
INTRODUCTION
Isopropyl methane sulfonate (IPMS), an alkyl sulfonate, is a potential genotoxic impurity (GTI) that can form as a byproduct during the synthesis of sulfonate salts when sulfonic acids react with isopropanol, a low molecular weight alcohol [1]. IPMS displays genotoxicity and carcinogenicity in in vitro and in vivo assays, and it is categorized as the most potent mutagen in the Ames and in vivo micronucleus assays [2-9]. Despite its hazardous profile, there has been little attention on IPMS compared to what is known about methyl methanesulfonate (MMS) and ethyl methanesulfonate (EMS), which are also potential GTIs. These alkyl sulfonates constitute a representative class of direct mutagens whose genotoxicity is attributed to their alkylating ability at the O6 position of dG [5, 10]. The genotoxicity of IPMS has been hypothesized to be attributed to the differences in the SN1/SN2 reaction type and the Swain Scott constants [11], as compared to MMS and EMS [5]. Although IPMS-mediated DNA adduct formation has been previously studied, it is important to determine its net biological effect (e.g. cytotoxicity and genotoxicity outcome), which is determined by the balance between the generation of DNA damage and the DNA repair efficiency. Understanding both the damage and repair aspects helps to more accurately interpret how individual alkylating agents induce genotoxicity. In this study, we conducted the DNA damage response (DDR) assay using isogenic chicken DT40 cell lines [12-14] to understand the repair or tolerant pathway activated in response to IPMS. DT40 cells originated from a chicken B-lymphocyte line derived from an avian leucosis virus-induced bursal lymphoma isolated in 1985 [15]. The isogenic DT40 cell lines in this study broadly probe biological targets, pathways and mechanisms in relation to genotoxicity and/or cytotoxicity endpoints for a large number of chemicals [16, 17]. The DDR assay, which examines cytotoxicity in DNA repair-deficient DT40 mutants versus the parental DT40 cells, is a rapid and simple method to evaluate the genotoxicity of xenobiotics.
Interestingly, small differences in chemical structure can drastically change genotoxicity. nPMS is an isomer of IPMS with a straight chain in the alkyl side chain structure, while IPMS has an isopropyl moiety. Despite the subtle change in structure, the genotoxic potential of nPMS is significantly weaker than IPMS [2, 4-6, 8, 9]. The difference in the activities of these two agents has not been adequately explained, but it is believed to be due to a combination of the DNA lesion-forming potential and repair or tolerance capability. A possible explanation for the different efficiencies in the formation of DNA adducts is that IPMS is able to form a carbonium ion (SN1) while the reactivity of nPMS occurs via a bimolecular nucleophilic displacement reaction (SN2). The SN1 reactivity of IPMS indicates that it possesses stronger reactivity at the O6 position of dG compared to nPMS [18]. As a result, IPMS is believed to generate more DNA adducts at the O6 position of dG than nPMS. Thus, the SN1/SN2 reaction type and the Swain Scott constants are useful values for predicting the potential for genotoxicity. However, as previously mentioned, genotoxicity is characterized not only by the generation of DNA damage but also the effect on DNA damage repair; therefore, it is important to characterize the changes in repair or tolerance capabilities after IPMS exposure, which have not been previously highlighted.
Alkylating agents predominantly form adducts at N- and O- atoms, and O-alkylations (e.g., O6-alkyl dG) are known to be highly mutagenic and clastogenic. Although its proportion is less than 10% of the total number of DNA methyl adducts, O-alkylations are stable and persist in the absence of the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) [19, 20]. The MGMT enzyme repairs these adducts by transferring the alkyl group from the oxygen in the guanine to a cysteine residue in the catalytic pocket of MGMT [21]. Exposure to IPMS results in bulkier adducts, which are slower to repair than those induced by ENU [22-24]. Dolan et al. reported that MGMT efficiently removes O6-n-propyl guanine, but its activity on O6-isopropyl dG is less extensive [25]. It is not clear how differences in MGMT repair efficiency impacts the cellular response. To address this point, we inhibited MGMT activity in cells, exposed these cells to IPMS, nPMS or MMS, and examined the cytotoxic or genotoxic activity. Because O6-n-propyl dG and O6-methyl dG are more effectively repaired by MGMT than O6-isopropyl dG, MGMT depletion would be expected to generate different responses to each chemical. In this study, cells were pre-incubated with O6-benzylguanine (OBG), a MGMT inhibitor, and the LC50 and MN induction in DT40 cells treated with IPMS, nPMS or MMS were measured as indices of cytotoxicity and genotoxicity.
Unrepaired O6-alkyl dG forms mispairings with deoxythymidine and is recognized by mismatch repair (MMR); however, MMR cannot repair O6-methylG/T mispairs, which eventually lead to double-strand breaks (DSBs). Because O6-isopropyl dG is poorly recognized by MGMT, it is highly likely to form mispairings and form DSBs. It has been reported that DSB repair pathways are activated by methylating agents [26]. Taking this into consideration, we measured γ-H2AX as an indicator for formation of DSBs. Wild-type and sensitive mutant cells were exposed to IPMS, and the population of γ-H2AX positive cells was measured to investigate whether the DSBs induced by IPMS causes the unique response found in the DDR assay.
RESULTS
Greater cytotoxicity of IPMS in FANC-deficient DT40 cells compared to MMS or EMS
To determine the repair or tolerance mechanism of IPMS-induce DNA damage, twenty-eight isogenic mutant cells were exposed to IPMS. The mutant cell lines cover a variety of DNA repair/tolerance pathways (e.g., BER, base excision repair; HEL, helicase; NER, nucleotide excision repair; NHEJ, non-homologous end-joining; TLS, translesion DNA synthesis; HR, homologous recombination; DDC, DNA damage checkpoint). Considering the weaker SN1-reactivity and stronger SN2-reactivity of MMS and EMS, we also exposed cells to these chemicals in order to see if the SN-1 dominant IPMS gave a different response from MMS and EMS. The LC50 in each mutant was compared to that in DT40 cells. The mean LC50’s in parental DT40 cells were 736 µM, 65.7 µM, and 1234 µM for IPMS, MMS and EMS, respectively. For the mutant cell lines, the average of 3 or more independent experiments are plotted as the relative LC50 (Figure 1). Among these cell lines, DT40 cells deficient in FANC genes showed greater sensitivity to IPMS than to MMS or EMS. The relative LC50 of MMS or EMS in FANC-deficient cells was approximately 60%, while the value dropped to a range of 10-40% in FANC-deficient cells treated with IPMS. On the other hand, IPMS showed moderate to no toxicity in the POLβ-, FEN1-, and PARP1-deficient DT40 cells, while MMS or EMS showed obvious toxicity in these cells, suggesting that the BER pathways are less involved in the repair of IPMS-induced DNA damage. For the other mutant cells, there was no difference in the relative LC50 among all of the three sulfonic acids tested. Interestingly, the LC50 values in MMS- and EMS-treated mutants were comparable, suggesting that the DDR to these two chemicals are quite similar.
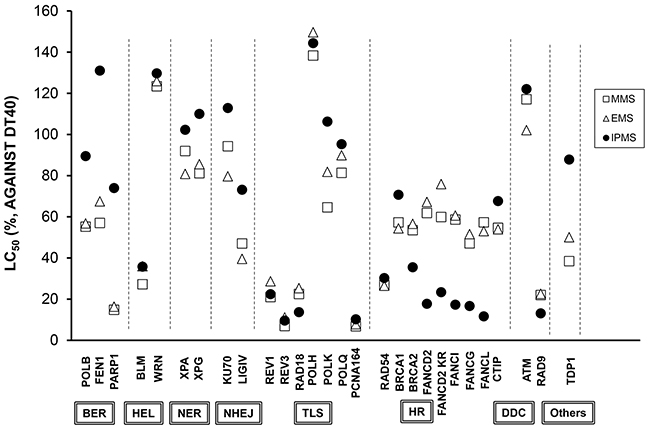
Figure 1: DT40 cell-based DNA damage response assay for IPMS, MMS and EMS. Each mutant cell line lacking a specific gene was exposed to serial dilutions of each chemical, and the relative LC50 was normalized based on the LC50 in DT40 wild-type cells. The data shows the mean of at least three independent experiments. See Table 1 for the abbreviation of the DNA repair or tolerance pathways.
Table 1: A list of examined DT40 mutant cell lines
DNA repair/tolerance pathway |
Genes |
|---|---|
Base Excision Repair (BER) |
POLβ, FEN1, PARP1 |
RecQ like Helicase |
BLM, WRN |
Nucleotide Excision Repair (NER) |
XPA, XPG |
Non-Homologous End-Joining (NHEJ) |
LIGIV, KU70 |
Homologous Recombination (HR) |
RAD54, BRCA1, BRCA2, FANCI, FANCG, FANCC, FANCL, FANCD2, FANCD2-K563R (monoubiquitination-deficient mutant), CTIP |
DNA damage checkpoint (DDC) |
RAD9, ATM |
Trans-Lesion Synthesis (TLS) |
REV1, REV3, POLH, POLK, POLQ, RAD18, PCNA K164R (monoubiquitination-deficient mutant) |
Other |
TDP1 |
The involvement of the FANC pathway in the repair and tolerance of IPMS-induced DNA damage
In order to confirm that the hypersensitivity of DT40 cells to IPMS results from a dysfunction in the FANC pathway, FANCD2-deficient cells were transfected with a functional FANCD2 plasmid and treated with IPMS, MMS or EMS. The sensitivity of these FANC2-deficient cells was compared with wild-type cells to determine if ectopic expression of FANC2 rescues its DNA damage repair function. After IPMS treatment, the survival curve of FANCD2-deficient cells transfected with the FANCD2 plasmid is similar to that of wild-type cells and is significantly different from the survival curve of FANCD2 mutant cells (Figure 2A). The obtained LC50 values were 75.99 µM, 253.4 µM and 355.1 µM in FANCD2, FANCD2+wt FANCD2 and wild-type cells, respectively. These results indicate that ectopic expression of FANCD2 restored the resistance to IPMS. In contrast, no obvious difference was observed on the survival curves between FANCD2 and FANCD2+wt FANCD2 cells treated with MMS and EMS (Figure 2B and 2C). IPMS also caused hyper-sensitivity to cells expressing monoubiquitination-deficient FANCD2 (FANCD2 K563R) (Figure 1). Thus, these results strengthen our findings that the FANCD2, or more specifically monoubiquitination of FANCD2 at lysine 563, is critical for tolerating IPMS-induced DNA damage. We then addressed whether human cells deficient in FANCD2 are also hyper-sensitive to IPMS. To test this question, FANCD2 gene was deleted in human TK6 lymphoblastoid cells (Supplementary Figure S1). As with the DT40 cells, TK6 FANCD2 knock-out cells were markedly sensitive to IPMS in terms of cell toxicity (Figure 2D). Together, these results suggest that the FANC pathway plays a pivotal role in the repair or tolerance of IPMS-induced DNA damage.
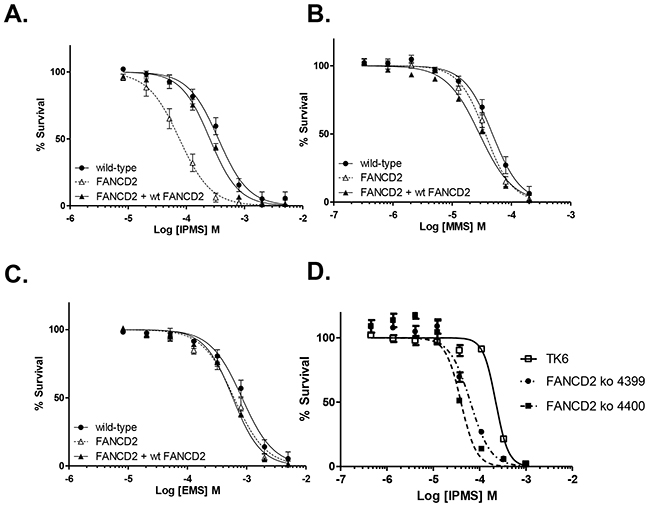
Figure 2: FANCD2 plays a critical role on cell survival in the presence of IPMS. FANCD2-deficient cells transfected with the FANCD2 plasmid were exposed to IPMS A. MMS B. or EMS C. at 8 concentrations in parallel with FANCD2 knock-out and wild-type cells. D. FANCD2 knock-out (two clones) and wild-type TK6 cells were treated with IPMS. After treatment, cell viability was evaluated with XTT, and the relative survival at each concentration was calculated. The data shows the mean and standard deviation of at least three independent experiments.
The isopropyl moiety plays a crucial role in DNA damage response to IPMS in the absence of FANC repair machinery
To understand how the subtle change in chemical structure between IPMS and nPMS gives different cellular responses, we examined the sensitivity of FANC-deficient cells to nPMS, which possesses the n-propyl moiety instead of the isopropyl moiety present in IPMS. The role of the FANC repair pathway in nPMS-derived DNA damage was investigated in comparison with IPMS-derived DNA damage. The survival curve of FANCD2 cells treated with IPMS is significantly different from that of wild-type cells while this difference was not observed when cells were treated with nPMS (Figure 3). The LC50 values were 1879 µM and 1421 µM in wild-type and FANCD2 cells treated with nPMS, whereas the values were 706 µM and 142 µM in wild-type and FANCD2 cells treated with IPMS. No obvious difference in sensitivity to nPMS was observed in FANCG, FANCI, and FANCL compared to that in wild-type cells, suggesting that the FANC repair pathway is not the primary repair or tolerance pathway activated in response to nPMS-induced DNA damage.
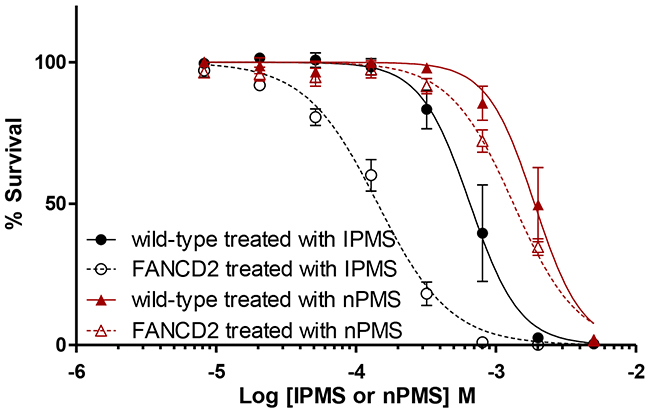
Figure 3: Differential toxicity of IPMS and nPMS in FANCD2-deficient DT40 cells. The cytotoxicity of IPMS and nPMS in FANCD2-deficient cells was examined. FANCD2-deficient cells and wild-type cells were exposed to IPMS and nPMS at 8 concentrations. After treatment, cell viability was evaluated with XTT, and the relative survival at each concentration was calculated. The data shows the mean and standard deviation of at least three independent experiments.
No involvement of the MGMT repair pathway in IPMS-derived DNA damage in DT40 cells
To investigate the involvement of the MGMT repair pathway on IPMS- and nPMS-derived DNA damage and consequent cell death, DT40 wild-type cells pretreated with O6-benzyl guanine (OBG), which inhibits MGMT activity, were exposed to IPMS and nPMS. To see if inactivation of the MGMT repair pathway further potentiates the cytotoxicity of alkylating agents, the LC50 values in the OBG pretreated group were compared with those in the untreated group. As a well-known alkylating agent, MMS was exposed in parallel. To determine if the FANC and MGMT repair pathways work independently, FANCI-, FANCG-, FANCL-, FANCC-, and FANCD2- deficient cells were also included in the assay. As a result, OBG pretreatment greatly reduced the LC50 values in all of the examined cells treated with nPMS and MMS, but not IPMS (Figure 4). These results are consistent with the fact that MGMT recognizes O6-methyl, ethyl, and propyl dG [25]. It is conceivable, therefore, that because O6-isopropyl dG is poorly recognized by MGMT, MGMT depletion does not change the LC50, while O6-n-propyl dG or O6-methyl dG, which are recognized by MGMT, greatly reduce the LC50 in the pretreated cells. The same pattern was observed in the FANC-deficient mutants.
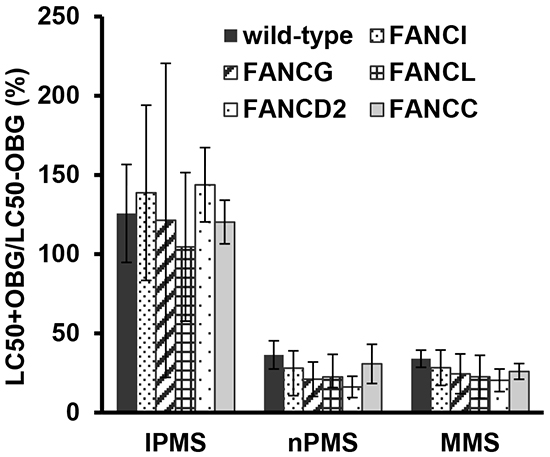
Figure 4: Impact of MGMT function on the toxicity of IPMS, nPMS, and MMS in DT40 cells deficient and proficient in FANC genes. Wild-type and FANC-deficient cells with or without O6-benzylguanine pre-treatment were exposed to IPMS, nPMS or MMS. After treatment, XTT was added, and cell viability was measured. Based on the cell viability data, the LC50 was calculated in each treatment condition. The LC50 with pre-treatment is divided by the LC50 without pre-treatment and expressed as percentage. The data shows the mean and standard deviation of at least three independent experiments.
Greater MN induction in FANC-deficient cells treated with IPMS
The MN assay with IPMS was conducted in wild-type and FANC-deficient cells to determine the impact of a dysfunction in the FANC pathway on MN induction. Results revealed MN induction by IPMS treatment in both wild-type and FANC-deficient mutant cells (Figure 5A). The point of departure (PoD) analysis revealed that the break point concentration of MN induction over the baseline level was lower in FANCG-, FANCI- and FANCL-deficient cells compared to that of wild-type cells, with the calculated PoD being 81 µM, 56 µM, and 51 µM in FANCG, FANCI and FANCL cells, respectively, as compared to 110 µM in DT40 wild-type cells. The PoD value indicates the concentration in which a biological effect is observed. Therefore, the lower PoD values observed in FANC-deficient cells indicate that the FANC pathway contributes to suppressing MN formation upon IPMS treatment. We also investigated the impact of MGMT inhibition on MN induction by IPMS or nPMS to examine whether the differential recognition between O6-isopropyl dG and O6-n-propyl dG by MGMT influences the MN results, as observed in the cytotoxicity assay. Cells pretreated with OBG were exposed to IPMS or nPMS, and the MN frequency was measured. As a result of two independent experiments, both chemicals increased the MN frequency in both wild-type and FANCD2-deficient cells (Figure 5B). The spontaneous MN frequency was higher in FANCD2 cells (0.4%) than in wild-type cells (0.1%), suggesting that the FANC pathway plays a protective role against spontaneously occurring DNA damage. The OBG pretreatment increased the MN frequency in wild-type cells treated with nPMS, but it did not increase the frequency in wild-type nor FANCD2-deficient cells treated with IPMS. The OBG pretreatment also reduced the relative population doubling in wild-type cells treated with nPMS, but not for IPMS-treated cells. In the FANCD2 cells treated with nPMS, OBG pretreatment slightly increased the MN frequency, but the effect was not significant. These results indicate that, in contrast to O6-isopropyl dG, MTMT removal of O6-n-propyl dG reduces MN formation.
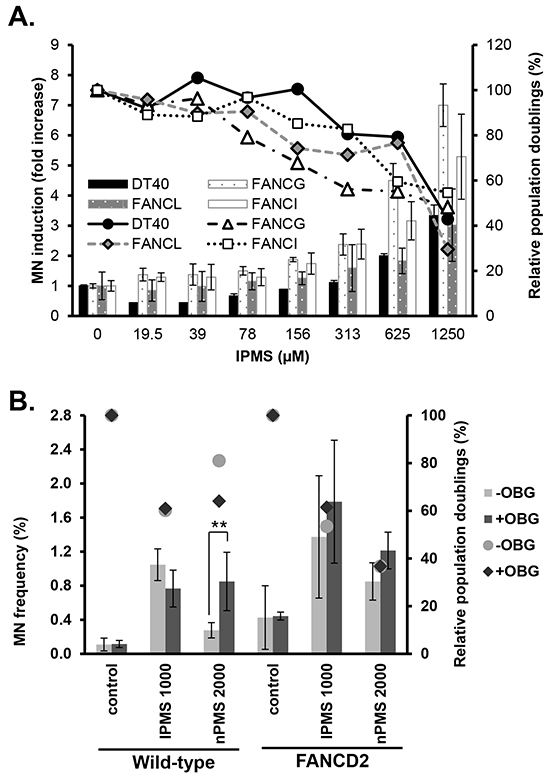
Figure 5: Micronucleus induction in wild-type and FANC-deficient cells treated with IPMS. A. The fold increase in MN frequency over the control in DT40 wild-type, FANCI, FANCG and FANCL mutants treated with IPMS for 16 hours is shown. The relative population doublings are also plotted. The data shows the mean and standard deviation of two independent experiments. B. This experiment was conducted to examine the effect of MGMT inhibition on micronucleus induction in wild-type and FANCD2-deficient cells treated with IPMS or nPMS. Wild-type or FANCD2-deficient cells with or without O6-benzylguanine pre-treatment were exposed to IPMS or nPMS. After 16 hours of treatment, cells were harvested and stained with fluorescence dye for the flow cytometry-based MN assay. Statistical analyses were conducted for the MN frequency comparing the pre-treated samples against the samples without pretreatment. The relative population doublings are also plotted. The data shows the mean and standard deviation of two independent experiments.
Induction of DSBs in DT40 and TK6 cells treated with IPMS
Next, we addressed whether O6-isopropyl dG generates DSBs and if the inability to repair DSBs in FANC-deficient cells results in an accumulation of DSBs, a higher incidence of cell death, and MN induction by IPMS. To do this, we analyzed the population of γH2AX-positive cells in wild-type and FANCD2-deficient cells. After a 4-hour treatment with IPMS, the population of γH2AX-positive cells in both wild-type and FANCD2-deficient cells showed a linear dose response at a concentration of 100 µM or higher (Figure 6A). In cells that were not treated with IPMS, 4.2 and 11.6% of wild-type and FANCD2-deficient cells were positive for γH2AX, respectively. The evidence for FANC involvement in DSB repair [27] is consistent with our observation that FANCD2-deficient cells display a higher incidence of γH2AX. To estimate the number of IPMS-induced γH2AX-positive cells, these baseline values were subtracted from the observed values at each time and dose point. Time course experiments revealed that there was an increase in γH2AX-positive cells within 1 hour of treatment in both wild-type and FANCD2-deficient cells treated with 400 µM IPMS. The number of γH2AX-positive cells peaked at 4 hours after treatment, and this peak was maintained thereafter. In FANCD2-deficient cells, the percentage of γH2AX-positive cells is almost twice that of wild-type cells at the 4- and 6-hour treatment time points (Figure 6B). We also examined nuclear γ-H2AX and 53BP1 foci formation in TK6 cells after 24-hour exposure to IPMS. A marked increase in both γ-H2AX and 53BP1 foci formation was observed in IPMS-exposed cells compared to control (Figure 7). To test if both γ-H2AX and 53BP1 are co-localized as foci, we performed co-immunostaining of TK6 cells with γ-H2AX and 53BP1. Many γ-H2AX foci were co-localized with 53BP1 foci, indicating that IPMS causes DSBs in human TK6 cells as well.
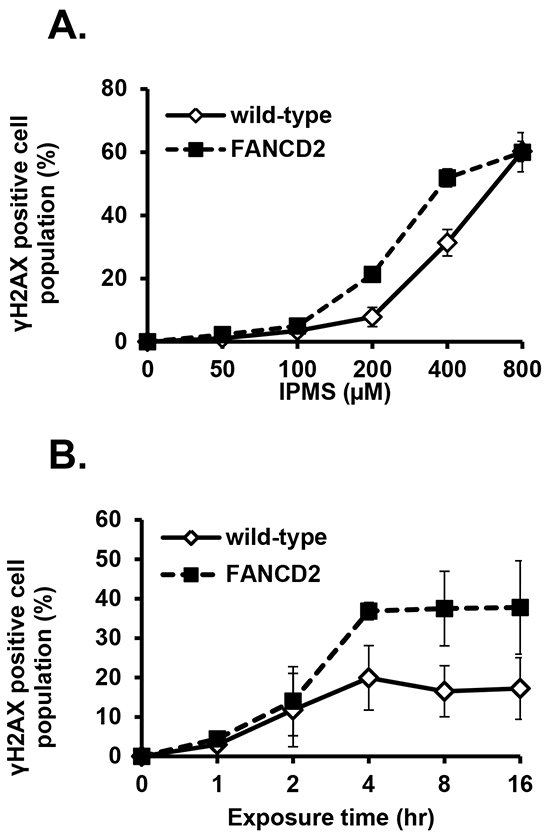
Figure 6: IPMS-induced γH2AX in DT40 cells with or without functional FANCD2. A. Dose response of γH2AX was examined in wild-type and FANCD2-deficient cells treated with IPMS. Cells were treated with IPMS for 4 hours, and the γH2AX expression was measured. The y-coordinate indicates the population of γH2AX-positive cells. Each measurement represents the mean and standard deviation of two independent experiments. B. Expression of γH2AX was examined in a time-dependent manner in wild-type and FANCD2-deficient cells treated with IPMS. The kinetics of γH2AX induction was investigated after treatment with 400 μM IPMS in wild-type and FANCD2-deficient cells. Cells were exposed to IPMS at different times at 39°C, and cells were harvested thereafter for immunostaining and flow cytometry-based analyses. Each measurement represents the mean of two independent experiments.
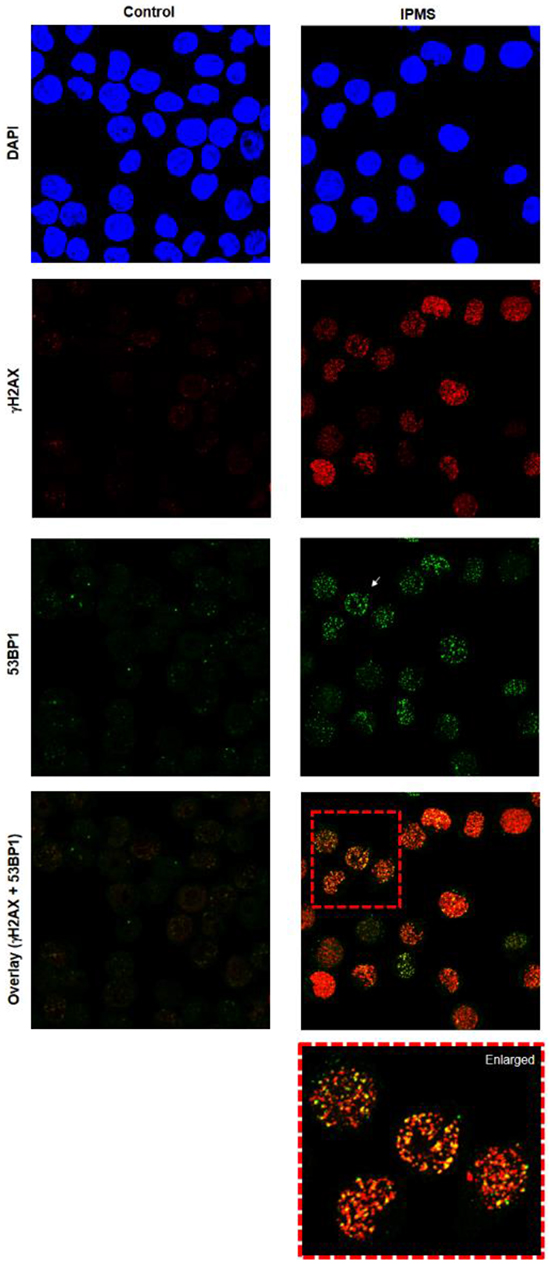
Figure 7: Induction of 53BP1 and γH2AX foci in TK6 cells following exposure to IPMS. TK6 cells were exposed to IPMS (750 µM) for 24h and co-immunostained with anti-53BP1 (green) and anti- γH2AX (red). Nuclei were counterstained with DAPI (blue). The images were captured using confocal microscopy and represents maximum intensity projections of z-stacks. Magnification, 600X.
DISCUSSION
The present study was conducted to elucidate the repair or tolerance pathway activated in response to IPMS and to understand how O6-isopropyl dG leads to cell death and/or genomic damage. To characterize cellular response to IPMS treatment, we compared the genotoxicity and cytotoxity potential between IPMS and nPMS, which is an isomer of IPMS that lacks the isopropyl moiety and exhibits weaker SN-1 reactivity and stronger SN-2 reactivity. Due to the higher SN1-reactivity of IPMS, we hypothesized a greater percentage of O6 adducts from IPMS treatment. Comparison of the sensitivity of wild-type and FANCD2-deficient cells to IPMS and nPMS revealed that the branched alkyl moiety of IPMS is a key structure that mediates the hypersensitivity of FANC-deficient cells to IPMS, and that the cellular response to nPMS is similar to that of MMS and EMS. Alkyl groups in O6-alkyl dG adducts are repaired by MGMT; however, the repair efficiency of this enzyme varies depending on the substrate. It has previously been seen that O6-isopropyl dG is less extensively repaired than O6-methyl dG or O6-ethyl dG [28], but it remained unclear how this difference in repair efficiency affects cellular responses (e.g. measured as cytotoxicity, mutagenicity and clastogenicity). To examine the impact of differences in efficiencies of the MGMT repair pathway on cell survival, wild-type and FANC-deficient cells were exposed to IPMS, nPMS or MMS with and without pretreatment with O6-benzyl guanine (OBG), an inhibitor of MGMT. Pretreatment with OBG did not change the LC50 values of IPMS in wild-type or FANC-deficient cells, while OBG potentiated the cytotoxicity of nPMS and MMS in all of the cells examined. These results suggest that MGMT is not involved in the repair of O6-isopropyl dG and does not rescue cells from death, but instead repairs O6-n-propyl dG and O6-methyl dG and promotes cell survival. These findings are consistent with the report that O6-n-propyl dG is recognized by MGMT as efficiently as O6-methyl guanine and O6-ethyl dG [25]. Taken together, the sensitivity of FANC-deficient cells to IPMS, nPMS, MMS and EMS is associated with MGMT recognition activity of the adduct type that is generated.
Consistent with the observation that O6-isopropyl dG escapes recognition and repair by MGMT, IPMS-treated cells accumulated more DNA damage than nPMS-, MMS- or EMS-treated cells and thus demonstrate higher cytotoxicity. These unrepaired DNA lesions are assumed to lead to MN formation. Our PoD analysis in wild-type and FANC-deficient cells revealed that the FANC pathway suppresses IPMS-induced MN formation. In the DDR assay using DT40 mutant cells, BRCA2, FANCD2, FANCI, FANCG, FANCL, REV1, REV3, RAD9, RAD18 and PCNA K164R mutants were hypersensitive to IPMS. Among these cells, the FANC pathway mutants were exclusively sensitive to IPMS while REV1, REV3, and PCNA K164R mutants were comparably sensitive to IPMS, MMS or EMS. These results suggest that the FANC repair pathway plays a pivotal role in repairing or tolerating IPMS-induced DNA damage compared to the other genes universally involved in repair or tolerance of alkylated DNA damages. Exogenous expression of the FANCD2 gene in FANCD2-deficient cells restored the resistance to IPMS. The FANC pathway is known to function in DNA cross-link repair, homologous recombination and resolution of the replication arrest [29, 30]. If O6-methyl dG is left unrepaired, DNA replication over the O6MeG results in an O6MeG:T mismatch or O6MeG:C ambiguous pair [31]. Subsequently, the mismatch repair pathway works to fix the mismatch. However, since O6MeG remains in the template, a futile repair loop can eventually result in highly toxic DSBs during DNA replication [32, 33]. The same cellular response likely takes place in IPMS-treated cells. IPMS may generate mismatch repair pathway-mediated and replication-associated DSBs, which appear to be repaired by the FANC pathway. Monoubiquitylation of FANCD2 and FANCI is critical for downstream FANC pathway activation in the repair and tolerance of crosslinking DNA damage [34]. Monoubiquitination of FANCD2 and FANCI is catalyzed by the FANC core complex, including FANCG and FANCC, through the ubiquitin ligase subunit FANCL [35]. Our results show that DT40 cells deficient in FANCD2, FANCI, FANCG, or FANCL are hyper-sensitive to IPMS as compared to MMS and EMS, and monoubiquitination-deficient FANCD2 mutant cells are also markedly sensitive to IPMS. This suggests that mismatch repair pathway-coupled O6-alkylguanine-mediated DSBs during DNA replication activates the Fanconi anemia core complex and leads to the monoubiquitination of FANCD2 and FANCI. DT40 mutants deficient in BRCA2 were also hyper-sensitive to IPMS, indicating that the monoubiquitination of FANCD2 and FANCI further stimulates downstream components of the FANC pathway to repair and tolerate DSBs caused by O6-alkylguanine. Interestingly, both RAD18 and RAD9-deficient DT40 cells showed a similar trend of toxicity with lesser extent than FANC-deficient cells, suggesting that these DDR pathways could involve the upstream events of FANC pathway. Indeed, the FANC pathway is activated by RAD18, which also interacts with ubiquitinated chromatin components and facilitates RAD9 recruitment to DNA DSBs [35, 36].
As unrepaired DSBs result in the formation of MN after cell division, we examined MN induction in cells treated with IPMS and nPMS in the presence and absence of OBG. The MN assay revealed that MGMT dysfunction elevated the MN induction frequency in wild-type cells treated with nPMS but not IPMS. MGMT did not affect MN frequency in FANCD2-deficient cells. These results indicate that O6-n-propyl dG accumulates in the absence of MGMT activity, but loss of MGMT activity does not change the number of O6-isopropyl dG adducts that remains on the DNA and eventually form MN or kill cells. An increase in MN frequency was not observed in OBG-pretreated FANCD2 cells after treatment with nPMS. High spontaneous MN frequency in FANCD2 cells, which is consistent with a previous report [37], may make it challenging to detect the small increases in MN frequency by MGMT inhibition. The increase in time-dependent accumulation of DSBs in FANCD2-deficient cells compared to wild-type cells may explain the stronger cytotoxicity and higher incidence of MN formation in FANC-deficient cells treated with IPMS. It is worthwhile to note that spontaneous MN frequency in FANCG-, FANCI-, and FANCL-deficient DT40 cells were 1.8, 3.4, and 6.6 times higher, respectively, than that in wild-type DT40 cells. Due to markedly high spontaneous MN frequency, the relative increase in MN frequency in FANCL-deficient DT40 cells is comparable to that in wild-type DT40 cells (Figure 5A). In contrast, the PoD concentration, which is indicative of a biologically significant increase, differed between wild type cells and FANCL-deficient cells. FANCL-deficient cells showed a lower value than that in wild type cells, which suggest FANCL-deficient cells are more sensitive to MN induction by IPMS.
The observation that the isopropyl moiety is a key structure that escapes recognition by MGMT can potentially be applied to cancer chemotherapy, since recent clinical trials have been undertaken to test DNA repair inhibitors that target PARP, BER, or MGMT in combination with alkylating agents [38]. In the case of OBG, a phase I clinical trial has defined the maximum tolerated dose of a single dose of TMZ when combined with OBG and has determined the dose of OBG that depletes tumor MGMT activity for 48 h [39]. Since IPMS causes MGMT-resistant O6-isopropyl dG and potential BER substrates such as N7-isopropyl dG and N3-isopropyl dA, co-treatment of IPMS and PARP inhibitor could cause synthetic lethality in HR-deficient tumor cells due to accumulation of DSBs caused by O6-isopropyl dG and induced by PARP1-DNA strand break complex during DNA replication. Therefore, incorporation of the isopropyl moiety may be worthwhile to test in future clinical trials, or test as a cancer chemotherapeutic approach, although the success of such approaches will of course depend on selective targeting of the tumor.
Overall, our findings in the present study reveal that IPMS-induced DNA damage escapes recognition by MGMT and generates DSBs. This leads to an activation of cell cycle checkpoints and results in FANC-mediated DSB repair.
MATERIALS AND METHODS
Chemicals
Isopropyl methanesulfonate (IPMS), ethyl methanesulfonate (EMS), methyl methanesulfonate (MMS), 2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino) carbonyl]-2H-tetrazolium hydroxide (XTT), 1-methoxy-5-methylphenazinium methyl sulfate (PMS), O6-benzylguanine and dimethylsulfoxide (DMSO) were obtained from Sigma. Propyl methanesulfonate (nPMS) was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Dulbecco’s phosphate buffered saline (PBS) was obtained from Life Technologies (Grand Island, NY, USA). All of the chemicals were dissolved in PBS, except XTT which was dissolved in DMSO.
DT40 cell culturing and maintenance
Fetal bovine serum (FBS) and penicillin/streptomycin were obtained from Atlanta Biologicals (Norcross, GA, USA) and Sigma-Aldrich (St. Louis, MO, USA), respectively. RPMI 1640 culture medium (+glutamine, –phenol red) and chicken serum (CS) were acquired from Life Technologies (Grand Island, NY, USA). FBS and CS were heat inactivated at 56°C for 30 min. DT40 cells were maintained as described in our previous report [13]. The DT40 cell line and its isogenic mutants were knocked out in specific DNA repair pathways (Supplementary Table S1).
Generation of FANCD2 knockout TK6 cell lines and cell cultivation
To disrupt the FANCD2 gene, the guide RNA sequence (5’-TTTGGAAAGTGGTTGCTTCC-3’) was designed against exon 2 and cloned into the pX330 (Addgene) plasmid after digestion with BbsI. To generate the targeting vectors, the left and right arms were amplified by polymerase chain reaction (PCR) using 5’-GCGAATTGGGTACCGGGCCCATTGAGTCTCAAATTTGGT-3’/5’-CTGGGCTCGAGGGGGGGCCAATATGGCAAATAGTAAAGG-3’ (for the left arm), and 5’-TGGGAAGCTTGTCGACTTAACAGAATCAACTAGGTAATAT-3’/5’-ACTAGTAGGCGCGCCTTAAGCTTCTTTTGGAAAGCTATT-3’ (for the right arm), respectively. The amplified fragments of the left and right arms were assembled by Seamless Cloning (A14606, Invitrogen) into the DT-ApA/NEOR (provided from the Laboratory for Animal Resources and Genetic Engineering, Center for Developmental Biology, RIKEN Kobe, http://www.cdb.riken.jp/arg/cassette.html) and DT-ApA/PUROR cassettes [40] which were digested with ApaI and AflII. The single underlines seen above indicate the homology of upstream and downstream from ApaI site. The double underlines above indicate the homology upstream and downstream from AflII site. The gene disruption was confirmed via immunoblot using an anti-FANCD2 antibody (sc-2022, Santa Cruz). β-actin (A5316, Sigma) was used as a loading control. TK6 cells were maintained in RPMI 1640 culture medium (+glutamine, –phenol red) supplemented with FBS (10%) and penicillin/streptomycin.
DT40 cell-based DNA damage response (DDR) assay
For the DDR assay, approximately 1,200 cells were seeded in each well of a 96-well plate in 75 μL of medium. The wild-type and mutant cells were seeded in one plate, and the cells were treated with IPMS, MMS or EMS in duplicate wells for each chemical. The outside 2 wells were used as blank controls. These plates were kept in an incubator at 39.5°C until the start of treatment. Separately, each chemical was serially diluted into eight concentrations. 8.35 µL of the chemical solutions or PBS (as a solvent control, 2 wells/row) were added to 2 rows of a 96-well plate. A preliminary dose finding test was conducted in DT40 wild type cells to select the appropriate concentration range, which included at least one concentration showing severe cytotoxicity and one concentration showing no cytotoxicity at a lower concentration.
After the chemical solutions were added, plates were incubated for at least 48 hours after which they were intermittently observed microscopically to determine the growth of the cells. Once the control cells covered 1/3 or more of a well bottom, cells were exposed to the XTT cocktail. The plates were returned to the incubator until the dye developed sufficient color for detecting absorbance on the plate reader, which normally takes 2-5 hours. Absorbance was measured at 450 nm with a reference of 620 nm using a Tecan Sunrise (Tecan Systems, San Jose, CA, USA) plate reader with XFluorTM software (Tecan, version 6.4). All data are summarized as the means of experiments done at least in triplicate.
Micronucleus assay
For the micronucleus assay, flow cytometry-based MN measurements were performed using an In Vitro MicroFlow™ Kit (Litron Laboratories, Rochester, NY, USA) according to the manufacturer’s instructions [41]. Briefly, 150 µL of DT40 cells adjusted to 2 × 105 cells/ml were seeded into 96-well plates at 39°C and 5% CO2 in a humid atmosphere. Chemical solutions were separately prepared, and 16.7 µL of the solutions were added to each well. Cells were cultured at 39°C for 16 hours, which corresponds to approximately two cell cycles. After treatment, the plate was centrifuged to collect cells, and the treatment medium was discarded. The plate was then placed on ice and incubated for 20 min, and 50 μL of Nucleic Acid DyeA working solution was added to each well. A light source was placed approximately 15 cm above the samples for 30 min. Subsequently, the samples were washed once with cold 1x Buffer Solution. Next, 100 μL of Complete Lysis Solution 1 (including Nucleic Acid DyeB) were added to each well, and the samples were incubated for 1 hour in the dark at 37°C. Subsequently, 100 μL of Complete Lysis Solution 2 (including Nucleic Acid DyeB) were added to each well, and the samples were incubated for 30 min in the dark at room temperature.
Samples for the MN assay were analyzed by flow cytometry (FACS Calibur or LSRII, BD Biosciences) using an excitation wavelength of 488 nm. The frequency of MN was determined by acquiring at least 10,000 gated nuclei per sample. The data is summarized as a mean of two or more independent experiments. The extent of survival in the treated cultures was evaluated as the reduction in population doubling (PD). This is described as relative population doubling (RPD), which is calculated using the following formula:
(Number of PD in treated cultures)/(Number of PD in control cultures) × 100,
where PD is calculated as
[log(post-treatment cell number/initial cell number)]/log 2.
Pretreatment with O6-benzylguanine
Cells were pretreated with O6-benzylguanine (OBG) and used for both the DDR and MN assays to see if MGMT inhibition changes the cellular response. Prior to treatment, cells were pre-incubated with 10 µM of OBG for 2 hours and exposed to chemicals. No additional OBG was supplemented during the exposure.
γH2AX assay
To determine the percentage of γH2AX-positive cells, IPMS-treated cells were harvested and stained with a FITC conjugated γH2AX antibody using an H2A.X phosphorylation Assay Kit for Flow Cytometry (Millipore Corporation, CA, USA) according to the manufacturer’s instructions. Briefly, 1 × 106 cells were exposed to chemicals and harvested after a 1, 2, 4, 8 or 16 hour treatment period. Cells were washed once with PBS and resuspended in 200 µL of 1X Fixation solution and stored on ice for 20 minutes. After fixation, cells were washed with PBS and resuspended in 100 µL of 1X Permeabilization solution, in which 3.5 µL of anti-phospho-Histone H2A.X (Ser139) was added. After incubation on ice for 20 minutes, 100 µL of 1X Wash solution was added. The cells were harvested by centrifugation and resuspended in 200 µL of PBS. The samples were analyzed by flow cytometry (FACS Calibur, BD Biosciences), and the data was analyzed by FlowJo (Ver. X, Tree Star, Inc.).
Immunofluorescence
TK6 cells (0.5 million/ml) were exposed to IPMS (750µM) for 24 hours. Cells were then fixed using 2% formaldehyde / PBS for 20 min, washed in PBS, and spotted on to double frosted microscopic glass slides (Fisher Scientific) at a concentration of 2 million/ml. Blocking was achieved using 5% (w/v) bovine serum albumin (BSA) in PBS-TT for 30 min. Cells were then incubated with 1:500 rabbit polyclonal anti-53BP1 antibody (Bethyl Laboratories, Montgomery, TX) and mouse γH2AX antibody (Millipore Corporation, CA, USA) in 1% BSA/PBS-TT for overnight at 4oC. Cells were then washed in PBS, incubated in 1:250 anti-mouse Alexa Fluor 555 (Life Technologies) and Alexa Fluor 488 antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) for 1h. After washing with PBS, slides were mounted with a cover slip using Vectashield with DAPI (Vector Laboratories) and sealed using nail polish. Slides were analyzed using confocal microscopy (Zeiss CLSM 700). Optical sections through the nuclei were captured at 0.5 µm intervals, and the images were obtained by maximum projection of the individual sections.
Data and statistical analysis
For DDR assay data sets, we calculated the lethal concentration 50 (LC50) values for each cell line using Graphpad Prism 5 (La Jolla, CA, USA). The obtained LC50 in each mutant was compared against the LC50 in wild-type cells.
In order to represent the point of departure values for concentration-response in the MN assay in wild-type and FANC-deficient cells, the benchmark dose (BMD) values (U.S.EPA/100/R-12/001 2012) were obtained using the EPA BMD software v 2.5. These values represent a one standard deviation departure from control values, and the BMDL refers to the corresponding lower limit of a one-sided 95% confidence interval on the BMD. A log10 (concentration+0.5) was used to avoid taking logarithms of zero control values. In each instance, the best fitting result from Hill, exponential, and polynominal response non-constant variance models was used.
Fisher’s exact test was used to examine whether the micronucleus frequency differed between the OBG pretreated cells and untreated cells following IPMS or nPMS treatment. The results were determined to be statistically significant when the P-value was less than 0.05.
ACKNOWLEDGMENTS
The authors thank Julian Sale, Hiroshi Arakawa, Takemi Enomoto, Masayuki Seki, Noritaka Adachi, Hideki Koyama, Douglas Bishop, Keizo Tano, Ken-ichi Yamamoto and Masahiko Kobayashi for providing DT40 mutant cells.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work was supported by the Center for Environmental Health and Susceptibility grant NIEHS P30-ES10126, Superfund Basic Research Program grant NIEHS P42-ES05948.
REFERENCES
1. Elder D, Facchine KL, Levey JN, Parsons R, Ridge D, Semo L and Teasdale A. An approach to control strategies for sulfonate ester formation in pharmaceutical manufacturing based on recent scientific understanding. Organic Process Research & Development. 2012; 16: 1707-1710.
2. Adler ID, Tinwell H, Kliesch U and Ashby J. Activity of iPMS and nPMS in mouse bone marrow micronucleus assays: comparison with mouse dominant lethal assay data. Mutation Research. 1996; 349: 241-247.
3. Coffing SL, Kenyon MO, Ackerman JI, Shutsky TJ and Dobo KL. Evaluation of the In Vivo Mutagenicity of Isopropyl Methanesulfonate in Acute and 28-Day Studies. Environmental and Molecular Mutagenesis. 2015; 56: 322-332.
4. Ehling UH and Neuhäuser-Klaus A. Induction of specific-locus and dominant lethal mutations in male mice by n-propyl and isopropyl methanesulfonate. Mutation Research. 1995; 328: 73-82.
5. Eder E, Kütt W and Deininger C. On the role of alkylating mechanisms, O-alkylation and DNA-repair in genotoxicity and mutagenicity of alkylating methanesulfonates of widely varying structures in bacterial systems. Chemico-Biological Interactions. 2001; 137: 89-99.
6. Glowienke S, Frieauff W, Allmendinger T, Martus HJ, Suter W and Mueller L. Structure-activity considerations and in vitro approaches to assess the genotoxicity of 19 methane-, benzene- and toluenesulfonic acid esters. Mutation Research. 2005; 581: 23-34.
7. Mattison JD, Penrose LB and Burlinson B. Preliminary results of ethylnitrosourea, isopropyl methanesulphonate and methyl methanesulphonate activity in the testis and epididymal spermatozoa of Muta Mice. Mutation Research. 1997; 388: 123-127.
8. Hakura A, Ninomiya S, Kohda K and Kawazoe Y. Studies on Chemical Carcinogens and Mutagens. XXVI. Chemical properties and mutagenicity of alkyl alkanesulfonates on Salmonella typhimurium TA100. Chemical & pharmaceutical bulletin. 1984; 32: 3626-3635.
9. Segal A, Seidman I and Melchionne S. Induction of thymic lymphomas and squamous cell carcinomas following topic application of isopropyl methanesulfonate to female Hsd:(ICR)BR mice. Cancer Research. 1987; 47: 3402-3405.
10. Hoppe H 4th, Skopeck TR, Liber HL and Thilly WG. Alkyl methane sulfonate mutation of diploid human lymphoblasts and Salmonella typhimurium. Cancer Research. 1978; 38: 1595-1600.
11. Li F, Solomon JJ, Mukai F and Segal A. In vitro reactions of isopropyl methanesulfonate with DNA and with 2’-deoxyribonucleosides. Cancer Biochemistry Biophysics. 1990; 11: 253-264.
12. Ridpath JR, Nakamura A, Tano K, Luke AM, Sonoda E, Arakawa H, Buerstedde JM, Gillespie DA, Sale JE, Yamazoe M, Bishop DK, Takata M, Takeda S, et al. Cells deficient in the FANC/BRCA pathway are hypersensitive to plasma levels of formaldehyde. Cancer Research. 2007; 67: 11117-11122.
13. Ridpath JR, Takeda S, Swenberg JA and Nakamura J. Convenient, multi-well plate-based DNA damage response analysis using DT40 mutants is applicable to a high-throughput genotoxicity assay with characterization of modes of action. Environmental and Molecular Mutagenesis. 2011; 52: 153-160.
14. Ji K, Kogame T, Choi K, Wang X, Lee J, Taniguchi Y and Takeda S. A novel approach using DNA-repair-deficient chicken DT40 cell lines for screening and characterizing the genotoxicity of environmental contaminants. Environmental Health Perspectives. 2009; 117: 1737-1744.
15. Baba TW, Giroir BP and Humphries EH. Cell lines derived from avian lymphomas exhibit two distinct phenotypes. Virology. 1985; 144: 139-151.
16. Yamamoto KN, Hirota K, Kono K, Takeda S, Sakamuru S, Xia M, Huang R, Austin CP, Witt KL and Tice RR. Characterization of Environmental Chemicals With Potential for DNA Damage Using Isogenic DNA Repair-Deficient Chicken DT40 Cell Lines. Environmental and Molecular Mutagenesis. 2011; 52: 547-561.
17. Attene-Ramos MS, Miller N, Huang R, Michael S, Itkin M, Kavlock RJ, Austin CP, Shinn P, Simeonov A, Tice RR and Xia M. The Tox21 robotic platform for the assessment of environmental chemicals-from vision to reality. Drug Discovery Today. 2013; 18: 716-723.
18. Vogel EW and Nivard MJ. International Commission for Protection Against Environmental Mutagens and Carcinogens. The subtlety of alkylating agents in reactions with biological macromolecules. Mutation Research. 1994; 305: 13-32.
19. Goth R and Rajewsky MF. Persistence of O6-ethylguanine in rat-brain DNA: correlation with nervous system-specific carcinogenesis by ethylnitrosourea. Proceedings of the National Academy of Sciences of the United States of America. 1974; 71: 639-643.
20. Kaina B, van Zeeland AA, de Groot A and Natarajan AT. DNA repair and chromosomal stability in the alkylating agent-hypersensitive Chinese hamster cell line 27-1. Mutation Research. 1990; 243: 219-224.
21. Kaina B, Christmann M, Naumann S and Roos WP. MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair; 2007; 6: 1079-1099.
22. Sega GA, Owens JG and Cumming RB. Studies on DNA repair in early spermatid stages of male mice after in vivo treatment with methyl-, ethyl-, propyl-, and isopropyl methanesulfonate. Mutation Research. 1976; 36: 193-212.
23. Morimoto K, Dolan ME, Scicchitano D and Pegg AE. Repair of O6-propylguanine and O6-butylguanine in DNA by O6-alkylguanine-DNA alkyltransferases from rat liver and E. coli. Carcinogenesis. 1985; 6: 1027-1031.
24. Pegg AE, Dolan ME, Scicchitano D and Morimoto K. Studies of the repair of O6-alkylguanine and O4-alkylthymine in DNA by alkyltransferases from mammalian cells and bacteria. Environmental Health Perspectives. 1985; 62: 109-114.
25. Dolan ME, Morimoto K and Pegg AE. Reduction of O6-alkylguanine-DNA alkyltransferase activity in HeLa cells treated with O6-alkylguanines. Cancer Research. 1985; 45: 6413-6417.
26. Roos WP, Nikolova T, Quiros S, Naumann SC, Kiedron O, Zdzienicka MZ and Kaina B. Brca2/Xrcc2 dependent HR, but not NHEJ, is required for protection against O(6)-methylguanine triggered apoptosis, DSBs and chromosomal aberrations by a process leading to SCEs. DNA Repair. 2009; 8: 72-86.
27. Houghtaling S, Newell A, Akkari Y, Taniguchi T, Olson S and Grompe M. Fancd2 functions in a double strand break repair pathway that is distinct from non-homologous end joining. Hum Mol Genet. 2005; 14: 3027-3033.
28. Pegg AE and Byers TL. Repair of DNA containing O6-alkylguanine. FASEB Journal. 1992; 6: 2302-2310.
29. Yamamoto K, Ishiai M, Matsushita N, Arakawa H, Lamerdin JE, Buerstedde JM, Tanimoto M, Harada M, Thompson LH and Takata M. Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Molecular and Cellular Biology. 2003; 23: 5421-5430.
30. Nakanishi K, Yang YG, Pierce AJ, Taniguchi T, Digweed M, D’Andrea AD, Wang ZQ and Jasin M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 1110-1115.
31. Chen HT, Bhandoola A, Difilippantonio MJ, Zhu J, Brown MJ, Tai X, Rogakou EP, Brotz TM, Bonner WM, Ried T and Nussenzweig A. Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science. 2000; 290: 1962-1965.
32. Margison GP and Santibáñez-Koref MF. O6-alkylguanine-DNA alkyltransferase: role in carcinogenesis and chemotherapy. BioEssays. 2002; 24: 255-266.
33. Kaina B, Fritz G and Coquerelle T. Contribution of O6-alkylguanine and N-alkylpurines to the formation of sister chromatid exchanges, chromosomal aberrations, and gene mutations: new insights gained from studies of genetically engineered mammalian cell lines. Environmental and Molecular Mutagenesis. 1993; 22: 283-292.
34. Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol. 2016; 17: 337-49.
35. Geng L, Huntoon CJ and Karnitz LM. RAD18-mediated ubiquitination of PCNA activates the Fanconi anemia DNA repair network. The Journal of Cell Biology. 2010; 191: 249-57.
36. Inagaki A, Sleddens-Linkels E, van Cappellen WA, Hibbert RG, Sixma TK, Hoeijmakers JH, Grootegoed JA and Baarends WM. Human RAD18 interacts with ubiquitylated chromatin components and facilitates RAD9 recruitment to DNA double strand breaks. PLoS One. 2011; 6: e23155.
37. Fenech M, Kirsch-Volders M, Natarajan AT, Surralles J, Crott JW, Parry J, Norppa H, Eastmond DA, Tucker JD and Thomas P. Molecular mechanisms of micronucleus, nucleoplasmic bridge and nuclear bud formation in mammalian and human cells. Mutagenesis. 2011; 26: 125-132.
38. Helleday T, Petermann E, Lundin C, Hodgson B and Sharma RA. DNA repair pathways as targets for cancer therapy. Nature Reviews Cancer. 2008; 8: 193-204.
39. Quinn JA, Desjardins A, Weingart J, Brem H, Dolan ME, Delaney SM, Vredenburgh J, Rich J, Friedman AH, Reardon DA, Sampson JH, Pegg AE, Moschel RC, et al. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. Journal of Clinical Oncology. 2005; 23: 7178-7187.
40. Keka IS, Mohiuddin, Maede Y, Rahman MM, Sakuma T, Honma M, Yamamoto T, Takeda S, Sasanuma H. Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Res. 2015; 43:6359-72.
41. Bryce SM, Avlasevich SL, Bemis JC, Lukamowicz M, Elhajouji A, Van Goethem F, De Boeck M, Beerens D, Aerts H, Van Gompel J, Collins JE, Ellis PC, White AT, et al. Interlaboratory evaluation of a flow cytometric, high content in vitro micronucleus assay. Mutation Research. 2008; 650: 181-195.