
DRUG RESISTANCE
Multiple myeloma (MM) is the second most common hematologic malignancy characterized by the infiltration of monoclonal malignant plasma cells (MM cells) at multiple sites within the bone marrow (BM) compartment [1]. The pathophysiology of MM depends both on several oncogenic events occurring in MM cells, i.e. genomic/chromosomal instability, gene mutations and chromosomal translocations, and on extracellular factors, i.e. dynamic interactions between MM cells and the BM microenvironment (BMME) in a reciprocal pro-survival loop [1]. Despite the significantly improved response rate and overall survival of MM patients since the advent of novel agents such as bortezomib, thalidomide, lenalidomide, and autologous stem cell transplantation, MM remains an incurable malignancy with a 5-years survival rate of around 40% [2]. Indeed, the majority of patients relapse or become refractory to therapies, implying that drug resistance (DR) prevents effective treatment of MM.
Resistance to chemotherapy can be acquired or de novo DR [3]. Acquired resistance develops gradually as a result of sequential genetic and epigenetic changes that ultimately confer the tumor cells a complex drug-resistant phenotype. Acquired resistance depends on multifactorial processes, namely decreased drug uptake, expression of new drug-efflux pumps, drug metabolism, repair of DNA damage, alterations of cell proliferation and/or apoptosis [4]. It is usually studied in vitro via prolonged exposure of cells to a cytotoxic agent until the drug resistant phenotype is acquired. De novo resistance is present before the drug exposure and then selected during the drug treatment [3]. Recent studies have demonstrated, by inter-phase fluorescence in situ hybridization (iFISH) [5] and flow cytometry [6], the heterogeneity of MM cells at genetic (chromosome number, genetic translocations and mutations), clonal and cell differentiation levels. In particular, flow cytometry analysis reveals the presence of multiple cell clones in patients at diagnosis that are selected by in vivo therapeutic pressure and induce a distinct phenotypic MM cell subclone with different clonogenic and cytogenetic profiles in minimal residual disease [6]. A form of de novo resistance is environment-mediated drug resistance (EMDR), in which the BMME protects tumor cells from chemotherapy, radiotherapy or receptor-targeting drugs [7]. Overall, these observations highlight that MM progression and drug resistance are multistep transformation processes regulated by a complex cross-talk between MM cells and the BMME.
Here we show the latest findings on EMDR in MM. In particular, we focus on the role of microRNAs (miRNAs/miRs), exosomes and cancer-associated fibroblasts (CAFs) as new BMME players contributing to EMDR.
THE BMME AS A NICHE FOR MM CELLS
The BMME includes a non-cellular compartment formed by extracellular matrix (ECM) proteins (laminin, fibronectin and collagen) and soluble factors (cytokines, growth factors, chemokines), and a rich cellular compartment constituted by hematopoietic cells (myeloid cells, T lymphocytes, B lymphocytes, NK cells) and non-hematopoietic cells (fibroblasts, osteoblasts, osteoclasts, endothelial cells (ECs), endothelial progenitor cells (EPCs), pericytes, mesenchymal stem cells, mesenchymal stromal cells) (Figure 1). All these cells form specialized microenvironment niches, the osteoblast/endosteal and vascular niches, that play a key role in MM cell growth, survival and DR [8]. The osteoblast niche is located in the endosteum, at the interface between trabecular bone and BM, and regulates hematopoietic stem cell quiescence and self-renewal, hence hematopoiesis. The blood vessel-rich vascular niche controls stem cell mobilization, proliferation and differentiation [9]. The osteoblast and vascular niches are adjacent and show a mutually related secretion of several cytokines/growth factors and/or expression of adhesion molecules, creating a permissive microenvironment in the BM, namely “MM niches” [9]. MM cells home to and reside in these niches where they are protected from apoptotic stimuli and acquire the DR phenotype. In the niches, MM cells suppress osteoblastic cells, leading to impaired bone formation and the development of osteolytic lesions, and enhance angiogenesis via angiogenic factors secreted by MM cells, ECs, and BM stromal cells, thus promoting disease progression. The inability of conventional anti-cancer drugs to cure MM has reinforced EMDR. During chemotherapy, the interactions of a small subset of tumor cells with the BMME allow them to survive in a quiescent and protected state, resulting in minimal residual disease that progressively develops the acquired-resistance phenotype [10]. Using intravital two-photon microscopy in live mice, Lawson et al. [11] demonstrated that a small subpopulation of MM cells colonizes BM sites close to collagen-expressing osteoblasts or bone-lining cells, and falls into a dormant state by down-regulating the expression of genes that govern the cell cycle. Dormant MM cells are resistant to melphalan; however, cells may be activated by microenvironmental signals that can switch cancer cell dormancy “on” or “off”. McMillin et al. [12] developed a tumor cell-specific in vitro bioluminescence imaging assay that analyzes the effect of drugs on tumor cell viability in the presence and absence of BM stromal cells. They identified a stroma-induced signature in tumor cells, including AKT, Ras, NF-kB, HIF-1α, Myc, hTert and IRF4 signaling pathways, which is correlated with an adverse clinical prognosis.
EMDR can be subdivided into: i) soluble factors-mediated resistance (SFM-DR), which relies on cytokines, chemokines and growth factors, and ii) cell adhesion-mediated resistance (CAM-DR) resulting from adhesion of tumor cells to BM stromal cells or to ECM components.
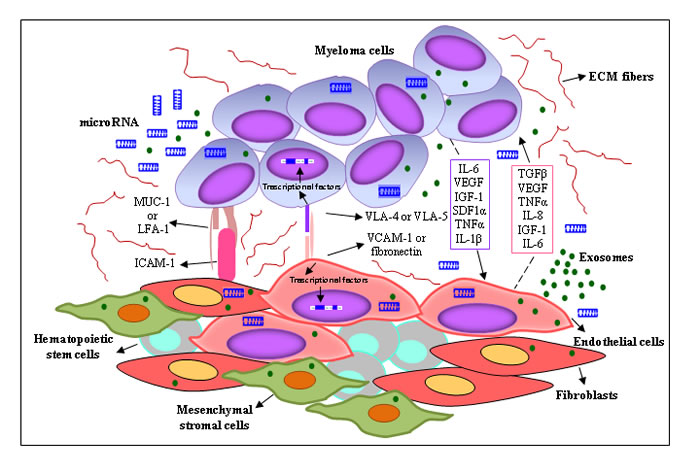
Figure 1: Interplay between MM cells and the surrounding microenvironment. MM cells are surrounded by a complex BMME composed of ECM proteins and several cell types, including BM stromal cells (ECs, mesenchymal stromal cells, CAFs). The cross-talk between MM cells and BM stromal cells is regulated by different mechanisms: (i) cell-to-cell adhesion between MM cells and ECM components/BM stromal cells; and (ii) soluble factors, i.e. cytokines, chemokines, growth factors, exosomes and miRNAs released by the BM stromal cells and MM cells, with autocrine and paracrine effects. Both mechanisms activate several signaling pathways in BM stromal cells and tumor cells, leading to MM drug resistance.
SOLUBLE FACTORS-MEDIATED DRUG RESISTANCE
The BMME contains soluble factors secreted by BM stromal cells or MM cells themselves, that play a multifaceted role in both MM onset and progression, and the development of DR. Since soluble factors can act on several cell targets, they regulate multiple processes such as cell growth and apoptosis, cell migration, adhesion, and angiogenesis. Notably, soluble factors cooperate among themselves through a complex communication network, creating the permissive BM niche.
The most important growth and survival factor is IL-6. It is secreted by both BM stromal cells and MM cells and inhibits MM apoptosis induced by serum starvation, dexamethasone and Fas ligand [13, 14]. Autocrine IL-6 production is associated with a malignant MM cells phenotype, i.e. a high proliferative index and resistance to dexamethasone-induced apoptosis [15]. By binding with its receptor, IL-6 triggers activation of the RAS/Raf/MEK/MAPK, JAK/STAT3 and PI3K/Akt signaling pathways [16]. Activation of the JAK/STAT3 pathway protects tumor cells from Fas-mediated apoptosis by upregulating the anti-apoptotic proteins Mcl-1, Bcl-XL and c-Myc. The IL-6/STAT3 signaling pathway is tightly controlled by the SOCS-1 protein, a negative regulator of cytokine signaling that is frequently inactivated by hypermethylation in MM patients, resulting in an uncontrolled IL-6 signaling [17]. Secretion of IL-6 by BM stromal cells is up-regulated by many molecules/cytokines (IL-1β, CD40, TNF-α, VEGF, TGF-β) and by MM cell adhesion via activation of the NF-kB pathway [8]. A major cytokine responsible for paracrine IL-6 production is the IL-1β secreted by MM cells, that increases the expression of adhesion molecules, stimulating IL-6 secretion by BM stromal cells [18]. All these data emphasize the key role of IL-6 in MM and explain why IL-1β and IL-6 may be useful targets in MM treatment [18]. However, several in vitro preclinical studies targeting the IL-6 signaling pathway, including antibodies against IL-6 and IL-6 receptors (CNTO328), IL-6 antisense oligonucleotides and IL-6 super antagonist Sant7, were unsuccessful due to autocrine IL-6 production, and/or activation of the IL-6/STAT3 and NF-κB pathways following MM cells-BM stromal cells interactions [19].
IL-6, in turn, enhances the secretion of VEGF, the most important angiogenic factor [20]. Angiogenesis is a constant hallmark in MM progression, and correlates with tumor growth, relapse and DR [21]. It is regulated by a balance between angiogenic and antiangiogenic molecules but this is lost in the angiogenic switch [20] that occurs during MM progression. The levels of VEGF, produced by both MM cells and BM stromal cells, increase during MM progression. FGF-2, TGF-β, and TNFα also stimulate its secretion [22]. By binding with its receptors, VEGF-R1 and VEGF-R2, VEGF stimulates vascular permeability, ECs proliferation, migration, invasion into surrounding tissue, and whole capillary tube formation [23]. It also governs the differentiation and survival of BM stromal cells [22]. Antivascular and antiangiogenic agents, including bortezomib and thalidomide, have cytotoxic effects on MM cells and ECs by inhibiting the secretion of multiple angiogenic factors including VEGF, HGF and FGF-2 [24, 25].
HGF is upregulated during MM progression. It enhances the expression of its receptor, cMET, and of other angiogenic factors, including VEGF and FGF-2, and suppresses the secretion of thrombospondin 1, an endogenous angiogenesis inhibitor [26]. The HGF/cMET pathway is involved in the MM pathogenesis: it is constitutively activated in MM cells [27] and ECs [28] from relapsed and resistant patients, and mediates DR [27, 28]. In ECs, the HGF/cMET loop enhances the expression of VEGF-A/VEGF-R2, sustaining MM angiogenesis [27]. In vitro and in vivo studies of a novel selective cMET inhibitor, SU11274, tested alone or in combination with bortezomib, lenalidomide and dexamethasone, suggest that the HGF/cMET pathway may be envisaged as a new therapeutic target for relapsed and refractory MM patients [28].
IGF-1 is produced by both MM cells and BM stromal cells, and contributes to MM pathobiology, inducing MM cell growth, survival, migration, MM-associated angiogenesis and osteolysis [29]. By binding its receptor, IGF-1R, it activates the PI3K/Akt and MEK/ERK signaling pathways, that induce overexpression of the anti-apoptotic proteins Bcl-XL, Bcl-2 and downregulation of the pro-apoptotic protein Bim [29, 30]. IGF-1R is overexpressed on MM cells: this aberrant expression associated with high IGF-1 levels has been related to disease progression and poor prognosis [29, 31]. In vitro and in vivo studies show that an increased expression and activation of the IGF-1/IGF-1R pathway in MM cells is associated with resistance to bortezomib [32]. Based on these observations, several therapeutic agents targeting the IGF-1/IGF-1R pathway have been developed and analyzed in preclinical studies [29]. Menu et al. [33, 34] demonstrated that picropodophyllin, a non-ATP-competitive IGF-1R tyrosine kinase inhibitor, inhibits tumor growth [33], MM-associated angiogenesis and bone disease [34] in 5T33MM-treated mice.
Another important soluble factor is IL-8 (CXCL8), a pro-inflammatory and angiogenic chemokine mainly secreted by BM stromal cells [35]. It contributes to tumor progression by inducing tumor cell proliferation, survival, migration and angiogenesis through the phosphorylation of VEGF-R2 [36, 37]. In vitro studies demonstrate that IL-8 contributes to BM stromal cells-induced NF-κB activity in MM cells and the consequent resistance to bortezomib [38].
Finally, the stromal cell-derived factor (SDF-1)/CXCR4 axis plays an important role in cell trafficking within the BM, favoring the formation of pro-metastatic BM niches and DR [39, 40]. SDF-1 (CXCL12) is constitutively expressed and released by BM stromal cells and fibroblasts, while its receptor CXCR4 is expressed by MM cells and ECs [40]. Activation of the SDF-1/CXCR4 axis promotes trans-endothelial migration, BM homing, migration and adhesion of MM cells. BM stromal cells-MM cells adhesion up-regulates SDF-1 secretion that, in turn, increases integrins expression and IL-6 and VEGF secretion, supporting MM cell growth and DR [41]. Using an in vivo murine xenografted mouse model, Roccaro et al. [42, 43] demonstrated that CXCR4 blockade by the monoclonal antibody Ulocuplumab, as well as neutralization of SDF-1 by Olaptesed-pegol (PEGylated mirror-image l-oligonucleotide) inhibits MM bone-to-bone cell dissemination and hence tumor progression.
Table 1: Adhesion molecules involved in MM drug resistance
PROTEINS |
LIGAND |
FUNCTION |
REFs |
Integrin β1 |
Laminin, Collagen type-VI, Fibronectin |
Cell protection from cell cycle-dependent drug therapies |
44, 45 |
Integrin β7 |
Fibronectin, E-cadherin |
Cell adhesion, migration, and homing |
44, 46 |
Integrin αvβ3 |
Vitronectin, Fibronectin |
Cell proliferation, protease secretion, invasion and spreading |
44, 47 |
1VLA-4 (α4β1) |
Fibronectin, 2VCAM-1 |
Cell adhesion, migration, homing and invasion, angiogenesis, cytokines secretion |
48, 49, 50, 51 |
1VLA-5 (α5β1) |
Fibronectin |
Cell homing and migration. Its down-expression correlates with MM progression |
44, 49 |
2VCAM- 1 |
1VLA-4 |
Cells migration, homing and invasion |
52 |
3LFA-1 |
5ICAM-1 |
Cell adhesion, proliferation and survival, angiogenesis, tumor dissemination |
48, 50, 53 |
4MUC-1 |
5ICAM-1 |
Cells adhesion, growth and survival, disease progression |
54 |
CD44 isoforms |
Hyaluronan |
Cell adhesion and invasion |
44, 50 |
CD138 (syndecan-1) |
Fibronectin |
Cell adhesion |
44 |
1VLA = Very Late Activation Antigen; 2VCAM = Vascular Cell Adhesion Molecule; 3LFA = Lymphocyte Function-Associated Antigen; 4MUC = Mucin-1 antigen; 5ICAM = Intercellular Adhesion Molecule.
CELL ADHESION-MEDIATED DRUG RESISTANCE
CAM-DR is a mechanism whereby MM cells escape the cytotoxic effects of anti-cancer therapy via adhesive interactions with BM stromal cells and/or ECM components. It is achieved through adhesion molecules of the integrin family [44-51], CD138 (syndecan-1) [44], CD44 [44, 50], Vascular Cell Adhesion Molecule-1 (VCAM-1) [52], Lymphocyte Function-Associated Antigen-1 (LFA-1) [48, 50, 53], Mucin-1 antigen (MUC-1) [54], and Intercellular Adhesion Molecule-1 (ICAM-1) [48, 50, 53, 54]. Among the integrins, major fibronectin receptors include the Very Late Activation Antigen (VLA)-4 (α4β1), VLA-5 (α5β1), αvβ3 and β7 integrins, that mediate MM cell trafficking and DR [44]. Damiano et al. [49] described, for the first time, CAM-DR as a reversible DR phenotype in fibronectin-adherent MM cells. The adhesive interactions between MM cells and BM stromal cells are complex because they involve several adhesion molecules expressed on both MM cells and BM stromal cells [48, 50-54]. MM cells-BM stromal cells adhesion triggers IL-6 secretion, NF-κB activation in stromal cells [51] and the up-regulation of many signaling pathways resulting in MM cell proliferation and survival [55].
Several studies [49, 51, 55, 56] have described CAM-DR to doxorubicin, melphalan, vincristine, dexamethasone and mitoxantrone in MM cell lines and patients primary MM cells due to their adhesion to fibronectin or BM stromal cells. Inhibition of cell adhesive interactions by shRNA-mediated knockdown of the α4 subunit of VLA-4 (CD49d) or by anti-α4 antibodies reverses CAM-DR, sensitizing MM cells to drug therapy [56]. VLA-4 is strongly expressed on MM cells and is the only integrin able to mediate both MM cell-ECM and MM cell-BM stromal cells interactions via separate binding sites [57]. Bortezomib overcomes CAM-DR to vincristine and dexamethasone by down-regulating VLA-4 expression on MM cells, thus inhibiting MM cell adhesion to fibronectin and BM stromal cells [56]. Other integrins involved in CAM-DR are VLA-5 and β7: the former supports cells survival by up-regulating Bcl-2 expression [58], the latter increases MM cells adhesion, migration, and homing into BM, and reduces bortezomib- and melphalan-induced apoptosis [46]. Moreover, β1-integrin-mediated CAM-DR protects cells from cell cycle-dependent drug therapies, such as the topoisomerase inhibitor etoposide, by up-regulating p27kip1 and halting the cell cycle [45]. In line with these observations, Paiva et al. [59] demonstrated that MM cells in minimal residual disease show an overexpression of integrins and adhesion molecules even compared to the cells at first diagnosis.
MM cells-BM stromal cells interactions are also mediated by Notch [60]. Binding of Notch receptors on MM cells with the specific ligands on BM stromal cells confers mitoxantrone and melphalan resistance. Indeed, Notch activation results in an increasing secretion of IL-6, IGF-1, and VEGF that, as previously described, contribute to create a permissive BMME [8]. Finally, a relevant consequence of MM cells-BM stromal cells adhesion is the IL-6 mediated up-regulation of PD-L1 (CD274 or B7-H1) expression on MM cells, that increases their proliferative ability, induces resistance to dexamethasone and melphalan, and down-regulates anti-tumor immune T cell responses [61].
All these findings demonstrate that CAM-DR influences the therapeutic response and suggest the importance of introducing anti-adhesion strategies in combination with chemotherapeutic drugs. Podar et al. [62] evaluated the effect of Natalizumab, a selective adhesion-molecule inhibitor, which binds α4 integrins and prevents MM cells interactions with ECM and BM stromal cells. They observed that Natalizumab inhibits MM cell proliferation, VEGF secretion and angiogenesis and enhances the anti-MM activity of bortezomib and dexamethasone, supporting the hypothesis that anti-adhesion treatment could improve the current therapeutic strategies for MM.
NEW PLAYERS: miRNAs, EXOSOMES AND CANCER-ASSOCIATED FIBROBLASTS
A new mechanism of intercellular communication between BM stromal cells and MM cells involved in MM pathogenesis and DR is shown to be accomplished by miRNAs, exosomes and CAFs.
miRNAs
miRNAs are endogenous, single-stranded, non-coding RNAs (19-25 nt), that regulate gene expression by targeting the 3’-untranslated region (3’-UTR) of mRNAs, and therefore inhibit protein translation, regulating a wide range of physiologic and pathologic cellular processes such as proliferation, differentiation, metabolism, aging and cell death [63, 64]. Deregulation of miRNAs has been described in various tumors including MM, where they can function either as tumor suppressors or oncogenes [65, 66].
In MM, a deregulated miRNAs expression in tumor cells has been associated to disease progression, pathobiology and DR, through modulating the expression of target genes involved in several pathways, such as p53, IGF-1/IGF-1R, VEGF/VEGF-R, NF-κB, IL-6-STAT3, SOCS1 [67-71]. In particular, miRNAs deregulation is correlated with MM clinical stages and/or MM molecular subtypes [67-70]. Over-expression of miR-21, -106b~25 cluster, -181a/b has been observed in MM and MGUS cells compared to healthy plasma cells, while a selective upregulation of miR-32 and -17~92 cluster has been demonstrated in MM cells and cell lines but not in MGUS cells [67]. Lionetti et al. [70], in an integrated analysis of miRNAs expression with genome-wide copy number variations and heterozygosity, noticed a strong correlation between miRNAs expression and IGH translocation and heterozygosity. Seckinger et al. [71] investigated miRNAs expression in 92 purified MM and MGUS cells and normal plasma cells using miChip-array, that analyzes 559 human miRNAs. Compared to normal plasma cells, MM cells showed 67 differentially expressed miRNAs, and MGUS cells 20. The authors found no correlation between miRNAs expression and MM molecular classifications. On the contrary, a deregulated expression of miR-135a, -135b, -200a, -200b and -596 was related with overall survival.
Emerging evidence relates miRNAs expression with anti-cancer drug activity or DR [69, 72]. The expression of miR-27a is associated with bortezomib resistance in MM patients [73]. Bortezomib treatment of MM cell lines significantly decreases the expression of miR-27a, whose gene target is the cyclin-dependent kinase 5 (CDK5), a major modulator of bortezomib sensitivity. These findings are in agreement with previous data [74] that demonstrate an increased bortezomib sensitivity of MM cell lines and patients tumor cells by down-regulating CDK5. miR-29 acts as tumor suppressor miRNA in several hematological malignancies [75]. It is down-regulated in patients MM cells [76, 77] and in MM cell lines with acquired resistance to bortezomib, carfilzomib and ixazomib [78]. Amodio et al. [76] described a miR-29b-Sp1 loop that explains the role of miR-29b in bortezomib-induced apoptosis. Indeed, Sp1, a transcription factor that regulates the cell cycle and apoptosis-related genes, whose expression is in turn governed by 26S proteasome [79], negatively regulates miR-29b expression. Bortezomib treatment up-regulates miR-29b levels through a down-regulation of Sp1, enhancing drug-induced apoptosis in patients MM cells and in MM cell lines. Enforced expression of miR-29b in MM cells enhances bortezomib-induced apoptosis through the reduction of proteasome activator PA200 levels [78].
Since the BMME plays a central role in determining MM progression and DR [7], the involvement of miRNAs in the cross-talk between the BMME and MM cells has been investigated. Roccaro et al. [80] described a MM-specific miRNAs signature in relapsed/refractory MM cells compared to their normal counterpart, characterized by a down-expression or the absence of miRs-15a and -16. MM-BM stromal cells interactions decrease miRs-15a and -16 expression in MM cells, inducing cell survival and IL-6-mediated bortezomib resistance [81, 82]. Enforced expression of pre-miRs-15a and -16 inhibits MM-BM stromal cells adhesion and MM cell proliferation, by inhibiting AKT3, ribosomal-protein-S6, MAP-kinases, NF-κB-activator MAP3KIP3, as well as limiting angiogenesis by decreasing VEGF secretion [80].
Adhesion of MM cells to BM stromal cells up-regulates miR-125a-5p [83] and -21 [84-86] levels in tumor cells. An increased expression of miR125a-5p in MM cells is associated to a subset of MM patients carrying the t(4;14) translocation [70]. Enforced expression of miR-125a mimics can downregulate the p53 pathway-related genes, i.e. TP53, BAX, MDM2, CDKN1A, leading to tumor growth by influencing cell proliferation, migration and apoptosis. In addition, it targets the TNFα-induced protein 3 (TNFAIP3) and activates the NF-κB pathways [87] suggesting that it may cause an aberrant NF-κB activation in bortezomib-resistant MM cells. miR-125a-5p reduces the expression of p53-responsive miR-192 and -194, which enhance cell cycle arrest and apoptosis. Based on these observations, Leotta et al. [83] suggested the combined use of miR-125 inhibitors and miR-192 and -194 mimics to treat MM. miR-21 promotes MM cell growth and viability. It is up-regulated following MM-BM stromal cells adhesion via the IL-6/STAT3 [84, 85] and NF-κB [86] pathways. MM cells overexpressing miR-21 are resistant to dexamethasone- and doxorubicin-induced apoptosis, and demonstrate a central role of miR-21 in CAM-DR. Targeting miR-21 inhibits MM cell growth, counteracting the protective effect of BM stromal cells and, in combination with dexamethasone and/or doxorubicin, synergistically triggers MM cell apoptosis [86].
miRNAs are secreted in biological fluids (e.g. plasma, serum, saliva, urine) [88] as nuclease resistant entities, packaged with RNA-binding proteins [89] or contained in microvesicles as exosomes (detailed below). Circulating miRNAs are fully functional and able to act as signaling molecules inside the recipient cells by modulating the expression of their target genes. Increasing evidence suggests that miRNAs could be diagnostic and prognostic markers in human tumors, including MM [90, 91]. Rocci et al. [91] analyzed the serum miRNAs levels of a large cohort of newly diagnosed MM patients and correlated the miRNAs levels with the clinical outcome to test their prognostic relevance. Among 800 miRNAs, they identified two circulating miR-16 and -25 whose serum levels were associated with overall survival. Nevertheless, they did not observe a correlation between miRNAs serum levels and miRNAs expression in MM cells, implying that circulating miRNAs do not resemble the miRNAs profile of tumor cells in the BM. Different results were illustrated by other authors [92-94]. Wang et al. [92] demonstrated a miRNAs signature in the extracellular BMME of MM patients that mirrored serum and plasma circulating miRNAs. A low expression of miR-let-7a, -15a and -106b was observed in the BMME and MM cells. Kubiczkova et al. [93] identified 5 circulating miRNAs (miR-34a, -130a, -744, let-7d, let-7e) that were differentially expressed in MM and MGUS serum compared to healthy subjects. The miR-744 and let-7e levels were correlated with overall survival. Navarro et al. [94] described a serum miRNAs signature (miR-16, -17, -19b, -20a and -660) as a potential diagnostic and prognostic tool in MM. Low levels of both miR-19b and -331 were shown to be a marker of short progression-free survival after autologous stem-cell transplantation.
Further studies of circulating miRNAs in MM are needed to elucidate their cell origin (cancer or normal, live or apoptotic cells), and their relationship with anti-MM drug activity or DR.
Exosomes
Exosomes are small membranous vesicles (40-100 nm) released in the extracellular milieu by several cell types [95-97] in physiological and pathological conditions. They mediate local and systemic cell-to-cell communication and regulate cell behavior by transferring mRNA, miRNAs and proteins, through fusion with the cell membrane or through endocytosis followed by internalization of recipient cells [98]. In the MM context, the exosomes-MM cells interaction is mediated by fibronectin binding to heparan sulfate, expressed on the surface of both exosomes and MM cells [99]. This binding activates p38 and pERK signaling and the expression of the downstream target genes DKK1 and MMP-9, and promotes MM progression by inducing tumor cell spread and ECs invasion [100]. The disruption of fibronectin-heparan sulfate interactions blocks exosome binding to MM cells or BM stromal cells, highlighting a specific cross-talk fostered by exosomes in the BMME [100]. The involvement of exosomes released by MM cells and BM stromal cells, as an active vehicle that can modulate the microenvironment and promote tumor progression and DR, has been investigated by in vitro and in vivo studies [101-103]. Exosomes derived from BM mesenchymal stromal cells of MM patients show a different functional activity compared to that of normal donors: the former facilitate MM progression and spread whereas the latter inhibit MM cell growth. Tissue-engineered bones loaded with MM cells alone, as control, and in the presence of either MM or normal BM-mesenchymal stromal cells-derived exosomes, were subcutaneously injected into SCID mice. Bioluminescence in vivo imaging showed a significantly higher tumor growth rate in mice transplanted with MM BM-mesenchymal stromal cells-derived exosomes than in those transplanted with normal BM-MSC-derived exosomes. Using in vivo confocal imaging, a higher ability of MM BM-mesenchymal stromal cells-derived exosomes to disseminate to the distant BM niches in vivo was demonstrated. These effects were related to a protein cargo of MM BM stromal cells-exosomes, i.e. high levels of IL-6, CCL2, γ-catenin, fibronectin, and to the absence of the tumor suppressor miR-15a [101]. The in vivo involvement of exosomes in the MM cells/BM stromal cells cross-talk has been demonstrated in the murine syngeneic 5T33MM model [102, 103]. Tumor exosomes contain multiple angiogenesis-related proteins, such as angiogenin, HGF, MMP-9, serpin E1, tissue inhibitor of metallopeptidase-1, thrombospondin 1 and VEGF, that promote ECs growth and angiogenesis. Furthermore, MM cells exosomes induce the growth of myeloid-derived suppressor cells and enhance their immunosuppressive capacity in vivo by up-regulating inducible nitric oxide synthase [102]. In turn, BM stromal cells-exosomes modulate the proliferation, survival, migration and bortezomib resistance of MM cells [103]. These effects are related to the exosome proteins content, activating several survival pathways, including c-Jun N-terminal kinase, p38, p53, AKT, and inhibiting MM cells bortezomib-induced apoptosis through the modulation of Bcl-2, caspase-9, -3 and PARP expression [103].
The exosome secretion, content and functionality depend on the tumor BMME and tumor phenotype. Umezu et al. [104] demonstrated that MM cells under hypoxia conditions produce more exosomes than the parental cells under normoxia, and that their content and function are different. Exosomes derived from hypoxic MM cells contain miR-135b whose target gene is the hypoxia-inducible factor-1α (HIF-1α) subunit inhibitor (FIH-1). Transfer of exosomal miR-135b to ECs reduces the expression of FIH-1 and increases HIF-1 transcriptional activity, accelerating angiogenesis both in vitro and in vivo. These observations may explain the HIF-1α protein expression and stabilization described in BM ECs from patients with relapsed/refractory MM [105].
Finally, higher exosomes levels have been observed in body fluids of cancer patients compared to healthy subjects, suggesting that exosomes could be used as biomarkers in the diagnosis and prognosis of several tumors including MM [106-109].
Cancer-associated fibroblasts
CAFs are the most populous cell type within the tumor microenvironment of many solid [110, 111] and hematological malignancies [112]. In MM patients BM, the CAFs population increases, and parallels the clinical stages [113]. According to literature [114], the BM CAFs exhibit phenotypic similarities to myofibroblasts or activated fibroblasts. They are CD45- cells expressing α-smooth muscle actin (α-SMA), fibroblast-specific protein-1 (FSP-1), fibroblast activation protein (FAP), and other markers as platelet-derived growth factor receptor α and β (PDGFR α/β), neuron-glial antigen2 (NG2), CD31, CD144, VEGF-R2, CD33, CD146 and CD90 that are specific for different cell types. This suggests that CAFs may derive from multiple cell lineages: resident fibroblasts, mesenchymal stem cells, via the mesenchymal transition [115], ECs and hematopoietic stem and progenitor cells, via the endothelial-mesenchymal transition [116].
In vitro and in vivo experiments highlight a mutual interplay between CAFs and tumor cells during MM onset and progression. MM cells induce and maintain the CAFs-activated phenotype, their proliferation and recruitment via TGF-β [113]. CAFs, in turn, modify the BM stroma and influence chemotaxis, adhesion, proliferation, and apoptosis of MM cells through cell-to-cell contact involving β3, β7, VLA-4, VLA-5 and αVβ3 integrins expressed on MM cells and β3 and β7 integrins expressed on CAFs, and the secretion of TGF-β, HGF, IGF-1, IL-1, IL-6 and SDF-1α by both cell types [113].
Recently, we demonstrated that BM CAFs from bortezomib-resistant patients are resistant in vitro to the drug and prevent the bortezomib-induced apoptosis of co-cultured MM cells [117]. This protection depends on the ability of bortezomib to foster bortezomib-resistant CAFs to secrete several anti-apoptotic cytokine/growth factors (as previously described), such as IGF-1, IL-6, IL-8, TGF-β, and exosomes. Preliminary data demonstrate a release of exosomes from bortezomib-treated CAFs that are swallowed by MM cells (Figure 2), thus preventing their bortezomib-induced apoptosis (unpublished data).
Proteomic and phospho-proteomic analyses reveal that the bortezomib-DR of MM CAFs is associated to cellular stress and activation of pro-survival autophagy mediated by the autocrine TGF-β pathway [117]. Indeed, blockade of the TGF-β pathway by a TβR-I/II inhibitor induces apoptosis of bortezomib-resistant CAFs, by inhibiting the Smad2/3 and autophagy signaling pathways, and overcomes bortezomib resistance of MM cells conferred by CAFs [117]. Therefore, CAFs are emerging as a novel potentially therapeutic target in MM by means of various strategies [112, 118] that act on both MM cells and CAFs or on CAFs alone. In particular, Öhlund et al. [118] identified four possible strategies to target the pro-tumorigenic effects of CAFs. These strategies include targeting the stromal barrier in order to increase the drugs delivery; the inhibition of CAFs-secreted factors that promote tumor progression and DR; the depletion or blockage of the ECM components and/or integrins, and, finally, the targeting of CAFs by deactivating the CAFs phenotype to that of quiescent, normal fibroblasts.
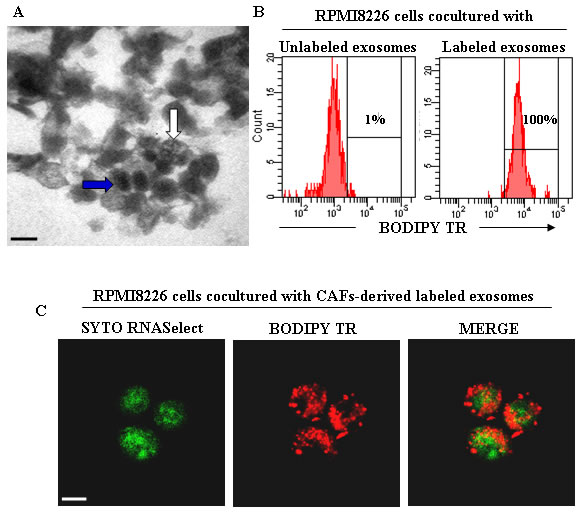
Figure 2: CAFs-derived exosomes and their uptake from MM cells. A. Transmission electron microscopy of exosomes isolated from BM CAFs of MM patients showing heterogeneous features of vesicles with an electrondense core (blue arrow) and multivesicular body (white arrow). Scale bar, 0.2 μm. B. Flow cytometry analysis of exosomes uptake by RPMI8226 cells. The RPMI8226 cells were co-cultured with unlabeled and BODIPY TR ceramide-labeled CAFs-derived exosomes. C. Confocal dual immunofluorescence images of RPMI8226 cells swallowed CAFs-derived exosomes labeled with SYTO RNASelect (green) and BODIPY TR ceramide (red), specific for RNAs and cell membranes, respectively. Scale bar, 7.5 μm.
CONCLUSIONS
A large volume of research in MM has highlighted the cardinal role of the BMME as a complex signaling molecules network, in which MM onset, progression and DR are regulated by a contact-dependent and -independent interplay between MM cells and their surrounding microenvironment. The role of the BMME network has been seen to be more complex since the discovery of the new players, miRNAs, exosomes and CAFs. The therapeutic failure of novel MM agents (first and second generation proteasome inhibitors, IMIDs, etc.) that target cell adhesion, cytokines secretion and survival pathways may be explained by their involvement. Notably, CAFs, miRNAs deregulation and/or the exosomes cargo (miRNAs/cytokines/proteins) could permit MM cells to achieve apoptotic escape and/or prosurvival autophagy by modulating alternative signaling pathways. Nevertheless, in-depth investigations are needed to better elucidate the role of miRNAs, exosomes and CAFs in affecting a tumor-prone BM niche.
In conclusion, MM drug resistance, being a multistep transformation process in which several players cooperate among themselves, provides the rationale for a multiple targets therapeutic approach with the aim of creating an unsupportive BMME and thereby enabling anti-MM therapies.
ACKNOWLEDGMENTs
The research leading to these results was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC), Investigator Grant (no. 14095 to AV); Special Program Molecular Clinical Oncology 5 per 1000 (no. 9965 to AV), Milan, IT; and the European Commission’s Seventh Framework Programme (EU FPT7) OVER-MyR (no. 278706 to AV).
Conflicts of interest
The authors declare no conflict of interest
REFERENCES
1. Anderson KC, Carrasco RD. Pathogenesis of myeloma. Annu Rev Pathol. 2011;6:249-74.
2. Brenner H, Gondos A, Pulte D. Recent major improvement in long-term survival of younger patients with multiple myeloma. Blood. 2008; 111:2521-6.
3. Hazlehurst LA, Dalton WS. De Novo and Acquired Resistance to Antitumor Alkylating Agents. Chapter 20 Cancer Drug Resistance Part of the series Cancer Drug Discovery and Development pp 377-389.
4. Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002; 53:615-27.
5. Zang M, Zou D, Yu Z, Li F, Yi S, Ai X, Qin X, Feng X, Zhou W, Xu Y, Li Z, Hao M, Sui W, et al. Detection of recurrent cytogenetic aberrations in multiple myeloma: a comparison between MLPA and iFISH. Oncotarget. 2015; 6:34276-87. doi: 10.18632/oncotarget.5371.
6. Paíno T, Paiva B, Sayagués JM, Mota I, Carvalheiro T, Corchete LA, Aires-Mejía I, Pérez JJ, Sanchez ML, Barcena P, Ocio EM, San-Segundo L, Sarasquete ME, et al. Phenotypic identification of subclones in multiple myeloma with different chemoresistant, cytogenetic and clonogenic potential. Leukemia. 2015; 29:1186-94.
7. Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008; 14:2519-26.
8. Manier S, Sacco A, Leleu X, Ghobrial IM, Roccaro AM. Bone marrow microenvironment in multiple myeloma progression. J Biomed Biotechnol. 2012; 2012:157496.
9. Ribatti D, Nico B, Vacca A. Multiple myeloma as a model for the role of bone marrow niches in the control of angiogenesis. Int Rev Cell Mol Biol. 2015; 314:259-82.
10. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009; 9:665-74.
11. Lawson MA, McDonald MM, Kovacic N, Hua Khoo W, Terry RL, Down J, Kaplan W, Paton-Hough J, Fellows C, Pettitt JA, Neil Dear T, Van Valckenborgh E, Baldock P, et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat Commun. 2015; 6:8983.
12. McMillin DW, Delmore J, Weisberg E, Negri JM, Geer DC, Klippel S, Mitsiades N, Schlossman RL, Munshi NC, Kung AL, Griffin JD, Richardson PG, Anderson KC, et al. Tumor cell-specific bioluminescence platform to identify stroma-induced changes to anticancer drug activity. Nat Med. 2010, 16:483-9.
13. Lichtenstein A, Tu Y, Fady C, Vescio R, Berenson J. Interleukin-6 inhibits apoptosis of malignant plasma cells. Cell Immunol. 1995; 162:248-55.
14. Chauhan D, Kharbanda S, Ogata A, Urashima M, Teoh G, Robertson M, Kufe DW, Anderson KC. Interleukin-6 inhibits Fas-induced apoptosis and stress-activated protein kinase activation in multiple myeloma cells. Blood. 1997; 89:227-34.
15. Frassanito MA, Cusmai A, Iodice G, Dammacco F. Autocrine interleukin-6 production and highly malignant multiple myeloma: relation with resistance to drug-induced apoptosis. Blood. 2001; 97:483-9.
16. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer. 2007; 7:585-98.
17. Galm O, Yoshikawa H, Esteller M, Osieka R, Herman JG. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood. 2003; 101:2784-8.
18. Lust JA, Lacy MQ, Zeldenrust SR, Dispenzieri A, Gertz MA, Witzig TE, Kumar S, Hayman SR, Russell SJ, Buadi FK, Geyer SM, Campbell ME, Kyle RA, et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc. 2009; 84:114-22.
19. Podar K, Chauhan D, Anderson KC. Bone marrow microenvironment and the identification of new targets for myeloma therapy. Leukemia. 2009; 23:10-24.
20. Vacca A, Ribatti D. Bone marrow angiogenesis in multiple myeloma. Leukemia. 2006; 20:193-9.
21. Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, Albini A, Bussolino F, Dammacco F. Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood. 1999; 93:3064-73.
22. Podar K, Anderson KC. The pathophysiologic role of VEGF in hematologic malignancies: therapeutic implications. Blood. 2005; 105:1383-95.
23. Vacca A, Ria R, Ribatti D, Semeraro F, Djonov V, Di Raimondo F, Dammacco F. A paracrine loop in the vascular endothelial growth factor pathway triggers tumor angiogenesis and growth in multiple myeloma. Haematologica. 2003; 88:176-85.
24. Vacca A, Scavelli C, Montefusco V, Di Pietro G, Neri A, Mattioli M, Bicciato S, Nico B, Ribatti D, Dammacco F, Corradini P. Thalidomide downregulates angiogenic genes in bone marrow endothelial cells of patients with active multiple myeloma. J Clin Oncol. 2005; 23:5334-46.
25. Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A, Dammacco F, Richardson PG, Anderson KC. Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res. 2006; 66:184-91.
26. Zhang YW, Su Y, Volpert OV, Vande Woude GF. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc Natl Acad Sci U S A. 2003 Oct 28;100:12718-23.
27. Moschetta M, Basile A, Ferrucci A, Frassanito MA, Rao L, Ria R, Solimando AG, Giuliani N, Boccarelli A, Fumarola F, Coluccia M, Rossini B, Ruggieri S, et al. Novel targeting of phospho-cMET overcomes drug resistance and induces antitumor activity in multiple myeloma. Clin Cancer Res. 2013; 19:4371-82.
28. Ferrucci A, Moschetta M, Frassanito MA, Berardi S, Catacchio I, Ria R, Racanelli V, Caivano A, Solimando AG, Vergara D, Maffia M, Latorre D, Rizzello A, et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clin Cancer Res. 2014; 20:5796-807.
29. Bieghs L, Johnsen HE, Maes K, Menu E, Van Valckenborgh E, Overgaard MT, Nyegaard M, Conover CA, Vanderkerken K, De Bruyne E. The insulin-like growth factor system in multiple myeloma: diagnostic and therapeutic potential. Oncotarget. 2016; 7(30):48732-48752. doi: 10.18632/oncotarget.8982.
30. De Bruyne E, Bos TJ, Schuit F, Van Valckenborg E, Menu E, Thorrez L, Atadja P, Jernberg-Wiklund H, Vanderkerken K. IGF-1 suppresses Bim expression in multiple myeloma via epigenetic and posttranslational mechanisms. Blood. 2010; 115:2430-40.
31. Chng WJ, Gualberto A, Fonseca R. IGF-1R is overexpressed in poor-prognostic subtypes of multiple myeloma. Leukemia. 2006; 20:174-6.
32. Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, Davis RE, Lin P, Wang H, Madden TL, Wei C, Baladandayuthapani V, Wang M, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood. 2012; 120:3260-70.
33. Menu E, Jernberg-Wiklund H, Stromberg T, De Raeve H, Girnita L, Larsson O, Axelson M, Asosingh K, Nilsson K, Van Camp B, Vanderkerken K. Inhibiting the IGF-1 receptor tyrosine kinase with the cyclolignan PPP: an in vitro and in vivo study in the 5T33MM mouse model. Blood. 2006; 107:655-60.
34. Menu E, Jernberg-Wiklund H, De Raeve H, De Leenheer E, Coulton L, Gallagher O, Van Valckenborgh E, Larsson O, Axelson M, Nilsson K, Van Camp B, Croucher P, Vanderkerken K. Targeting the IGF-1R using picropodophyllin in the therapeutical 5T2MM mouse model of multiple myeloma: beneficial effects on tumor growth, angiogenesis, bone disease and survival. Int J Cancer. 2007; 121:1857-61.
35. Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008; 14:6735-41.
36. Lazennec G, Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol Med. 2010; 133-44.
37. Petreaca ML, Yao M, Liu Y, Defea K, Martins-Green M. Transactivation of vascular endothelial growth factor receptor-2 by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. Mol Biol Cell. 2007; 18:5014-23.
38. Markovina S, Callander NS, O’Connor SL, Xu G, Shi Y, Leith CP, Kim K, Trivedi P, Kim J, Hematti P, Miyamoto S. Bone marrow stromal cells from multiple myeloma patients uniquely induce bortezomib resistant NF-kappaB activity in myeloma cells. Mol Cancer. 2010; 9:176.
39. Menu E, Asosingh K, Indraccolo S, De Raeve H, Van Riet I, Van Valckenborgh E, Vande Broek I, Fujii N, Tamamura H, Van Camp B, Vanderkerken K. The involvement of stromal derived factor 1alpha in homing and progression of multiple myeloma in the 5TMM model. Haematologica. 2006; 91:605-12.
40. Bianchi G, Kumar S, Ghobrial IM, Roccaro AM. Cell trafficking in multiple myeloma.. Open J Hematol. 2012; 3(Suppl 1). pii: 4.
41. Alsayed Y, Ngo H, Runnels J, Leleu X, Singha UK, Pitsillides CM, Spencer JA, Kimlinger T, Ghobrial JM, Jia X, Lu G, Timm M, Kumar A, et al. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007; 109:2708-17.
42. Roccaro AM, Mishima Y, Sacco A, Moschetta M, Tai YT, Shi J, Zhang Y, Reagan MR, Huynh D, Kawano Y, Sahin I, Chiarini M, Manier S, et al. CXCR4 regulates extra-medllary myeloma through epithelial mesenchymal-transition-like transcriptional activation. Cell Rep. 2015; 12:622-35.
43. Roccaro AM, Sacco A, Purschke WG, Moschetta M, Buchner K, Maasch C, Zboralski D, Zöllner S, Vonhoff S, Mishima Y, Maiso P, Reagan MR, Lonardi S, et al. SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Rep. 2014; 9:118-28.
44. Katz BZ. Adhesion molecules- The lifelines of multiple myeloma cells. Semin Cancer Biol. 2010; 20:186-95.
45. Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene. 2000; 19:4319-27.
46. Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, Lin C, Duggan P, Tassone P, Mansoor A, Stewart DA, Boise LH, Ghobrial IM, et al. Integrin β7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. 2011; 117:6202-13.
47. Ria R, Vacca A, Ribatti D, Di Raimondo F, Merchionne F, Dammacco F. Alpha(v)beta(3) integrin engagement enhances cell invasiveness in human multiple myeloma. Haematologica. 2002; 87:836-45.
48. Barker HF, Ball J, Drew M, Hamilton MS, Franklin IM. The role of adhesion molecules in multiple myeloma. Leuk Lymphoma. 1992; 8:189-96.
49. Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS. Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood. 1999; 93:1658-67.
50. Vacca A, Di Loreto M, Ribatti D, Di Stefano R, Gadaleta-Caldarola G, Iodice G, Caloro D, Dammacco F. Bone marrow of patients with active multiple myeloma: angiogenesis and plasma cell adhesion molecules LFA-1, VLA-4, LAM-1, and CD44. Am J Hematol. 1995; 50:9-14.
51. Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, Anderson KC. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood. 1996; 87:1104-12.
52. Okada T, Hawley RG, Kodaka M, Okuno H. Significance of VLA-4-VCAM-1 interaction and CD44 for transendothelial invasion in a bone marrow metastatic myeloma model. Clin Exp Metastasis. 1999; 17:623-9.
53. Schmidmaier R, Mandl-Weber S, Gaul L, Baumann P, Bumeder I, Straka C, Emmerich B. Inhibition of lymphocyte function associated antigen 1 by LFA878 induces apoptosis in multiple myeloma cells and is associated with downregulation of the focal adhesion kinase/phosphatidylinositol 3 kinase/Akt pathway. Int J Oncol. 2007;31: 969-76.
54. Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12: 335-48.
55. Hideshima T, Bergsagel PL, Kuehl WM, Anderson KC. Advances in biology of multiple myeloma: clinical applications. Blood. 2004; 104:607-18.
56. Noborio-Hatano K, Kikuchi J, Takatoku M, Shimizu R, Wada T, Ueda M, Nobuyoshi M, Oh I, Sato K, Suzuki T, Ozaki K, Mori M, Nagai T, et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene. 2009; 28:231-42.
57. Pulido R, Elices MJ, Campanero MR, Osborn L, Schiffer S, García-Pardo A, Lobb R, Hemler ME, Sánchez-Madrid F. Functional evidence for three distinct and independently inhibitable adhesion activities mediated by the human integrin VLA-4. Correlation with distinct alpha 4 epitopes. J Biol Chem. 1991; 266:10241-5.
58. Zhang Z, Vuori K, Reed JC, Ruoslahti E. The alpha 5 beta 1 integrin supports survival of cells on fibronectin and up-regulates Bcl-2 expression. Proc Natl Acad Sci U S A. 1995; 92:6161-5.
59. Paiva B, Corchete LA, Vidriales MB, Puig N, Maiso P, Rodriguez I, Alignani D, Burgos L, Sanchez ML, Barcena P, Echeveste MA, Hernandez MT, García-Sanz R, et al. Phenotypic and genomic analysis of multiple myeloma minimal residual disease tumor cells: a new model to understand chemoresistance. Blood. 2016; pii: blood-2015-08-665679.
60. Nefedova Y, Cheng P, Alsina M, Dalton WS, Gabrilovich DI. Involvement of Notch-1 signaling in bone marrow stroma-mediated de novo drug resistance of myeloma and other malignant lymphoid cell lines. Blood. 2004; 103:3503-10.
61. Tamura H, Ishibashi M, Yamashita T, Tanosaki S, Okuyama N, Kondo A, Hyodo H, Shinya E, Takahashi H, Dong H, Tamada K, Chen L, Dan K, et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia. 2013; 27:464-72.
62. Podar K, Zimmerhackl A, Fulciniti M, Tonon G, Hainz U, Tai YT, Vallet S, Halama N, Jäger D, Olson DL, Sattler M, Chauhan D, Anderson KC. The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: therapeutic implications. Br J Haematol. 2011; 155:438-48.
63. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281-97.
64. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004; 5:522-31.
65. Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6:857-66.
66. Dimopoulos K, Gimsing P, Grønbæk K. Aberrant microRNA expression in multiple myeloma. Eur J Haematol. 2013; 91:95-105.
67. Pichiorri F, Suh SS, Ladetto M, Kuehl M, Palumbo T, Drandi D, Taccioli C, Zanesi N, Alder H, Hagan JP, Munker R, Volinia S, Boccadoro M, et al. MicroRNAs regulate critical genes associated with multiple myeloma pathogenesis. Proc Natl Acad Sci U S A. 2008; 105:12885-90.
68. Bi C, Chng WJ. MicroRNA: important player in the pathobiology of multiple myeloma. Biomed Res Int. 2014; 2014:521586.
69. Munker R, Liu CG, Taccioli C, Alder H, Heerema N. MicroRNA profiles of drug-resistant myeloma cell lines. Acta Haematol. 2010; 123:201-4.
70. Lionetti M, Biasiolo M, Agnelli L, Todoerti K, Mosca L, Fabris S, Sales G, Deliliers GL, Bicciato S, Lombardi L, Bortoluzzi S, Neri A. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood. 2009; 114:e20-6.
71. Seckinger A, Meißner T, Moreaux J, Benes V, Hillengass J, Castoldi M, Zimmermann J, Ho AD, Jauch A, Goldschmidt H, Klein B, Hose D. miRNAs in multiple myeloma--a survival relevant complex regulator of gene expression. Oncotarget. 2015; 6:39165-83. doi: 10.18632/oncotarget.5381.
72. Zheng T, Wang J, Chen X, Liu L. Role of microRNA in anticancer drug resistance. Int J Cancer. 2010; 126:2-10.
73. Ballabio E, Armesto M, Breeze CE, Manterola L, Arestin M, Tramonti D, Hatton CS, Lawrie CH: Bortezomib action in multiple myeloma: microRNA-mediated synergy (and miR-27a/CDK5 driven sensitivity)? Blood Cancer J. 2012; 2:e83.
74. Zhu YX, Tiedemann R, Shi CX, Yin H, Schmidt JE, Bruins LA, Keats JJ, Braggio E, Sereduk C, Mousses S, Stewart AK. RNAi screen of the druggable genome identifies modulators of proteasome inhibitor sensitivity in myeloma including CDK5. Blood. 2011; 117:3847-57.
75. Amodio N, Rossi M, Raimondi L, Pitari MR, Botta C, Tagliaferri P, Tassone P. miR-29s: a family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget. 2015; 6:12837-61. doi: 10.18632/oncotarget.3805.
76. Amodio N, Di Martino MT, Foresta U, Leone E, Lionetti M, Leotta M, Gullà AM, Pitari MR, Conforti F, Rossi M, Agosti V, Fulciniti M, Misso G, et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis. 2012; 3:e436.
77. Saha MN, Abdi J, Yang Y, Chang H. miRNA-29a as a tumor suppressor mediates PRIMA-1Met-induced anti-myeloma activity by targeting c-Myc. Oncotarget. 2016; 7:7149-60. doi: 10.18632/oncotarget.6880.
78. Jagannathan S, Vad N, Vallabhapurapu S, Vallabhapurapu S, Anderson KC, Driscoll JJ. MiR-29b replacement inhibits proteasomes and disrupts aggresome+autophagosome formation to enhance the antimyeloma benefit of bortezomib. Leukemia. 2015; 29:727-38.
79. Liu S, Wu LC, Pang J, Santhanam R, Schwind S, Wu YZ, Hickey CJ, Yu J, Becker H, Maharry K, Radmacher MD, Li C, Whitman SP, et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell. 2010; 17:333-47.
80. Roccaro AM, Sacco A, Thompson B, Leleu X, Azab AK, Azab F, Runnels J, Jia X, Ngo HT, Melhem MR, Lin CP, Ribatti D, Rollins BJ, et al. MicroRNAs 15a and 16 regulate tumor proliferation in multiple myeloma. Blood. 2009; 113:6669-80.
81. Hao M, Zhang L, An G, Sui W, Yu Z, Zou D, Xu Y, Chang H, Qiu L. Suppressing miRNA-15a/-16 expression by interleukin-6 enhances drug-resistance in myeloma cells. J Hematol Oncol. 2011; 4:37.
82. Hao M, Zhang L, An G, Meng H, Han Y, Xie Z, Xu Y, Li C, Yu Z, Chang H, Qiu L. Bone marrow stromal cells protect myeloma cells from bortezomib induced apoptosis by suppressing microRNA-15a expression. Leuk Lymphoma. 2011; 52:1787-94.
83. Leotta M, Biamonte L, Raimondi L, Ronchetti D, Di Martino MT, Botta C, Leone E, Pitari MR, Neri A, Giordano A, Tagliaferri P, Tassone P, Amodio N. A p53-dependent tumor suppressor network is induced by selective miR-125a-5p inhibition in multiple myeloma cells. J Cell Physiol. 2014; 229:2106-16.
84. Leone E, Morelli E, Di Martino MT, Amodio N, Foresta U, Gullà A, Rossi M, Neri A, Giordano A, Munshi NC, Anderson KC, Tagliaferri P, Tassone P. Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clin Cancer Res. 2013; 19:2096-106.
85. Löffler D, Brocke-Heidrich K, Pfeifer G, Stocsits C, Hackermüller J, Kretzschmar AK, Burger R, Gramatzki M, Blumert C, Bauer K, Cvijic H, Ullmann AK, Stadler PF, et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood. 2007; 110:1330-3.
86. Wang X, Li C, Ju S, Wang Y, Wang H, Zhong R. Myeloma cell adhesion to bone marrow stromal cells confers drug resistance by microRNA-21 up-regulation. Leuk Lymphoma. 2011; 52:1991-8.
87. Kim SW, Ramasamy K, Bouamar H, Lin AP, Jiang D, Aguiar RC. MicroRNAs miR-125a and miR-125b constitutively activate the NF-κB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc Natl Acad Sci U S A. 2012; 109:7865-70.
88. Weber JA, Baxter DH, Zhang S, Huang DY, Huang KH, Lee MJ, Galas DJ, Wang K. The microRNA spectrum in 12 body fluids. Clin Chem. 2010; 56:1733-41.
89. Liang H, Zhang J, Zen K, Zhang CY, Chen X. Nuclear microRNAs and their unconventional role in regulating non-coding RNAs. Protein Cell. 2013; 4:325-30.
90. Kosaka N, Iguchi H, Ochiya T. Circulating microRNA in body fluid: a new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010; 101:2087-92.
91. Rocci A, Hofmeister CC, Geyer S, Stiff A, Gambella M, Cascione L, Guan J, Benson DM, Efebera YA, Talabere T, Dirisala V, Smith EM, Omedè P, et al. Circulating miRNA markers show promise as new prognosticators for multiple myeloma. Leukemia. 2014; 28:1922-6.
92. Wang W, Corrigan-Cummins M, Barber EA, Saleh LM, Zingone A, Ghafoor A, Costello R, Zhang Y, Kurlander RJ, Korde N, Roccaro AM, Ghobrial IM, Landgren O, et al. Aberrant Levels of miRNAs in Bone Marrow Microenvironment and Peripheral Blood of Myeloma Patients and Disease Progression. J Mol Diagn. 2015; 17:669-78.
93. Kubiczkova L, Kryukov F, Slaby O, Dementyeva E, Jarkovsky J, Nekvindova J, Radova L, Greslikova H, Kuglik P, Vetesnikova E, Pour L, Adam Z, Sevcikova S, et al. Circulating serum microRNAs as novel diagnostic and prognostic biomarkers for multiple myeloma and monoclonal gammopathy of undetermined significance. Haematologica. 2014; 99:511-8.
94. Navarro A, Díaz T, Tovar N, Pedrosa F, Tejero R, Cibeira MT, Magnano L, Rosiñol L, Monzó M, Bladé J, Fernández de Larrea C. A serum microRNA signature associated with complete remission and progression after autologous stem-cell transplantation in patients with multiple myeloma. Oncotarget. 2015; 6:1874-83. doi: 10.18632/oncotarget.2761.
95. van Niel G, Porto-Carreiro I, Simoes S, Raposo G. Exosomes: a common pathway for a specialized function. J Biochem. 2006; 140:13-21.
96. Théry C, Regnault A, Garin J, Wolfers J, Zitvogel L, Ricciardi-Castagnoli P, Raposo G, Amigorena S. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein hsc73. J Cell Biol. 1999; 147:599-610.
97. Blanchard N, Lankar D, Faure F, Regnault A, Dumont C, Raposo G, Hivroz C. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J Immunol. 2002; 168:3235-41.
98. Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013; 200:373-83.
99. Purushothaman A, Bandari SK, Liu J, Mobley JA, Brown EE, Sanderson RD. Fibronectin on the surface of myeloma cell-derived exosomes mediates exosome-cell interactions.. J Biol Chem. 2016; 291:1652-63.
100. Thompson CA, Purushothaman A, Ramani VC, Vlodavsky I, Sanderson RD. Heparanase regulates secretion, composition, and function of tumor cell-derived exosomes. J Biol Chem. 2013; 288: 10093-9.
101. Roccaro AM, Sacco A, Maiso P, Azab AK, Tai YT, Reagan M, Azab F, Flores LM, Campigotto F, Weller E, Anderson KC, Scadden DT, Ghobrial IM. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013; 123:1542-55.
102. Wang J, De Veirman K, Faict S, Frassanito MA, Ribatti D, Vacca A, Menu E. Multiple myeloma exosomes establish a favorable bone marrow microenvironment with enhanced angiogenesis and immunosuppression. J Pathol. 2016; 239:162-73.
103. Wang J, Hendrix A, Hernot S, Lemaire M, De Bruyne E, Van Valckenborgh E, Lahoutte T, De Wever O, Vanderkerken K, Menu E. Bone marrow stromal cell-derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. 2014; 124:555-66.
104. Umezu T, Tadokoro H, Azuma K, Yoshizawa S, Ohyashiki K, Ohyashiki JH. Exosomal miR-135b shed from hypoxic multiple myeloma cells enhances angiogenesis by targeting factor-inhibiting HIF-1.Blood. 2014; 124:3748-57.
105. Ria R, Catacchio I, Berardi S, De Luisi A, Caivano A, Piccoli C, Ruggieri V, Frassanito MA, Ribatti D, Nico B, Annese T, Ruggieri S, Guarini A,et al. HIF-1α of bone marrow endothelial cells implies relapse and drug resistance in patients with multiple myeloma and may act as a therapeutic target. Clin Cancer Res. 2014; 20:847-58.
106. Zhang X, Yuan X, Shi H, Wu L, Qian H, Xu W. Exosomes in cancer: small particle, big player. J Hematol Oncol. 2015; 8:83.
107. Ghosh AK, Secreto CR, Knox TR, Ding W, Mukhopadhyay D, Kay NE. Circulating microvesicles in B-cell chronic lymphocytic leukemia can stimulate marrow stromal cells: implications for disease progression. Blood. 2010; 115:1755-64.
108. Caivano A, Laurenzana I, De Luca L, La Rocca F, Simeon V, Trino S, D’Auria F, Traficante A, Maietti M, Izzo T, D’Arena G, Mansueto G, Pietrantuono G, et al. High serum levels of extracellular vesicles expressing malignancy-related markers are released in patients with various types of hematological neoplastic disorders. Tumour Biol. 2015; 36:9739-52.
109. Di Noto G, Paolini L, Zendrini A, Radeghieri A, Caimi L, Ricotta D. C-src enriched serum microvesicles are generated in malignant plasma cell dyscrasia. PLoS One. 2013; 8:e70811.
110. Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6:392-401.
111. Shimoda M, Mellody KT, Orimo A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumor progression. Semin Cell Dev Biol. 2010; 21:19-25.
112. Raffaghello L, Vacca A, Pistoia V, Ribatti D. Cancer associated fibroblasts in hematological malignancies. Oncotarget. 2015; 6:2589-603. doi: 10.18632/oncotarget.2661.
113. Frassanito MA, Rao L, Moschetta M, Ria R, Di Marzo L, De Luisi A, Racanelli V, Catacchio I, Berardi S, Basile A, Menu E, Ruggieri S, Nico B, et al. Bone marrow fibroblasts parallel multiple myeloma progression in patients and mice: in vitro and in vivo studies. Leukemia. 2014; 28:904-16.
114. Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006; 5:1640-6.
115. Kalluri R, Weinberg RA.The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420-8.
116. Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007; 67:10123-8.
117. Frassanito MA, De Veirman K, Desantis V, Di Marzo L, Vergara D, Ruggieri S, Annese T, Nico B, Menu E, Catacchio I, Ria R, Racanelli V, Maffia M, et al. Halting pro-survival autophagy by TGFβ inhibition in bone marrow fibroblasts overcomes bortezomib resistance in multiple myeloma patients. Leukemia. 2016; 30:640-8.
118. Öhlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014; 211:1503-23.