
INTRODUCTION
Telomeres protect chromosomal ends and are comprised of tracts of G-rich nucleotide repeats bound by a protein complex, shelterin, containing TRF1 and TRF2 (telomeric repeat binding factors 1 and 2), POT1 (protection of telomeres 1), TIN2 (TRF1-interacting protein 2), TPP1 (POT1–TIN2 organizing protein, also known as ACD) and RAP1 (repressor-activator protein 1) [1, 2].
The protective function of telomeres requires a sufficient amount of telomeric repeats and integrity of the shelterin complex. Telomere shortening due to incomplete replication of telomeric DNA, as well as shelterin defects, result in telomere dysfunction, which can lead to chromosomal and genomic instability. However, cells with dysfunctional telomeres are removed via senescence or apoptosis, and the p53 protein, a guardian of genomic integrity, regulates this physiological response. hTERT (human telomerase reverse transcriptase) is a key component of telomerase, which can regenerate and stabilize shortened telomeres, enabling unlimited cell proliferation. Telomere dysfunction, p53 deficiency and hTERT activation cooperate in oncogenesis, promoting genetically unstable and immortal tumor cell clones [3–5].
In chronic lymphocytic leukemia (CLL), telomere shortening and hTERT expression are generally observed in patients with advanced disease and/or high-risk biological features, such as unmutated immunoglobulin heavy chain variable (IGHV) genes, but some early-stage patients can also display short telomeres and high hTERT. Telomere shortening and hTERT expression have been also related to poor outcomes [6–10]. The shelterin complex is globally dysregulated at the transcriptional level [11, 12] and the presence of dysfunctional telomeres has been correlated with a downregulation of shelterin genes [11, 13], but the relationship between shelterin status and the clinico-biological parameters and outcomes of CLL remain unknown.
Previous reports suggest that CLL patients with TP53 loss, due to del(17p), may show excessive telomere shortening compared to other cytogenetic subgroups [12, 14–16]. Deletion of 17p and also TP53 mutations in the absence of del(17p) are found in 10–15% of untreated CLL patients, and represent the strongest adverse prognostic factor to predict early resistance to standard genotoxic agents and a poor outcome [17, 18]. Given the high prognostic significance of TP53 and telomere status and their cooperation in oncogenesis, we investigated the potential link between TP53 gene alterations and impaired telomeres in CLL. We performed a comprehensive study of deletions and mutations of the TP53 gene and of telomere parameters, including assessment of telomeres as well as hTERT and shelterin gene expression in 115 CLL patients.
RESULTS
TP53 mutations and deletions are associated with short telomeres and hTERT overexpression
TP53 mutations were found in 20 patients (17.4%): of these, 10 also had a del(17p). Four additional cases had a del(17p) only. We found that telomere length and hTERT expression did not differ between patients with a del(17p) and those carrying a TP53 mutation in the absence of a 17p deletion (Figure 1A-1B). In contrast, telomere length in these cases was considerably shorter and hTERT expression was higher compared to that in patients with wild-type (wt) TP53. In further analyses, we considered TP53 mutated and/or deleted cases as a single TP53-disrupted subgroup (n=24).
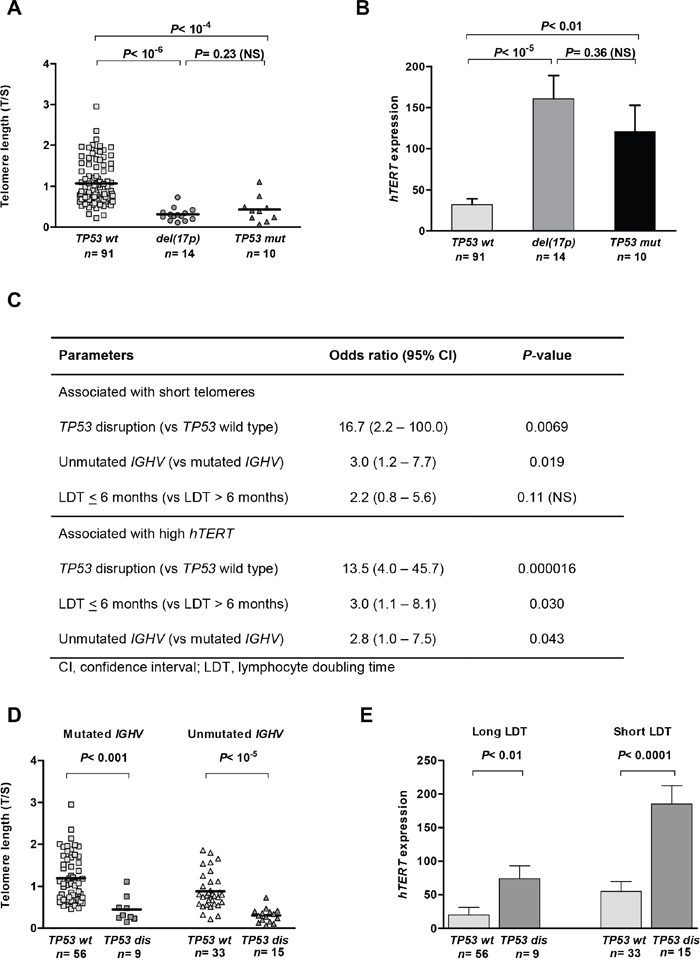
Figure 1: TP53 loss and mutation are equally associated with telomere shortening and hTERT overexpression in chronic lymphocytic leukaemia (CLL). Mean telomere length A. and hTERT expression B. in patients with wild-type (wt) TP53 and in patients with either del(17p) or TP53 mutations (mut) in the absence of del(17p) (Kruskal–Wallis H test). The impact of TP53 disruption (dis) was independent of IGHV status and lymphocyte doubling time (LDT), as shown in the multiple logistic regression, with an association between shorter (<0.925, mean) telomeres and higher (>55.5, mean) hTERT C. This is illustrated by comparing telomere lengths in patients with mutated and unmutated IGHV D. and hTERT expression to patients with long and short LDTs E. Bars correspond to the standard error of the mean.
We then investigated the impact of TP53 status on telomeres in relation to other adverse biological features of CLL, which are known to correlate with telomere length and hTERT expression [14, 19]. In our cohort, short telomere length was, as expected, associated with unmutated IGHV genes (P= 0.00096) and short lymphocyte doubling time (LDT; P= 0.00083). hTERT overexpression was found in patients with unmutated IGHV (P= 0.0032) and short LDTs (P= 0.0016). Although all these characteristics increased the risk of having short telomere length and high hTERT expression, TP53 disruption had the strongest impact in the multiple logistic regression analyses (Figure 1C). This is illustrated in Figure 1D, showing that telomere length was significantly shorter in patients with a TP53-disrupted status compared to a TP53 wild-type status, irrespective of the IGHV mutational status. Similarly, hTERT expression was significantly higher in patients with TP53 disruption compared to those with a wild-type TP53 status in the subgroups that had either a long or a short LDT (Figure 1E).
We also compared telomere lengths and hTERT-expression levels between cytogenetic CLL subgroups. There was gradual and significant telomere shortening, and an increase in hTERT from low-risk subgroups to a TP53-disrupted subgroup (Supplementary Figure S1A-S1B). In addition, the mean number of karyotype aberrations increased gradually and was greatest in patients with disrupted TP53 (Supplementary Figure S1C).
Telomere abnormalities in metaphase from TP53-disrupted cases
We performed pantelomeric FISH analysis on six TP53-disrupted cases and six TP53 wild-type cases (Figure 2). The mean number of telomere signal losses, representative of severe telomere erosion (Figure 2B), was significantly higher in TP53-disrupted than in wild-type TP53 cases (5.6 vs 1.1, respectively, t-test, P <0.01). In contrast, no difference was observed in the frequency of multiple telomeric signals, which are markers of telomere replication defects: 0.7 in TP53-disrupted cases vs 0.65 in wild-type TP53 cases, NS. In addition, nine out of 24 TP53-disrupted cases had complex karyotypes, and four of these showed telomeric fusions (Figure 2B), which are characteristic of extreme telomere erosion, thus highlighting the association between telomeric and chromosomal instability.
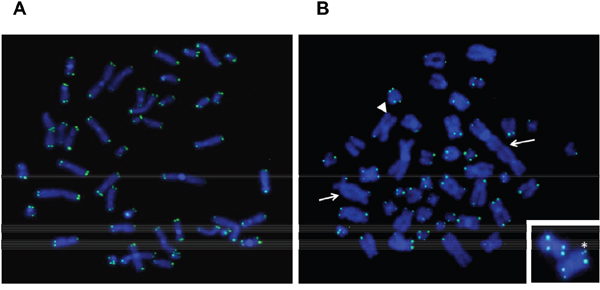
Figure 2: Pantelomeric FISH of representative metaphases from wild-type (wt) TP53 A. and disrupted TP53 B. TP53 disruption was associated with telomeric fusions that led to dicentric chromosomes (arrows) and frequent loss of telomeric signals (one of them is shown with an arrowhead). A duplication of a telomeric signal is shown with an asterisk.
Low shelterin gene expression in patients with TP53 disruption
We first performed unsupervised hierarchical clustering analysis that included TP53 status and telomere characteristics: i.e. shelterin and hTERT gene-expression levels and relative telomere length (Figure 3A). This approach identified three distinct groups. Cluster #1 (n= 34) was characterized by shortened telomeres, high hTERT and low shelterin gene expression. This cluster included 22 of the 24 patients with TP53 disruption. Interestingly, we also observed a smaller group of patients with wild-type TP53 and without hTERT overexpression and telomere shortening, but with downregulated shelterin genes (cluster #2, n= 24). Finally, there was a large group (cluster #3, n= 55) that had heterogeneous telomere lengths, low hTERT and relatively high levels of shelterin gene expression. Overall, TP53 disruption was clearly associated with low shelterin gene expression, but a similar downregulation was also found in a proportion of wild-type TP53 cases.
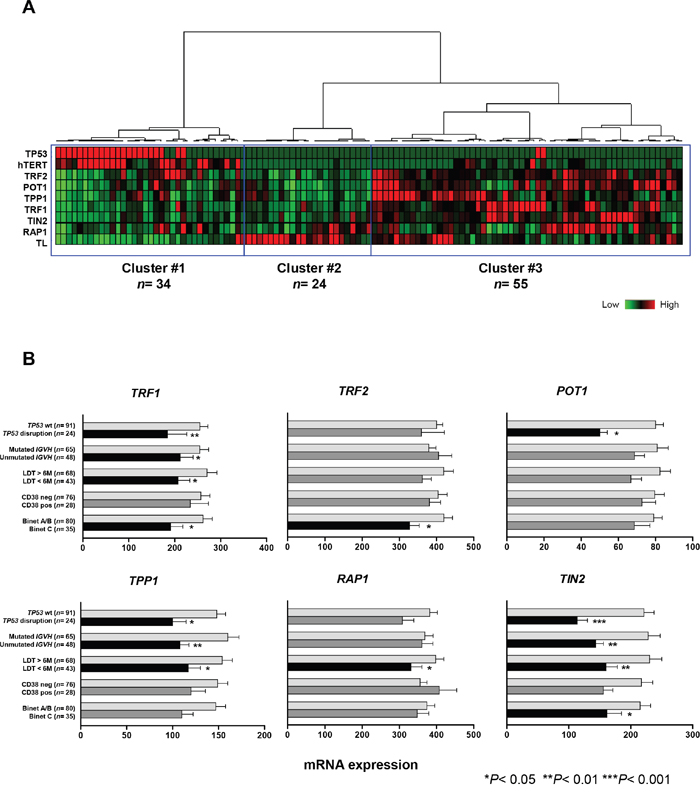
Figure 3: Low shelterin-gene expression in CLL patients with disrupted TP53 and in patients with adverse prognostic factors. A. Hierarchical clustering of 115 CLL patients according to TP53 status, telomere length and expression levels of hTERT and shelterin genes (TRF1, TRF2, POT1, TPP1, RAP1, TIN2). Patients with disrupted TP53 (red) are clustered into a distinct group with short telomeres, high hTERT and low levels of shelterin genes. B. Mean expression levels of shelterin genes in different CLL subgroups according to TP53 status, IGHV mutation profile, CD38 expression, lymphocyte doubling time (LDT) and Binet stage (H test). Bars correspond to the standard error of the mean.
The expression of shelterin genes has not yet been correlated with established CLL prognostic factors; we performed this analysis in our series (Figure 3B). Significant downregulation of several shelterin genes were found in patients in Binet C stage (TRF1, TRF2 and TIN2). A short LDT was associated with significant downregulation of all the shelterin genes except for TRF2 and POT1. Expression of TRF1, TPP1 and TIN2 was lower in cases with unmutated IGHV. TP53-disruption was also significantly associated with lower expression of TRF1, POT1, TPP1 and TIN2. This was independent of the type of TP53 alteration (data not shown).
Regarding the correlation with cytogenetic subgroups, the changes to shelterin gene expression were not consistent, except from TIN2, which showed highly significant gradual reduction from a normal/del(13q) pattern to a TP53-disruption subgroup, in parallel with telomere shortening (Supplementary Figure S2).
Prognostic value of telomere length and shelterin gene expression in treated patients with wild-type TP53
As expected, survival analyses in treated patients confirmed the significantly shorter progression-free survival (PFS) of patients with disrupted TP53 compared to patients with wild-type TP53 (P= 0.0033; Figure 4A). In addition, telomere length, hTERT and three shelterin-gene (POT1, TPP1, TIN2) transcript levels were also significant predictors of PFS (data not shown), but these parameters were strongly correlated with TP53 status. We therefore investigated whether these telomere characteristics had an additional prognostic value beyond classifying patients into TP53-disrupted or TP53-wild-type subgroups. We performed survival analysis among patients with wild-type TP53 and found that short telomeres (P= 0.00047, Figure 4B), low POT1 (P= 0.0039; Figure 4C), low TPP1 (P= 0.0047; Figure 4D) and low TIN2 (P= 0.0051; Figure 4E) identified subsets of patients with significantly shorter survival times. These parameters were also associated with a higher risk of progression in the univariate Cox’s analysis (Figure 4F). A multivariate Cox’s analysis of known poor prognostic CLL features (IGHV status, LDT, CD38, Binet stage) showed that unmutated IGVH genes significantly and independently predicted PFS in this patient subgroup with the hazard ratio of 2.2 (95% CI: 1.0 to 4.3, P= 0.022). When adjusted for IGHV mutation status and telomere length, shelterin transcript levels remained significant predictors of PFS in patients with wild-type TP53 (Figure 4F).
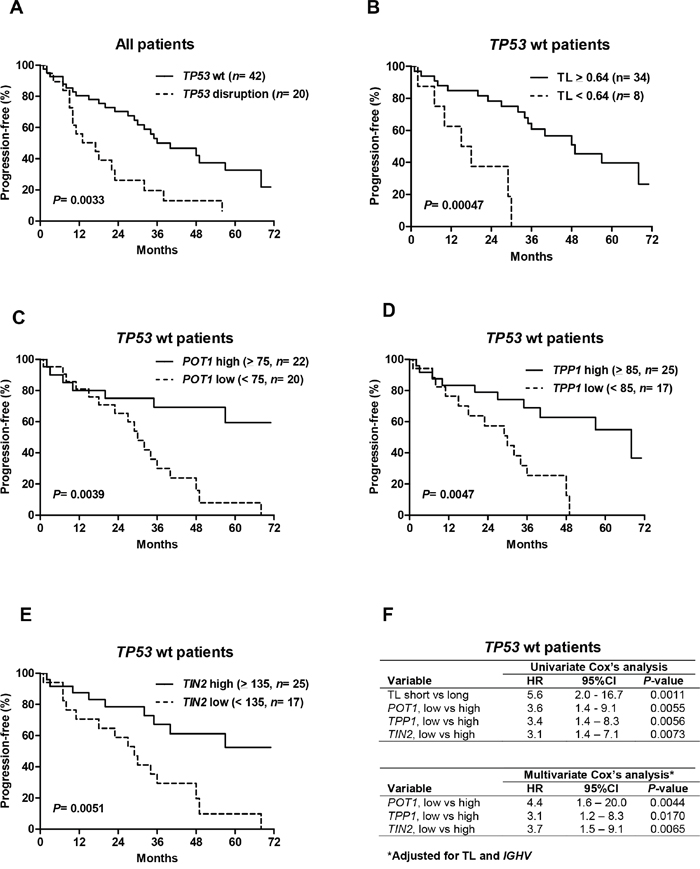
Figure 4: Telomere length and shelterin expression predict progression-free survival (PFS) in patients with wild-type (wt) TP53. A. Kaplan–Meier estimates of PFS in the whole patient population as a function of TP53 status and the wild-type TP53 cohort according to telomere length B. POT1 C. TPP1 D. and TIN2 E. levels. The optimal cut-off values were determined using recursive partitioning. CLL patients with short telomeres and low POT1, TPP1 and TIN2 levels had significantly shorter PFSs. F. Corresponding hazard ratios (HR) and 95%-confidence intervals (95%CI) are presented. When adjusted for telomere length and IGHV mutation profile, low POT1, TPP1 and TIN2 expression remained significant predictors for a shorter PFS.
DISCUSSION
We have shown that CLL patients with disrupted TP53 have severe telomere shortening associated with hTERT overexpression and downregulation of shelterin genes. This telomere profile was found in patients with TP53 mutations in the absence of del(17p) and in patients with concomitant TP53 mutations and del(17p) or del(17p) alone: this suggests that any alteration to TP53 leads to important telomere dysfunction in CLL.
Both TP53 alteration and short telomeres have been previously correlated with progressive CLL, and with unfavorable biological factors such as unmutated IGHV and a high proliferation rate [14, 19, 20]. We have demonstrated that the association of TP53 disruption with short telomeres and high hTERT was an independent factor in this adverse biological profile, and that TP53 was the strongest determinant of telomere status and hTERT expression. We have also shown that patients with disrupted TP53 had the shortest telomeres among the cytogenetic subgroups; this association has been previously described for a 17p deletion [16], but has not yet been reported for mutated TP53. Patients with disrupted TP53 also had the highest levels of hTERT among the subgroups.
Using pantelomeric FISH, we found that TP53 disruption was associated with increased numbers of telomeric deletions, and we also observed chromosomal end-to-end fusions in patients with disrupted TP53 and complex karyotypes. These findings illustrate that impaired p53 pathways can allow continuous cell division despite telomere shortening, and the accumulation of cells with critically eroded telomeres, thus leading to chromosomal fusions and genomic instability. In addition, wild-type p53 is a powerful inhibitor of hTERT, and loss of this function can also enhance expression of hTERT, which is essential for survival and replication of tumor cells [21]. Telomere attrition and TP53 disruption may thus represent two important cooperating events of tumor progression in CLL, as has been previously shown in epithelial tumor models [4, 22].
Telomere shortening in CLL patients with disrupted TP53 was also associated with low expression of TRF1, POT1, TPP1 and TIN2 shelterin genes. Using clustering analysis, we found that almost all cases of disrupted TP53 were within a distinct subset that had short telomeres and downregulated shelterin genes. The abnormal expression of shelterin genes, in the context of p53-deficiency and short telomeres, may have contributed to the higher levels of genomic instability in this CLL subset.
Clustering analyses indicated that some patients with wild-type TP53 also showed shelterin downregulation, but this low expression was independent of telomere shortening. Moreover, we also found significantly lower shelterin gene expression in cases with adverse clinical and biological parameters, such as advanced Binet stage, unmutated IGHV and short LDTs. Interestingly, shelterin status was not consistently correlated with the cytogenetic subgroups, with the exception of TIN2. These results highlight the heterogeneity of shelterin status in CLL and suggest the complexity that underlies these mechanisms.
The factors regulating shelterin genes remain unknown. Nevertheless, some events that could affect their function and expression in cancer, such as mutation and DNA methylation, have been described. Somatic mutations of POT1 have been reported in CLL, and this represents the first description of a shelterin complex-gene mutation in human cancer [23]. CLL cells with POT1 mutations display multiple telomeric and chromosomal aberrations, suggesting that these mutations and the subsequent alterations to shelterin can favor the acquisition of the malignant features seen in CLL cells.
In breast-cancer cell lines, a significant reduction in POT1 mRNA level was associated with POT1 promotor methylation [24]. Interestingly, in the clonal evolution of CLL, a POT1 mutation seems to precede the appearance of alterations to TP53 [26]. In addition, downregulation of shelterin genes can occur prior to telomere shortening in early CLL and so contribute to the reduction in telomere protection [13]. Sequential analyses of patients with low levels of gene expression of shelterin would be of interest to investigate the evolution of telomere status and the timing of TP53 appearance and other alterations, as well as the onset of chromosomal instability.
Finally, we found a significant adverse impact of short telomeres and low POT1, TPP1 and TIN2 on PFS of the CLL patients in our cohort, who had been treated mainly with standard genotoxic agents. This impact was found in the whole population and also in patients with wild-type TP53, emphasizing the additional prognostic value of these parameters. In particular, the significant impact of shelterin gene expression on survival in CLL has not been described previously. Of note, these shelterin components regulate telomerase recruitment via POT1, and TIN2 recruits the TPP1–POT1 complex, constituting a bridge between the different shelterin components [2]. Unlike POT1, mutated TIN2 and TPP1 have not been observed in tumors, but germline mutations of these genes have been reported in human genetic diseases leading to bone-marrow failure [26, 27]. Taken together, these observations suggest that POT1, TIN2 and TPP1 could be key-components in the shelterin complex implicated in telomere dysfunction and adverse outcomes in CLL.
Further studies of patients with downregulated shelterin genes are warranted for mechanistic understanding of the role of telomere abnormalities in CLL. In addition to TP53 disruption, these investigations have to take into account other recurrent CLL mutations with an emphasis on telomeres, such as altered ATM and SF3B1 genes and, as stated above, the POT1 gene. Mutations in the ATM and SF3B1 genes are known to be associated with shorter telomeres in CLL cells [28, 16]. ATM, a major DNA damage sensor, appears to play a complex role in telomere protection and length regulation trough interactions with the shelterin complex [29]. A possible telomeric role of the SF3B1 spliceosome protein was recently revealed by the transcriptomic study of SF3B1-mutated CLL cells, showing an overexpression of the TERC gene encoding telomerase RNA template, which could lead to aberrant telomerase regulation [30].
In conclusion, we have demonstrated that CLL patients with disrupted TP53 show severe dysfunction of telomeres, involving telomeric DNA, hTERT and shelterin. This telomeric dysfunction can contribute to enhanced genomic instability and, consequently, to treatment resistance in CLL. Further studies are required to evaluate the impact of TP53 in the context of other molecular alterations, in particular ‘multiple hit’ events described recently in resistant CLL [31]. Finally, this telomeric profile could be tested as a predictive marker in CLL patients treated with new non-genotoxic therapies.
MATERIALS AND METHODS
Patients and samples
After informed consent according to the Declaration of Helsinki, peripheral blood samples were collected from 115 CLL patients at the Clermont-Ferrand University Hospital (France) between 2007 and 2012. The patients’ characteristics at the time of sampling are shown in Supplementary Table S1. Diagnosis and treatment requirements were based on the guidelines of the International Workshop on Chronic Lymphocytic Leukemia [32]. A total of 62 patients required treatment, consisting of immunochemotherapy (fludarabine, cyclophosphamide, rituximab n= 44, other regimens n= 6), alemtuzumab (n= 9) or alkylating agent-based regimens (n= 3). Median follow-up time from the start of treatment was 41 months. Peripheral blood samples were obtained before treatment. Peripheral blood lymphocytes were isolated by Lymphoprep™. The proportion of tumor cells obtained was >85%.
Cytogenetic analyses
Analyses were performed at the time of sampling. Karyotype was performed after immuno-stimulation of cell cultures with CpG-oligonucleotide and interleukin 2. Chromosome preparation and staining were done according to standard protocols. Twenty metaphases were analyzed. Complex karyotype was defined as ≥3 abnormalities. Locus-specific fluorescence in situ hybridization (FISH) was performed on interphase nuclei and/or metaphases, following standard procedures and using specific probes (co-hybridized with a control probe) to detect deletions of 13q14.3, 11q22.3 and 17p13 (LSI D13S319, LSI ATM and LSI TP53). Trisomy 12 was studied using the centromeric probe D12Z3. All probes were from Vysis (Abbott Molecular, Rungis, France) and used in accordance with the manufacturer’s recommended procedures. At least 200 interphase nuclei were examined. The cut-off levels for the presence of aberrations were 7% of positive cells for deletions and 5% for trisomy 12. Patients were classified according to previously reported hierarchical patterns [31]. Pantelomeric FISH was performed with a Telomere PNA FISH Kit (DAKO, Glostrup, Denmark), used in accordance with the manufacturer’s recommended procedures. Results were recorded using an Axioplan2 imaging fluorescence microscope (ZEISS, Göttingen, Germany) fitted with appropriate filters, a CCD camera, and the digital-imaging software, Isis, v3.8.8 (Metasystems, Althussheim, Germany).
TP53 mutational analyses
Genomic DNA was extracted using a Nucleospin Blood L (Macherey-Nagel) kit. Mutation analyses of TP53 exons 2–11 were done by DNA direct sequencing using a 3130XL sequencer (Applied Biosystems, Villebon-sur-Yvette, France) and analyzed with Seqscape software. The genomic reference sequence used was IARC NC_000017 v9. The primers were designed to cover all coding exons and intron–exon boundaries. The characteristics and distributions of mutations were assessed using the IARC TP53 Mutation Database and the Ensembl Database. TP53 gene mutations found in our CLL cohort are detailed in Supplementary Table S2. Most were deleterious missense mutations located in the DNA-binding domain and have been previously observed in other cancer types.
Assessment of telomere length using quantitative PCR
Average telomere length was evaluated with quantitative real-time DNA-PCR in a LightCycler 480 System (Roche Diagnostics, Meylan, France) using SYBR Green I technology (SYBR Green Kit, Roche Diagnostics), as described elsewhere [15]. This method measures the relative telomere length as the T/S ratio between the template amounts of telomere repeat (T) and a reference single-copy gene (S) [33]. We used GAPDH as a reference gene. Using this approach, we have previously shown that the T/S ratio in tumor cells was proportional to average telomere length as assessed by classical telomere restriction-fragment analyses [15].
Quantitative RT-PCR for hTERT and shelterin complex gene expression
Total RNA was extracted using TRIzol (Fisher Scientific, Illkirch, France) according to standard procedures. Total RNA was converted to cDNA by reverse transcription using Superscript II reverse transcriptase (Invitrogen, Cergy-pontoise, France), according to the manufacturer’s instructions. The expression of hTERT and the shelterin complex genes TRF1, TRF2, POT1, TPP1, RAP1 and TIN2 were quantified using real-time RT-PCR in a LightCycler 480 System (Roche Diagnostics, Meylan, France), as described previously [11, 15]. The normalized copy numbers were expressed as the ratio between the numbers of transcript copies of the target and control (GUSB) genes, multiplied by 100.
Statistical analyses
Comparisons of quantitative variables were performed using standard tests. As is usual in exploratory studies, we chose not to adjust probabilities by the Bonferroni method. While decreasing the rate of false positives, this method also increases in a similar proportion the rate of false negatives. Unsupervised hierarchical clustering analysis was performed to identify subclasses with distinct expression of shelterin genes and telomeric-length profiles. Progression-free survival (PFS) was calculated from the date of starting treatment until a relapse, progression or death from any cause. Survival curves were established using the Kaplan–Meier method and compared with the log-rank test. The univariate and multivariate analyses was performed according Cox’s regression model. The data were analyzed using SEM software [34].
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005; 19:2100-2110.
2. Martínez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer. 2011; 11:161-176.
3. Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999; 97:527-538.
4. Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000; 406:641-645.
5. Pantic M, Zimmermann S, El Daly H, Opitz OG, Popp S, Boukamp P, Martens UM. Telomere dysfunction and loss of p53 cooperate in defective mitotic segregation of chromosomes in cancer cells. Oncogene. 2006; 25:4413-4420.
6. Damle RN, Batliwalla FM, Ghiotto F, Valetto A, Albesiano E, Sison C, Allen SL, Kolitz J, Vinciguerra VP, Kudalkar P, Wasil T, Rai KR, Ferrarini M et al. Telomere length and telomerase activity delineate distinctive replicative features of the B-CLL subgroups defined by immunoglobulin V gene mutations. Blood. 2004; 103:375-382.
7. Tchirkov A, Chaleteix C, Magnac C, Vasconcelos Y, Davi F, Michel A, Kwiatkowski F, Tournilhac O, Dighiero G, Travade P. hTERT expression and prognosis in B-chronic lymphocytic leukemia. Ann Oncol. 2004; 15:1476-1480.
8. Terrin L, Trentin L, Degan M, Corradini I, Bertorelle R, Carli P, Maschio N, Bo MD, Noventa F, Gattei V, Semenzato G, De Rossi A. Telomerase expression in B-cell chronic lymphocytic leukemia predicts survival and delineates subgroups of patients with the same igVH mutation status and different outcome. Leukemia. 2007; 21:965-972.
9. Lin TT, Norris K, Heppel NH, Pratt G, Allan JM, Allsup DJ, Bailey J, Cawkwell L, Hills R, Grimstead JW, Jones RE, Britt-Compton B, Fegan C et al. Telomere dysfunction accurately predicts clinical outcome in chronic lymphocytic leukaemia, even in patients with early stage disease. Br J Haematol. 2014; 167:214-223.
10. Strefford JC, Kadalayil L, Forster J, Rose-Zerilli MJJ, Parker A, Lin TT, Heppel N, Norris K, Gardiner A, Davies Z, Gonzalez de Castro D, Else M, Steele AJ et al. Telomere length predicts progression and overall survival in chronic lymphocytic leukemia: data from the UK LRF CLL4 trial. Leukemia. 2015; 29:2411-2414.
11. Poncet D, Belleville A, t’kint de Roodenbeke C, Roborel de Climens A, Ben Simon E, Merle-Beral H, Callet-Bauchu E, Salles G, Sabatier L, Delic J, Gilson E. Changes in the expression of telomere maintenance genes suggest global telomere dysfunction in B-chronic lymphocytic leukemia. Blood. 2008; 111:2388-2391.
12. Hoxha M, Fabris S, Agnelli L, Bollati V, Cutrona G, Matis S, Recchia AG, Gentile M, Cortelezzi A, Morabito F, Bertazzi PA, Ferrarini M, Neri A. Relevance of telomere/telomerase system impairment in early stage chronic lymphocytic leukemia. Genes Chromosomes Cancer. 2014; 53:612-621.
13. Augereau A, T’kint de Roodenbeke C, Simonet T, Bauwens S, Horard B, Callanan M, Leroux D, Jallades L, Salles G, Gilson E, Poncet D. Telomeric damage in early stage of chronic lymphocytic leukemia correlates with shelterin dysregulation. Blood. 2011; 118:1316-1322.
14. Roos G, Kröber A, Grabowski P, Kienle D, Bühler A, Döhner H, Rosenquist R, Stilgenbauer S. Short telomeres are associated with genetic complexity, high-risk genomic aberrations, and short survival in chronic lymphocytic leukemia. Blood. 2008; 111:2246-2252.
15. Véronèse L, Tournilhac O, Callanan M, Prie N, Kwiatkowski F, Combes P, Chauvet M, Davi F, Gouas L, Verrelle P, Guièze R, Vago P, Bay JO et al. Telomeres and chromosomal instability in chronic lymphocytic leukemia. Leukemia. 2013; 27:490-493.
16. Mansouri L, Grabowski P, Degerman S, Svenson U, Gunnarsson R, Cahill N, Smedby KE, Geisler C, Juliusson G, Roos G, Rosenquist R. Short telomere length is associated with NOTCH1/SF3B1/TP53 aberrations and poor outcome in newly diagnosed chronic lymphocytic leukemia patients. Am J Hematol. 2013; 88:647-651.
17. Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, Bühler A, Edelmann J, Bergmann M, Hopfinger G, Hensel M, Hallek M, Döhner H et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010; 28:4473-4479.
18. Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, Dearden CE, Richards SM, Catovsky D, Morgan GJ. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011; 29:2223-2229.
19. Rampazzo E, Bonaldi L, Trentin L, Visco C, Keppel S, Giunco S, Frezzato F, Facco M, Novella E, Giaretta I, Del Bianco P, Semenzato G, De Rossi A. Telomere length and telomerase levels delineate subgroups of B-cell chronic lymphocytic leukemia with different biological characteristics and clinical outcomes. Haematologica. 2012; 97:56-63.
20. Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S, De Paoli L, Spina V, Gattei V, Capello D, Forconi F, Lauria F, Gaidano G. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res. 2009; 15:995-1004.
21. Xu D, Wang Q, Gruber A, Björkholm M, Chen Z, Zaid A, Selivanova G, Peterson C, Wiman KG, Pisa P. Downregulation of telomerase reverse transcriptase mRNA expression by wild type p53 in human tumor cells. Oncogene. 2000; 19:5123-5133.
22. Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010; 31:9-18.
23. Ramsay AJ, Quesada V, Foronda M, Conde L, Martínez-Trillos A, Villamor N, Rodríguez D, Kwarciak A, Garabaya C, Gallardo M, López-Guerra M, López-Guillermo A, Puente XS, et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat Genet. 2013; 45:526-530.
24. Motevalli A, Yasaei H, Virmouni SA, Slijepcevic P, Roberts T. The effect of chemotherapeutic agents on telomere length maintenance in breast cancer cell lines. Breast Cancer Res Treat. 2014; 145:581-591.
25. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Böttcher S, Carter SL, Cibulskis K, Mertens D et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015; 526:525-530.
26. Dokal I. Dyskeratosis congenita. Hematol Educ Program Am Soc Hematol Am Soc Hematol Educ Program. 2011; 2011:480-486.
27. Guo Y, Kartawinata M, Li J, Pickett HA, Teo J, Kilo T, Barbaro PM, Keating B, Chen Y, Tian L, Al-Odaib A, Reddel RR, Christodoulou J et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood. 2014; 124:2767-2774.
28. Britt-Compton B, Lin TT, Ahmed G, Weston V, Jones RE, Fegan C, Oscier DG, Stankovic T, Pepper C, Baird DM. Extreme telomere erosion in ATM-mutated and 11q-deleted CLL patients is independent of disease stage. Leukemia. 2012; 26:826-830.
29. Diotti R, Loayza D. Shelterin complex and associated factors at human telomeres. Nucleus. 2011; 2:119-35.
30. Fan J, Wang L, Brooks AN, Wan Y, Neuberg DS, Rassenti LZ, Ghia EM, Kipps TJ, Brown JR, Li S, Livak KJ, Meyerson MM, Kharchenko PV, Wu CJ. Comprehensive bulk and single cell transcriptomic characterization of SF3B1 mutation reveals its pleiotropic effects in chronic lymphocytic leukemia. Blood. 2015; 126:2906-2906.
31. Guièze R, Robbe P, Clifford R, Guibert S de, Pereira B, Timbs A, Dilhuydy M-S, Cabes M, Ysebaert L, Burns A, Nguyen-Khac F, Davi F, Véronèse L et al. Presence of multiple recurrent mutations confers poor trial outcome of relapsed/refractory CLL. Blood. 2015; 126:2110-2117.
32. Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, Kipps TJ. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008; 111:5446-5456.
33. Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002; 30:e47.
34. Kwiatkowski F, Girard M, Hacene K, Berlie J. [Sem: a suitable statistical software adaptated for research in oncology]. Bull Cancer. 2000; 87:715-721.