et al. CDX2 as a Prognostic Biomarker in Stage II and Stage III Colon Cancer. New Engl J Med. 2016; 374:211–222.
20. Dunne PD, Dasgupta S, Blayney JK, McArt DG, Redmond KL, Weir JA, Bradley CA, Sasazuki T, Shirasawa S, Wang TT, Srivastava S, Ong CW, Arthur K, et al. EphA2 Expression Is a Key Driver of Migration and Invasion and a Poor Prognostic Marker in Colorectal Cancer. Clin Cancer Res. 2016; 22:230–242.
21. Goos JA, de Cuba EM, Coupe VM, Diosdado B, Delis-Van Diemen PM, Karga C, Belien JA, Menke-Van der Houven van Oordt CW, Geldof AA, Meijer GA, Hoekstra OS, Fijneman RJ, DeCoDe PETG. Glucose Transporter 1 (SLC2A1) and Vascular Endothelial Growth Factor A (VEGFA) Predict Survival After Resection of Colorectal Cancer Liver Metastasis. Ann Surg. 2016; 263:138–145.
22. Xing X, Cai W, Shi H, Wang Y, Li M, Jiao J, Chen M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: a meta-analysis. Br J Cancer. 2013; 108:2542–2548.
23. Linhares MM, Affonso RJ, Viana LD, Silva SRM, Denadai MVA, de Toledo SRC, Matos D. Genetic and Immunohistochemical Expression of Integrins ITGAV, ITGA6, and ITGA3 As Prognostic Factor for Colorectal Cancer: Models for Global and Disease-Free Survival. Plos One. 2015; 10.
24. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013; 13:11–26.
25. McBride WH, Iwamoto KS, Syljuasen R, Pervan M, Pajonk F. The role of the ubiquitin/proteasome system in cellular responses to radiation. Oncogene. 2003; 22:5755–5773.
26. Ostman A, Hellberg C, Bohmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006; 6:307–320.
27. Lan YT, Yang SH, Chang SC, Liang WY, Li AF, Wang HS, Jiang JK, Chen WS, Lin TC, Lin JK. Analysis of the seventh edition of American Joint Committee on colon cancer staging. Int J Colorectal Dis. 2012; 27:657–663.
28. Marshall JL, Haller DG, de Gramont A, Hochster HS, Lenz HJ, Ajani JA, Goldberg RM. Adjuvant Therapy for Stage II and III Colon Cancer: Consensus Report of the International Society of Gastrointestinal Oncology. Gastrointest Cancer Res. 2007; 1:146–154.
29. Benson AB 3rd, Schrag D, Somerfield MR, Cohen AM, Figueredo AT, Flynn PJ, Krzyzanowska MK, Maroun J, McAllister P, Van Cutsem E, Brouwers M, Charette M, Haller DG, et al. American Society of Clinical Oncology recommendations on adjuvant chemotherapy for stage II colon cancer. J Clin Oncol. 2004; 22:3408–3419.
30. Gray R, Barnwell J, McConkey C, Hills RK, Williams NS, Kerr DJ. Adjuvant chemotherapy versus observation in patients with colorectal cancer: a randomised study. Lancet. 2007; 370:2020–2029.
31. Webber EM, Lin JS, Evelyn PW. Oncotype DX tumor gene expression profiling in stage II colon cancer. Application: prognostic, risk prediction. PLoS currents. 2010; 2.
32. Tan IB, Tan P. Genetics: an 18-gene signature (ColoPrint(R)) for colon cancer prognosis. Nat Rev Clin Oncol. 2011; 8:131-133.
33. Hu H, Zhang H, Ge W, Liu X, Loera S, Chu P, Chen H, Peng J, Zhou L, Yu S, Yuan Y, Zhang S, Lai L, et al. Secreted protein acidic and rich in cysteines-like 1 suppresses aggressiveness and predicts better survival in colorectal cancers. Clin Cancer Res. 2012; 18:5438–5448.
34. James G, Witten D, Hastie T, Tibshirani R. An Introduction to Statistical Learning with Applications in R. Springer-Verlag New York. 2013.
35. Cho HJ, Yu A, Kim S, Kang J, Hong SM. Robust likelihood-based survival modeling with microarray data. J Stat Softw. 2009; 29:1–16.
36. Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:14863–14868.
37. Simon R, Lam A, Li MC, Ngan M, Menenzes S, Zhao Y. Analysis of gene expression data using BRB-ArrayTools. Cancer Inform. 2007; 3:11–17.
38. Zou KH, O’Malley AJ, Mauri L. Receiver-operating characteristic analysis for evaluating diagnostic tests and predictive models. Circulation. 2007; 115:654–657.