
INTRODUCTION
The sympathetic nervous system cancer neuroblastoma (NB) is the most common extra-cranial solid tumor in childhood. NB is characterized by a broad range of clinical behaviors and biological heterogeneity. Despite intensive treatments that include chemotherapy, radiation, surgery, stem cell transplant and immunotherapy, and an increased understanding of molecular characteristics of NB, more than 50% of children with metastatic NB relapse, and survival from recurrent NB is less than 10% [1,2]. Thus novel therapies are needed for patients with newly diagnosed and relapsed NB. Repurposing existing medications prescribed for non-malignant diseases has been successfully used to identify active agents for pre-clinical testing for cancers including NB. FDA-approved drugs have been identified to have anti-cancer properties by unbiased high-throughput cell based screening assays using drug libraries or studies of candidate drugs with proposed mechanisms of actions that may target tumor promoting signaling pathways [3-6]. In pediatrics a recent systematic review of repurposing drugs found that almost 10% of drugs approved for primary use in children have been repurposed for new indications for pediatric oncology or other pediatric indications [7]. In contrast to development of new compounds repurposing approved drugs may result in faster or more streamlined initiation of clinical trials. In order to identify novel therapies for relapsed NB we previously conducted high-throughput screens with the Prestwick library (Prestwick Chemical, Inc) of 1,120 FDA approved drugs using a panel of NB cell lines [6]. In addition to chemotherapies with known efficacy in NB, we identified additional compounds with anti-NB activity including propranolol hydrochloride, a nonselective beta-blocker that competitively inhibits the action of epinephrine (EPI) and norepinephrine (NE) on β1- and β2-adrenoreceptors (AR). Although originally used for the treatment of hypertension and other cardiovascular disorders, recently propranolol has been shown to be effective and safe for the treatment of large hemangiomas in infants [8].
Additional rationale supports potential anti-cancer, and specifically anti-NB effects for β-blockers. Catecholamines and their metabolites increase proliferation of several different cancer cell types in vitro [9,10] and patients with NB often have elevated serum and urinary catecholamines [11]. Anti-tumor activity of propranolol in vitro has also been demonstrated for many cancer cell lines including pancreatic, breast, gastric, head and neck squamous cell carcinoma and leukemia [12-17]. Furthermore, retrospective epidemiology studies suggest that cancer patients treated with beta-blockers have improved outcomes [18-21]. Based on these clinical findings, the pro-proliferative effects of catecholamines, and the safety profile for propranolol in children we hypothesized that the beta blocker propranolol may have potential efficacy in NB [22,23].
In this study we demonstrate that propranolol reduces the viability of human NB cell lines through the inhibition of proliferation and induction of apoptosis. The β2-AR is expressed on NB cell lines and primary tumor tissue, and higher levels of expression correlate with improved survival. However, the level of expression does not correlate with sensitivity to propranolol. Propranolol treatment in vitro is associated with induction of apoptosis and the pro-apoptotic p53 family proteins p53 and p73. Propranolol treatment at doses similar to those used to treat infants with hemangiomas also results in growth inhibition of NB xenografts and induction of p53 in vivo. Our findings suggest that propranolol, alone or in combination with chemotherapy, may be an effective agent in NB.
RESULTS
Propranolol inhibits NB growth, viability, and proliferation
Propranolol is a non-selective β-blocker that competitively inhibits the action of EPI and NE on β1- and β2-AR. To determine the effect of propranolol on NB, a panel of fifteen established human NB cell lines representing a range of genetic profiles (eg. status of MYCN amplification, p53 mutation, 1p and 11q LOH) were treated with increasing doses of propranolol to determine the half-maximal inhibitory concentration (IC50) using alamarBlue, an indicator of metabolic activity and cellular health (Figure 1A). The IC50s ranged from 114 μM to 218 μM (Figure 1B), doses similar to those reported for propranolol in non-NB cancer cell lines, which range from 100-300 µM [12-17]. The IC50 for human umbilical vein endothelial cells (HUVEC) was similar to the IC50 measured for the majority of NB cells (Figure 1C). In contrast to the growth inhibition observed in response to treatment with β-antagonists we detected an increase in proliferation of NB cells in response to β-agonist EPI, at levels consistent with those reported for other non-NB cell types [10,24] (Figure 1D).
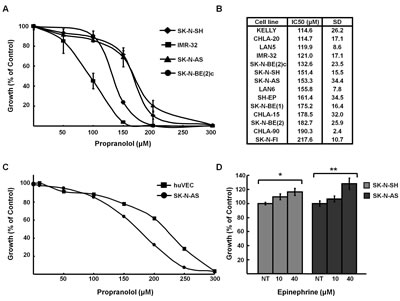
Figure 1: Propranolol inhibits neuroblastoma growth. A, Growth curves of NB cell lines treated with indicated doses of propranolol for 72h. B, IC50 values for fifteen NB cell lines. IC50 measurement was performed with alamarBlue (triplicate). Results represent the average of three experiments expressed as a mean percentage of untreated control cells. C, HUVEC cells were treated with indicated concentrations of propranolol and IC50 was determined with alamar blue (triplicate). Results represent the average of three experiments expressed as a mean percentage of untreated control cells. D, SK-N-SH and SK-N-AS cells were stimulated with indicated doses of EPI for 24h. Cell growth was measured using alamarBlue (triplicate) and normalized to untreated controls. Data shown are representative of three independent experiments and are expressed as means of triplicates ± s.e. *p=0.04, **p=0.011
We next asked whether growth inhibition in response to propranolol was due to changes in viability and/or proliferation. Decreased viability and proliferation were detected by trypan blue (Figure 2A) and BrdU incorporation assays (Figure 2B), respectively. To determine whether topoisomerase I inhibitors such as irinotecan, which is commonly used to treat NB relapse, result in enhanced efficacy cells were treated with the combination of propranolol and the irinotecan active metabolite SN-38. In comparison to cells treated with either drug alone a greater decrease in cell viability was detected for cells treated with the combination (Figure 2C). This effect was synergistic based on the combination index (CI) of 0.719 calculated by the Chou-Talalay method. Propranolol is rapidly metabolized and thus, usually delivered in at least two divided doses per day. For our in vitro assays a single dose was delivered prior to performing specific growth or proliferation assays. In order to determine longer-term effects of lower doses of treatment in vitro we used a focus formation assay that assesses self-renewal capacity, in which cells were treated for 14 days with propranolol replaced daily. There was a significant decrease in the number of foci or colonies in a dose dependent manner following 14 days of treatment (Figure 2D). Compared to control cells, foci were reduced by 50% following treatment with 25μM propranolol and 84% with 50μM propranolol.
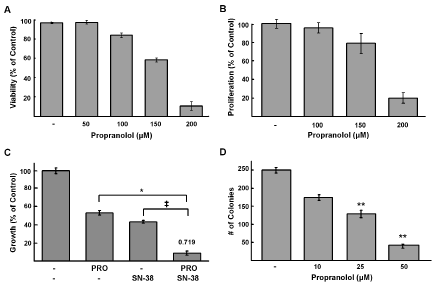
Figure 2: Propranolol inhibits NB viability and proliferation and is synergistic with SN-38. A, SK-N-AS cells were incubated with indicated doses of propranolol for 24h and cell viability was determined using trypan blue exclusion method. Results are expressed as a mean percentage of trypan blue negative cells (live cells); data shown are representative of three independent experiments and are expressed as means of triplicates ± s.d. B, SK-N-SH cells were treated with indicated doses of propranolol for 24h. Cell proliferation was assessed by BrdU incorporation. Results are a mean percentage of control cells. C, alamarBlue assays were performed on SK-N-AS cells treated with increasing doses of propranolol and SN-38 at a ratio of 10,000:1 (100μM propranolol: 0.01μM SN-38) for 48hr. Results are expressed as percentage compared to controls. The combination index (CI) was determined based on the Chou-Talalay method is shown on the top of the bar representing combination treatment * p= 0.008, ‡ p= 0.0009. D, Concentration-dependent effects of propranolol on clonogenic growth. SK-N-AS cells were treated for 14 days with propranolol at indicated doses or media alone (control); colonies were detected by staining with crystal violet. Bars represent the mean number of colonies of triplicate wells from three independent experiments ± s.d; ** p <0.001 students t-test.
Growth inhibition is specific to β2-blockers and β2-AR are expressed in NB
To determine whether the growth inhibitory effects of propranolol were a result of its inhibitory effects on β1- or β2-AR, SK-N-BE(2)c cells were treated with (±)-propranolol HCl, the β1-specific antagonist metoprolol tartrate or the β2-specific antagonist ICI 118,551 hydrochloride. While the β1-specific antagonist had no effect on viability, the β2-specific antagonist ICI118,551 was slightly more potent than propranolol, suggesting that propranolol may induce cell death in NB via β2-AR specific antagonism (Figure 3A). We also tested three different enantiomers of propranolol HCl ((S)-(−)-propranolol HCl, (R)-(+)-propranolol HCl, (±)-propranolol HCl) and detected no significant difference in potency between the racemic mixture and the two enantiomers (Figure 3A). We next asked whether NB cells express β2-AR mRNA (ADRB2) and whether the levels observed vary in cell lines with differing sensitivity to propranolol. All NB cell lines tested expressed ADRB2 and there was no direct correlation between the IC50 and the level of ADRB2 mRNA detected (Figure 1 and Figure 3B). Most cultured NB cell lines are isolated from patients with metastatic NB. To determine whether the levels of ADRB2 in primary NB tumors correlate with prognosis or known biological risk factors we examined expression data in publically available databases [25-29]. Using data from the R2: microarray analysis and visualization platform (http://r2.amc.nl), which includes annotated data on 88 patients (Versteeg dataset) [29], a univariate analysis showed that higher levels of ADRB2 mRNA correlate with improved survival (Figure 3C). These findings were confirmed in a second cohort, Neuroblastoma Oncogenomics Database (http://pob.abcc.ncifcrf.gov/cgi-bin/JK) (data not shown). In contrast, mRNA levels of the β1-AR ADRB1 did not predict differences in prognosis (Figure 3D). Furthermore, in comparison to the expression of ADRB2 in normal adrenal tissue the relative expression of ADRB2 is higher in NB tumors (Figure 3E). ADRB2 is detected in tumors from patients with all stages of disease, but relatively higher levels were observed in patients without MYCN amplification (Figure 3F) and those <1 year of age (data not shown).
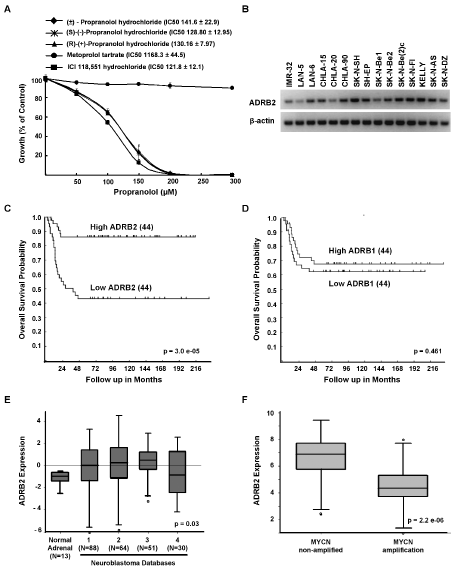
Figure 3: Growth inhibition is specific to β2-blockers and β2-AR is expressed in NB. A, SK-N-SH was treated with increasing doses of either (±)-propranolol HCl enantiomers and a β1-AR specific antagonist, metoprolol tartrate, or a β2-AR specific antagonist, ICI 118,551 HCl for 72 hours. Results are expressed as percentages of growth compared with untreated controls. Data shown are representative of three independent experiments and are expressed as means of triplicates ± s.d. For each β-AR antagonist, the IC50 of each cell line is shown with the corresponding legend. B, β2-AR (ADRB2) expression in NB cell lines as demonstrated by semi-quantitative PCR. C-D, Cumulative overall survival probability curves of patients with neuroblastoma based on expression of ADRB2 mRNA [C] or ADRB1 mRNA [D] (N=88 patients, Versteeg dataset). The cut-off for these analyses is the median expression level, high and low represent levels of expression for 44 patients in each group relative to the mean. E, Relative ADRB2 mRNA expression in normal adrenal tissue and neuroblastoma samples. F, Relative ADRB2 mRNA expression in MYCN non-amplified and amplified neuroblastoma samples. Kaplan-Meier curves and Box blots were generated from the R2: microarray analysis and visualization platform (http://r2.amc.nl), which includes data submitted by different investigators (Databases: 1, Versteeg; 2, Delattre; 3, Hiyama; 4, Lastowska).
Propranolol induces apoptosis in neuroblastoma
To determine if the decreases in NB cell growth and viability following propranolol treatment was due to induction of cell death, we performed three assays to detect apoptosis. First, we detected an 8- to 15-fold increase in caspase 3 and 7 activity (Figure 4A). In response to propranolol treatment there was a 2.5 fold increase in the number of live NB cells undergoing apoptosis as detected by AnnexinV positivity (Figure 4B). Finally, the levels of poly-ADP-ribose polymerase (PARP) cleavage increased in both a dose- and time-dependent manner (Figure 4C).
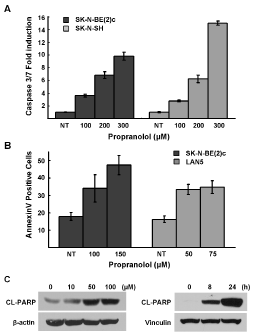
Figure 4: Propranolol induces apoptosis. A, SK-N-SH and SK-N-BE(2)c were treated with indicated doses of propranolol for 5h and cellular caspase 3/7 activities were analyzed. Results are representative of three independent experiments, performed in triplicate and are expressed as mean fold-induction over the control cells ± s.d. B, LAN5 and SK-N-BE(2)c cells were treated with indicated doses (24 hours) and live cells were analyzed for induction of annexinV positivity by flow cytometry. Results are expressed as mean fold-induction over the untreated control ± s.d. C, SK-N-BE(2)c cells were treated with indicated doses for 48h and CHLA-20 cells were treated with 150 μM of propranolol for indicated durations and then analyzed for PARP cleavage by immunoblot analysis.
Propranolol increases p53 and TAp73 and pro-apoptotic target genes
We next addressed potential downstream apoptotic signaling mechanisms involved in propranolol induced NB cell death. A number of signaling pathways have been implicated in EPI-mediated activation of β2-AR, including activation of mitogen-activated protein kinase/extracellular signal-related kinase (MAPK/ERK) and changes in p53. Hara and colleagues recently demonstrated that in response to catecholamines including EPI and NE, β2-AR signaling led to DNA-damage and activation of β-arrestin-1 as well as AKT, which phosphorylates HDM2 (pHDM2), resulting in p53 degradation [30]. Therefore, we predicted that inhibition of the β2-AR with propranolol may result in increased levels of p53, which is required for apoptosis in response to common chemotherapies as well as other drugs studied in preclinical NB models [31,32]. Following treatment with propranolol, we detected an increase in p53 as well as a time-dependent decrease in the levels of the phosphorylated form of the p53 negative regulator HDM2 (pHDM2) (Figure 5A). Some of the NB cell lines that are sensitive to propranolol express mutant and/or inactive p53 proteins (eg. SK-N-B(E)2, SK-N-AS, SK-N-FI). We and others have shown that the p53 paralogue p73 can activate p53 target genes that mediate apoptosis in response to a number of chemotherapeutic agents [33]. Thus, we asked whether the protein levels of the pro-apoptotic p73 isoform TAp73β changed in response to propranolol. Following propranolol treatment TAp73β isoform levels increased (Figure 5B). In addition, the protein levels of the p53 and TAp73 downstream pro-apoptotic target gene proteins NOXA and p57kip2 also increased (Figure 5C, 5D). We next examined whether activation of p53 and p73 might be due to DNA damaging effects of propranolol. As expected levels of γH2AX, a marker for double strand DNA breaks, increased in response to doxorubicin; however, no increase in γH2AX was detected following propranolol treatment (Supplemental Figure 1). We examined other downstream pathways previously implicated in the proliferative response to EPI in other cell types. Specifically, in response to propranolol treatment we observed decreases in levels of cyclo-oxygenase-2 (COX-2) (Figure 5E). Since we and others have demonstrated anti-NB activity for the COX-2 inhibitor celecoxib via effects of p53/p73 and VEGF signaling [31,34,35] the effects of celecoxib in combination with propranolol were assessed. Interestingly, celecoxib was synergistic with propranolol based on the combination index (CI) of 0.604 (Figure 5F).
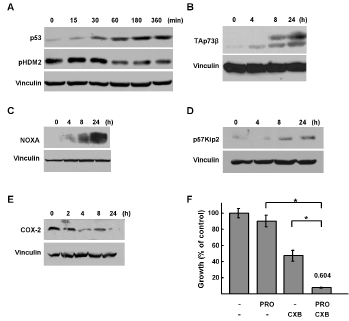
Figure 5: Regulation of p53 family signaling by propranolol. A, p53 and pHDM2 western immunoblots of lysastes from SK-N-SH cells treated with propranolol (100 μM) for the indicated durations. B-E, TAp73β, NOXA and p57kip2, COX-2, western immunoblots of lysates from SK-N-AS cells treated with 150 μM of propranolol for the indicated durations. F, alamarBlue assays were performed on SK-N-AS cells treated with increasing doses of propranolol and Celecoxib (CXB) at a ratio of 2:1 (150μM propranolol: 75 μM CXB) for 48hr. Results are expressed as percentage compared to controls. The combination index (CI) was determined based on the Chou-Talalay method is shown on the top of the bar representing combination treatment * p<0.0001
Propranolol inhibits growth in vivo
The doses of propranolol required to inhibit NB growth in short term assays in vitro were higher than those predicted to be achievable in vivo. However, since focus formation assays demonstrated efficacy with lower doses of propranolol, we next investigated the effects of propranolol on growth of SK-N-AS NB xenografts. When tumors reached 50mm3, mice were injected subcutaneously with 2mg/kg/day (1mg/kg/day twice a day) for fourteen days. The tumors of mice treated with propranolol demonstrated slower growth as compared to PBS vehicle-treated control mice. The mean tumor volume of propranolol-treated group was lower than the control group on day 14: 197.6 ± 44.3 mm3 versus 414.5 ±76.6 mm3, unpaired t-test p = 0.0246) (Figure 6A, 6B). Notably, the mean body weight of propranolol-treated mice was not statistically significantly lower that that of the vehicle treated (control) mice, suggesting that propranolol exposure was not resulting in significant short-term toxicity (Figure 6A). Mice treated with propranolol also had a prolonged survival in comparison to control mice (Figure 6C). Importantly, similar to our in vitro results we also detected induction of p53 and cleaved PARP in lysates from xenografts of mice treated with propranolol (Figure 6D).
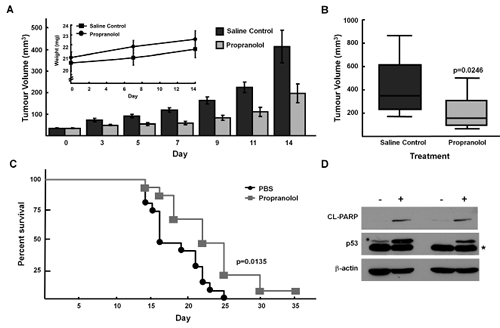
Figure 6: Propranolol inhibits neuroblastoma tumor growth in vivo. NOD/SCID mice with SK-N-AS xenografts were treated with propranolol (1mg/kg bid) by subcutaneous injection for 14 days. A, Tumour volumes measured at indicated days post-injection of cells. Inset, absolute mean body weight, in grams, after the beginning of the treatment. Columns depict mean values; bars, SE. B, Box plot depicting mean tumour volumes of control (vehicle) treated and propranolol treated tumours at day 14. Each treatment group was composed of 10 mice (p<0.05). Bars depict mean values and error bars represent 95% confidence intervals. p values (2-tailed) were calculated using students t-test and are compared to control groups at indicated time points. C, Kaplan–Meier survival curve of NOD/SCID mice from control-treated (black, circle; N=15) or propranolol-treated (grey, square; N=15) tumours. Wilcoxon: p= 0.0135, Log-Rank: p=0.0062. D, p53 and cleaved-PARP western immunoblots of lysates from SK-N-AS xenografts harvested day 14 (lanes 1,2) and day 22 (lanes 3,4) post vehicle control or propranolol (1mg/kg/dose bid). Note: asterisk refers to a faster migrating non-specific band detected in p53 immunoblot of SK-N-AS xenograft lysates.
DISCUSSION
In our study, we found that the non-selective β2-AR antagonist propranolol inhibits NB growth and that β2, but not β1, antagonists were active against a panel of human NB cell lines at doses comparable to those previously reported in vitro for other cancer cell types. Propranolol induced apoptosis that correlated with induction of p53 and the p53 paralogue TAp73 as well as activation of downstream p53/p73 target genes. In addition, propranolol was synergistic with the topoisomerase inhibitor SN-38 and the COX-2 inhibitor celecoxib, and had efficacy in vivo against NB xenografts.
Although initially used in the treatment of hypertension and arrhythmias in adults and children, recently propranolol has become standard therapy in infants with large hemangiomas, proliferative lesions of vascular endothelial cells [8,36]. β-ARs and their antagonists have also been shown to have efficacy in several pre-clinical cancer models. Catecholamines including EPI and NE as well as the β-AR agonist isoproterenol have pro-proliferative effects on cancer cell lines derived from lung, pancreatic, and oral squamous cell carcinomas [9,10,24,37-40]. β-ARs are expressed on multiple malignant cell types and treatment with propranolol has been reported to reduce viability, and in some cases induce apoptosis, of cancer cell lines including pancreatic, gastric, leukemia, melanoma, and oral squamous cell carcinoma [12-14,17,18,41]. Recent epidemiology studies provide further evidence for the potential anti-cancer effects of β-blockers. A ten year longitudinal study found that women with breast cancer receiving β-blocker therapy for hypertension had fewer metastases, recurrences and decreased mortality [16]. In a second retrospective study of breast cancer patients, women receiving the nonselective β-blocker propranolol, but not those receiving β1-specific antagonists, were significantly less likely to present with advanced stage tumors [19]. Similar trends have been reported in retrospective analyses of patients with other types of tumors including melanoma [18,42,43].
Rationale for the use of β-AR antagonist propranolol specifically in NB include the observation that the majority of NB tumors produce elevated levels of catecholamines including EPI and NE, both of which have been shown to have pro-proliferative effects when added to cells in culture [9,10,24,37-40]. We also detected enhanced growth of NB cell lines in response to EPI as well as growth inhibition in response to propranolol treatment in 15 NB cell lines at concentrations similar to those reported in cell lines derived from other cancers. Although MYCN status has been linked to the sensitivity of NB cell lines to several drugs including aurora kinase inhibitors we did not detect a difference in the IC50 values based on MYCN amplification [44,45]. The cellular signaling events that mediate apoptosis in response to propranolol are poorly understood. Recent data demonstrated that in primary and transformed cells, β2-AR signaling activated by catecholamines leads to βarrestin-1 activation, which results in enhanced AKT-mediated phosphorylation and inactivation of HDM2. In response to β2-AR blockade with propranolol we detected decreased levels of the phosphorylated form of HDM2 (pHDM2) and increased p53 protein. We further found that treatment with propranolol resulted in induction of TAp73. The full-length form of the p53 paralogue p73, TAp73, can also mediate apoptotic responses to chemotherapies as well as other anti-cancer agents with efficacy in NB cells, including chemotherapies, aurora kinase inhibitors, and COX-2 inhibitors, particularly when p53 is inactivated [33,35]. Accordingly, we found induction of TAp73β and activation of p53/p73 pro-apoptotic genes NOXA and PUMA as well as p57KIP2, a cyclin-dependent kinase inhibitor, the promoter for which has previously been identified to contain p53 and p73 response elements [46-48]. Furthermore, recent reports support a role for p53 family proteins in the regulation of genes involved in angiogenesis, raising the possibility that previously reported anti-angiogenic effects of propranolol may be in part mediated by p53 or p73 [49]. The mechanism for induction of TAp73 in response to propranolol is not clear, but is not associated with DNA-damage as measured by γH2AX foci. Instead, the increased TAp73 but may be in part related to the decreased levels of COX-2 observed following propranolol treatment since COX inhibitors have been shown to induce TAp73β protein and activity [35].
β2-specific AR antagonists, but not β1-specfic AR antagonists, inhibited the growth of NB cell lines in vitro; suggesting that expression of the β2-AR may be required for propranolol-induced cell death. Interestingly, the specific RNA levels of the β2-AR did not correlate with propranolol sensitivity in vitro, but were fairly similar in the 15 cell lines that we tested. β2-AR mRNA (ADRB2) is expressed in the majority of NB tumors, as well as cell lines derived from metastatic tumors, at levels that are higher than those detected in normal adrenal tissue. Interestingly, relative levels of ADRB2, but not ADRB1, correlate with favorable prognosis. The observation that high levels of ADBR2 are associated with more favorable prognosis is similar to findings for patients with other types of cancers, including oral squamous cell carcinoma (OSCC), leukemia, and prostate cancer [50-52]. Our findings demonstrate that the majority of patients have tumors that express β2-AR and thus, if expression is required for response, we would predict that most NB tumors would be sensitive to propranolol. However, it is also possible that growth inhibitory effects of propranolol may not be due specifically to inhibition of the β2-AR and may instead be due to off-target effects.
Although the dose required to induce apoptosis in vitro was high (100μM range), tumor inhibition in SK-N-AS xenografts was observed at doses similar to those used to treat infants with hemangiomas (1mg/kg/day bid). Interestingly, the IC50 for hemangioma-derived cells in vitro (100μM) is similar to those for NB cells in vitro [53]. Pharmacokinetic data suggests that in patients treated with propranolol the peak serum concentrations range from 200-400 ng/ml, which is equivalent to 0.77-1.5µM [54]. There are many possible explanations for this significant discrepancy between in vitro and in vivo effects. First, propranolol has been reported to inhibit pro-angiogenic factors, such as VEGF, and thus, in vivo tumor inhibition may be due to effects on endothelial cells in the blood vessels that surround the tumor. Although we did not detect significant difference in the IC50 for HUVEC endothelial cells in comparison to NB cells in vitro, which is supported by previous reports [55], a recent publication by Pasquier and colleagues demonstrates that propranolol can potentiate the anti-angiogenic effects vincristine in vivo , which included decreased capillary vessel formation [56]. Another potential mechanism may involve effects on the immune system. The β-adrenergic agonist metoproterenol inhibits Natural killer (NK) cell function, and its effects are reversible by treatment with propranolol [57]. Thus, future studies examining effects of propranolol on the tumor microenvironment or anti-tumor NK function may be warranted.
Despite the discrepancy between in vitro and in vivo concentrations the doses effective against NB SK-N-AS xenografts were similar to those used in regimens for hypertension and those reported for children with hemangiomas (2-3 mg/kg/day) [8,22,36,58]. The in vitro synergy with the irinotecan metabolite SN-38 and COX-2 inhibitors also supports potential combination therapy with topoisomerase inhibitors, such as irinotecan or topotecan, or COX inhibitors such as celecoxib in relapsed patients including those with central nervous system (CNS) metastases since propranolol, which is highly lipophilic, is known to cross the blood brain barrier and concentrate in the central nervous system (CNS) [59]. In a recent study by Pasquier and colleagues propranolol alone did not demonstrate single agent efficacy when administered to TH-MYCN transgenic mice, but was synergistic with vincristine and vinblastine, two agents commonly used for relapsed NB [60].
Taken together our results suggest that targeting the β2-AR with propranolol, which has been used in children for almost 50 years and more recently in infants with large hemangiomas, may be effective in children with NB. NB cell lines derived from metastases are sensitive to propranolol mediated growth inhibition. Although additional studies are required to determine whether β2-AR protein levels or other biomarkers predict which tumors are most sensitive to beta blockade in vivo our results suggest the majority of NB tumors express ADRB2 and thus may respond to β-AR antagonists. Therefore, together with recent retrospective studies that demonstrate improved outcomes for adult cancer patients receiving β-blockers, our data suggests that propranolol should be considered in combination with chemotherapies for the treatment of patients with relapsed or refractory neuroblastoma and possibly for neuroblastoma patients requiring anti-hypertensive therapy. Furthermore, early phase clinical trials for refractory neuroblastoma utilize tumor biopsies for expression profile analyses in an effort to identify FDA-approved drug combinations that may be effective for individual patients, and thus, our findings may provide additional rationale to include propranolol in regimens for subsets of these patients [61,62].
MATERIALS AND METHODS
Cell culture and Drugs
Established human neuroblastoma cell lines, KELLY, CHLA-20, LAN-5, IMR-32, SK-N-BE1, SK-N-BE(2), SK-N-BE(2)c, SK-N-SH, SK-N-AS, LAN-6, SH-EP, CHLA-15, CHLA-90, SK-N-FI were cultured in RPMI containing 10% fetal bovine serum (Invitrogen, Burlington, ON, Canada) and obtained from American Type Culture Collection and Dr. Patrick Reynolds (Children’s Oncology Cell Bank). Cells were incubated at 37°C and 5% CO2 tissue culture incubator. Drugs: (±)-Propranolol hydrochloride, (S)(−)-propranolol HCl, (R)(+)-propranolol HCl, Metoprolol tartrate, ICI 118,551 hydrochloride and Epinephrine-HCL (Sigma, St. Louis, MO, USA) were dissolved in water; SN-38 (Tocris Bioscience, Bristol, UK) and Celecoxib (Toronto Research Chemicals, Toronto, ON, CA) were dissolved in DMSO.
Apoptosis assays
Cells were seeded into a 96-well plate at 1.5x104 cells per well and treated 24 hours later with propranolol for five hours. The combined caspase-3 and caspase-7 activity was measured using the Apo-ONE caspase 3/7 Assay Kit, according to manufacturer’s instruction (Promega, Madison, WI, USA). Fluorescence was read using a luminometer (Spectra MAX Gemini EM, Molecular Devices) at 499/521 nM wavelengths. For annexinV assays cells treated with propranolol for 24 h were harvested and stained with propidium iodide (PI) and APC-AnnexinV (Dead Cell Apoptosis kit, Invitrogen) according to the manufacturer’s instructions. The PI negative population was selected and analyzed for AnnexinV staining using a FACS Canto II flow cytometry (Beckton Dickinson).
Cell Viability, Proliferation and Focus formation Assays
IC50 values were determined with alamarBlue assay (Invitrogen, Burlington, ON, Canada). 3-6 x103 cells were plated in 96-well plates and treated with indicated doses of propranolol for 72 hours. At 48 hours, alamarBlue was added (10% of total volume) and then incubated overnight. The fluorescence was measured using a spectrophotometer at excitation 530 nm and emission 590 nm (Spectra MAX Gemini EM, Molecular Devices). Cell proliferation was assessed using Bromodeoxyuridine (BrdU) (Cell Signaling, Danvers, MA, USA). 4000 cells were treated with indicated drug and then incubated with BrdU (10μM) for 4h and detected using anti-BrdU antibody. The absorbance was measured at 450nM using a spectrophotometer plate reader (VersaMax tunable microplate reader, Molecular Devices). Trypan blue exclusion assay (Gibco, St. Louis, MO, USA) was used to measure viability following 24 hours of propranolol treatment. Live and dead cells (trypan blue positive) were counted in triplicate using a hemocytometer. For the colony or foci formation assay cells were seeded (102/well in six-well plates) in 2 ml of growth medium and incubated overnight at 37°C. Propranolol or media was added at the specified concentrations. Every 24h fresh propranolol and growth medium was added, and the plates were incubated at 37°C. Fourteen days after seeding, colonies were fixed in 70% ethanol and stained with 10% methylene blue. Colonies of 50 cells were counted.
Western immunoblot analysis
Whole cell extracts were prepared using EBC buffer (50mM Tris (pH 8.0), 120 mM NaCl, 0.5% NP-40) with protease inhibitors (Roche Diagnostics, Laval, QC, Canada) and total protein concentration was determined using Bradford reagent (Bio-Rad laboratories, Hercules, CA, USA). Equal amounts were resolved by SDS-PAGE and transferred to nitrocellulose membrane. The membranes were blocked in 5% milk/tris-buffered saline with tween (TBST), probed with indicated primary antibodies, horseradish peroxidase-conjugated secondary antibodies (Thermo Fisher Scientific, Rockford, IL, USA). Protein was isolated from tumors by homogenization in RIPA buffer containing protease inhibitors. Proteins were detected by an enhanced chemiluminescene system (Super Signal West Pico, Thermo Fisher Scientific, Rockford, IL, USA). Primary antibodies include: Cl-PARP, pHDM2ser116 (Cell Signaling Technology, Beverly, MA, USA), TAp73/GC-15, p53/DO-1/AB-6 (Oncogene, La Jolla, CA, USA), Vinculin (Upstate, Lake Placid, NY, USA), NOXA/114C307 (Novus Biological LLC, Littleton, CO, USA), p57kip2 /c-20 (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
γH2AX immunostaining
SK-N-AS cells seeded in uncoated Labtek chamber slides (Nunc) (80,000 cells/chamber), were grown for 24 hrs and treated for an additional 24 hrs with 0.5mL media containing the indicated. Cells were fixed with 4% PFA, permeabilized with 0.2% Triton X-100 for 5 min and blocked with 6% Normal Goat Serum in 0.5% BSA for 1 hr. Slides were stained with primary antibody Phospho-Histone H2AX (Ser139) (20E3) Rabbit mAB (Cell Signaling) (1:400) overnight at 4ºC, secondary Alexa Fluor 488 Goat Anti-Rabbit IgG (H+L) (Life Technologies) (1:5000) for 1hr at room temperature and DAPI (1:5000) and visualized using spinning disc confocal microscopy. 50 fields-of-view (FOV) were captured and cells were scored positive for the presence of γH2AX foci and compared to the number of foci in untreated cells.
Semi-quantitative PCR
Total RNA was extracted using TRIzol (Invitrogen). cDNA was generated using the Omniscript RT Kit (Qiagen, Mississauga, ON, Canada) and amplified by semi- using the Taq DNA polymerase Kit (Qiagen). RT-PCR conditions: initial denaturation (95°C/3min), 40 cycles of denaturation (94°C/1min), annealing (55°C/1min) and extension (72°C/1min), final extension (72°C/ 10min). Primer sequences for ADRB2 are: 5’-GAGCAAAGCCCTCAAGAC-3’ and 5’-TGGAAGGCAATCCTGAATC-3’, β-actin 5’-CTGGAACGGTGAAGGTGACA-3’ and 5’-AAGGGACTTCCTGTAACAATGCA-3’. The RT-PCR products were resolved using a 1.5% agarose gel.
Combination Index and Statistical analyses
For combination treatment, cells treated in a ratio of 10,000:1 (Propranolol:SN-38) or 2:1 (Propranolol:CXB) for 48 hours were subjected to alamarBlue assay. Using CalcuSyn Software (Biosoft, Cambridge, UK) the combination index (CI) was calculated [63,64]. CI<1 and CI>1 indicated synergism and antagonism, respectively, and CI=1 indicates an additive effect. Comparisons between 2 groups were done using unpaired Student t-test with the Graphpad Prism software (GraphPad Software, Inc., version 3.0). The Kaplan–Meier method was used to determine survival of mice. All p values < 0.05 were considered statistically significant.
Xenograft studies
NOD/SCID (non-obese diabetic/severe combined immunodeficiency) mice were injected with 1.5x106 SK-N-AS cells in PBS and Matrigel Basement Membrane Matrix (100μM) (BD biosciences, Franklin Lakes, NJ USA) subcutaneously in the left flank. When xenografts reached 50mm3 mice were treated with propranolol (Sigma, Israel) dissolved in phosphate buffered saline (PBS) or PBS alone (control) administrated subcutaneously at a dose of 1 mg/kg twice daily for up to 35 days. Tumor growth was monitored three times a week and tumor volume (mm3) was calculated. Mice were sacrificed when tumors were greater 500mm3. Tumor volumes were compared on Day 14 in two independent experiments (N=5 and N=10 per group). Survival curves were generated by combining two experiments (N=15 per group).
ACKNOWLEDGMENTS
This work was supported by Canadian Cancer Society Research Institute Grant to MSI, and funds from the James Birell Fund for Neuroblastoma Research and Lilah’s Fund. DRK and MSI are Canada Research Chairs.
References
1. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer 2003; 3: 203–216
2. Maris JM. Recent advances in neuroblastoma. N Engl J Med 2010; 362: 2202–2211
3. Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DVS, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam GS. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discov 2011; 10: 188–195
4. Spagnuolo PA, Hu J, Hurren R, Wang X, Gronda M, Sukhai MA, Di Meo A, Boss J, Ashali I, Beheshti Zavareh R, Fine N, Simpson CD, Sharmeen S, Rottapel R, Schimmer AD. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood 2010; 115: 4824–4833
5. Yip KW, Mao X, Au PYB, Hedley DW, Chow S, Dalili S, Mocanu JD, Bastianutto C, Schimmer A, Liu F-F. Benzethonium chloride: a novel anticancer agent identified by using a cell-based small-molecule screen. Clin Cancer Res 2006; 12: 5557–5569
6. Smith KM, Datti A, Fujitani M, Grinshtein N, Zhang L, Morozova O, Blakely KM, Rotenberg SA, Hansford LM, Miller FD, Yeger H, Irwin MS, Moffat J, Marra MA, Baruchel S, Wrana JL, Kaplan DR. Selective targeting of neuroblastoma tumour-initiating cells by compounds identified in stem cell-based small molecule screens. EMBO Mol Med 2010; 2: 371–384
7. Blatt J, Corey SJ. Drug repurposing in pediatrics and pediatric hematology oncology. Drug Discovery Today 2013; 18: 4–10
8. Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo J-B, Taïeb A. Propranolol for severe hemangiomas of infancy. N Engl J Med 2008; 358: 2649–2651
9. Bernabe DG, Tamae AC, Biasoli ÉR, Oliveira SHP. Stress hormones increase cell proliferation and regulates interleukin-6 secretion in human oral squamous cell carcinoma cells. Brain Behavior and Immunity 2011; 25: 574–583
10. Liu X, Wu WKK, Yu L, Sung JJY, Srivastava G, Zhang ST, Cho CH. Epinephrine stimulates esophageal squamous-cell carcinoma cell proliferation via beta-adrenoceptor-dependent transactivation of extracellular signal-regulated kinase/cyclooxygenase-2 pathway. J Cell Biochem 2008; 105: 53–60
11. Eldeeb BB, Hammond EM, Worthington DJ, Mann JR. Urinary catecholamines and their metabolites in management of neuroblastoma. Pediatr Hematol Oncol 1988; 5: 229–237
12. Hajighasemi F, Mirshafiey A. In vitro sensitivity of leukemia cells to propranolol. J Clin Med Res 2009; 1: 144–149
13. Zhang D, Ma Q-Y, Hu H-T, Zhang M. β2-adrenergic antagonists suppress pancreatic cancer cell invasion by inhibiting CREB, NFκB and AP-1. Cancer Biol Ther 2010; 10: 19–29
14. Zhang D, Ma Q, Shen S, Hu H. Inhibition of pancreatic cancer cell proliferation by propranolol occurs through apoptosis induction: the study of beta-adrenoceptor antagonist’s anticancer effect in pancreatic cancer cell. Pancreas 2009; 38: 94–100
15. Liao X, Che X, Zhao W, Zhang D, Bi T, Wang G. The β-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor κB signaling. Oncol Rep 2010; 24: 1669–1676
16. Powe DG, Voss MJ, Zänker KS, Habashy HO, Green AR, Ellis IO, Entschladen F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. 2010; 1: 628–638
17. Wolter NE, Wolter JK, Enepekides DJ, Irwin MS. Propranolol as a novel adjunctive treatment for head and neck squamous cell carcinoma. J Otolaryngol Head Neck Surg 2012; 41: 334–344
18. De Giorgi V, Grazzini M, Gandini S, Benemei S, Lotti T, Marchionni N, Geppetti P. Treatment with β-blockers and reduced disease progression in patients with thick melanoma. Arch Intern Med 2011; 171: 779–781
19. Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population- based study. Journal of Clinical Oncology 2011; 29: 2635–2644
20. Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD, Hortobagyi GN, Gonzalez-Angulo A-M. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. Journal of Clinical Oncology 2011; 29: 2645–2652
21. Diaz ES, Karlan BY, Li AJ. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol Oncol 2012; 127: 375–378
22. Pickoff AS, Zies L, Ferrer PL, Tamer D, Wolff G, Garcia O, Gelband H. High-dose propranolol therapy in the management of supraventricular tachycardia. The Journal of Pediatrics 1979; 94: 144–146
23. Love JN, Sikka N. Are 1–2 tablets dangerous? Beta-blocker exposure in toddlers. J Emerg Med 2004; 26: 309–314
24. Huang X-Y, Wang H-C, Yuan Z, Huang J, Zheng Q. Norepinephrine stimulates pancreatic cancer cell proliferation, migration and invasion via β-adrenergic receptor-dependent activation of P38/MAPK pathway. Hepatogastroenterology 2012; 59: 889–893
25. Asgharzadeh S, Pique-Regi R, Sposto R, Wang H, Yang Y, Shimada H, Matthay K, Buckley J, Ortega A, Seeger RC. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. JNCI Journal of the National Cancer Institute 2006; 98: 1193–1203
26. Fix A, Lucchesi C, Ribeiro A, Lequin D, Pierron G, Schleiermacher G, Delattre O, Janoueix-Lerosey I. Characterization of amplicons in neuroblastoma: high-resolution mapping using DNA microarrays, relationship with outcome, and identification of overexpressed genes. Genes Chromosomes Cancer 2008; 47: 819–834
27. Ohtaki M, Otani K, Hiyama K, Kamei N, Satoh K, Hiyama E. A robust method for estimating gene expression states using Affymetrix microarray probe level data. BMC Bioinformatics 2010; 11: 183
28. Lastowska M, Cotterill S, Pearson AD, Roberts P, McGuckin A, Lewis I, Bown N. Gain of chromosome arm 17q predicts unfavourable outcome in neuroblastoma patients. U.K. Children’s Cancer Study Group and the U.K. Cancer Cytogenetics Group. European Journal of Cancer 1997; 33: 1627–1633
29. Molenaar JJ, Koster J, Zwijnenburg DA, van Sluis P, Valentijn LJ, van der Ploeg I, Hamdi M, van Nes J, Westerman BA, van Arkel J, Ebus ME, Haneveld F, Lakeman A, Schild L, Molenaar P, Stroeken P, van Noesel MM, Ora I, Santo EE, Caron HN, Westerhout EM, Versteeg R. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012; 483: 589–593
30. Hara MR, Kovacs JJ, Whalen EJ, Rajagopal S, Strachan RT, Grant W, Towers AJ, Williams B, Lam CM, Xiao K, Shenoy SK, Gregory SG, Ahn S, Duckett DR, Lefkowitz RJ. A stress response pathway regulates DNA damage through β2-adrenoreceptors and β-arrestin-1. Nature 2011; 477: 349–353
31. Lau L, Hansford LM, Cheng LS, Hang M, Baruchel S, Kaplan DR, Irwin MS. Cyclooxygenase inhibitors modulate the p53/HDM2 pathway and enhance chemotherapy-induced apoptosis in neuroblastoma. Oncogene 2007; 26: 1920–1931
32. Wolter J, Angelini P, Irwin M. p53 family: Therapeutic targets in neuroblastoma. Future Oncol 2010; 6: 429–444
33. Irwin MS, Kondo K, Marin MC, Cheng LS, Hahn WC, Kaelin WG. Chemosensitivity linked to p73 function. Cancer Cell 2003; 3: 403–410
34. Subbaramaiah K. Inhibition of Cyclooxygenase-2 Gene Expression by p53. Journal of Biological Chemistry 1999; 274: 10911–10915
35. Lau LMS, Wolter JK, Lau JTML, Cheng LS, Smith KM, Hansford LM, Zhang L, Baruchel S, Robinson F, Irwin MS. Cyclooxygenase inhibitors differentially modulate p73 isoforms in neuroblastoma. Oncogene 2009; 28: 2024–2033
36. Cushing SL, Boucek RJ, Manning SC, Sidbury R, Perkins JA. Initial Experience With a Multidisciplinary Strategy for Initiation of Propranolol Therapy for Infantile Hemangiomas. Otolaryngology -- Head and Neck Surgery 2010; 144: 78–84
37. Shang ZJ, Liu K, Liang DF. Expression of beta2-adrenergic receptor in oral squamous cell carcinoma. J Oral Pathol Med 2009; 38: 371–376
38. Pérez Piñero C, Bruzzone A, Sarappa MG, Castillo LF, Lüthy IA. Involvement of α2- and β2-adrenoceptors on breast cancer cell proliferation and tumour growth regulation. Br J Pharmacol 2011; 166: 721–736
39. Chan C, Lin H-J, Lin J. Stress-associated hormone, norepinephrine, increases proliferation and IL-6 levels of human pancreatic duct epithelial cells and can be inhibited by the dietary agent, sulforaphane. int J Oncol 2008; 33: 415–419
40. Park PG, Merryman J, Orloff M, Schuller HM. Beta-adrenergic mitogenic signal transduction in peripheral lung adenocarcinoma: implications for individuals with preexisting chronic lung disease. Cancer Research 1995; 55: 3504–3508
41. Weddle DL, Tithoff P, Williams M, Schuller HM. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis 2001; 22: 473–479
42. Wang HM, Liao ZX, Komaki R, Welsh JW, O’Reilly MS, Chang JY, Zhuang Y, Levy LB, Lu C, Gomez DR. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann Oncol 2013; 24: 1312–1319
43. Lemeshow S, Sørensen HT, Phillips G, Yang EV, Antonsen S, Riis AH, Lesinski GB, Jackson R, Glaser R. β-Blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomarkers Prev 2011; 20: 2273–2279
44. Faisal A, Vaughan L, Bavetsias V, Sun C, Atrash B, Avery S, Jamin Y, Robinson SP, Workman P, Blagg J, Raynaud FI, Eccles SA, Chesler L, Linardopoulos S. The aurora kinase inhibitor CCT137690 downregulates MYCN and sensitizes MYCN-amplified neuroblastoma in vivo. Molecular Cancer Therapeutics 2011; 10: 2115–2123
45. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, Berwanger B, Eilers M. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009; 15: 67–78
46. Blint E, Phillips AC, Kozlov S, Stewart CL, Vousden KH. Induction of p57(KIP2) expression by p73beta. Proc Natl Acad Sci USA 2002; 99: 3529–3534
47. Sasaki Y, Ishida S, Morimoto I, Yamashita T, Kojima T, Kihara C, Tanaka T, Imai K, Nakamura Y, Tokino T. The p53 family member genes are involved in the Notch signal pathway. J Biol Chem 2002; 277: 719–724
48. Gonzalez S, Perez-Perez MM, Hernando E, Serrano M, Cordon-Cardo C. p73beta-Mediated apoptosis requires p57kip2 induction and IEX-1 inhibition. Cancer Research 2005; 65: 2186–2192
49. Farhang Ghahremani M, Goossens S, Haigh JJ. The p53 family and VEGF regulation: “It’s complicated”. Cell Cycle 2013; 12: 1331–1332
50. Bravo-Calderón DM, Oliveira DT, Marana AN, Nonogaki S, Carvalho AL, Kowalski LP. Prognostic significance of beta-2 adrenergic receptor in oral squamous cell carcinoma. Cancer Biomark 2011; 10: 51–59
51. Pottier N, Paugh SW, Ding C, Pei D, Yang W, Das S, Cook EH, Pui C-H, Relling MV, Cheok MH, Evans WE. Promoter polymorphisms in the β-2 adrenergic receptor are associated with drug-induced gene expression changes and response in acute lymphoblastic leukemia. Clin Pharmacol Ther 2010; 88: 854–861
52. Yu J, Cao Q, Mehra R, Laxman B, Yu J, Tomlins SA, Creighton CJ, Dhanasekaran SM, Shen R, Chen G, Morris DS, Marquez VE, Shah RB, Ghosh D, Varambally S, Chinnaiyan AM. Integrative genomics analysis reveals silencing of beta-adrenergic signaling by polycomb in prostate cancer. Cancer Cell 2007; 12: 419–431
53. Ji Y, Li K, Xiao X, Zheng S, Xu T, Chen S. Effects of propranolol on the proliferation and apoptosis of hemangioma-derived endothelial cells. J Pediatr Surg 2012; 47: 2216–2223
54. Dey M, Brisson J, Davis G, Enever R, Pray K, Zaim B, Dvornik D. Relationship between plasma propranolol concentration and dose in young, healthy volunteers. Biopharm Drug Dispos 1986; 7: 103–111
55. Xie W, Xie H, Liu F, Li W, Dan J, Liu L, Dan L, Xiao X, Li J, Chen X. Propranolol induces apoptosis of human umbilical vein endothelial cells through down-regulation of CD147. Br J Dermatol 2012;
56. Pasquier E, Street J, Pouchy C, Carre M, Gifford AJ, Murray J, Norris MD, Trahair T, Andre N, Kavallaris M. β-blockers increase response to chemotherapy via direct antitumour and anti-angiogenic mechanisms in neuroblastoma. Br J Cancer 2013;
57. Shakhar G, Ben-Eliyahu S. In vivo beta-adrenergic stimulation suppresses natural killer activity and compromises resistance to tumor metastasis in rats. J Immunol 1998; 160: 3251–3258
58. Potter DE, Schambelan M, Salvatierra O, Orloff S, Holliday MA. Treatment of high-renin hypertension with propranolol in children after renal transplantation. The Journal of Pediatrics 1977; 90: 307–311
59. Neil-Dwyer G, Bartlett J, McAinsh J, Cruickshank JM. Beta-adrenoceptor blockers and the blood-brian barrier. Br J Clin Pharmacol 1981; 11: 549–553
60. Morgenstern DA, Baruchel S, Irwin MS. Current and Future Strategies for Relapsed Neuroblastoma: Challenges on the Road to Precision Therapy. J Pediatr Hematol Oncol 2013; inpress
61. Saulnier Sholler G. A Pilot Trial Testing the Feasibility of Using Molecular-Guided Therapy in Patients with Recurrent Neuroblastoma. Future of clinical genomics in pediatric oncology 2012; 03: 602–612
62. Janeway KA, Place AE, Kieran MW, Harris MH. Future of clinical genomics in pediatric oncology. Journal of Clinical Oncology 2013; 31: 1893–1903
63. Chou TC, Talalay P. Generalized equations for the analysis of inhibitions of Michaelis-Menten and higher-order kinetic systems with two or more mutually exclusive and nonexclusive inhibitors. Eur J Biochem 1981; 115: 207–216
64. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22: 27–55