
Introduction
Porcine reproductive and respiratory syndrome virus (PRRSV) is the etiologic agent of PRRS that causes reproductive failure in sows, severe respiratory distress and high morbidity and mortality in piglets [1]. PRRSV is a single positive-strand RNA virus of the Arteriviridae family that replicates its RNA genome on the endoplasmic reticulum derived membrane structure [2, 3]. PRRSV infection induces mitochondrial dysfunction, leading a collapse of the mitochondrial transmembrane potential and raises the content of reactive oxygen species [4].
Mitochondrial homeostasis is maintained by two interlinked processes, mitochondrial dynamics and mitophagy [5]. Mitochondria are highly dynamic organelles that undergo constant fission and fusion. Mitochondrial fusion involves two sets of key GTPase proteins: the outer membrane mitofusins (Mfn1 and Mfn2) and the inner membrane optic atrophy 1 (OPA1) [6, 7]. Mitochondrial fission is mediated by the recruitment of cytosolic dynamic relative protein 1 (Drp1) to mitochondrial surface [8]. Specifically, Drp1 recruitment to mitochondria is regulated by phosphorylation at Ser616[9]. Besides, mitochondrial fission is promoted by several mitochondrial outer membrane proteins, including mitochondrial fission factor (Mff), mitochondrial fission protein 1 (Fis1), mitochondrial dynamic proteins of 49 and 51kDa (MiD49 and MiD51, respectively) [10-12]. These proteins are coordinate to recruit Drp1 to mitochondrial surface.
In human, the mutations of phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) and Parkin are linked to autosomal recessive forms of Parkinson’s disease [13, 14]. Later researches have uncovered their functional role in selective elimination of damaged mitochondria, termed as mitophagy [15]. PINK1 is maintained at low level through rapid degradation by mitochondrial protease in the N-terminal rule [16]. However, during mitochondrial stress, PINK1 stabilizes on the mitochondrial outer membrane and recruits cytosolic Parkin to mitochondria [17]. Once translocated to mitochondrial surface, Parkin mediates the engulfment and subsequent degradation of mitochondria by autophagosome.
In this study, we investigated the involvement of mitochondrial fission and mitophagy in PRRSV infection. Our results demonstrate that PRRSV infection enhances mitochondrial fission and promotes selective autophagic degradation of damaged mitochondria via mitophagy. PRRSV infection stimulated mitochondrial fission by promoting Drp1 Ser616 phosphorylation and its mitochondrial translocation. PRRSV infection facilitated the elimination of dysfunctional mitochondria via mitophagy. Furthermore, we demonstrated that inhibition of mitochondrial fission and mitophagy by silencing Drp1 and Parkin results in the hindrance of PRRSV replication and enhanced apoptosis. These observations unambiguously implicate the functional relevance of mitochondrial dynamics and mitophagy in PRRSV replication.
Results
PRRSV infection promotes mitochondrial swelling and fragmentation
PRRSV infection induces oxidative stress of endoplasmic reticulum that causes mitochondrial dysfunction and damage [18, 19]. To investigate the morphological alteration of mitochondria after PRRSV infection, Marc145 cells were infected with PRRSV at a MOI of 1 and were subjected to transmission electron microscopy analysis. As shown in Figure 1A, distinct fragmented mitochondria were observed in PRRSV-infected cells, in contrast to uninfected cells, which displayed elongated mitochondria (Figure 1A). Mitochondrial injury characterized by mitochondrial swelling and cristae distortion was observed after PRRSV infection. A quantitative analysis of relative mitochondrial length after PRRSV infection is presented in Figure 1B. The results from confocal microscopy further substantiated the occurrence of mitochondrial fission in PRRSV-infected cells, compared with uninfected cells (Figure 1C). Together, these results demonstrate that PRRSV infection induces mitochondrial fission.
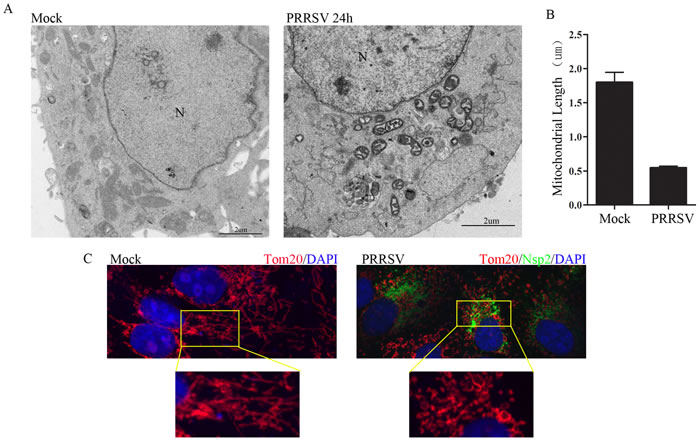
Figure 1: PRRSV infection induces mitochondrial fission. A. Electron microscopy analysis showing mitochondrial fission in PRRSV-infected cells. At 24h post-infection, PRRSV (MOI, 1) infected Marc145 cells and uninfected cells were examined by electron microscope. In the zoomed images, elongated mitochondria in uninfected cells and fragmented mitochondria with defective cristae in infected cells are shown. B. Quantification of mitochondrial diameter is shown. More than 50 cells were calculated for quantification. N, nucleus. C. At 24h post-infection, Marc145 cells were immunostained with antibodies specific to tom20 (red), Nsp2 (green). The zoomed images show the typical tubular mitochondria in uninfected cells and fragmented mitochondria in PRRSV infected cells.
PRRSV enhances Drp1 Ser616 phosphorylation and its mitochondrial translocation
Drp1 recruitment to mitochondria is modulated by phosphorylation at Ser616 by cyclin B/cyclin-dependent kinase 1 (Cdk1) [9, 20]. To investigate whether PRRSV infection enhances Drp1 Ser616 phosphorylation and its mitochondrial location, PRRSV-infected cells were observed by confocal microscopy. We found a significant elevated phosphorylated Drp1 in PRRSV-infected cells as compared to uninfected cells (Figure 2A). Confocal microscopy results also revealed that most of the Ser616 phosphorylated Drp1 translocated to the mitochondria (Figure 2D). PRRSV infection increased the expression of Drp1 and Mff at mRNA level (Figure 2B). However, Fis1 expression was decreased by PRRSV infection (Figure 2B). Although, the total Drp1 protein expression has no change, the Ser616 phosphorylated Drp1 was robustly elevated (Figure 2C). To further substantiate the observation that PRRSV induced Drp1 translocation to mitochondria, we extracted mitochondria from PRRSV-infected and uninfected Marc145 cells. Western Blot analysis of subcellular fractions demonstrated that phosphorylated Drp1 is dramatically enriched in mitochondrial fraction in PRRSV infected cells (Figure 2E). Together, these results indicate that PRRSV promotes mitochondrial fission via Drp1 phosphorylation and its mitochondrial translocation.
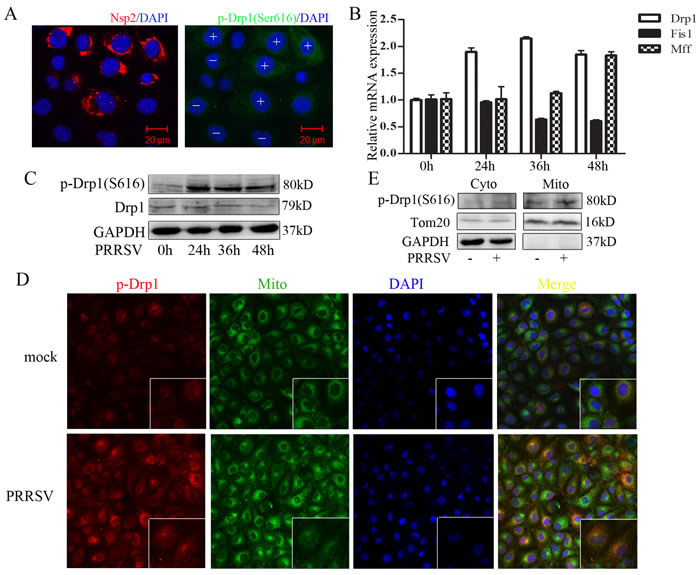
Figure 2: PRRSV infection enhances Drp1 phosphorylation and its mitochondrial translocation. A. Immunofluorescence analysis showing the induction of drp1 Ser616 phosphorylation in PRRSV-infected cells. +, PRRSV-infected cells; -, uninfected cells. B.-C. Marc145 cells were infected with PRRSV for 0h, 24h, 36h, 48h, RNA and protein were collected for q-PCR and Weatern Blot analysis. D. Marc145 cells were infected with PRRSV for 36h and mitochondria were extracted. Phosphorylated Drp1 of subcellular fraction was determined by Western Blot. Cyto, cytosolic fraction; Mito, mitochondria fraction. Tom20, mitochondria marker; GAPDH, cytoplasmic marker. E. Marc145 cells were infected with PRRSV for 36h and location of phosphorylated Drp1was analyzed by confocal microscope. In the zoomed images, the yellow spots indicate the merge of phosphorylated Drp1 with mitochondria. Data are expressed as means with SEM of three independent experiments.
PRRSV induces mitochondrial DNA (mtDNA) depletion and Parkin-dependent mitophagy
To investigate the possible effect of PRRSV infection on mtDNA copy, we quantified the relative mtDNA copy number as the ratio of mtDNA cytochrome c oxidase subunit I (COI) to 18S rDNA [21]. As shown in Figure 3A, the total number of mitochondria was drastically diminished in PRRSV-infected cells as compared to the uninfected group, which is in line with the result from Tom20 expression. Considering that mitochondria are excluded from cell by mitophagy, we further investigated whether PRRSV stimulates the expression of mitophagy-related genes. PRRSV infection modestly but significantly increased the expression of Parkin and PINK1 at both mRNA and protein levels (Figure 3B and 3C). We have previously shown that PRRSV up-regulated LC3 II expression [22], which is a reliable indicator of autophagic process. Activating transcription factor 4 (ATF4) was also stimulated by PRRSV infection (Figure 3B). ATF4 is a transcriptional factor that triggers Parkin expression via unfolded protein response (UPR) [23]. Parkin translocates to mitochondria is a well-characterized event that triggers mitophagy [24]. To further assess PRRSV-induced mitophagy, we utilized purified mitochondria and cytosolic fraction of PRRSV-infected and uninfected-Marc145 cells to evaluate Parkin translocation. Western Blot analysis of these fractions is presented in Figure 3D. Parkin was markedly enriched in the mitochondrial fraction of PRRSV-infected cells. To monitor the complete mitophagy in PRRSV-infected cells, a dual fluorescence vector encoding a mitochondrial targeting sequence fused with mRFP and EGFP was used. We observed predominantly a yellow color in uninfected cells indicating the mitochondria in cytoplasm, whereas in PRRSV-infected cells, most of the mitochondria displayed red color which is the representative of the delivery of damaged mitochondria to lysosome (Figure 3E). Relative quantification of the co-localization of mRFP and EGFP is shown in Figure 3F. To further substantiate that PRRSV-induced mitophagy is Parkin-dependent, we assessed LC3 II expression in cytosolic and mitochondrial fraction in Parkin silencing cells. The results showed that LC3 II markedly decreased in mitochondrial fraction in Parkin silencing cells (Figure 3G). These results suggest that Parkin-dependent mitophagy was induced by PRRSV infection.
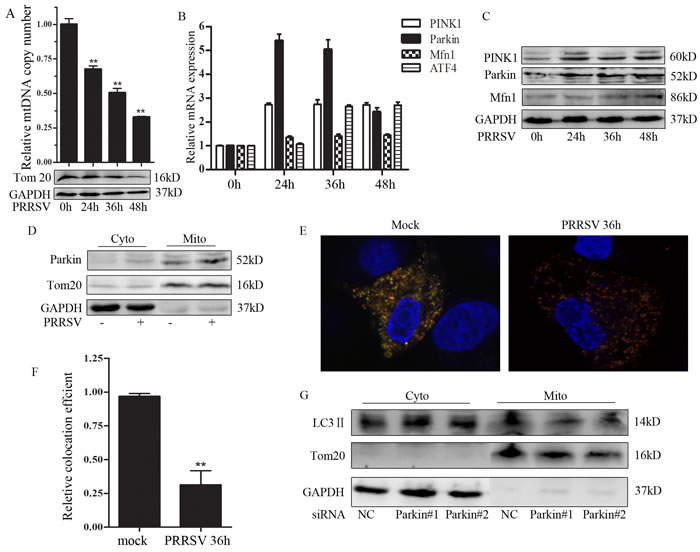
Figure 3: PRRSV induces mtDNA depletion and Parkin-dependent mitophagy. A. Marc145 cells were infected with PRRSV, DNA and protein were collected at different time point and subjected to q-PCR and Western Blot analysis. B. and C. Marc145 cells were infected with PRRSV, RNA and protein were collected at different time point and subjected to q-PCR and Western Blot analysis, respectively. D. Western Blot analysis the expression of Parkin of the subcellular fraction from uninfected- and PRRSV-infected Marc145 cell. E. Marc145 cells were transfected with mito-mRFP-EGFP and infected with PRRSV for 36h. Confocal image analyzed the level of mitophagy. F. Quantitative analysis of the fluorescence signal in F. More than 50 cells were calculated for the quantification. G. Marc145 cells were transfected with siParkin and infected with PRRSV for 36h. Then mitochondria were separated and the expression of LC3 II was verified by Western Blot. Cyto, cytosolic fraction; Mito, mitochondria fraction. Tom20, mitochondria marker; GAPDH, cytosolic marker. Data are expressed as means with SEM of three independent experiments.
Drp1 and Parkin deficiency affects mtDNA copy and depresses PRRSV replication
To evaluate the functional role of Drp1-dependent mitochondrial fission and Parkin-dependent mitophagy in mtDNA copy and PRRSV replication, we employed the siRNA strategy to silence the expression of Drp1 and Parkin. We first verified the silencing efficiency of Drp1 and Parkin by Western Blot (Fig 4A). Among the three siRNAs of Parkin, siRNA #1 and siRNA #2 were effective, so we chose them for further investigation. It has been shown that PRRSV infection resulted in the depletion of mtDNA copies (Figure 3A). Here, we further examined the effect of silencing Drp1 or Parkin on PRRSV-induced mtDNA depletion. Interference of Drp1 expression profoundly exacerbated PRRSV-induced mtDNA depletion (Figure 4B). Whereas, silencing of Parkin robustly blocked PRRSV-induced mtDNA depletion (Figure 4C). To characterize the functional significance of Drp1 and Parkin in PRRSV replication, we determined PRRSV replication in Drp1- or Parkin-silencing cells. As shown in Figure 4D and 4E, intracellular viral non-structural protein 2 (Nsp2) was decreased in Drp1- or Parkin-deficient cells, indicating the functional role of Drp1-dependent mitochondrial fission and Parkin-dependent mitophagy in PRRSV replication.
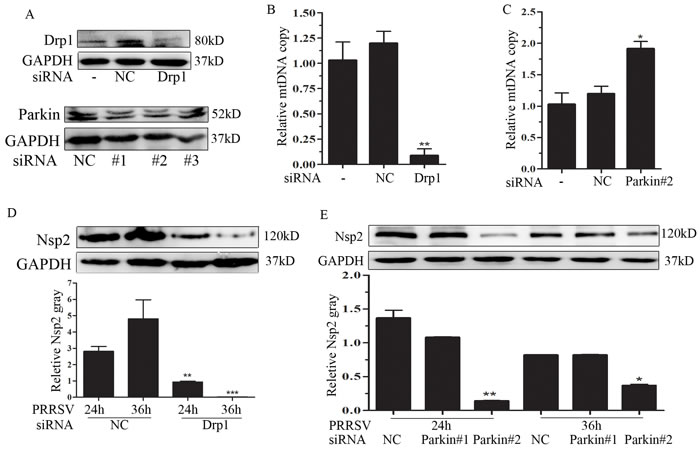
Figure 4: Drp1 and Parkin affects mitochondrial copy and PRRSV replication. A. Marc145 cells were transfected with negative control (NC) or specific siRNA targeting Drp1 or Parkin. At 24h post-transfection, the silencing effect was determined. B. and C. The DNA were extracted from Mock-, NC-, Drp1- and Parkin-transfected cells and subjected to mitochondrial copy determination by qPCR. D. and E. The effect of Drp1 and Parkin silencing on PRRSV replication were determined by Western Blot. Nsp2 protein gray was quantified by Image J. Data are expressed as means with SEM of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Disruption of mitochondrial fission and mitophagy induces apoptosis
Mitochondrial dynamic is closely linked to apoptosis [25, 26]. Caspases are a family of cysteine proteases that play a vital role in apoptosis and inflammation, demonstrated by their cleavage activation [27]. Depletion of Drp1 or Parkin induced robust activation of caspase 3 and substantial cleavage of poly (ADP-ribose) polymerase (PARP), a substrate of caspase 3 (Figure 5A). Induction of apoptosis was also substantiated by caspase 3 activity assay (Figure 5B). In order to exclude the impact of siRNAs on cell viability, cell viability was tested by trypan blue exclusion. The results indicated that there was no significant difference on cell viability between NC-transfected cells and Drp1- or Parkin-transfected cells (Figure 5C). Together, these results strongly suggest that PRRSV-mediated induction of mitochondrial fission and mitophagy, although serving as a quality control mechanism to eliminate dysfunctional mitochondria, also protects PRRSV-infected cells from apoptotic cell death, and facilitating persistent viral infection.
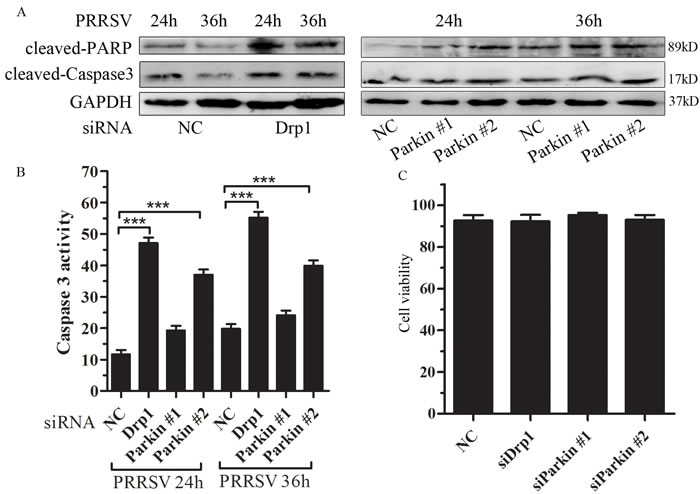
Figure 5: Disruption of mitochondrial fission and mitophagy lead to induction of apoptosis. A. and B. Drp1 and Parkin silencing accelerates PRRSV induced apoptosis. Marc145 cell were transfected with negative control (NC) or specific targeting Drp1 or Parkin and infected with PRRSV. At 24h and 36h postinfection, apoptosis was analyzed by Western Blot (A) and Caspase 3 activity (B). C. Marc145 cells were seeded in 12-well plate for 24h and transfected with indicated siRNAs for other 24h, cells were collected and detected cell viability by trypan blue straining. Data are expressed as means with SEM of three independent experiments. ***, P < 0.001.
Discussion
The mitochondrial dynamics and mitophagy play crucial roles in metabolism, cellular differentiation and neurodegeneration [28, 29]. In the past, the aspects of mitochondrial dynamics and mitophagy were broadly studied in the context of neurodegenerative disorders like Parkinson’s and Huntington’s diseases [28]. The study of the essential role of mitochondrial dynamic and mitophagy in virus infection is in its infancy. Our study characterized for the first time that PRRSV usurps Drp1-mediated mitochondrial fission to promote viral replication. A similar result was also observed in Hepatitis C virus [30] and Hepatitis B virus [31]. It may provide a potential mechanism by which PRRSV exploit mitochondrial fission in favor to viral replication. However, the molecular mechanism by which PRRSV induces mitochondrial fission needs further investigation.
Many DNA and RNA viruses have been shown to tightly regulate the autophagy process to prevent their clearance by host cell, to evade host immune response, or to favor its replication [32]. Recently, several researches including our group have been shown that PRRSV facilitates bulk autophagy to favor its replication [22, 33-35]. Several viruses including Hepatitis C virus [36], Oncolytic measles virus [37] and Newcastle disease virus [38] have been confirmed to subvert mitophagy to facilitate their own proliferation. Here, we found that PRRSV infection induced Parkin-dependent mitophagy (Figure 3). siRNA-mediate interference of Parkin mitigated PRRSV proliferation (Figure 4E). Our data suggests a functional interaction between PRRSV and autophagy pathway providing evidence that selective autophagy is probably involved in promoting PRRSV replication. Previous study has demonstrated that NOD-RIP2 signaling was inducible during PRRSV infection [39]. The research from Lupfer et al. have indicated that NOD-RIP2 signaling play an essential role in regulation of Influenza A virus-induced mitophagy, which exclude the role of Parkin in this process [40]. This suggests that diverse mitophagy signaling is exploited by viruses to their own benefits. Illumination of the functional relevance of mitophagy and virus replication will provide new therapeutic strategies to resist virus infection.
Apoptosis is considered to be an essential mechanism of host defense to limit viral replication [41]. The results of the study reported herein support the idea that mitochondrial dynamics and mitophagy attenuate apoptosis during virus infection. Our results demonstrated that PRRSV-induced apoptosis was drastically elevated when the cell suffered from mitochondrial fission or mitophagy depletion (Figure 5). One possible mechanism is that the segregation and degradation of damaged mitochondria resulting from PRRSV infection by mitochondrial fission and mitophagy may delay the induction of apoptosis by preventing the release of cytochrome C. In summary, this study provides evidence that PRRSV hijacks host signaling pathway by inducing mitochondrial fission and mitophagy to facilitate persistent viral infection which extend our knowledge about the pathology of PRRSV infection.
Materials and methods
Cells and virus
Marc145 cells that is a clone of the African green monkey kidney cell line MA-104, were obtained from China Center for type Culture Collection (CCTCC) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 g/ml streptomycin at 37 °C in a humidified 5% CO2 incubator. PRRSV strain WUH3, which was adapted to Marc145 cells at a MOI of 0.5. Virus stocks of PRRSV were prepared in Marc145 cells and infectious virus titer was analyzed in Marc145 cells using the Reed and Muench method [42]. For virus infection, cells were initially incubated with PRRSV for 1 h at 37 °C. After 1 h of adsorption, cells were exchanged to the medium containing 5% FBS and cultured for indicated time.
DNA constructs
To construct mito-mRFP-EGFP plasmid, the sequence of human cytochrome c oxidase subunit VIII (COXVIII) was inserted N terminally in frame into pMCS-mRFP-EGFP (Yrgene). The primer used for amplification is as follows: COXVIII former primer 5’- GAAGATCTATCATGTCCGTCCTGACGCCGCTGC-3’, COXVIII reverse primer 5’- CGGAATTCACACTCTGGCCTCCTGTAGGTCTCC-3’.
Quantitative real-time PCR
Total cellular RNA were extracted from cells using Total RNAeasy kit (Omega, R6834-01), subsequently, complementary DNAs (cDNAs) were synthesized by using First-Strand Synthesis System (Thermo Scientific, #00238582) according to the manufacturer’s instructions. To analyze the expression level of mitochondrial DNA, total cellular DNA was extracted from the cells using DNA kit (Qiagen, M1116) and subsequently quantified by qPCR. All PCR was performed under the following conditions: 95°C for 3 min followed by 40 cycles of 95°C for 10s, 60°C for 20s. SYBR Premix ExTaqTM kit (Bio-Rad, #172-5121) was used according to the manufacturer’s instructions, and the real-time PCR was performed on an iQ5 Real-Time PCR system (Bio-rad). The real-time PCR results were analyzed and expressed as relative expression of CT (threshold cycle) valuing the 2−ΔΔCt method. All primer pairs used for RT-PCR are presented in Table 1.
Table 1: Primers used for quantitative real-time PCR
gene name |
primer name |
primer sequence (5’-3’) |
Drp1 |
F1 |
TGGGCGCCGACATCA |
R1 |
GCTCTGCGTTCCCACTACGA |
|
Fis1 |
F1 |
GTCTGTGGAGGACCTGCTGA |
R1 |
CACGATGCCTTTACGGATG |
|
Mff |
F1 |
GCACTGAAAACACCACCTCG |
R1 |
TCTGCCAACTGCTCGGATT |
|
Parkin |
F1 |
GCCACCTACCCAGTGACCAT |
R1 |
TCGCTTAGCAACCACCTCCT |
|
PINK1 |
F1 |
GGCTTGTCAATCCCTTCTACG |
R1 |
AGCACATTTGCGGCTACTC |
|
ATF4 |
F1 |
CGATTCCAGCAAAGCACC |
R1 |
ATCCACAGCCAGCCATTC |
|
COI |
F1 |
CTAACAGACCGCAACCTCAAC |
R1 |
TCCGAAGCCTGGTAGGATAAG |
|
18S rRNA |
F1 |
TGTGATGCCCTTAGATGTCC |
R1 |
TGGGGTTCAGCGGGTTAC |
|
Beta actin |
F1 |
AGCAAGCAGGAGTATGACGAGT |
R1 |
CAAGAAAGGGTGTAACGCAACT |
Western blot
The whole cell lysate were extracted with RIPA buffer containing 1% protease inhibitor PMSF, 1% cocktail (SIGMA, P8340) and 1% phosphatase inhibitor (SIGMA, P0040). Mitochondria were separated by using a Mitochondria/Cytosol Fractionation Kit (Beyotime, C3601), according to the instruction. 10-12% SDS-PAGE gels was used to separate proteins and then transferred to polyvinylidene fluoride (PVDF) membranes (Bio-rad). The membranes were blocked with 5% nonfat milk and incubated with the primary antibody overnight at 4°C. Membranes were washed and incubated with HRP-conjugated secondary antibody. The proteins were detected by the enhanced chemiluminescence (ECL) system (Bio-Rad) following the manufacturer’s instructions. The relative expression normalized GAPDH was calculated in Image J software (NIH).
Confocal fluorescence microscopy
The Marc145 cells were grown on glass cover slips and transfected with indicated plasmid DNA, fixed in 4% paraformaldehyde, washed, and then permeabilized with 0.1% TritonX-100. The cells were incubated with the indicated antibodies. Wherever indicated, nuclei are stained with DAPI. Images were visualized under a 63× or 100× oil objective using a confocal microscope.
Electron microscopy
Briefly, Marc145 cells and PRRSV-infected cells grown in 35mm dishes were washed and fixed with fixative containing 2% glutaraldehyde in 0.1M sodiun cacodylate buffer. Cell pellets were embedded in 2% agarose, post-fixed with 1% osmium tetroxide, and dehydrated with an ethanol series. Samples were infiltrated, embedded in Durcupan and polymerized at 60°C for 48h. Ultrathin sections were prepared and examined using Tecnai transmission electron microscope.
Cell viability assay
Cell viability was determined by trypan blue exclusion assay. Marc145 cells were transfected with indicated siRNA. At 24h post-transfection, cells were harvested by Trypsin/EDTA and stained 0.4% trypan blue staining solution.
Reagents and antibodies
Chemical reagents used in this study were Mito Green Tracker (Beyotime, C1048). Primary antibodies used in this study include the following: rabbit polyclonal anti-Drp1 (Proteintech, #12957-1-AP, 1: 500 dilution); rabbit polyclonal anti-Drp1(Ser616) (Cell Signaling, #3455, 1:1000 dilution); rabbit polyclonal anti-PINK1 (Cell Signaling, #6946, 1:1000 dilution); rabbit monoclonal anti-Parkin (abcam, ab15954, 1:1000 dilution); rabbit polyclonal anti-Mfn1 (Proteintech, #13798-1-AP, 1: 500 dilution); rabbit monoclonal anti-LC3 II (Cell Signaling, #3868, 1:1000 dilution); rabbit anti-cleaved Caspase3 (Cell Signaling, #9664, 1:500 dilution); rabbit anti-cleaved PARP (Cell Signaling, #9509, 1:500 dilution); mouse monoclonal anti-GAPDH (Proteintech, #60004-1-lg, 1:1000 dilution); rabbit polyclonal anti-Tom20 (Proteintech, #11802-1-AP, 1:1000 dilution); mouse polyclonal anti-PRRSV Nsp2 has been described previously [39]. The secondary antibodies used for immunoblot analysis were HRP-conjugated anti-mouse IgG (BOSTER), HRP-conjugated anti-rabbit IgG (Cell Signaling). The secondary antibodies used for immunofluorescence was Cy3-conjugated anti-mouse IgG (BOSTER), FITC-conjugated anti-mouse IgG (BOSTER).
Transfection of siRNA
SiRNA was transfected into Marc145 cells at the concentration of 100pM with Lipofectamine 2000 (Invitrogen, 11668-019) according to the manufacturer’s protocol. SiRNA is synthesized by Gene-Pharma (Shanghai, China). The siRNA sequences are as follows: siDrp1: 5’-AACGCAGAGCAGCGGAAAGAG-3’ [43]; siParkin #1: 5’-GGUAGAUCAAUCUACAACATT-3’; siParkin #2: 5’-CCUGAUCGCAACAAAUAGUTT-3’; siParkin #3: 5’-CCUUCUGCCGGGAAUGUAATT-3’; Negative control (NC): 5’-UUCUCCGAACGUGUCACGUTT-3’.
Statistical analysis
All experiments were performed independently three times, and data were presented as the means with SEM. Data were analyzed by One-way ANOVA followed by Tukey’s Honest Significant difference test with SPSS version 16.0 (SPSS, Chicago, IL, USA). P value < 0.05 was considered as statistically significant.
Acknowledgments
We thank Professor Liurong Fang for providing us the Nsp2 antibody, Professor Changchun Li for providing help for cell culture and Pei Zhang from The Core Facility and Technical Support, Wuhan Institute of Virology, for her help with producing EM micrographs.
Conflicts of interests
The authors declare that they have no competing interests.
Grant support
This work was supported by the Natural Science Foundation of China (31272427 and 31572367) and Hubei Province International Cooperation Project (36215001).
References
1. Rossow K. Porcine reproductive and respiratory syndrome. Veterinary Pathology Online. 1998; 35:1-20.
2. Huang Y and Meng X. Novel strategies and approaches to develop the next generation of vaccines against porcine reproductive and respiratory syndrome virus (PRRSV). Virus research. 2010; 154:141-149.
3. Pedersen KW, van der Meer Y, Roos N and Snijder EJ. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. Journal of Virology. 1999; 73:2016-2026.
4. Lee SM and Kleiboeker SB. Porcine reproductive and respiratory syndrome virus induces apoptosis through a mitochondria-mediated pathway. Virology. 2007; 365:419-434.
5. Twig G and Shirihai OS. The interplay between mitochondrial dynamics and mitophagy. Antioxidants & redox signaling. 2011; 14:1939-1951.
6. Cerveny KL, Tamura Y, Zhang Z, Jensen RE and Sesaki H. Regulation of mitochondrial fusion and division. Trends in cell biology. 2007; 17:563-569.
7. Song Z, Chen H, Fiket M, Alexander C and Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. The Journal of cell biology. 2007; 178:749-755.
8. Smirnova E, Griparic L, Shurland DL and Van Der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Molecular biology of the cell. 2001; 12:2245-2256.
9. Taguchi N, Ishihara N, Jofuku A, Oka T and Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. Journal of Biological Chemistry. 2007; 282:11521-11529.
10. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ and Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. The Journal of cell biology. 2010; 191:1141-1158.
11. Palmer CS, Elgass KD, Parton RG, Osellame LD, Stojanovski D and Ryan MT. Adaptor proteins MiD49 and MiD51 can act independently of Mff and Fis1 in Drp1 recruitment and are specific for mitochondrial fission. Journal of Biological Chemistry. 2013; 288:27584-27593.
12. Losón OC, Song Z, Chen H and Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Molecular biology of the cell. 2013; 24:659-667.
13. Moore DJ, West AB, Dawson VL and Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005; 28:57-87.
14. Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR and Healy DG. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004; 304:1158-1160.
15. Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W and Ko HS. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proceedings of the National Academy of Sciences. 2010; 107:378-383.
16. Eiyama A and Okamoto K. PINK1/Parkin-mediated mitophagy in mammalian cells. Current opinion in cell biology. 2015; 33:95-101.
17. Narendra D, Tanaka A, Suen DF and Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology. 2008; 183:795-803.
18. Huo Y, Fan L, Yin S, Dong Y, Guo X, Yang H and Hu H. Involvement of unfolded protein response, p53 and Akt in modulation of porcine reproductive and respiratory syndrome virus-mediated JNK activation. Virology. 2013; 444:233-240.
19. Lee S-M and Kleiboeker SB. Porcine arterivirus activates the NF-B pathway through IB degradation. Virology. 2005; 342:47-59.
20. Kashatus DF, Lim KH, Brady DC, Pershing NL, Cox AD and Counter CM. RALA and RALBP1 regulate mitochondrial fission at mitosis. Nature cell biology. 2011; 13:1108-1115.
21. Jin R, Zhu W, Cao S, Chen R, Jin H, Liu Y, Wang S, Wang W and Xiao G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE. 2013; 8:e52909.
22. Li S, Zhou A, Wang J and Zhang S. Interplay of autophagy and apoptosis during PRRSV infection of Marc145 cell. Infection, Genetics and Evolution. 2016; 39:51-54.
23. Bouman L, Schlierf A, Lutz A, Shan J, Deinlein A, Kast J, Galehdar Z, Palmisano V, Patenge N and Berg D. Parkin is transcriptionally regulated by ATF4: evidence for an interconnection between mitochondrial stress and ER stress. Cell Death & Differentiation. 2011; 18:769-782.
24. Youle RJ and Narendra DP. Mechanisms of mitophagy. Nature reviews Molecular cell biology. 2011; 12:9-14.
25. Suen DF, Norris KL and Youle RJ. Mitochondrial dynamics and apoptosis. Genes & development. 2008; 22:1577-1590.
26. Youle RJ and Karbowski M. Mitochondrial fission in apoptosis. Nature reviews Molecular cell biology. 2005; 6:657-663.
27. Fan TJ, Han LH, Cong RS and Liang J. Caspase family proteases and apoptosis. Acta biochimica et biophysica Sinica. 2005; 37:719-727.
28. Chen H and Chan DC. Mitochondrial dynamics-fusion, fission, movement, and mitophagy-in neurodegenerative diseases. Human molecular genetics. 2009; 18:R169-R176.
29. Zorzano A, Liesa M and Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. The international journal of biochemistry & cell biology. 2009; 41:1846-1854.
30. Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG and Siddiqui A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:6413-6418.
31. Kim SJ, Khan M, Quan J, Till A, Subramani S and Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013; 9:e1003722.
32. Chiramel AI, Brady NR and Bartenschlager R. Divergent roles of autophagy in virus infection. Cells. 2013; 2:83-104.
33. Zhou A, Li S, Khan FA and Zhang S. Autophagy postpones apoptotic cell death in PRRSV infection through Bad-Beclin1 interaction. Virulence. 2016; 7:98-109.
34. Liu Q, Qin Y, Zhou L, Kou Q, Guo X, Ge X, Yang H and Hu H. Autophagy sustains the replication of porcine reproductive and respiratory virus in host cells. Virology. 2012; 429:136-147.
35. Sun MX, Huang L, Wang R, Yu YL, Li C, Li PP, Hu XC, Hao HP, Ishag HA and Mao X. Porcine reproductive and respiratory syndrome virus induces autophagy to promote virus replication. Autophagy. 2012; 8:1434-1447.
36. Kim SJ, Syed GH and Siddiqui A. Hepatitis C virus induces the mitochondrial translocation of Parkin and subsequent mitophagy. PLoS Pathog. 2013; 9:e1003285.
37. Xia M, Gonzalez P, Li C, Meng G, Jiang A, Wang H, Gao Q, Debatin K-M, Beltinger C and Wei J. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. Journal of virology. 2014; 88:5152-5164.
38. Meng G, Xia M, Wang D, Chen A, Wang Y, Wang H, Yu D and Wei J. Mitophagy promotes replication of oncolytic Newcastle disease virus by blocking intrinsic apoptosis in lung cancer cells. Oncotarget. 2014; 5:6365-6374. doi: 10.18632/oncotarget.2219.
39. Jing H, Fang L, Wang D, Ding Z, Luo R, Chen H and Xiao S. Porcine reproductive and respiratory syndrome virus infection activates NOD2-RIP2 signal pathway in MARC-145 cells. Virology. 2014; 458:162-171.
40. Lupfer C, Thomas PG, Anand PK, Vogel P, Milasta S, Martinez J, Huang G, Green M, Kundu M, Chi H, Xavier RJ, Green DR, Lamkanfi M, Dinarello CA, et al. Receptor interacting protein kinase 2-mediated mitophagy regulates inflammasome activation during virus infection. Nature immunology. 2013; 14:480-488.
41. Everett H and McFadden G. Apoptosis: an innate immune response to virus infection. Trends in microbiology. 1999; 7:160-165.
42. Reed LJ and Muench H. A simple method of estimating fifty per cent endpoints. American journal of epidemiology. 1938; 27:493-497.
43. Sugioka R, Shimizu S and Tsujimoto Y. Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. Journal of Biological Chemistry. 2004; 279:52726-52734.