
INTRODUCTION
Multiple myeloma (MM) is a plasma cell malignancy, which annually is diagnosed in approximately 30,300 patients and accounts for about 12,600 deaths in the U.S, amounting to 2% of all cancer-related mortality. Overall, the disease ranks second - first among African-Americans - in terms of incidence among all hematological cancers [1]. Although males are afflicted more than females (59% vs. 41%), death rates by sex for the disease are nearly equal. Reflecting recent advances made in treatment, five-year survival rates for MM have nearly doubled since the 1980’s - 49% in the 2005-2011 period vs. 27% for 1987-1989 [2]. The course of MM almost always includes an asymptomatic pre-malignant stage known as monoclonal gammopathy of undetermined significance (MGUS) which occurs in about 2.3% of Caucasians and 3.7% of African-Americans over the age of 50 [3]. The term smoldering MM (SMM) often is used to describe an asymptomatic state intermediate between MGUS and MM, accounting for about 15% of patients newly diagnosed with MM [4]. For several decades MM has been characterized by the classic tetrad of hypercalcemia, renal failure, anemia, and bone lesions (CRAB). Another feature of MM is the presence in the blood or urine of free light chains, regarded as a major contributor to renal damage [5], and monoclonal protein (M protein). One protein in particular, serum beta2-microglobulin, is considered a major prognostic indicator of patient survival [6]. In 2014, the International Myeloma Working Group (IMWG) issued a consensus update expanding the diagnostic criteria for MM to include validated biomarkers predictive of myeloma-related organ damage within two years prior to their manifestation [7]. About two-thirds of MM cases are found in persons over the age of 65 and, as the population ages and the new IMWG criteria are applied, the number of cases of MM is expected to double in the next 15 years [8].
For several decades the standard drugs for treating MM were the alkylating agents, primarily melphalan, in combination with corticosteroids. Over the past decade, this paradigm has shifted dramatically with the introduction of the immunomodulators (thalidomide, lenalidomide, and pomalidomide), proteasome inhibitors (bortezomib, carfilzomib, and ixazomib), and the histone deacetylase (HDAC) blocker panobinostat [9, 10]. Supplementation of MM therapy with autologous stem cell transplantation has further extended the range of options for combatting this disease. In late 2015, the U.S. Food and Drug Administration (FDA) approved for use in MM two monoclonal antibodies, daratumumab and elotuzumab, both directed against glycoproteins found on the surface of myeloma cells. The former targets CD38 (cyclic ADP ribose hydrolase) while the latter is directed against signaling lymphocytic activation molecule F7 (SLAMF7) [11, 12]. These developments, coupled with the sequencing of the myeloma genome and the identification of several genes, including those encoding kinases, critical for myeloma cell survival have given impetus for new lines of research in the quest for more effective drugs against a disease that heretofore has been considered fatal and mostly incurable [13, 14].
In addition to those noted above, a number of other molecular targets have been investigated in the search for new agents to treat MM [15]. These include inhibitors of a wide array of kinases known to be involved in cellular signaling pathways wherein aberrant functioning is believed to drive the malignant state. Although small-molecule kinase inhibitors have had a major impact on the treatment of several cancer types, including hematologic malignancies such as chronic myelogenous leukemia (ABL kinase inhibitors imatinib, dasatinib, and nilotinib) and mantle cell lymphoma (Bruton’s tyrosine kinase inhibitor ibrutinib) [16], to date, no kinase inhibitors have been approved for use in MM. The objective of this review is to examine the current status of small-molecule kinase inhibitors for potential use in MM therapy.
Complicating the therapy of MM are the interactions between myeloma cells and the surrounding bone marrow microenvironment [17, 18]; for example, myeloma cells suppress bone-forming osteoblasts while also promoting bone-resorbing osteoclasts [19]. In this regard bisphosphonates have proven useful in reducing myeloma-induced bone fractures [20]. These effects and other aspects of disease progression are further favored by stroma-secreted cytokines, such as transforming growth factor (TGF)beta [21], insulin-like growth factor 1 (IGF1) [22], interleukin 6 (IL-6) [23], hepatocyte growth factor (HGF) [24], tumor necrosis factor (TNF)alpha [25], Fms-like tyrosine kinase 3 (FLT3) [26], endoglin (CD105) [27], and vascular endothelial growth factor (VEGF) [28], each of whose corresponding tyrosine kinase (TK) receptors serve as targets for several compounds investigated for their anti-myeloma activity.
With a few notable exceptions, most of the anti-kinase work in MM drug development has focused on protein kinase targets. The human genome encodes a total of 518 different protein kinases [29], each of which operates by transferring the gamma-phosphate group of ATP to hydroxyl groups of proteins. The protein kinases, which represent key components of cellular signal transduction pathways by regulating cell proliferation, survival, and migration, are broadly divided into two classes according to the amino acids that are phosphorylated. Many of the tyrosine kinases (TKs) are primarily found as components of the cytosolic domains of plasma membrane receptors while dual function serine/threonine kinases (S/TKs) may be located in the plasma membrane but most are generally dispersed throughout the cytosol and other cellular compartments. The major non-protein kinase targets described in this review are phosphoinositide 3-kinase (PI3K) and sphingosine kinase, which target lipid-based substrates. For a broad overview of work on the kinome as relates to cancer and the development of kinase inhibitors influencing research in several disease states, including cancer, the reader is referred to the excellent recent reviews of Fleuren et al. [30] and Rask-Andersen et al. [31].
Receptor TK (RTK) inhibitors
Aberrant activation of the TK function of the IGF1 receptor is known to play a key role in MM both in vivo and in vitro [22, 32]. This receptor is the major target for linsitinib (OSI-906), an orally bioavailable TK inhibitor that is currently the subject of a clinical trial (NCT01672736) in combination with bortezomib and dexamethasone for the treatment of relapsed or refractory MM. A preclinical study [33] had earlier shown that the addition of linsitinib to bortezomib-based therapy delayed emergence of resistance to the latter. In addition to linsitinib, a number of other IGF1 TK inhibitors have been reported to have anti-MM activity (Table 1).
Table 1: RTK Inhibitors With Anti-MM Activity
Compound |
Route |
Structure |
Kinase(s) Inhibited (IC50 in nM)a |
References/ Clinical Trials |
GSK1838705A* |
Oral |
|
IGF1R (2) |
[37] |
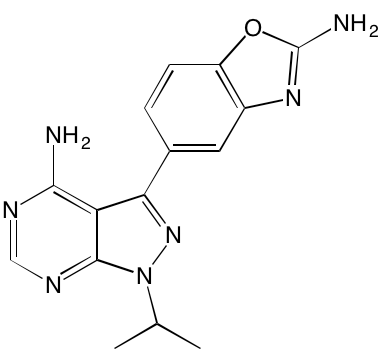
GSK1904529A* |
Oral |
|
IGF1R (27) |
[231] |
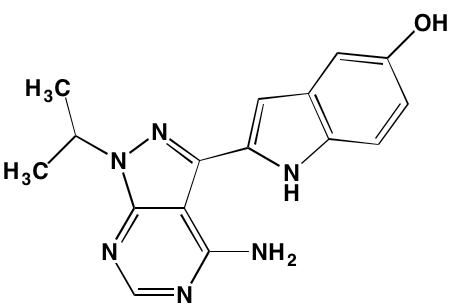
GTX-134 |
I.P. |
|
IGF1R (97) |
[232] |
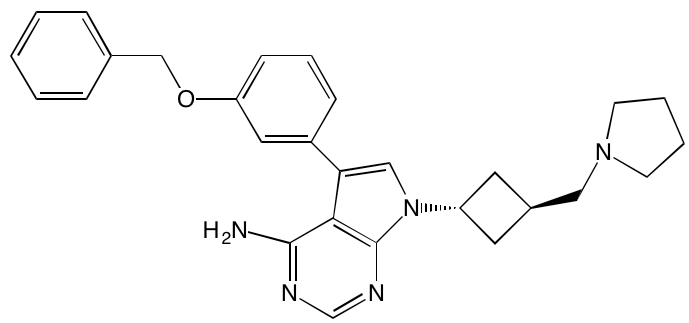
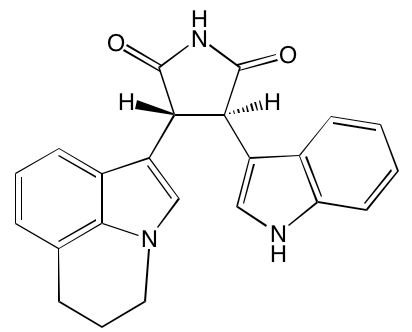
Linsitinib (OSI-906)* |
Oral |
|
IGF1R (35) |
|
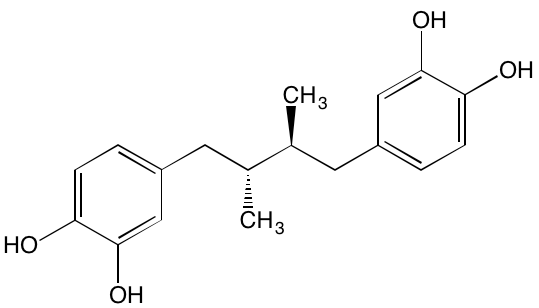
Masoprocol (Nordihydro-guaiaretic acid)† |
Oral |
|
IGF1R (NA) |
[234] |
NVP-ADW742* |
Oral |
|
IGF1R (170) |
|
Picropodophyl-lotoxin (AXL1717)† |
Oral |
|
IGF1R (1) |
|
Amuvatinib* |
Oral |
|
KIT (10); MET (NA); PDGFRalpha (40) |
[43] |
Imatinib* |
Oral |
|
KIT (100); BCR-ABL (25) |
|
Masitinib* |
Oral |
|
KIT (200); PDGFRalpha/beta (540/800) |
[241]; NCT01470131; NCT00866138 |
Cabozantinib* |
Oral |
|
MET (1.3); VEGFR2 (0.035) |
|
SU11274* |
Oral |
|
MET (10) |
|
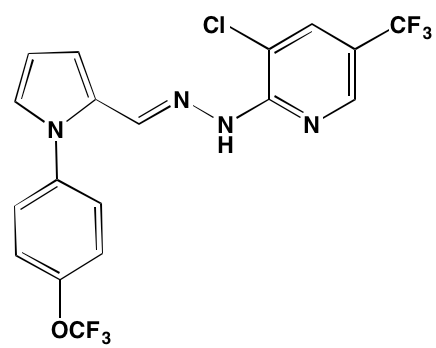
Tivantinib† |
Oral |
|
MET (355b) |
[245]; NCT01447914 |
AZ8010* |
Oral |
|
FGFR1/2/3 (8/1/17) |
[46] |
Dovitinib* |
Oral |
|
FGFR1/3(8/9); VEGFR1/2/3 (10/13/8); PDGFRalpha/beta (210/27) |
|
LY2874455* |
Oral |
|
FGFR1/2/3/4 (2.8/2.6/6.4/6.0) |
[248] |
NF449† |
NA |
|
FGFR3 (200-500) |
[227] |
PD173074* |
Oral |
|
FGFR1/3 (21/NA) |
|
SU5402* |
NA |
|
FGFR1/3(30/NA); VEGFR1/2 (30/20) |
|
TAS-120‡ |
Oral |
Undisclosed |
FGFR (NA) |
[253]; NCT02052778 |
GW654652 |
Oral |
|
VEGFR1/2/3 (12/2.3/2.5) |
|
Nintedanib (BIBF 1120)* |
Oral |
|
VEGFR1/2/3 (34/21/13); FGFR1/2/3 (69/37/108); PDGFRalpha/beta (59/65) |
|
Pazopanib (GW786034B)* |
Oral |
|
VEGFR1/2/3 (10/30/47); KIT (140) |
NCT00256880 |
Semaxanib (SU5416)* |
Infusion |
|
VEGFR2 (1230) |
[260]; NCT00006013 |
Sorafenib (BAY 43-9006)* |
Oral |
|
VEGFR2 (90); PDGFRbeta (57) |
|
Sunitinib (SU11248)* |
Oral |
|
VEGFR2 (80); PDGFRbeta (2) |
NCT00514137 |
Tamibarotene (AM80) |
Oral |
|
VEGFR (NA) |
[265] |
Vandetanib (ZD6474)* |
Oral |
|
VEGFR2 (40) |
|
Vatalanib (PTK787)* |
Oral |
|
VEGFR1/2 (77/37); KIT (73) |
[268]; NCT00165347 |
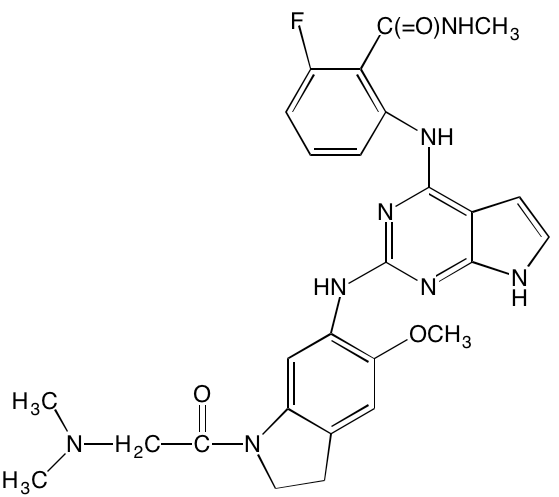
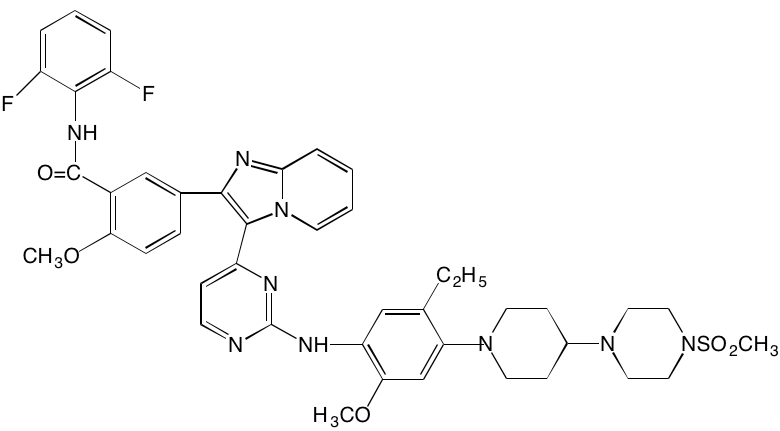
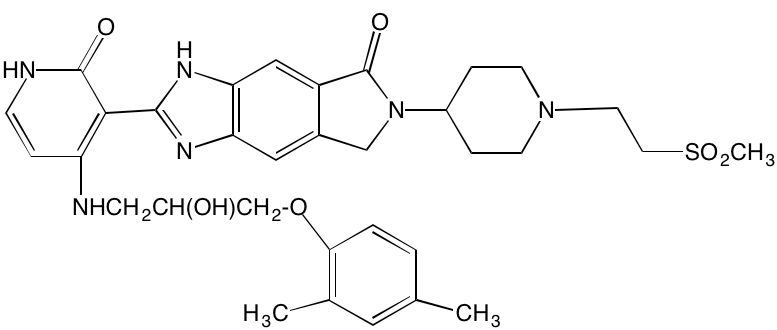
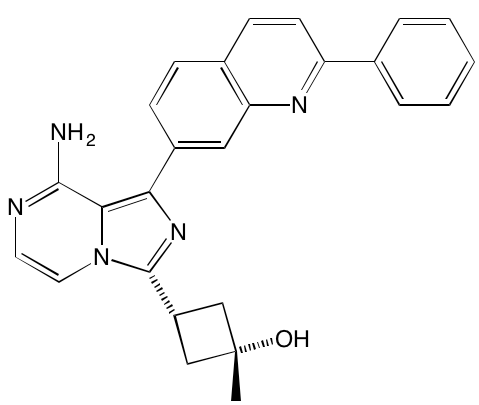
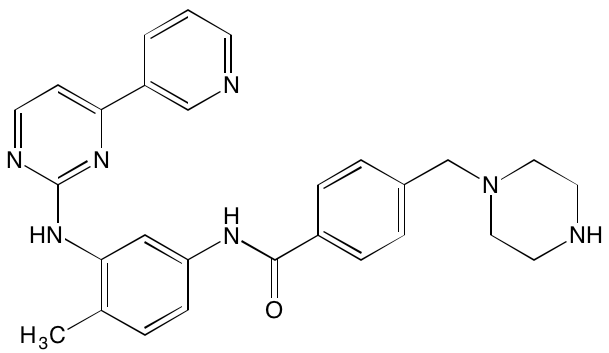
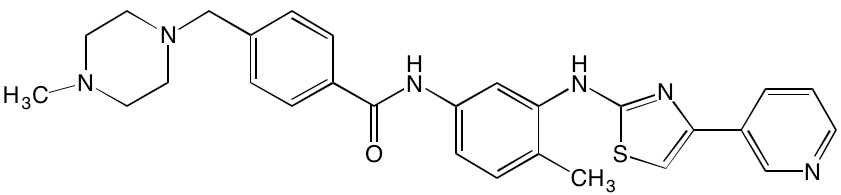
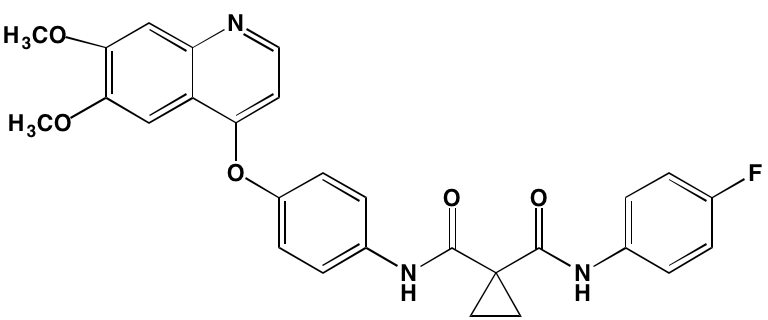
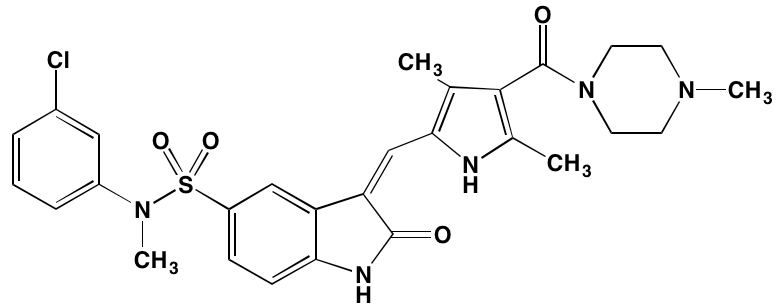
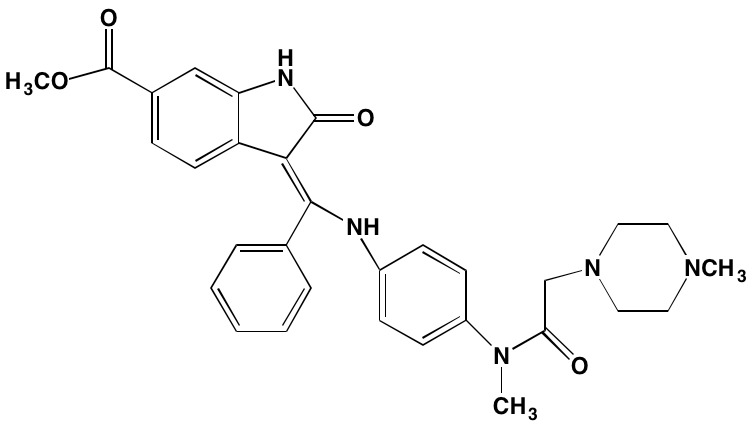
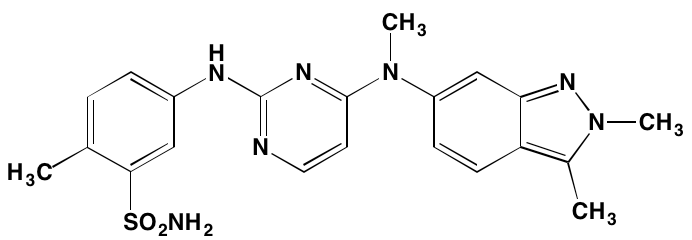
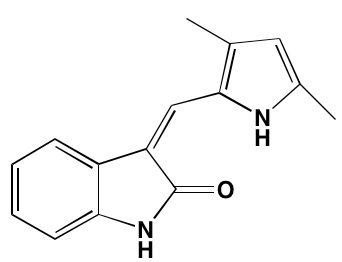
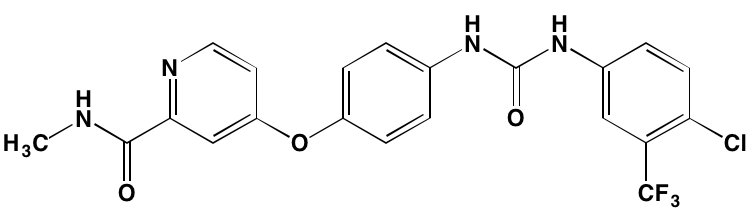
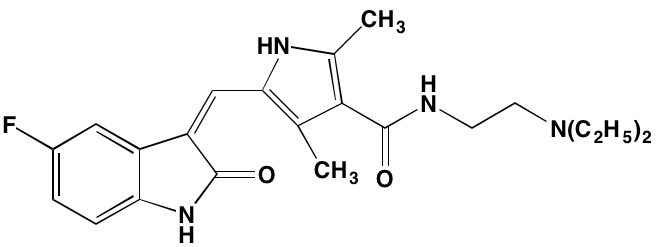
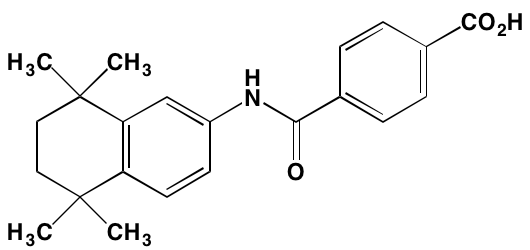
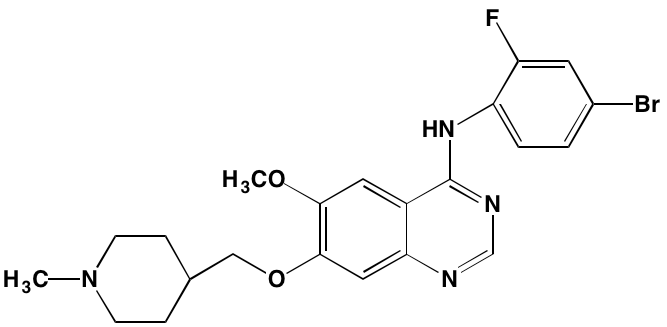
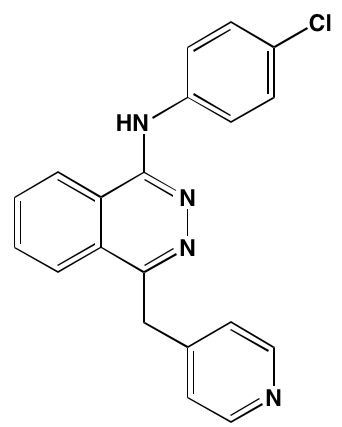
NA = Information not available; *ATP-competitive inhibitor; †allosteric inhibitor; ‡irreversible inhibitor; aas determined in cell-free assays; bKi in nM
A potential drawback to the development of IGF1 small molecule inhibitors in cancer therapy derives from the finding that the receptors for both IGF1 and insulin exhibit close sequence homology and identical ATP-binding clefts [34]. In addition, IGF1 and insulin act in concert to regulate blood glucose levels [35]. Thus, inhibition of the IGF1 TK domain might be expected to affect insulin receptors as well with consequences for glucose homeostasis in patients. Consistent with this expectation, hyperglycemia has been noted as an adverse event in clinical studies with the anti-IGF1 monoclonal antibody figitumumab in cancer patients, including those with MM [36]. On the other hand, the small molecule TK inhibitor GSK1838705A, which inhibits growth of MM cell lines at concentrations below one micromolar, has been reported to inhibit both IGF1 and insulin receptors (in addition to another RTK, anaplastic lymphoma kinase) at comparable IC50 values (2.0 and 1.6 nanomolar, respectively) but elicited only minimal effects on glucose levels in mice [37]. Moreover, the IGF1 TK inhibitor NVP-AEW541 reportedly produced significant decreases in blood glucose in non-tumor bearing nude mice [38].
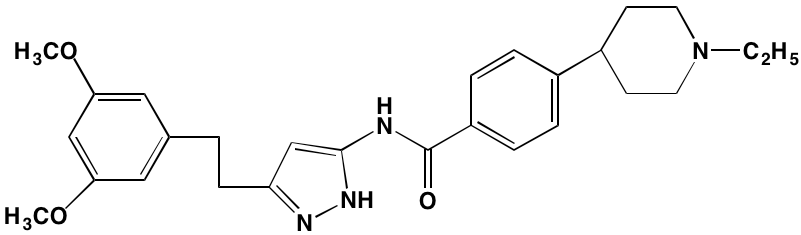
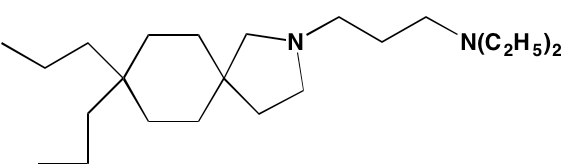
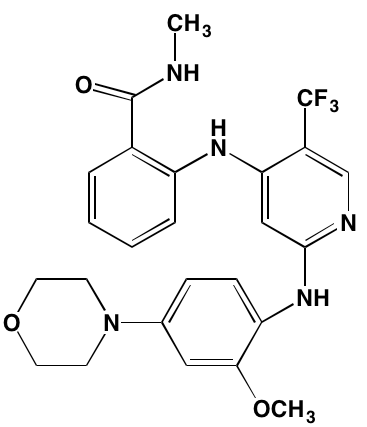
The KIT TK surface receptor for the stem cell growth factor (SCGF) is absent in normal plasma cells but expressed in about one-third of myeloma cells and thus represents an attractive target for MM. Masitinib, an oral multi-targeted TK inhibitor primarily affecting KIT, as well as the receptors for fibroblast growth factor (FGF)3 and platelet-derived growth factor (PDGF), has been the subject of two clinical trials in MM (NCT01470131 and NCT00866138). Imatinib, whose primary targets are KIT and the oncogenic BCR-ABL fusion TK, produced poor outcomes in a Phase II trial of patients with relapsed or refractory MM [39].
MET, an oncoprotein that serves as the receptor TK for HGF, has been implicated in a number of cancers, including MM [40, 41]. Tivantinib (ARQ197), a non-ATP competitive oral inhibitor of both constitutive and ligand-induced phosphorylation of MET, is the subject of a Phase II trial in relapsed/refractory MM (NCT01447914). This agent works by binding to and thereby stabilizing the inactive conformation of MET. In pre-clinical work, tivantinib was found to be equally effective in inducing apoptosis in chemotherapy-refractory myeloma cell lines and in cells co-cultured with a protective bone marrow microenvironment [42]. The orally bioavailable ATP-competitive MET inhibitor amuvatinib, currently in trials for the treatment of solid tumors, has been shown to result in cell death in myeloma cells known to be dependent on HGF/MET signaling [43]. Amuvatinib also is known to inhibit other TKs, including KIT, PDGFRalpha, and FLT3. SU11274 is another oral ATP-competitive MET inhibitor with much less or no activity against other RTKs. It has been shown to block the autocrine HGF/MET loop that sustains MM angiogenesis [44]. Cabozantinib, which is FDA-approved for medullary thyroid cancer and renal cell carcinoma, is yet another oral ATP-competitive MET inhibitor; it is now under clinical study for relapsed/refractory MM (NCT01866293).
The FGF family of transmembrane receptors exist as different isoforms FGFR1-4, all of which feature an immunoglobulin-like extracellular domain and a cytoplasmic TK domain (Figure 1). FGFR mediates a number of signal transduction pathways in the cell, most notably those involving RAS-MAPK, PI3K-AKT, and phospholipase C-gamma, impacting cell proliferation, survival, and invasion, as well as angiogenesis and drug resistance [45]. FGFR is overexpressed in a number of tumor types, including gastric, breast, and urothelial cancers and MM [46]. Translocations in the immunoglobulin heavy chain region of chromosome 14q32, are found in 40-60% of MM patients [47]. Particularly noteworthy is the t(4;14) translocation of FGFR3 and MMSET (Multiple Myeloma Set domain), which occurs in approximately 15% of MM patients and is associated with poor prognosis and resistance to chemotherapeutic measures [48]. Moreover, FGFR3 has been cited as one of the most significantly mutated genes in MM [49]. FGFR inhibitors with reported anti-myeloma activity are shown in Table 1, including two experimental FGFR3 inhibitors, SU5402 and PD173074, that have demonstrated activity against t(4;14)-positive myeloma cell lines [50].
MM was the first hematological cancer in which enhanced angiogenesis was discovered [51] and several subsequent studies have demonstrated that the growth of MM cells is highly dependent on angiogenesis [52]. VEGF, a glycoprotein principally secreted in response to hypoxia and belonging to the platelet-derived growth factor (PDGF) superfamily is the key mediator of angiogenesis. In mammals, VEGF is comprised of five members - VEGFA (the best characterized), VEGFB, VEGFC, VEGFD, and placenta growth factor. These act as ligands for three types of VEGF TK plasma membrane receptors, designated VEGFR1 (or FLT1), VEGFR2 (also known as FLK1/KDR; the key mediator of angiogenesis induced by VEGF), and VEGFR3 (the primary regulator of lymphangiogenesis), that activate downstream signaling pathways that result in vascular endothelial cell proliferation, migration, and survival [53].
Myeloma cells are known to produce VEGF through both autocrine and paracrine signaling mechanisms. Moreover, MM cells stimulate bone marrow stromal cells to produce VEGF [54, 55]. A number of VEGFR TK inhibitors have been the subject of both preclinical and clinical studies in MM, although none of these compounds has advanced beyond Phase II (Table 1). The major site of action of these compounds is VEGFR2, consistent with reports that the effects of VEGF in MM are primarily due to binding to VEGFR2 [55].
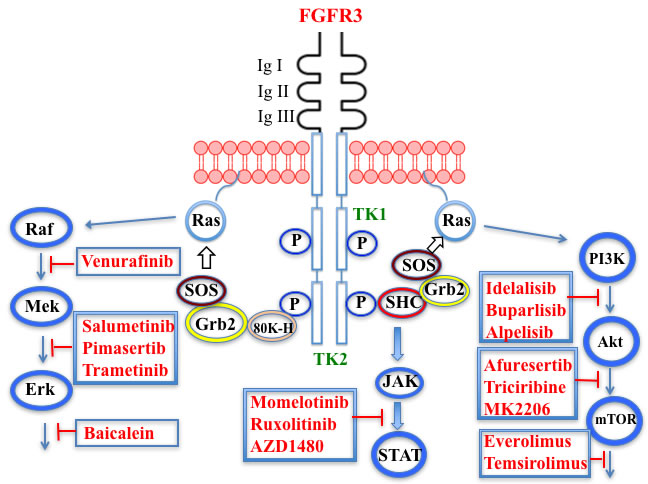
Figure 1: Putative sites of action of several kinase inhibitors with demonstrated anti-myeloma activity using activation of the FGFR3 receptor as an example. Three signal transduction pathways that are common features of receptor tyrosine kinase signaling are shown: JAK/STAT; PI3K/Akt/mTOR; and Raf/Mek/Erk. The FGFR3 receptor possesses three extracellular immunoglobulin domains (Ig I-III), a single transmembrane domain, and a split tyrosine kinase domain (TK1 and TK2). Also shown are adaptor proteins linking the TK domain with Ras activation: SRC-homology-2-domain-containing (SHC), growth factor receptor-bound protein 2 (GRB2), son of sevenless (SOS) adaptor proteins, and 80K-H (a protein kinase C substrate).
Non-receptor TK inhibitors
The role of IL-6 in the growth and survival of MM cells is well-documented and elevated IL-6 serum levels are considered evidence of poor prognosis in the disease [56]. IL-6 binds to its transmembrane receptor and induces a cascading sequence of tyrosine phosphorylation reactions starting with Janus kinase (JAK) and leading to the activation of signal transduction and activator of transcription (STAT)3. Following activation, STAT3 translocates to the nucleus where it targets genes involved in survival, proliferation, and apoptosis. The JAK family is comprised of four protein kinases, designated JAK1, JAK2, JAK3, and tyrosine kinase, nonreceptor, 2 (TYK2). Seven members of the STAT family are known: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6 [57]. Patients with MM show increased expression of anti-apoptotic proteins, such as BCL-2, BCL-XL, and MCL1, linked to constitutive expression of STAT3 [58, 59]. Inhibitors of the JAK/STAT pathway that have demonstrated activity against myeloma cells are shown in Table 2.
Bruton’s TK (BTK), a non-receptor member of the TEC family plays a key role in the development of B cells [60] and demonstrates robust expression in malignant plasma cells in MM patients [61]. Ibrutinib, an irreversible orally bioavailable BTK inhibitor, currently approved for mantle cell lymphoma, chronic lymphocytic leukemia, and Waldenstrom’s macroglobinemia, also is undergoing study for potential use in MM (NCT01962792). CC-292, another oral BTK inhibitor, in combination with the proteasome inhibitor carfilzomib, was found to augment the osteoclast inhibitory effects of the latter in an in vivo MM mouse model [62].
Sulzmaier et al. [63] have recently reviewed the complex roles played by the cytoplasmic TK focal adhesion kinase (FAK), which associates not only with plasma membrane receptors but also with nuclear protein complexes, to promote tumor progression and metastasis. However, the part played by FAK in specifically driving MM has received little attention in the literature until relatively recently. For example, Wang et al. [64] found that higher expression levels of FAK correlated with lower levels of the tumor suppressor phosphatase and tensin homolog (PTEN) in MM patients with extramedullary disease. Furthermore, Ko et al. [65] have suggested that bortezomib and possibly other proteasome inhibitors may exert their effects in MM by suppressing FAK expression. In addition, the naturally-occurring triterpene asiatic acid, which down-regulates FAK expression, causes G2/M phase arrest in myeloma cells [66]. Proline-rich TK2 (PYK2), another non-receptor member of the FAK family having substantial homology with FAK, also has been reported to play a role in MM progression [67]. This kinase, in contrast to the ubiquitous expression of FAK, is restricted to the endothelium, central nervous system, and hematopoietic lineages. PYK2 appears to work by modulating the Wnt/beta-catenin signaling pathway in myeloma cells. The FAK/PYK2 inhibitors VS-4718 and VS-6062 have been reported to inhibit MM cell growth both in vitro and in vivo [67, 68].
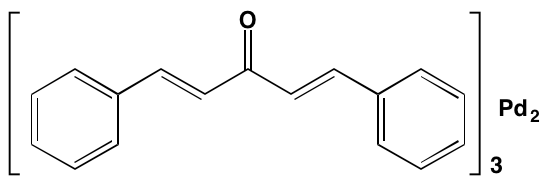
SRC, a non-receptor protein TK, which is known to regulate a number of key cellular processes, such as cell survival, differentiation, growth, and migration, is constitutively activated in both MM cell lines and patient tumors [69]. The SRC inhibitor tris (dibenzylideneacetone) dipalladium (Tris DBA) was recently reported to induce G1 arrest and apoptosis and thereby reduce proliferation of MM cells. Moreover, this agent reversed the hypoxia-inducing properties associated with resistance to proteasome inhibitors [70]. Dasatinib is an orally effective inhibitor of receptor and non-receptor TKs, including BCR-ABL, KIT, PDGFR, and SRC family kinases. Although this drug has been studied in several hematological malignancies and is approved for the treatment of chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphocytic leukemia, results with MM, either as a single agent or in combination with immunomodulators have been disappointing [71, 72].
Another intracellular TK that has received attention in hematological cancers is spleen TK (SYK), which, following activation by SRC family kinases, phosphorylates a number of target kinases, including those in the PI3K and mitogen-activated protein kinase (MAPK) pathways [73]. In pre-clinical studies SYK signaling has emerged as an important target for B-cell malignancies [74] and inhibitors of this pathway have been shown to have therapeutic potential in MM [75].
Table 2: Non-Receptor TK Inhibitors With Anti-MM Activity
Compound |
Route |
Structure |
Kinase(s) Inhibited (IC50 in nM)a |
References/ Clinical Trials |
Atiprimod |
Oral |
|
STAT3 (NA) |
[269] |
AZD1480* |
Oral |
|
JAK2 (0.26b); STAT3 (350) |
|
Brevilin A (6-O-Angeloylplenolin) |
NA |
|
JAK2 (NA); STAT3 (NA) |
[272] |
Compound K |
NA |
|
JAK1 (NA); STAT3 (NA) |
[273] |
Ergosterol peroxide |
NA |
|
JAK2 (NA); STAT3 (NA) |
[274] |
Farnesol |
NA |
|
JAK1/2 (NA); STAT3 (NA) |
[275] |
Icaritin |
NA |
|
JAK2 (NA); STAT3 (NA) |
[276] |
INCB20* |
NA |
Undisclosed |
JAK1/2/3 (0.9/0.5/0.5); TYK2 (0.3) |
[277] |
INCB16562* |
Oral |
|
JAK1/2/3 (2.2/0.3/10) |
|
Momelotinib (CYT387)* |
Oral |
|
JAK1/2/3 (11/18/155) |
|
Piceatannol* |
Oral |
|
JAK1 (NA); STAT3 (NA); SYK (NA) |
|
Pyridone-6* |
NA |
|
JAK1/2/3 (15/1/5); TYK2 (1) |
|
Ruxolitinib (INCB018424)* |
Oral |
|
JAK1/2 (3.3/2.8) |
|
SC99 |
NA |
|
JAK2 (NA); STAT3 (NA) |
[289] |
TG101209* |
Oral |
|
JAK2 (6) |
|
TM-233 |
NA |
|
JAK2 (NA); STAT3 (NA) |
[292] |
Tyrphostin (AG490)* |
NA |
|
JAK2 (100); STAT3 (NA) |
|
Decursin |
NA |
|
STAT3 (NA) |
|
8-Hydrocalamenene |
NA |
|
STAT3 (NA) |
[297] |
5-Hydroxy-2-methyl-1,4-naphthoquinone (plumbagin) |
NA |
|
STAT3 (NA) |
[298] |
Acalabrutinib (ACP-196) ‡ |
Oral |
|
BTK (3) |
[299]; NCT02211014 |
CC-292‡ |
Oral |
|
BTK (<0.5) |
|
Ibrutinib‡ |
Oral |
|
BTK (0.5) |
NCT01962792 |
Asiatic acid |
NA |
|
FAK (NA); PYK2 (NA) |
[66] |
VS-4718 (PND-1186)* |
Oral |
|
FAK (1.5); PYK2 (85) |
|
VS-6062 (PF562271)* |
Oral |
|
FAK (1.5); PYK2 (13) |
|
Dasatinib* |
Oral |
|
SRC (0.8); ABL (0.6); KIT (79) |
|
Tris(dibenzylidene-acetone) dipalladium |
NA |
|
SRC (NA) |
[70] |
BAY-61-3606* |
Oral |
|
SYK (10) |
|
R406 (Fostamatinib active metabolite)* |
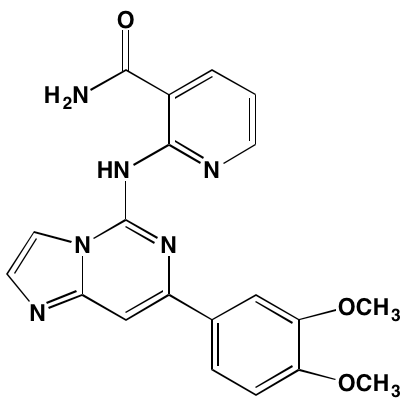
Oral |
|
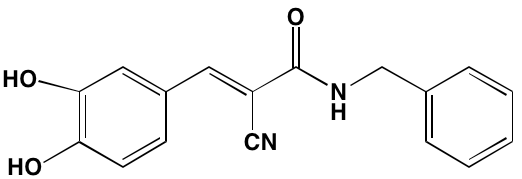
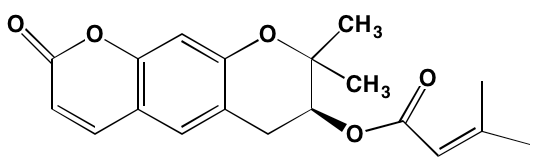
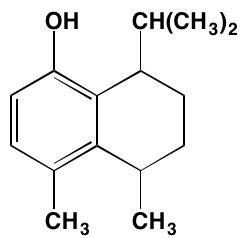
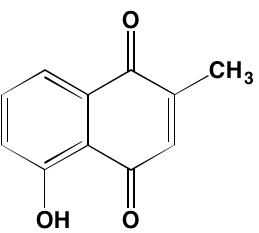
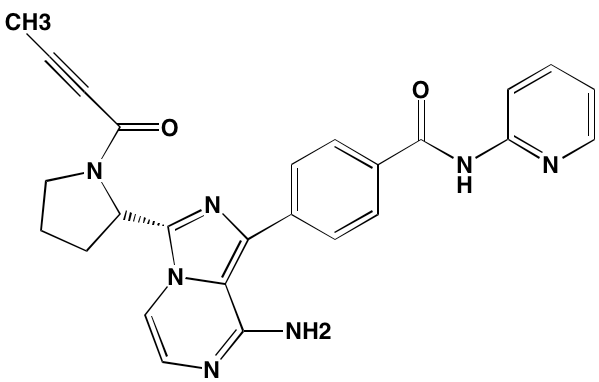
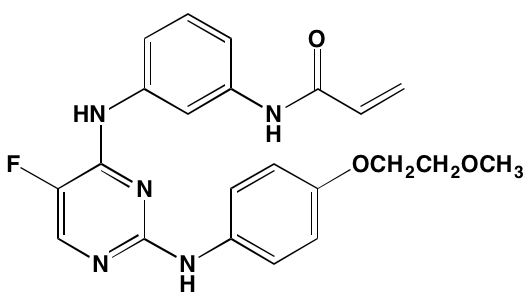
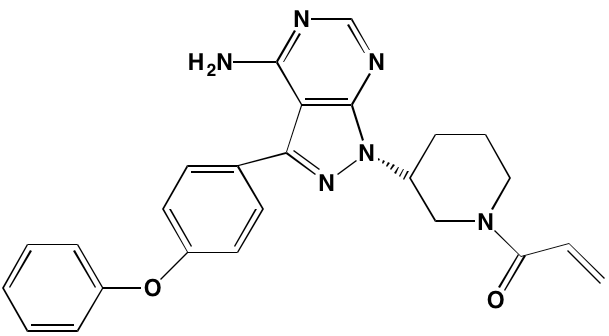
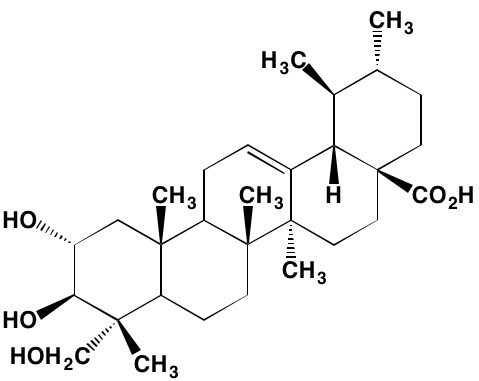
SYK (30b) |
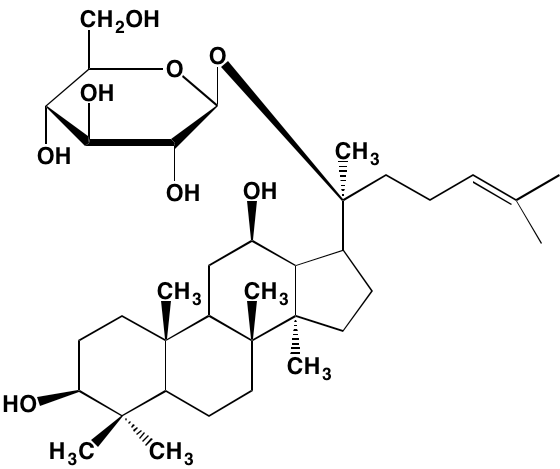
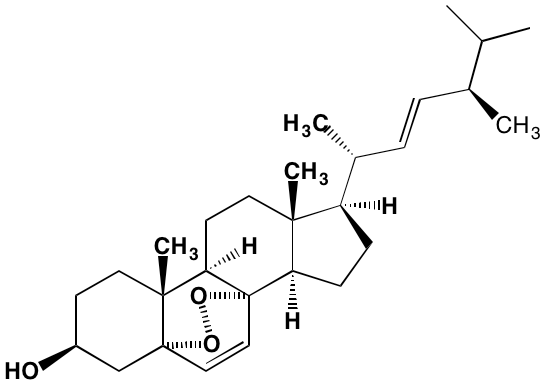
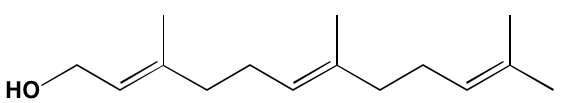
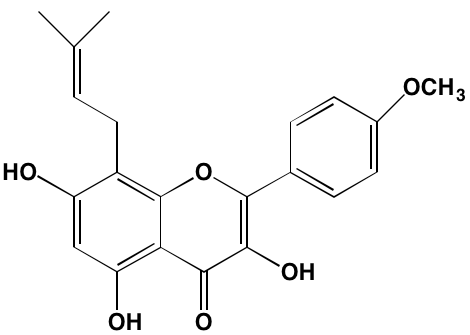
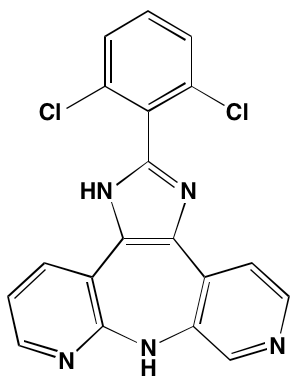
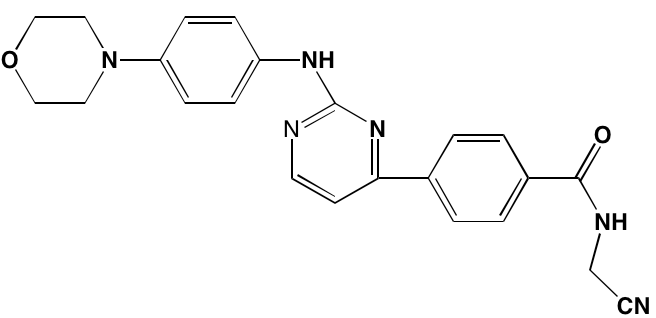
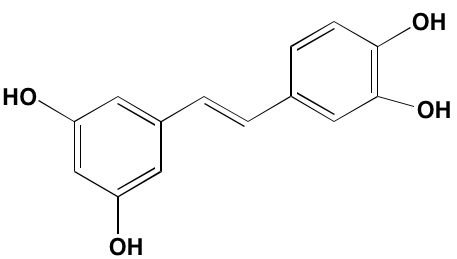
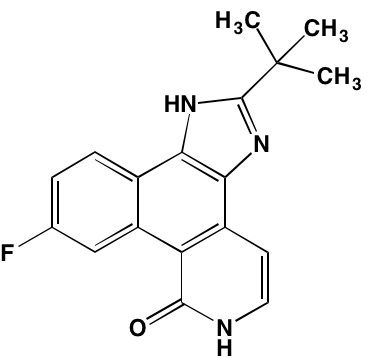
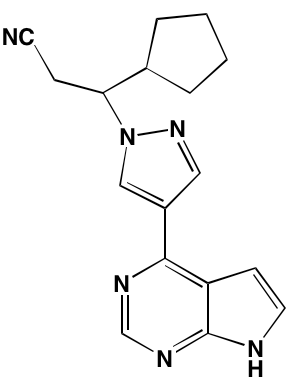
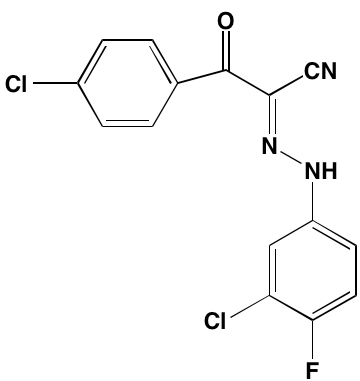
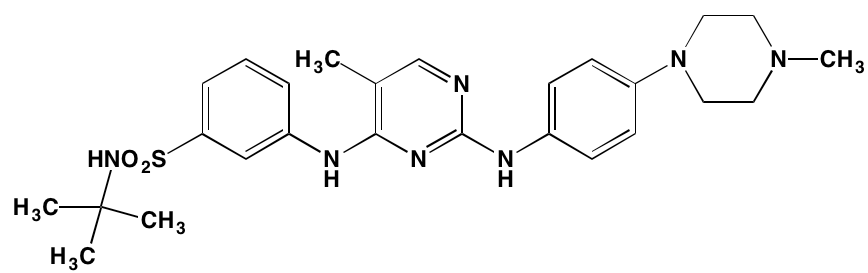
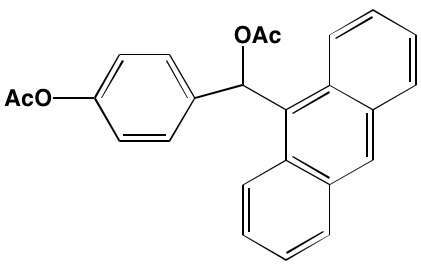
NA = Information not available; *ATP-competitive inhibitor; ‡irreversible inhibitor;
aas determined in cell-free assays; bKi in nM
Receptor S/TK inhibitors
The receptors for TGFbeta are single-pass plasma membrane proteins containing a cytosolic S/TK domain. They exist as homodimers of two types (type I and type II), which form an active tetrameric complex following phosphorylation. The complex phosphorylates and thereby activates transcription factors of the SMAD family, which enter the nucleus to control transcription of target genes [76]. TGFbeta acts as an inhibitor of proliferation in many cells, including normal plasma cells. However, TGFbeta signaling is suppressed in MM cells. Moreover, TGFbeta produced in the bone microenvironment enhances secretion of various cytokines (e.g., IGF1, basic FGF, and IL-6) that stimulate MM cell growth. A negative balance in bone turnover occurs in MM with osteoclast bone resorption strongly outweighing osteoblast differentiation and maturation. The effect on osteoblasts is primarily due to Wnt signal inhibitors constitutively secreted by MM cells, both in myeloma cell lines and from MM patients. TGFbeta also plays a role by suppressing matrix mineralization during the later stages of osteoblast maturation. Moreover, bone resorption in MM causes release of TGFbeta from the matrix to further exacerbate loss of osteoblast activity [21, 76].
Most of the impetus for developing TGFbeta inhibitors for use in MM derives from their potential role in treating MM-induced bone disease. The TGFbeta type I receptor kinase inhibitors Ki26894 (structure undisclosed) and SB431542 were found to facilitate osteoblast differentiation while also preventing bone loss in MM cells in vivo although neither compound was cytotoxic to myeloma cells [77]. Another type I inhibitor, SD-208, has been reported to abrogate secretion of IL-6 and VEGF from myeloma-derived bone marrow stromal cells while also reducing tumor cell growth [78].
SB431542 SD-208
Non-receptor serine/threonine kinase (S/TK) inhibitors
Dysregulation of the PI3K/AKT/mTOR pathway is a hallmark of many malignancies, including MM [79]. In this pathway, PI3K is attracted to the cytosolic TK domains of the activated receptors for the cytokine IL-6 [80] and certain growth factors, notably, PDGF [69] and IGF1 [22], which are overexpressed in MM. PI3K, a non-protein kinase, specifically phosphorylates the 3-position of the plasma membrane lipid phosphatidylinositol-4,5-bisphosphate (PIP2) to form the corresponding trisphosphate (PIP3), which serves as a docking site for AKT via the latter’s pleckstrin homology (PH) domain. This docking causes a conformational change in the AKT molecule such that T308 and S473 become available for phosphorylation by phosphoinositide-dependent protein kinase-1 (PDPK1), which likewise uses a PH domain to target AKT. PI3K also is activated by plasma membrane-tethered RAS.
Three classes of PI3Ks, designated I, II, and III, are known. Members of Class I, which primarily are involved in regulating cell growth, proliferation, and survival, are subdivided further into Classes IA and IB. Those in Class IA are activated by RTKs and RAS while Class IB members are regulated by G protein-coupled receptors. Class I PI3Ks are characterized by their heterodimeric structures, consisting of a p110 catalytic subunit and a p85 regulatory subunit. Four isoforms of p110 are known - p110alpha, beta, and delta found in Class IA and p110gamma, which is confined to Class IB [81]. The alpha and beta isoforms are widely distributed in mammalian tissues while the gamma and delta subunits are mostly found in leukocytes, which makes the latter pair attractive targets for therapy of leukemias, lymphomas, and MM with reduced systemic effects [82]. The delta isoform in particular has been identified by Ikeda et al. [83] as an especially attractive target for MM. Mutations in the PIK3CA gene, which codes for the p110alpha isoform are noted in a number of cancers [84]. Two hotspots are primarily associated with these gain-of-function mutations: E545K and E542K in the helical region and H1047R in the kinase domain, although none of these mutations appear to be associated with MM [85].
The complex downstream effects of AKT include: 1) blockage of apoptosis through inhibition of the pro-apoptotic factors BCL-2-associated death promoter (BAD), caspase-9, and the transcription factor Foxhead box O1 (FOXO1), as well as by activation of the antiapoptotic proteins IkappaB kinase (IKK) and MDM2; 2) blockage of the anti-proliferative effects of glycogen synthase kinase (GSK)-3beta, FOXO4, and p21Cip1; and 3) inhibition of the anti-growth properties of tuberous sclerosis protein 2 (TSC2), a tumor suppressor gene product, whose phosphorylation by AKT results in mammalian target of rapamycin (mTOR) activation. mTOR has myriad downstream effects, including modulation of cell growth, angiogenesis, and autophagy. In addition, mTOR and AKT are capable of reciprocal phosphorylation.
PI3K has received much attention in recent years as a target for new anticancer drug design and development and MM is no exception [86-88]. High throughput virtual screening was used to identify two potent blockers of the ATP binding site of Class I PI3Ks, designated C96 [89] and PIK-C98 [90], that demonstrated good activity against MM in preclinical studies. C96 showed a preference for the alpha and delta p110 isoforms. Pictilisib (GDC 0941), another inhibitor of both alpha and delta isoforms, has been found effective in both MM cell lines and in patient myeloma cells [91]. The delta-selective PI3K blockers idelalisib and IC488743 have demonstrated cytotoxicity in murine myeloma models [83] while copanlisib (BAY 80-6946), which is selective for the alpha isoform, has been shown to be effective against four different MM cell lines [92].
In some cases, PI3K inhibition has been combined in the same molecule with chemical moieties that target other cellular pathways relevant in MM. An example of this is CUDC-907, an orally bioavailable inhibitor of all four p110 isoforms of PI3K, as well as both Classes I and II HDACs. Its molecular design combines structural elements found in other PI3K inhibitors with the hydroxamic acid functionality common to many HDAC inhibitors. CUDC-907 currently is in Phase I studies in lymphoma and MM patients (NCT01742988) [93, 94]. In this context, it is noteworthy, as cited above, that the FDA only recently approved the HDAC inhibitor panobinostat for MM. Dactolisib (BEZ235) [95] and BGT226 [96] are dual pan-PI3K/mTOR inhibitors that have demonstrated oral activity against MM in mouse models. PI-103, another such dual inhibitor, was found to cause cell cycle arrest in MM cells having t(4;14) and t(14;16) transversions that are often associated with poor treatment outcomes [97].
AKT, which occupies the central portion of this signaling pathway and is classed with the AGC superfamily of protein kinases [98], exists in three different isoforms in mammals: AKT1, AKT2, and AKT3. Three functional domains comprise each isoform in mammals. The N-terminus contains the PH domain, which uses a flexible linker to connect to the central kinase domain bearing T308. The 21-residue regulatory C-terminus has a hydrophobic motif and bears the S473 phosphorylation site [99, 100]. In its inactive state the AKT PH domain shields the two phosphorylation sites of AKT (“PH-in” conformation) from phosphorylation by PDPK1. When PI3K is activated, the resulting PIP3 serves as a docking site for the PH domain of AKT, thus exposing the two phosphorylation sites to PDPK1. This docking causes a conformational change (“PH-out” conformation) in the AKT molecule such that T308 and S473 become available for phosphorylation by PDPK1 and mTOR complex 2 (mTORC2), respectively. Phosphorylation at these two sites serves a dual purpose: 1) opening up of the enzyme’s active site by forming a binding pocket for ATP proximate to the T308 site and 2) reducing accessibility of phosphatases. Transfer of a phosphate from ATP to substrate and the consequent formation of ADP exposes both threonine and serine phosphorylated residues to phosphatases and a return to the inactive form of the enzyme [101]. ATP-competitive inhibitors of AKT not only bar ATP from the active site but also lock the AKT into a conformation that disallows phosphatase access, thus maintaining AKT in an inactive hyperphosphorylated state [102]. Furthermore, phosphorylation of AKT at S473 by mTORC2 leads to inactivation of BAD, a pro-apoptotic protein [103].
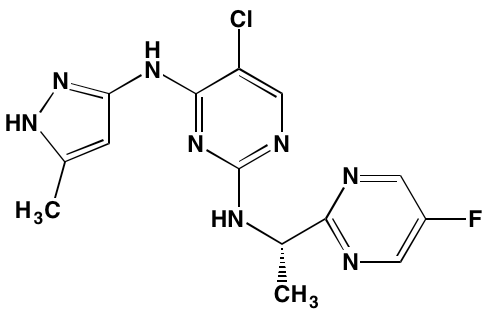
Afuresertib (GSK2110183), an orally active ATP-competitive inhibitor of all three isoforms of AKT, has exhibited a favorable activity profile in bortezomib-refractory MM patients [104]. The closely related pyrazole derivative uprosertib (GSK2141795) combined with MEK inhibitor trametinib is under study for patients with solid tumors and MM (NCT01951495) [105].
The high degree of homology between AKT isoforms, as well as with other AGC family members, poses a major challenge to design of isozyme-specific AKT inhibitors and has stimulated an allosteric approach to the design of AKT inhibitors. This group of inhibitors binds to regions of the AKT molecule distinct from the active site, preventing the activating phosphorylations at T308 and S473 by PDPK1 and mTORC2 [106]. MK-2206, an AKT allosteric inhibitor, which is in Phase I studies for solid tumors, has exhibited promising activity in myeloma cell lines, but as yet has not been the subject of a myeloma-based clinical trial [107]. In addition, both MK-2206 and the nucleoside analog triciribine have shown synergistic activity against MM cells in combination with the farnesyltransferase inhibitor tipifarnib [108]. Triciribine, which is activated by intracellular conversion to its triphosphate, is known to target the PH domains of AKT1 and AKT2, thus competing with AKT for the PIP3 binding site [109].
Another compound that interacts with the AKT PH domain to block its interaction with the plasma membrane is perifosine, an orally bioavailable alkylphospholipid [110]. Although AKT appears to be its primary target, perifosine also has been shown to affect other kinases, including the Jun N-terminal kinase (JNK), whose activation by the agent results in apoptosis [111]. These and other facets of perifosine’s mechanism of action have been reviewed recently by Fensterle et al. [112]. Perifosine has been evaluated clinically in several tumor types, including MM. In a Phase I study with lenalidomide and dexamethasone perifosine demonstrated favorable activity in relapsed and relapsed/refractory MM [113]. Phase I and II results, in combination with bortezomib and dexamethasone, were especially encouraging [114]; however, a Phase III trial of the latter combination was terminated when perifosine failed to provide progression-free survival benefit compared to bortezomib and dexamethasone alone (NCT01002248).
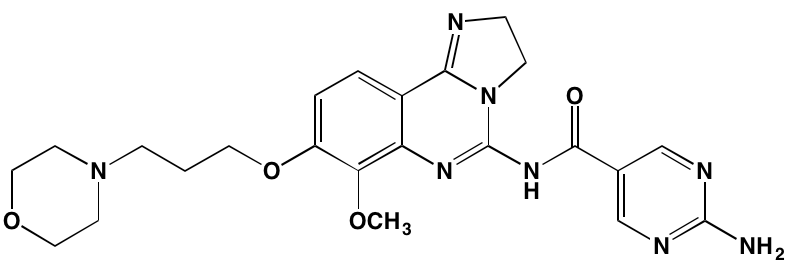
PDPK1, like its phosphorylation target AKT, belongs to the AGC family of kinases. PDPK1 also is known to phosphorylate at least 22 other proteins [98]. Although much evidence implicates PDPK1 in cancer [115], including MM [116], few specific inhibitors of this enzyme have been developed. The most advanced of these is AR-12/OSU-03012, an orally bioavailable celecoxib derivative devoid of cyclooxygenase-2 inhibitory activity and, based on promising preclinical studies [117], including demonstration of cytotoxicity to myeloma cells [118], has been the subject of a Phase I trial in patients with advanced solid tumors (NCT00978523).
mTOR plays a central role in two multiprotein complexes of which it is a component: mTOR Complex 1 (mTORC1), a rapamycin-sensitive complex which stimulates cell growth and in which mTOR associates with raptor, among other proteins; and rapamycin-refractory mTORC2, in which one of the protein partners is rictor. Downstream targets of mTORC1 include p70 ribosomal protein S6 kinase (P70S6K) and translation initiation regulator 4E binding protein (4E-BP1). Thus, inhibitors of mTORC1 would be expected to block protein synthesis by operating at different levels of the translational process.
Regulation of the complex interplay of downstream effects of mTOR and their relationship to cancer have been the subject of intense study in recent years. For example, constitutive activation of mTORC1 by AKT is known to occur in a two-step process. First, phosphorylation by AKT of the GTPase-activating protein TSC2, causes inactivation of TSC2. This, in turn, leads to activation of RHEB (ras homolog enriched in brain), a RAS-related GTPase and subsequent phosphorylation of mTOR and/or raptor at a number of sites. In addition, activation of mTORC1 inhibits AKT by a negative feedback mechanism [119].
The rapalogs everolimus and temsirolimus are orally-effective specific allosteric inhibitors of mTORC1 that work by binding to the intracellular protein FKBP (FK506 binding protein)12. The complex thus formed has a strong affinity for mTORC1, blocking downstream signaling. Preclinical work with these agents demonstrated anti-MM activity [120] and prompted their employment in clinical trials as single agents, albeit with only modest results [121]. Subsequently, Ghobrial et al. [122] in a Phase I/II study concluded that temsirolimus in combination with bortezomib may have a role in the treatment of relapsed or refractory MM without the addition of steroids. A Phase I study of everolimus in combination with lenalidomide drew a similar conclusion [123]. Recently, Li et al. [124] extensively reviewed mTOR signaling as a therapeutic target in MM.
It has been speculated that one reason for the generally poor clinical results seen with mTORC1 inhibitors in MM relates to the feedback activation of AKT. This has spurred the development of dual mTORC1/C2 inhibitors on the premise that prevention of AKT activation will remove the anti-apoptotic effect of this kinase [125]. For example, Maiso et al. [126] reported that the ATP-competitive dual inhibitor sapanisertib (INK128) is much more active against myeloma cell lines than is rapamycin alone. Another dual inhibitor, AZD8055, unlike rapamycin, produced apoptosis in MM cells [127]. PI3K/AKT/mTOR pathway inhibitors that have shown anti-myeloma activity are shown in Tables 3 and 4.
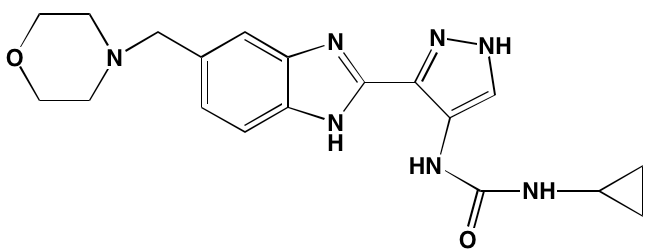
The proviral insertion site of Moloney murine leukemia virus (PIM) kinases recently have received attention as potential targets for new drug development in the autoimmune and cancer fields [128]. The three PIM kinase isoforms (PIM1, PIM2, and PIM3), which lack a regulatory domain are thus constitutively active, share a high degree of sequence homology and are important downstream effectors of JAK/STAT, ABL, and FLT3. They also have in common a feature unique among kinases - the presence of proline in the enzyme’s hinge region, resulting in a single hydrogen bond site, rather than two donor points, for ATP binding, a finding that offers the potential for designing highly selective PIM kinase inhibitors [129]. The PIM kinases phosphorylate a broad range of substrates involved in cell cycle progression and apoptosis, as well as in gene transcription and protein synthesis. Among the specific phosphorylation targets of PIM2, which is highly expressed in myeloma cells, is TSC2, a known modulator of mTORC1 whose activation promotes cell proliferation [130]. A number of small molecule PIM kinase inhibitors have demonstrated anti-tumor activity in myeloma cell lines (see Table 4).
Table 3: PI3K/AKT/mTOR Pathway Inhibitors With Anti-MM Activity
Compound |
Route |
Structure |
Kinase(s) Inhibited (IC50 in nM)a |
References/ Clinical Trials |
Alpelisib (BYL719)* |
Oral |
|
PI3Kalpha (5) |
|
BENC-511 |
Oral |
|
PI3K (non-selective); AKT (non-selective) |
[308] |
BGT226 (NVP-BGT226)* |
Oral |
|
PI3Kalpha/beta/ gamma (4/63/38); mTOR (NA) |
|
Buparlisib (BKM120)* |
Oral |
|
PI3Kalpha/beta/ gamma/delta (52/166/262/116) |
|
Copanlisib (BAY 80-6946) |
IV |
|
PI3Kalpha/beta/ gamma/delta (0.5/ 3.7/6.4/0.7) |
|
CUDC-907 |
Oral |
|
PI3Kalpha/beta/ gamma/delta (19/54/311/ 39) |
|
C96 |
NA |
|
PI3Kalpha/delta (5410/7050) |
[89] |
Dactolisib (BEZ235)* |
Oral |
|
PI3Kalpha/beta/ gamma/delta (4/75/5/7); mTOR (6) |
|
IC488743 |
Oral |
Not disclosed |
PI3Kdelta (5) |
[83] |
Idelalisib* (CAL-101) |
Oral |
|
PI3Kdelta (2.5) |
NCT00710528 |
LY294002 (SF1101)* |
NA |
|
PI3Kalpha/beta/delta (500/970/570) |
|
PI-103* |
NA |
|
PI3Kalpha/beta/ gamma/delta (2/3/ 15/3); mTOR (30); DNA-PKcs (23) |
[97] |
Pictilisib (GDC-0941)* |
Oral |
|
PI3Kalpha/delta (3/3) |
|
PIK-C98* |
NA |
|
PI3Kalpha/beta/ gamma/delta (590/ 1640/740/3650) |
[90] |
SF1126 (LY294002 prodrug)* |
IV |
|
PI3K (non-selective); mTOR |
|
S14161 |
Oral |
|
PI3K (non-selective) |
|
Afuresertib* (GSK2110183) |
Oral |
|
AKT1/2/3 (0.08/2/2.6) d |
|
MK2206† |
Oral |
|
AKT1/2/3 (8/12/65) |
|
Perifosine† |
Oral |
|
AKT (4700b) |
[113, 114]; NCT00401011; NCT00375791; NCT00415064; NCT01002248 |
Triciribine |
IV |
|
AKT (130c) |
|
Uprosertib* (GSK2141795) |
Oral |
|
AKT (non-selective) |
[329]; NCT01951495 |
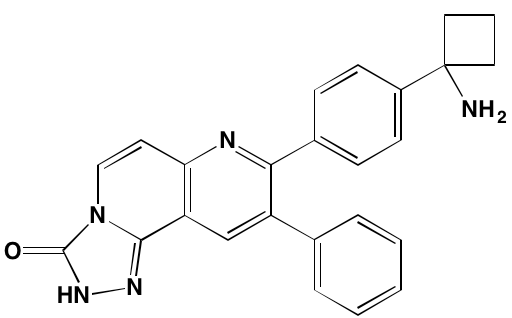
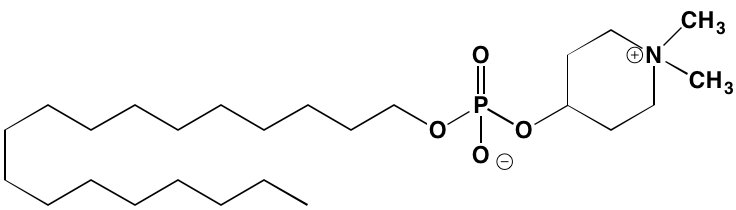
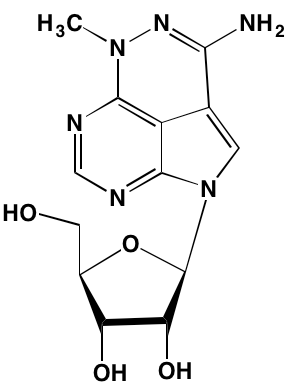
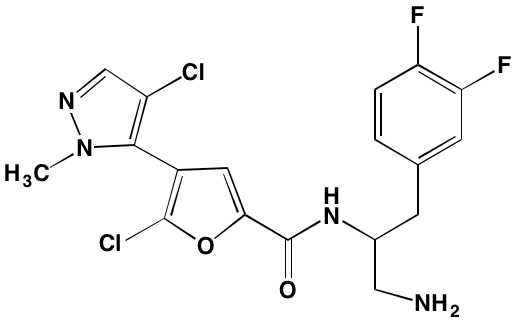
AR-12/OSU-03012* |
Oral |
|
PDPK1 (5000) |
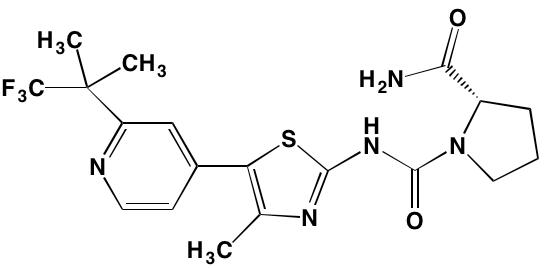
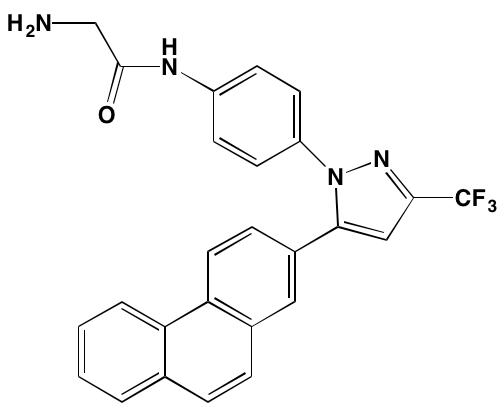
NA = Information not available; *ATP-competitive inhibitor; †allosteric inhibitor;
aas determined in cell-free assays, unless otherwise indicated; bMM.1S cells; cPC3 cells;
dKi expressed in nM
Table 4: PIMK and Additional mTOR Inhibitors With Anti-MM Activity
Compound |
Route |
Structure |
Kinase(s) Inhibited (IC50 in nM)a |
References/ Clinical Trials |
AZD8055 * |
Oral |
|
mTOR (0.8; 1.3)b |
|
CC-223* |
Oral |
|
mTORC1; mTORC2 |
[336]; NCT01177397 |
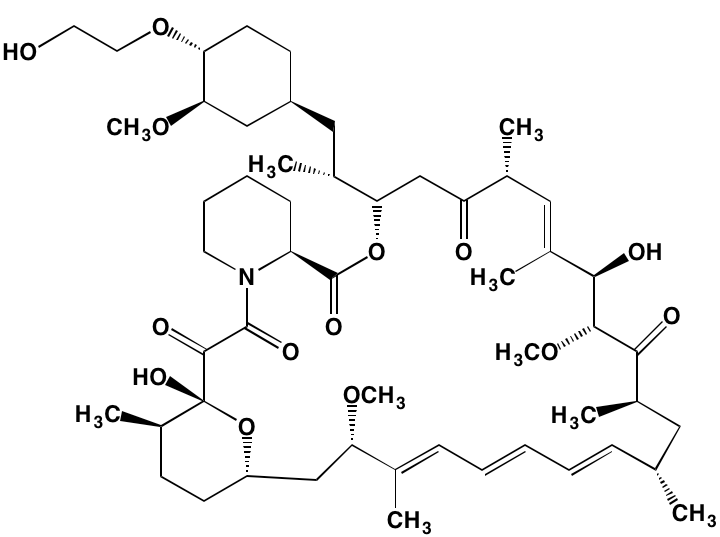
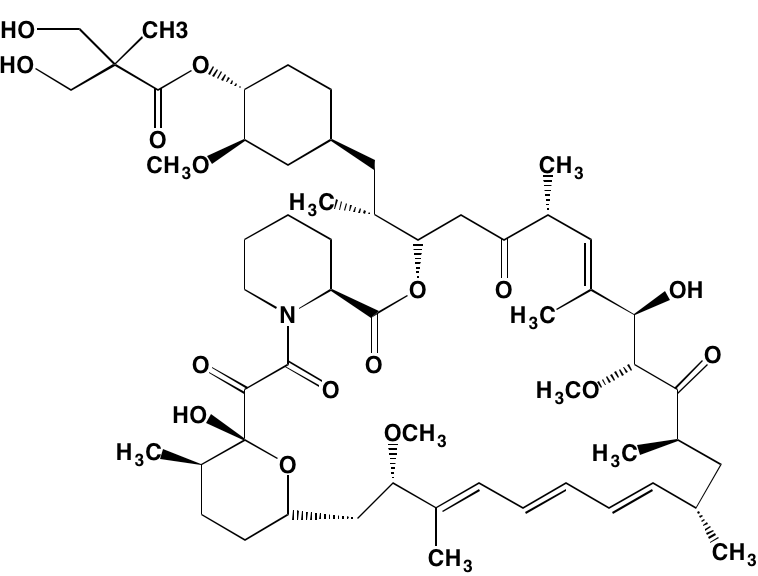
Everolimus† |
Oral |
|
mTOR (1.6-2.4) |
NCT00474929; NCT00618345; NCT00918333; NCT01234974; NCT01889420 |
Sapanisertib (INK128, MLN0128)* |
Oral |
|
mTOR (1.4b) |
[126] |
PP242* |
Oral |
|
mTOR (8) |
|
SC06 |
Oral |
|
mTORC1; mTORC2 |
[339] |
Temsirolimus† |
Oral |
|
mTOR (1760) |
NCT00079456; NCT00398515;NCT00422656;NCT00483262;NCT00693433 |
Torin 1* |
Oral |
|
mTORC1 (2); mTORC2 (10) |
|
WYE-354* |
Oral |
|
mTOR (5) |
|
Cnicin |
Oral |
|
PIMK (non-selective) |
[345] |
LGB321* |
Oral |
|
PIMK1/2/3 (1/2.1/0.8)c |
|
LGH447 (PIM447)* |
Oral |
|
PIMK1/2/3 (5.8/18.0/9.3)c |
[347]; NCT01456689 |
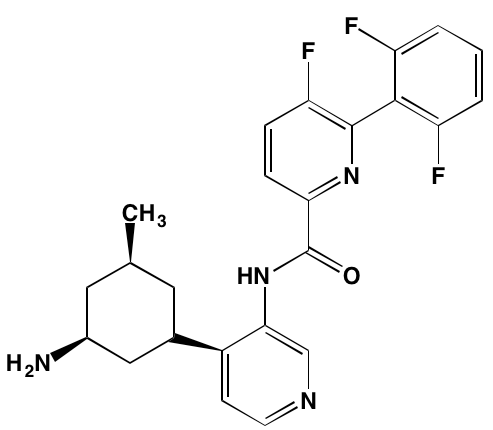
SGI-1776* |
Oral |
|
PIMK1 (7) |
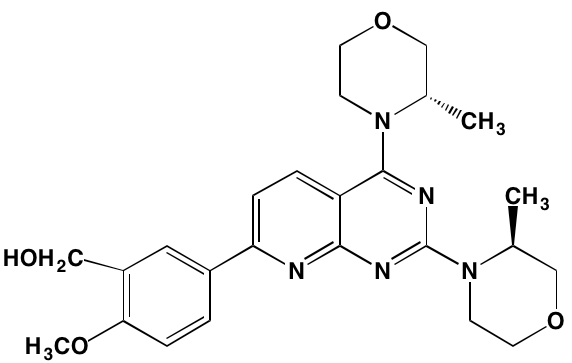
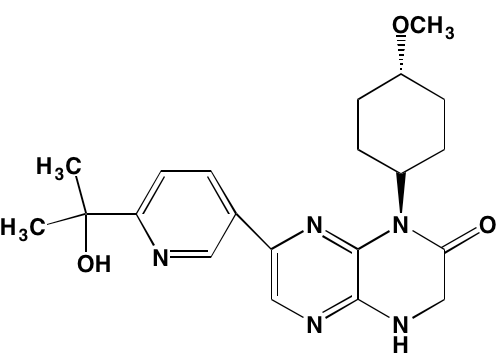
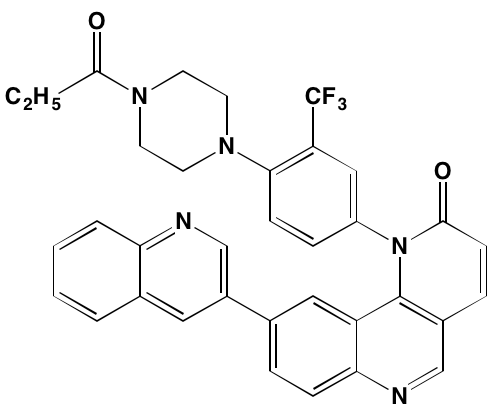
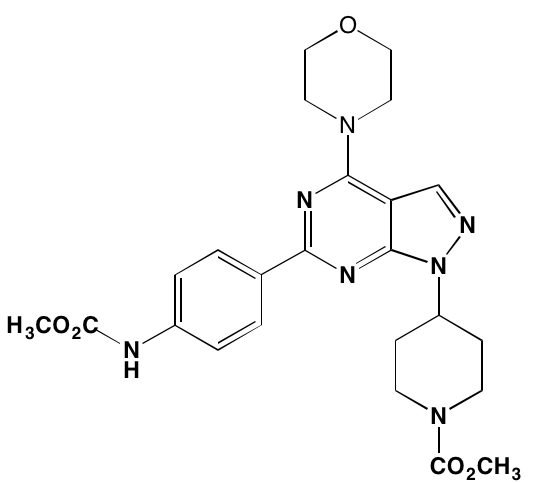
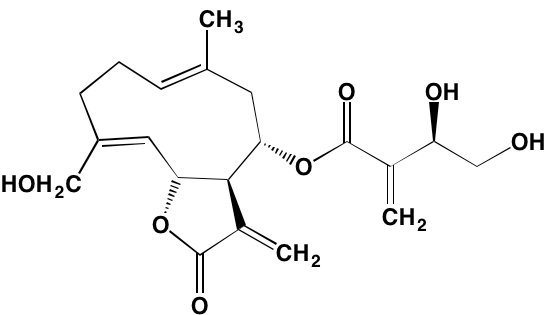
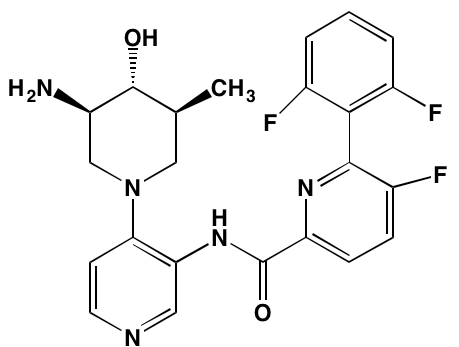
NA = Information not available; *ATP-competitive inhibitor; †allosteric inhibitor;
aas determined in cell-free assays; bKi expressed in nM; cKi expressed in pM
Cell cycle control S/TK
Progression through the cell cycle requires replication of the genome with exquisite fidelity and total segregation of the duplicated chromosomes between the two daughter cells during mitosis. The cellular machinery controlling sequential entry into and exit from the cycle’s four major phases (G1, S, G2, and M) is governed to a major extent by a series of kinases whose levels and extent of activation fluctuate as required according to the phase of the cycle. The relevance of several of the kinases described below to cell functioning in general and to transformed cells specifically has been reviewed by Malumbres [131].
Members of the Aurora kinase (AURK) family perform several functions in cell cycle regulation, particularly mitosis, where they coordinate events from G2 through to cytokinesis. The C-terminal catalytic domains in the family’s three members - AURKA, B, and C - are highly conserved while their regulatory N-termini exhibit little sequence homology. The ATP-binding sites in all three are identical [132]. AURKC is specifically expressed in testis where it plays a role in meiosis during spermatogenesis; its role in cancer development is not as clear as it is in the case of the other two family members, which are overexpressed in many different cancers, including hematological malignancies such as MM [133].
AURKA and B differ from each other in terms of their subcellular localization sites during mitosis and their timing of activation during the cell cycle. AURKA localizes to the duplicated centrosomes, as well as to the spindle poles during mitosis, while AURKB is found in the chromosome arms and inner centromeres from prophase to metaphase and in the central spindle in anaphase. During cytokinesis, both AURKA and B relocate to the spindle mid-zone. In addition to autophosphorylation, AURKA phosphorylates polo-like kinase 1 (PLK1), which, in turn promotes binding of AURKA to centrosomin, an important step in mitotic spindle assembly. AURKA also plays a key role in the G2/M transition by phosphorylating CDC (cell division cycle)25B, which is responsible for the activation of CDK1 (cyclin-dependent kinase1)-cyclin B1 [133]. Other AURKA phosphorylation targets include TPX2 (targeting protein for Xklp2), LIM protein, p53, and BRCA1 [134]. AURKB, a member of the chromosomal passenger complex that also includes survivin, borealin, and inner centromere protein (INCENP) [135], assists in chromosome condensation, segregation and microtubule attachment to the kinetochore and central spindle assembly. Its targets include histones H3 and H2A, MCAK (mitotic centromere-associated kinesin), topoisomerase II, INCENP, survivin, centromere protein A (CENP-A), and MYB-binding protein 1A (MYBBP1A) [136]. A number of AURK inhibitors have demonstrated activity against MM in both preclinical and clinical studies [133, 137]. These agents are summarized in Table 5.
Table 5: Cell Cycle Control S/TK Inhibitors With Anti-MM Activity
Compound |
Route |
Structure |
Kinase(s) Inhibited (IC50 in nM)a |
References/ Clinical Trials |
Alisertib (MLN8237)* |
Oral |
|
AURKA/B (1.2/396.5) |
|
AT9283* |
IV |
|
AURKA/B(3.0/3.0); JAK2/3 (1.2/1.1) |
|
Barasertib (AZD1152)* |
Oral |
|
AURKA/B (1368/ 0.37) |
|
Danusertib (PHA739358)* |
IV |
|
AURKA/B (13/79); ABL (25); FGFR1 (47) |
|
ENMD-2076 |
Oral |
|
AURKA/B (15/290); FLT3 (1.86); VEGFR2 (58.2) |
|
Tozasertib (VE-465, VX-680, MK 0457)* |
IV |
|
AURKA/B (0.6/18)b; BCR-ABL (30)b; FLT3 (30)b |
|
Alvocidib (Flavopiridol)* |
IV |
|
CDK1/2/4/6 (40/40/ 40/40) |
|
AT7519* |
Oral |
|
CDK1/2/4/5/6/9 (210/47/100/13/170/<10) |
|
Dinaciclib* |
IV |
|
CDK1/2/5/9 (3/1/1/4) |
|
LCQ195/ AT9311 |
NA |
|
CDK1/2/3/5/9 (2/2/ 42/1/15) |
[372] |
P276-00* |
IV |
|
CDK1/4/9 (79/63/20) |
|
Palbociclib* |
Oral |
|
CDK4/6 (11/16) |
NCT02030483; NCT00555906 |
PHA-767491* |
NA |
|
CDK9 (34); CDC7 (10) |
|
RGB-286638 |
IV |
|
CDK1/2/3/4/5/6/7/9 (2/3/5/4/5/55/44/1); JAK2 (50); AMPK (41); GSK3beta (3) |
[378] |
Seliciclib (CYC202, R-roscovitine)* |
Oral |
|
CDK2/5 (700/160) |
|
SNS-032 |
IV |
|
CDK2/7/9 (38/62/4) |
|
TG02 (SB1317) |
Oral |
|
CDK2 (13); JAK2 (73); FLT3 (56); ERK5 (NA) |
|
89S |
Oral |
|
CDC7 (2) |
[153] |
BI 2536 |
NA |
|
PLK1/2/3 (0.83/3.5/ 9.0) |
|
Scytonemin |
NA |
|
PLK1 (2000) |
|
KU55933* |
NA |
|
ATM (12.9) |
|
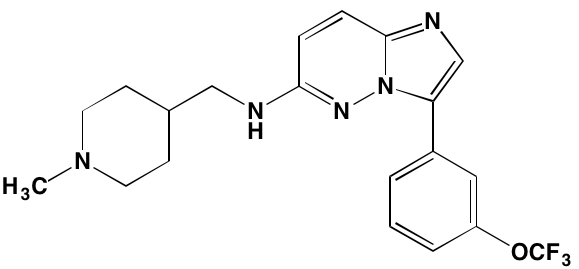
AZD7762* |
IV |
|
CHK1/2 (5/<10) |
|
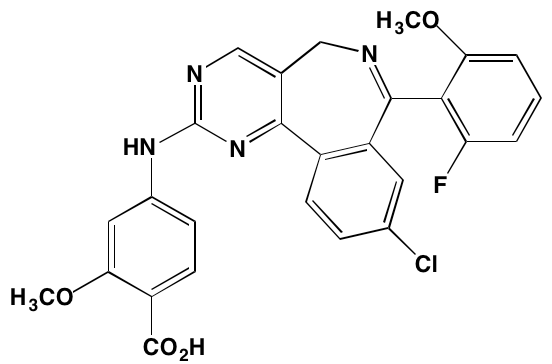
NU7026* |
NA |
|
DNA-PKcs (230) |
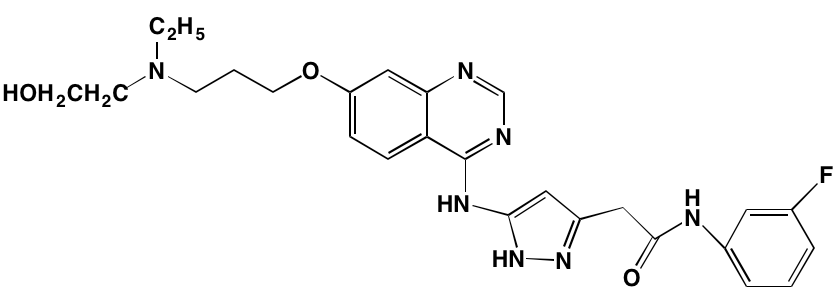
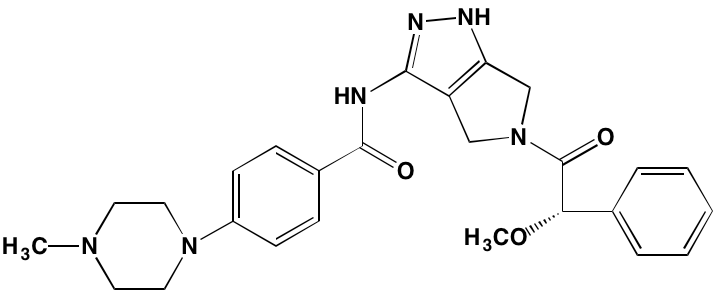
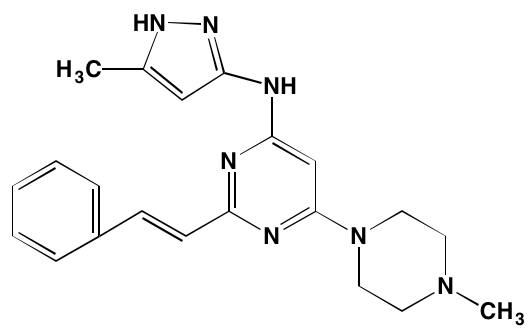
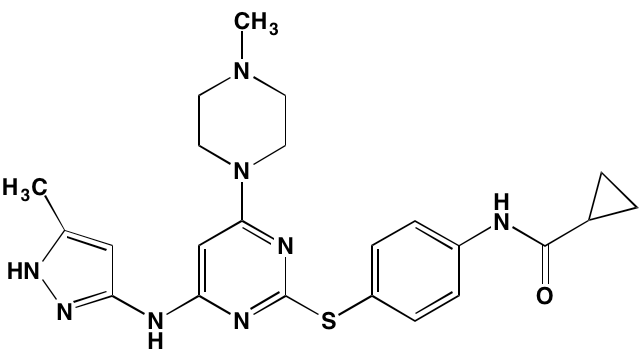
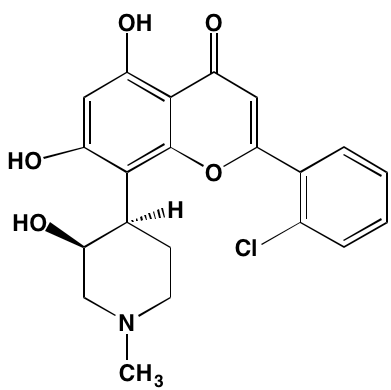
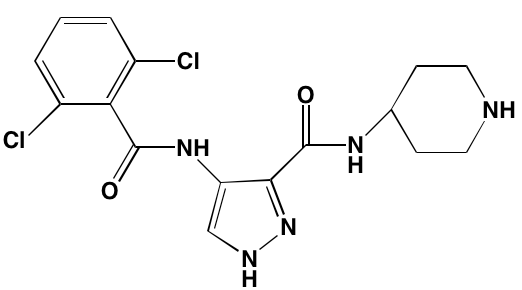
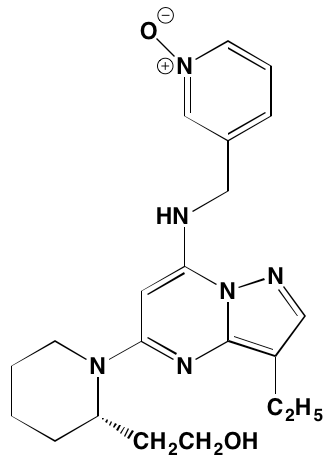
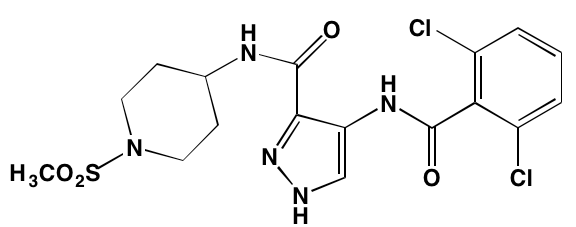
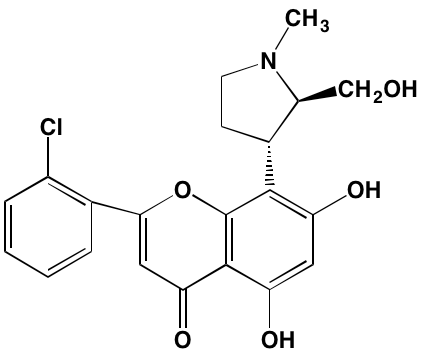
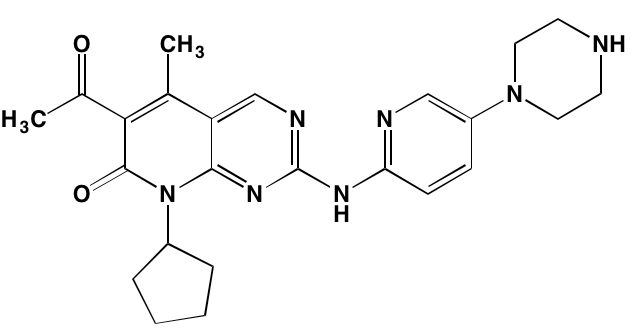
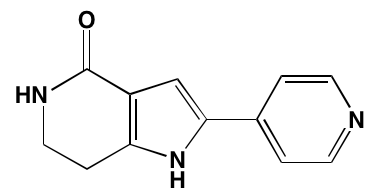
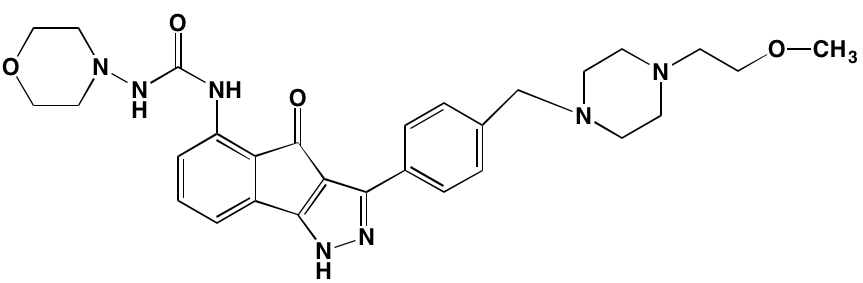
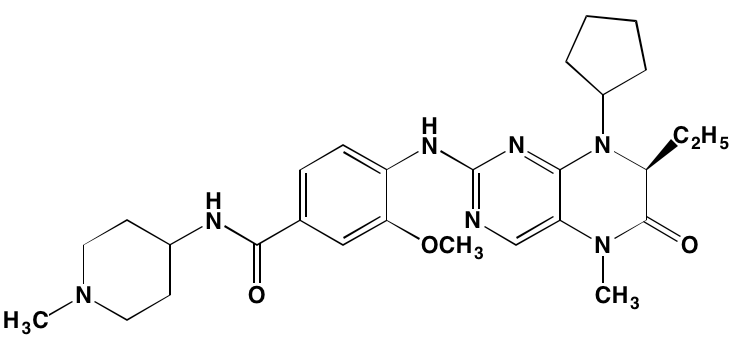
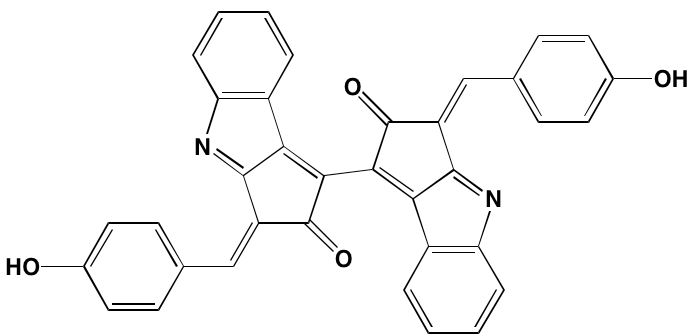
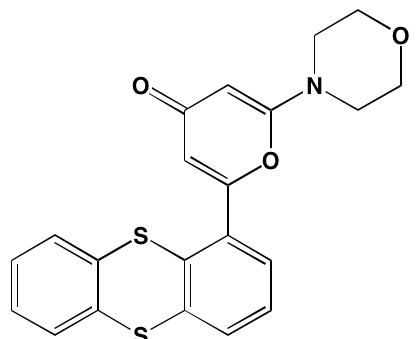
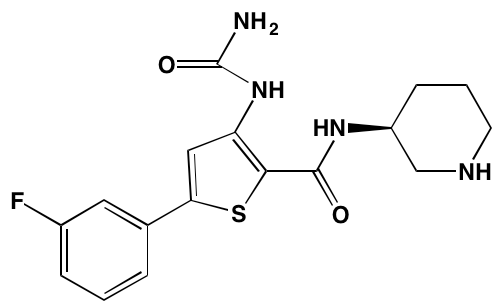
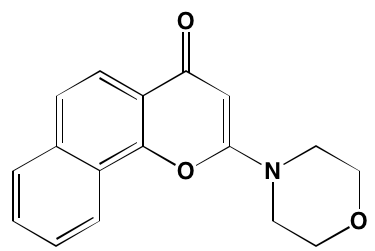
NA= Information not available; *ATP-competitive inhibitor; aas determined in cell-free assays; bKi in nM
Table 6: MAPK Pathways Inhibitors with Anti-MM Activity
Compound |
Route |
Structure |
Kinase(s) Inhibited (IC50)a |
References/ Clinical Trials |
Vemurafenib (PLX4032, RG7204)* |
Oral |
|
B-RAF V600E (31) |
|
PD184352 |
Oral |
|
MEK1/2 (17/17) |
|
PD325901 |
Oral |
|
MEK1/2 (0.33) |
|
Pimasertib (AS703026)† |
Oral |
|
MEK1/2 (NA) |
[396] |
Selumetinib (AZD6244, ARRY 142886)† |
Oral |
|
MEK1 (14); ERK1/2 (10c) |
|
Trametinib† |
Oral |
|
MEK1/2 (0.92/1.8) |
|
Baicalein |
Oral |
|
ERK1/2 (NA) |
[400] |
BI-D1870* |
NA |
|
RSK1/2/3/4 (31/24/18/15) |
|
FMK‡ |
NA |
|
RSK2 (15) |
[187] |
RMM46* |
NA |
|
RSK2 (12) |
|
SL0101-1* |
NA |
|
RSK2 (89) |
|
SP600125* |
NA |
|
JNK1/2/3 (40/40/90) |
|
Ralimetinib (LY2228820)* |
Oral |
|
p38alpha MAPK (7) |
|
SB202190* |
NA |
|
p38alpha/beta MAPK (50/100) (38d) |
|
SB203580* |
Oral |
|
p38alpha/beta MAPK (300-500)b |
|
SD-169* |
Oral |
|
p38alpha/beta MAPK (3.2/122) |
|
SD-282* |
Oral |
|
p38alpha MAPK (1.1) |
[414] |
Talmapimod (SCIO-469)* |
Oral |
|
p38alpha MAPK (9) |
|
VX-745* |
Oral |
|
p38alpha MAPK (35) |
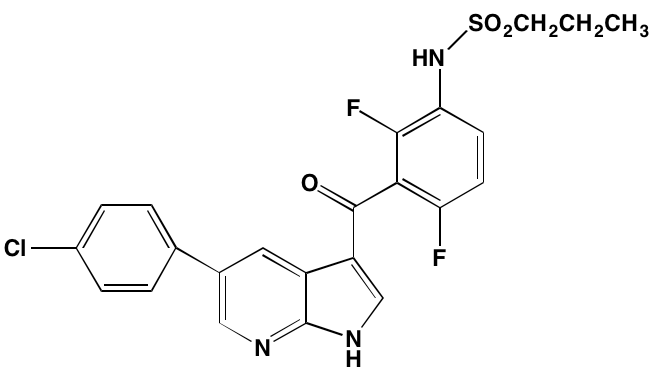
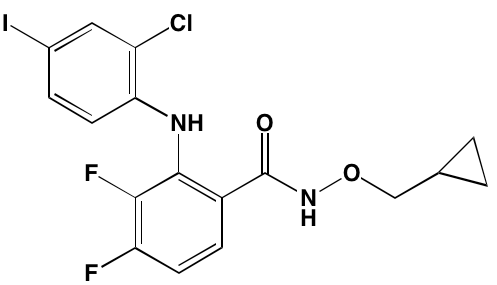
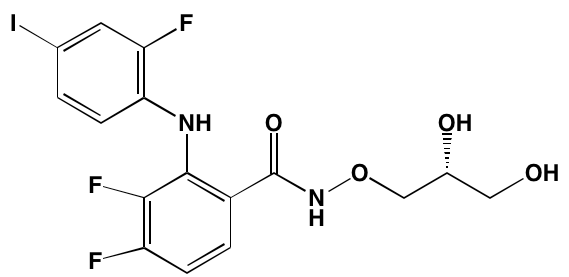
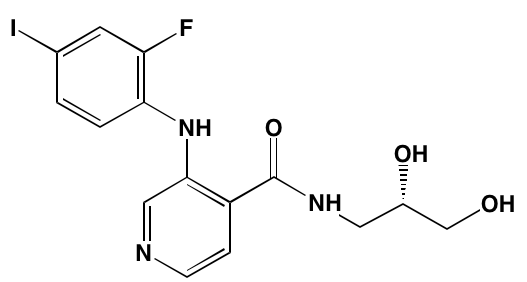
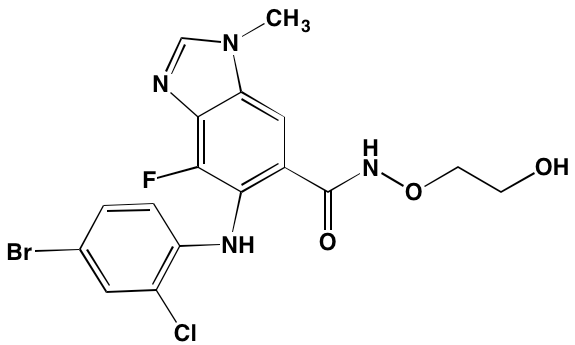
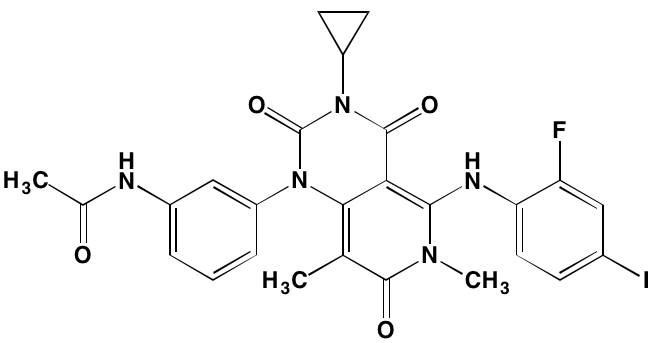
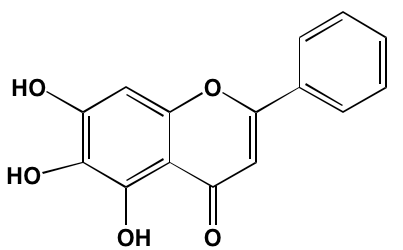
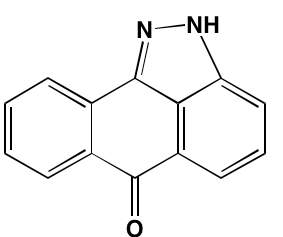
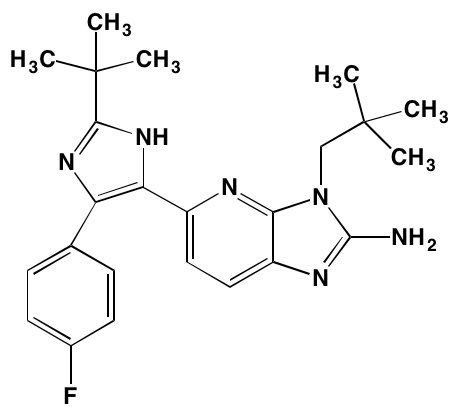
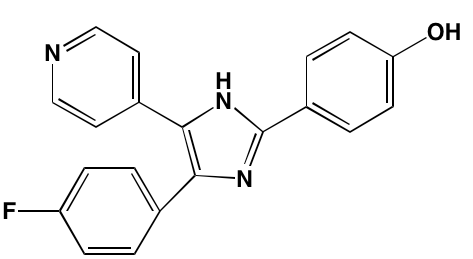
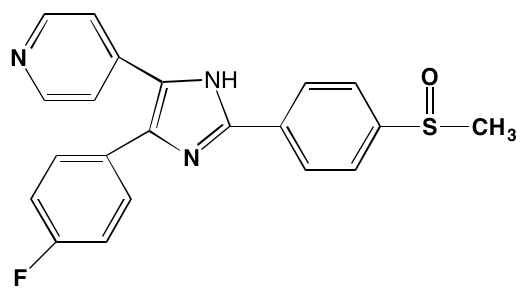
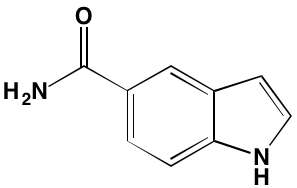
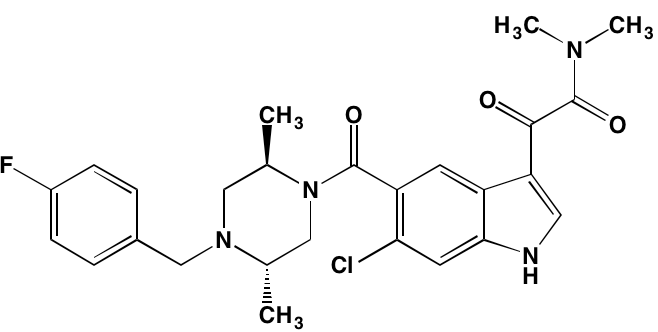
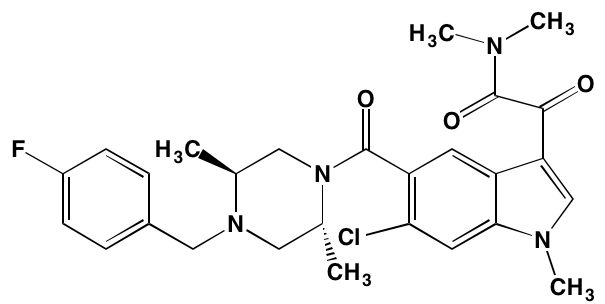
NA = Information not available; *ATP-competitive inhibitor; †allosteric inhibitor; ‡irreversible inhibitor; a as determined in cell-free assays, unless otherwise indicated; bTHP-1 cells; cMalme-3M cells; dKd in nM
Cyclin-dependent kinases (CDKs) act in association with regulatory partner proteins known as cyclins. The latter are subject to oscillating expression at various points in the cell cycle to exercise regulatory control of the cycle. Specific CDK/cyclin complexes control progression through the cell cycle at five key points: G0/G1 transition (cyclin C/CDK3); G1 phase (cyclin D/CDK4/6), G1/S transition (cyclin E/CDK2), S phase (cyclin A/CDK1/2), and M phase (cyclin B/CDK1) [138]. Over-expression of CCND1, the gene that codes for cyclin D1, one of the three isoforms of cyclin D, is found in over 80% of MM cases [139], making this cyclin a favored target for anti-myeloma drug development. Recent evidence points to the cyclin D1 G870A polymorphism as being associated with an increased risk of MM. Cyclin D1 catalyzes the phosphorylation of Rb, a tumor suppressor protein, which releases the transcription factor 2EF, thus activating downstream genes required for progression through the cell cycle. Several of the CDK inhibitors shown in Table 5 are active by virtue of their ability to compete with ATP at the enzyme’s active site [140, 141].
Some CDK family members are atypical in that they are neither associated with cell cycle regulation nor controlled by cyclins. One such example is CDK5, found primarily in terminally-differentiated neurons and regulated by non-cyclin subunits p35 and p39 [142]. This kinase also has been found to play important roles in extra-neuronal tissues, as well as in several cancers [143]. Silencing CDK5 has been demonstrated to sensitize MM cells to treatment with bortezomib and CDK5 inhibitors, such as seliciclib and dinaciclib, have been shown to enhance bortezomib-induced cytotoxicity in myeloma cells [144-146]. A recent report that dinaciclib sensitizes MM cells to the effects of poly (ADP-ribose) polymerase (PARP)1/2 inhibitors discloses another possible drug combination opportunity [147]. Other CDK family members play key roles in transcription. Chief among these are CDK7/cyclin H and CDK9/cyclin T, which activate the largest subunit of RNA polymerase II through phosphorylation at specific serine residues [148].
Cell division cycle 7 (CDC7) is a highly conserved kinase that plays an important role in regulation of normal cell cycle progression. The activity of CDC7 is modulated by fluctuating levels of DBF4 and DRF1, proteins which bind to the kinase, enabling phosphorylation of several sites on the DNA helicase subunit MCM2 (microchromosome maintenance protein 2) along with maintenance of replication forks, all critical events required for DNA synthesis, G1/S phase transition, and progression through the S phase [149]. CDC7 depletion by siRNA causes a number of tumor cell lines to undergo p53-independent apoptosis while normal cells experience cell cycle arrest with no effect on cell viability [150]. Moreover, upregulation of CDC7 has been observed in numerous cancer cell lines making this kinase an attractive chemotherapeutic target with potential selectivity [151]. However, only two reports have appeared in the literature concerning CDC7 inhibition in myeloma cells. In one, PHA-767491, an inhibitor of both CDC7 and CDK9, demonstrated anti-myeloma activity both in cells obtained from patients and in cell cultures [152]. Another compound, the related pyrrolopyridinone (designated 89S), inhibited two myeloma cell lines at low micromolar concentrations [153].
PLK1 is a principal regulator of the cell cycle, promoting entry into mitosis, spindle formation, sister chromatid segregation, and cytokinesis [154]. In addition, it acts as an inhibitor of apoptosis and its overexpression in various tumors has been linked with poor prognosis [155]. Although a number of small molecule inhibitors of PLK1 have been studied as anticancer agents [156], until recently, PLK1 has received little attention as a potential target in the therapy of MM. The PLK1 inhibitor BI 2536 has been reported to cause cell death in both MM cell lines and patient samples [157, 158]. Also, scytonemin, a pigment produced by cyanobacteria, has been found to induce cell cycle arrest and block cell growth in MM cells through inhibition of PLK1 [159].
An important aspect of cell cycle control is the ability to repair DNA damage prior to chromosome duplication. A number of S/TKs with possible links to MM play key roles in the DNA damage response (DDR). The ATM gene, whose mutation was originally found to be responsible for the autosomal recessive disease ataxia-telangiectasia (A-T), encodes a kinase known to play a central role in the DDR. DNA double-strand breaks initiate a signaling pathway that begins with recruitment to the damage site of ATM or the related kinase ATR (AT and Rad-3 related protein) in the case of single-strand breaks occurring during DNA replication stress. Both ATM and ATR, along with DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and mTOR, are often grouped together as the phosphatidylinositol 3-kinase related kinase (PIKK) family [160]. Phosphorylation of ATR and ATM activates the checkpoint kinases CHK1 and CHK2, respectively. Both CHK1 and CHK2 phosphorylate p53, which causes p53 to dissociate from its inhibitor MDM2. In the nucleus, the activated p53 promotes transcription of the p21 gene, whose protein product binds to and inactivates CDK complexes causing cell cycle arrest at G1/S and G2/M interfaces.
Little is known concerning the roles played by these DDR kinases in MM. A recent study of MM patients found that ATM was either lost or mutated in 1.3% and 3%, respectively, and that ATR deletions or mutations were present in 1.5% of samples. In all, mutations in TP53, ATM, and ATR were found in 17% of cases with poor outcomes. The inability to produce an effective apoptotic DDR was the most significant prognostic mutational indicator in this study [161].
A number of DDR inhibitors have been studied in MM as single agents, as well as for their ability to enhance the antitumor activity of conventional DNA-damaging agents, such as alkylating agents and topoisomerase II inhibitors [162-164]. For example, the ATM inhibitor KU55933 was reported to augment the activity of topoisomerase inhibitors in cultured myeloma cells [165]. Also, the CHK1 inhibitor AZD7762 was found to enhance the cytotoxic effects of bendamustine, melphalan, and doxorubicin in MM cell lines deficient in p53. Furthermore, AZD7762 and a set of three thienopyridinone-based CHK1 inhibitors potentiated the anti-myeloma activity of melphalan in p53-mutated cultured cells although they showed little effect on cell proliferation as single agents [162].
Atypical protein kinase inhibitors
The protein kinase C (PKC) family is a member of the AGC superfamily of kinases and includes at least 13 isoforms, four of which are grouped together as the classical PKCs - PKCalpha, PKCbetaI and betaII (representing two alternatively spliced forms), and PKCgamma [166]. These conventional and other isoforms are calcium-dependent and are activated by plasma membrane-bound diacylglycerol, which is formed by phospholipase C cleavage of phosphatidylinositol-4,5-bisphosphate [(PI(4,5)P2]. A number of studies have implicated PKC isoforms in cellular growth regulation in hematopoietic cancers although only a few PKC small molecule inhibitors have been studied in MM [167]. Enzastaurin (LY317615), an orally administered acyclic bisindolylmaleimide PKCbeta blocker, was reported to suppress cell proliferation in a panel of several human MM cell lines [168]. However, a Phase II trial (NCT00718419) in pre-treated MM patients found enzastaurin to be ineffective, although well-tolerated [169]. Midostaurin (PKC412), an oral multi-kinase inhibitor of PKCalpha, beta, and gamma isoforms and AKT, as well as of the TK receptors PDGFR, VEGFR, and FLT3, is known to induce apoptosis in human myeloma cells ([170]. Although no further work has been reported in MM, this agent has been the subject of a Phase I study with azacitidine in acute myeloid leukemia patients [171]. Down-regulation by rottlerin of PKCdelta, which is commonly expressed in MM, has been shown to produce apoptosis in myeloma cells [172].
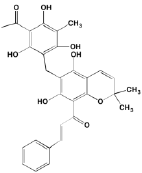
Enzastaurin Midostaurin Rottlerin
AMP-activated protein kinase (AMPK) and its upstream activator, the tumor suppressor liver kinase B1 (LKB1), are both allosterically activated - by AMP in the case of AMPK and by the pseudokinase STRAD and the adaptor protein MO25 in the case of LKB1. AMPK activation also may occur in response to energy and ATP depletion as a consequence of hypoxia and glucose deprivation. In this regard AMPK acts as an intracellular energy sensor to maintain the cell’s energy balance. AMPK, which has several downstream targets, plays a key role in the control of lipid and glucose metabolism. Among its effects are increased glycolysis, lipolysis, and fatty acid oxidation, as well as inhibition of lipogenesis, cholesterol formation, and protein synthesis. AMPK’s role in cancer is controversial, appearing to act as either a tumor promoter or a tumor suppressor depending on the context [173]. In the case of MM, AMPK activation by 5-aminoimidazole carboxamide riboside (AICAR) inhibited cell growth in myeloma cell lines [174] whereas AMPK inhibition by BML-275 (Compound C) induced apoptosis in these same cells [175].
Type 2 diabetes mellitus has been associated with increased risk of MM, as well as of other cancers [176]. Interestingly, the widely used antidiabetic agent metformin, an AMPK activator, has been associated with reduced risk of disease recurrence, overall mortality, and cancer-specific mortality in different cancer patient cohorts, including those with MM [177]. Zi et al. [178] have reported that in in vitro and in vivo xenograph models metformin inhibited MM cell proliferation in synergy with dexamethasone through induction of apoptosis and G0/G1 cell cycle arrest, suggesting the combined use of these two agents as a treatment option in MM.
![]()
AICAR BML-275 Metformin
Constitutive activation of RAS, leading to downstream dysregulation of the mitogen-activated protein (MAP) kinase (RAF/MEK/ERK) pathway, is associated with MM (Figure 1) [179]. BRAF, cited as one of the most significantly mutated genes in MM [161], has received considerable attention in cancer treatment, particularly in melanoma patients harboring the V600E mutation. Approximately 4% of MM patients also have been shown to have a BRAF mutation, associated with an aggressive clinical course [180-182]. Significantly, vemurafenib, a B-RAF V600E mutation inhibitor approved for melanoma, also has clinical activity in MM [183]. Ribosomal S6 kinase 2 (RSK2), a phosphorylation target of ERK, is responsible for activation of additional downstream targets associated with cell metabolism, cell survival, and cell cycle regulation [184, 185]. In MM RSK2 signaling has been reported to be associated with FGFR3 (t;4,14) activation [186, 187] and a number of RSK2 inhibitors have been shown to have anti-myeloma activity (Table 6).
The S/TK p38 MAPK, which exists in four different isoforms, alpha, beta, gamma, and delta, and is activated by a number of factors, including stress and inflammation [188], is known to be constitutively active in MM [189]. Such activation has been linked to osteolytic bone destruction [190] in MM, as well as to anti-myeloma drug resistance [191]. For example, talmapimod, which predominately blocks the p38alpha isoform, has been shown to enhance bortezomib-induced apoptosis in MM cells [192].
Other inhibitors of relatively understudied kinases
The NFkappaB family of transcription factors plays an important role in hematological malignancies, including MM in which it is overexpressed [193, 194]. Two NFkappaB pathways have been described in many cell types, including plasma cells (Figure 2). The canonical pathway is activated by stimulation of, among others, TNFalpha receptors leading to phosphorylation of IkappaB kinase (IKK), an intracellular complex comprised of two kinase subunits, alpha and beta, along with a regulatory subunit NEMO (NFkappaB essential modulator). IKKbeta phosphorylates IkappaB(inhibitor of kappa B), which in turn, leads to the ubiquitination and proteasomal degradation of IkappaB, causing its release from binding to NFkappaB, which is thereby freed to form dimers that translocate into the nucleus where transcription of genes participating in cell growth and adhesion, immune and inflammatory responses, and anti-apoptosis, are activated. In the non-canonical pathway NFkappaB is activated through a different set of receptors, such as CD40 ligand and B-cell activating factor (BAFF), is dependent on homodimeric IKKalpha and independent of IKKbeta and NEMO, and is regulated upstream of IKK by NFkappaB-inducing kinase (NIK). Blockage of the proteosomal degradation of IkappaB is believed to play a major role in the mechanism of bortezomib in MM therapy. The roles of and complex interactions between the two pathways in MM have been reviewed by Staudt [195] and by Gardam and Beyaert [196]. Although many IKKbeta inhibitors have been studied, none has reached clinical development because of liver toxicity concerns observed in knockout mice. A number of IKK and NIK blockers that have shown activity against MM cells are shown in Table 7.
Table 7: IKK and NIK Inhibitors as Anti-MM Agents
Compound |
Route |
Structure |
Kinase(s) Inhibited (IC50 in nM)a |
References |
Angelicin* (AS602868) |
Oral |
|
IKKbeta (20) |
|
BAY 11-7082‡ |
NA |
|
IKKalpha/beta (10000 c) |
[420] |
BAY 11-7085‡ |
NA |
|
IKKalpha/beta (10000 c) |
|
BMS-345541† |
Oral |
|
IKKbeta (300) |
|
DETT |
Oral |
|
IKKalpha/beta (NA) |
[423] |
MLN 120B* |
Oral |
|
IKKbeta (45b) |
[424] |
PS-1145* |
Oral |
|
IKKbeta (88) |
|
AM-0216* |
NA |
|
NIK (2b) |
[426] |
AM-0561* |
NA |
|
NIK (0.3b) |
[426] |
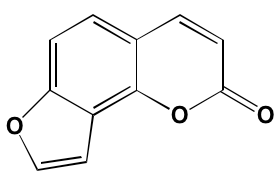
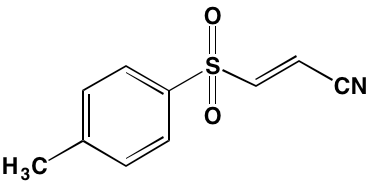
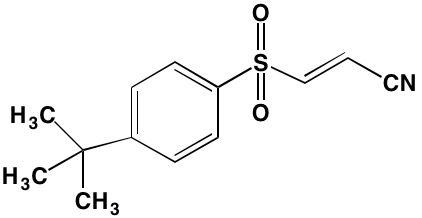
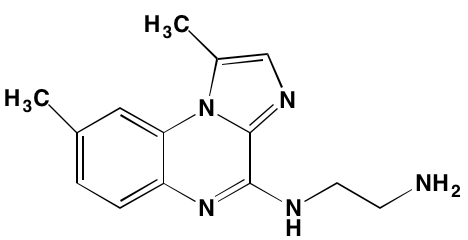
NA = Information unavailable; *ATP-competitive inhibitor; †allosteric inhibitor; ‡irreversible inhibitor; aas determined in cell-free assays, unless otherwise indicated; bKi in nM; chuman umbilical vein endothelial cells (HUVEC)
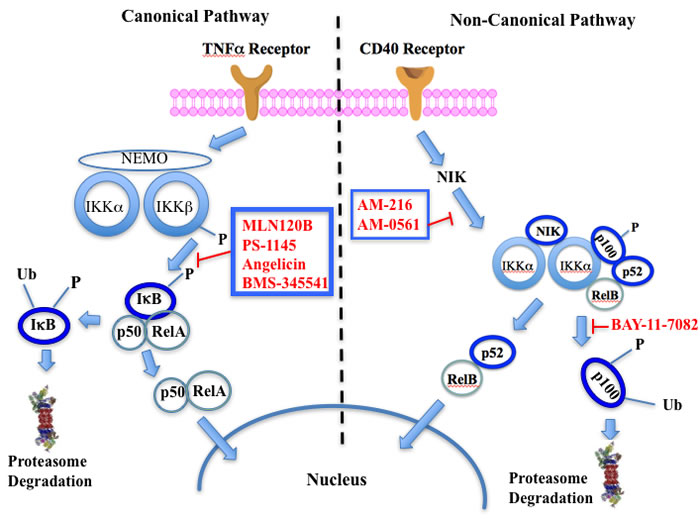
Figure 2: Putative sites of action of representative NFkappaB kinase inhibitors with anti-myeloma activity. Both canonical and non-canonical pathways resulting in NFkappaB activation are shown. The former results from stimulation by cytokines, such as tumor necrosis factor alpha (TNFalpha), leading to phosphorylation of the beta subunit of inhibitor of kappa B kinase (IKK), which in addition to an alpha subunit contains a regulatory subunit, NEMO (NFkappaB essential modifier). Subsequent phosphorylation of IkappaB by IKKbeta leads to ubiquitination and proteasome degradation of IkappaB, releasing the p50-RelA heterodimer, an NFkappaB family member, which translocates to the nucleus to activate the transcription of target genes. In the non-canonical pathway activation results from a different set of receptors, such as CD40 (shown here) and B-cell activating factor (BAFF), causing activation of NFkappaB-inducing kinase (NIK), which forms a complex with IKKalpha, p100, p52, and RelB. IKKalpha phosphorylation, ubiquitination, and proteasomal degradation of p100 frees the p52-RelB dimer, another NFkappaB family member, to enter the nucleus to activate its own set of target genes.
Glycogen synthase kinase (GSK) exists in two different isoforms - alpha and beta, which have 98% sequence identity in their kinase domains but only 38% in their carboxyl termini. Although originally identified as playing an important role in glycogen synthesis by phosphorylating and inactivating glycogen synthase, this kinase now has been implicated in a number of human diseases, including cancer, neurological disorders, diabetes, and cardiovascular disease. The multiple physiological roles in diverse signaling pathways played by this kinase, as well as its potential as a target for cancer treatment has recently been reviewed by McCubrey et al. [197]. In a specific focus on MM, Piazza, et al. [198] cited a number of studies implicating both GSK isoforms as pro-survival kinases in malignant plasma cells and MM-associated bone disease. SB-216763 and SB-415286, maleimide-derived (and closely related to enzastaurin) ATP-competitive inhibitors of both GSK isoforms, have been shown to activate the intrinsic apoptotic pathway in both MM cell lines and in patient-derived cells and to enhance bortezomib-induced cytotoxicity [199]. Moreover, the osteoprotective effect of the beta-selective GSK inhibitor 6-bromoindirubin-3’-oxime (BIO) has been cited as a potentially feasible approach to adjunct therapy of MM [200].
SB216763 SB415286 BIO
Casein kinase 2 (CK2) is involved in the phosphorylation of numerous cellular targets important to hematopoietic cell survival signaling pathways and is constitutively expressed in many cell types even under normal conditions. It is among the most pleiotropic of all S/TKs in the cell, accounting for the generation of about 20% of the human phosphoproteome [201]. The enzyme is comprised of two catalytic subunits (alpha and alpha’) and two regulatory beta subunits. In normal cells the enzyme is ubiquitously located in both the cytoplasm and the nucleus, shifting to primarily the nuclear compartment in cancer cells. CK2 overexpression has been found in many hematological cancers, including MM, and is associated with poor prognosis [202]. Silmitasertib (CX-4945), an orally bioavailable ATP-competitive inhibitor of both catalytic subunits of CK2 [203], currently is the subject of a Phase I study (NCT01199718) in patients with relapsed or refractory MM [202]. Another CK2 inhibitor that has shown anti-myeloma activity is the flavonoid apigenin, which additionally targets HSP90 and the co-chaperone CDC37 to induce apoptosis in MM cells [204].
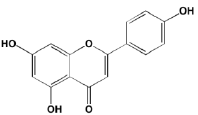
Silmitasertib Apigenin
Integrin-linked kinase (ILK) is found in focal adhesions. It also serves as an adapter protein interacting with integrin subunits to transmit extracellular signals from integrins and growth factors to the cell interior. ILK plays a role in regulating cell survival, cell cycle progression, angiogenesis, and PI3K-dependent signaling pathways. Overexpression of ILK is associated with several malignancies and correlates with poor prognosis [205]. Wang et al. [206] reported ILK expressed in myeloma cell lines could be inhibited by QLT0267, reducing invasiveness as well as VEGF and IL-6 secretion in co-cultured bone marrow stem cells.
Sphingosine kinase (SPHK), which catalyzes the phosphorylation of the primary OH of sphingosine to yield sphingosine-1-phosphate (S1P), exists as two distinct isoforms - SPHK1 and SPHK2. Although these isozymes share 80% similarity in their amino acid sequences, there are substantial differences in their biological roles and cellular localization [207]. Activation of any of five different extracellular G-protein-coupled receptors by S1P stimulates a number of cancer-promoting processes, such as cell proliferation, angiogenesis, and apoptosis blockade [208]. Several literature reports have suggested that SPHK1 regulates a rheostat balancing the effects of proapoptotic ceramide and sphingosine with those of the antiapoptotic S1P [209]. Numerous studies have demonstrated overexpression of SPHK1 in several cancers [210, 211]; however, associations with MM are less well documented. Tsukamoto et al. [212] recently reported a marked increase in SPHK1 in both patient and human cell line myeloma cells. The role played by SPHK2 in cancer is not well characterized and contradictory functions have been proposed for this isoform [213]. Significantly, fingolimod (FTY720) and ABC294640, specific inhibitors of SPHK1 and SPHK2, respectively, have been shown to induce apoptosis in MM cells [207, 214]. Fingolimod has been approved in the U.S for the treatment of multiple sclerosis.
![]()
![]()
Fingolimod ABC294640
Activation of protein kinase RNA-like endoplasmic reticulum kinase (PERK) results in phosphorylation of eukaryotic translation-initiation factor 2 (eIF2)alpha and general reduction of translation activity, decreasing the load of proteins translocated into the endoplasmic reticulum (ER) in an effort to lessen the stress. If the abundance of misfolded proteins in the ER greatly exceeds ER capacity, the resulting stress causes the unfolded protein response (UPR) to switch from pro-survival mode to stimulation of apoptosis [215].
Sunitinib has been shown to inhibit both the kinase and ribonuclease activities of inositol-requiring enzyme-1alpha (IRE1alpha) in myeloma cell lines [216] and Newbatt et al. [217] have developed a high-throughput screening assay to identify inhibitors of IRE1alpha autophosphorylation as potential anti-myeloma agents. The screen identified JNJ7706621, a previously known kinase inhibitor, as one compound active at submicromolar concentrations. GSK2656157 [218] and GSK2606414 [219] have been reported as orally available PERK inhibitors with anti-MM activity.
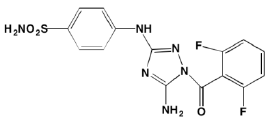
JNJ7706621 GSK2656157 GSK2606414
CONCLUSIONs
This review has focused on the role of the kinase family of enzymes in driving the malignant state in MM and efforts to design and develop small molecule kinase inhibitors to treat the disease. Therapeutic efforts aimed at combatting MM have been bolstered considerably in the past several years as new drug classes, including immunomodulators, proteasome inhibitors, HDAC inhibitors, and monoclonal antibodies, in conjunction with bone marrow transplantation, have entered clinical practice. Although five-year survival rates have approximately doubled to the 50% range over the past three decades, MM is still regarded as incurable and resistance continues to account for most patient setbacks.
Preclinical exploration of the kinome for possible therapeutic targets with the potential to combat MM has yielded much in the way of tangible results even though no kinase inhibitors have received FDA approval for MM therapy. While a number of kinase inhibitors are or have been included in MM clinical trials, at present no agent of this drug class appears to be on the verge of approval. This stands in contrast to the successes realized in developing small molecule protein kinase inhibitors to treat a number of other malignancies, including melanomas, renal cell carcinoma, chronic myelogenous leukemia, and cancers of the lung and thyroid.
In a recent analysis of the druggable genome Rask-Andersen and colleagues [220] identified 93 (64.1%) out of 145 different protein kinases - the largest category of targets - among the gene-encoded targets of agents in clinical trials for all disease categories but unrepresented in currently marketed pharmaceuticals. Most of these novel targets belong to the S/TK group with lesser numbers comprising TKs. Their study also revealed that a substantial majority of the 518 protein kinases known to be encoded in the human genome remain unexplored as possible candidate targets for new drug development. The problem of narrowing the field of potential targets for MM new drug development has been facilitated by the recent work of Tiedemann et al. [13] in which 57 genes were cataloged as potent MM survival genes with druggable potential. Included in this number were the genes for six kinases: PLK1, AURKB, CDK11A, CDK11B, TYK2, and unc-51-like kinase 3 (ULK3). As described in this review, inhibitors of half of these (PLK1, TYK2 and AURKB) have been studied to one degree or another as potential targets in MM while the other three represent fertile ground for new anti-myeloma development.
Combination chemotherapy to overcome drug resistance is a mainstay in the approach to MM [221, 222]. A review of the clinical trials.gov website identified a total of 51 recently initiated, ongoing, or completed MM studies in either Phase I/II or Phase II that include at least one kinase inhibitor in various combinations. Published results from most of these trials reveals that, although the agents with some exceptions are well-tolerated, in most cases they fail to demonstrate significant clinical benefit in MM. One exception to this result is a published report [223] of a Phase II trial in which the CDK4/6 inhibitor palbociclib demonstrated efficacy in 20% of patients. A few clinical studies combining two kinase inhibitors working by different mechanisms in MM have been initiated. These include simultaneous blockade of PIMK and PI3K using LGH447 and BYL719, respectively (NCT02144038); however, results of this completed trial have yet to be reported. In March 2016, participant recruitment for a Phase II study (NCT01951495) in relapsing/recurring MM patients employing AKT inhibitor GSK2141795 and MET blocker trametinib was suspended without explanation. To date, only two kinase inhibitors have been advanced to Phase III trials for the disease. One such study (NCT01002248) combining perifosine with bortezomib and dexamethasone was terminated because of failure to demonstrate efficacy [224], in spite of encouraging results seen in an earlier Phase II trial (NCT00401011). No published results are available for the second study (NCT01470131) involving dexamethasone and bortezomib with and without the oral agent masitinib in patients with relapsing MM.
As noted in Tables 1-7, a number of the kinase inhibitors with anti-myeloma activity compete with ATP, the common substrate for all protein kinases, for the enzyme’s active site. The observation that these sites are quite similar structurally, not to mention the high intracellular concentration of ATP relative to inhibitor, might appear to be a daunting challenge to target-specific design. However, the fact that a number of small molecule kinase inhibitors that take advantage of subtle ATP binding site differences have been developed and are used clinically [225], albeit often accompanied by off-target effects, to treat a number of different cancers lends credence to the validity of the use of ATP-competitive inhibitors in cancer treatment. On the other hand, design of small molecule ATP-non-competitive kinase inhibitors, acting either allosterically, irreversibly through covalent attachment, tight-binding within the active site, or by blocking critical upstream kinase-activating protein-protein interactions, have been advocated as valid approaches to kinase inhibitor design applicable to the treatment of cancer and other diseases wherein kinases play key roles [30, 225-230]. As the viability of these strategies to new drug design and development gains traction in the future and the underlying molecular mechanisms and critical biomarkers for the disease are identified, MM may yet be added to the list of diseases treatable with this class of drugs.
ACKNOWLEDGMENTS
The author wishes to thank Professor Anjaneyulu Kowluru for his many helpful discussions and review of this manuscript. Gratitude also is expressed to the late Professor Richard L. Slaughter, to whom this manuscript is dedicated, for his many years of leadership and staunch support.
conflicts of interest
The author has no conflict of interest with this work.
REFERENCES
1. Waxman AJ, Mink PJ, Devesa SS, Anderson WF, Weiss BM, Kristinsson SY, McGlynn KA and Landgren O. Racial disparities in incidence and outcome in multiple myeloma: a population-based study. Blood. 2010; 116:5501-5506.
2. Siegel RL, Miller KD and Jemal A. Cancer statistics, 2016. CA. 2016; 66:7-30.
3. Landgren O, Graubard BI, Katzmann JA, Kyle RA, Ahmadizadeh I, Clark R, Kumar SK, Dispenzieri A, Greenberg AJ, Therneau TM, Melton LJ, 3rd, Caporaso N, Korde N, et al. Racial disparities in the prevalence of monoclonal gammopathies: a population-based study of 12,482 persons from the National Health and Nutritional Examination Survey. Leukemia. 2014; 28:1537-1542.
4. Blade J, Dimopoulos M, Rosinol L, Rajkumar SV and Kyle RA. Smoldering (asymptomatic) multiple myeloma: current diagnostic criteria, new predictors of outcome, and follow-up recommendations. Journal of clinical oncology. 2010; 28:690-697.
5. Dimopoulos MA, Kastritis E, Rosinol L, Blade J and Ludwig H. Pathogenesis and treatment of renal failure in multiple myeloma. Leukemia. 2008; 22:1485-1493.
6. Cuzick J, De Stavola BL, Cooper EH, Chapman C and MacLennan IC. Long-term prognostic value of serum beta 2 microglobulin in myelomatosis. British journal of haematology. 1990; 75:506-510.
7. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P, Landgren O, Paiva B, Dispenzieri A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. The lancet oncology. 2014; 15:e538-548.
8. Smith BD, Smith GL, Hurria A, Hortobagyi GN and Buchholz TA. Future of cancer incidence in the United States: burdens upon an aging, changing nation. Journal of clinical oncology. 2009; 27:2758-2765.
9. Mimura N, Hideshima T and Anderson KC. Novel therapeutic strategies for multiple myeloma. Experimental hematology. 2015; 43:732-741.
10. Mina R, Cerrato C, Bernardini A, Aghemo E and Palumbo A. New pharmacotherapy options for multiple myeloma. Expert opinion on pharmacotherapy. 2015:1-12.
11. Lonial S, Weiss BM, Usmani SZ, Singhal S, Chari A, Bahlis NJ, Belch A, Krishnan A, Vescio RA, Mateos MV, Mazumder A, Orlowski RZ, Sutherland HJ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016; 387:1551-1560.
12. Lonial S, Dimopoulos M, Palumbo A, White D, Grosicki S, Spicka I, Walter-Croneck A, Moreau P, Mateos MV, Magen H, Belch A, Reece D, Beksac M, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. The New England journal of medicine. 2015; 373:621-631.
13. Tiedemann RE, Zhu YX, Schmidt J, Shi CX, Sereduk C, Yin H, Mousses S and Stewart AK. Identification of molecular vulnerabilities in human multiple myeloma cells by RNA interference lethality screening of the druggable genome. Cancer research. 2012; 72:757-768.
14. Tiedemann RE, Zhu YX, Schmidt J, Yin H, Shi CX, Que Q, Basu G, Azorsa D, Perkins LM, Braggio E, Fonseca R, Bergsagel PL, Mousses S, et al. Kinome-wide RNAi studies in human multiple myeloma identify vulnerable kinase targets, including a lymphoid-restricted kinase, GRK6. Blood. 2010; 115:1594-1604.
15. Mina R, Cerrato C, Bernardini A, Aghemo E and Palumbo A. New pharmacotherapy options for multiple myeloma. Expert opinion on pharmacotherapy. 2016; 17:181-192.
16. Hannah AL. Kinases as drug discovery targets in hematologic malignancies. Current molecular medicine. 2005; 5:625-642.
17. Palumbo A and Anderson K. Multiple myeloma. The New England journal of medicine. 2011; 364:1046-1060.
18. Noll JE, Williams SA, Tong CM, Wang H, Quach JM, Purton LE, Pilkington K, To LB, Evdokiou A, Gronthos S and Zannettino AC. Myeloma plasma cells alter the bone marrow microenvironment by stimulating the proliferation of mesenchymal stromal cells. Haematologica. 2014; 99:163-171.
19. Kawano Y, Moschetta M, Manier S, Glavey S, Gorgun GT, Roccaro AM, Anderson KC and Ghobrial IM. Targeting the bone marrow microenvironment in multiple myeloma. Immunological reviews. 2015; 263:160-172.
20. Ashcroft AJ, Davies FE and Morgan GJ. Aetiology of bone disease and the role of bisphosphonates in multiple myeloma. The lancet oncology. 2003; 4:284-292.
21. Matsumoto T and Abe M. TGF-beta-related mechanisms of bone destruction in multiple myeloma. Bone. 2011; 48:129-134.
22. Qiang YW, Yao L, Tosato G and Rudikoff S. Insulin-like growth factor I induces migration and invasion of human multiple myeloma cells. Blood. 2004; 103:301-308.
23. Guo Y, Xu F, Lu T, Duan Z and Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer treatment reviews. 2012; 38:904-910.
24. Alexandrakis MG, Passam FH, Sfiridaki A, Kandidaki E, Roussou P and Kyriakou DS. Elevated serum concentration of hepatocyte growth factor in patients with multiple myeloma: correlation with markers of disease activity. American journal of hematology. 2003; 72:229-233.
25. Hideshima T, Chauhan D, Schlossman R, Richardson P and Anderson KC. The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene. 2001; 20:4519-4527.
26. Kokonozaki M, Tsirakis G, Devetzoglou M, Kyriakaki S, Antonakis A, Vyzoukaki R, Pappa CA, Tzardi M and Alexandrakis MG. Potential role of FLT3-ligand in the angiogenic process of multiple myeloma. Leukemia research. 2015; 39:1467-1472.
27. Pappa CA, Alexandrakis MG, Boula A, Psarakis FE, Kolovou A, Bantouna V, Stavroulaki E and Tsirakis G. Emerging roles of endoglin/CD105 and angiogenic cytokines for disease development and progression in multiple myeloma patients. Hematological oncology. 2013; 31:201-205.
28. Kumar S, Witzig TE, Timm M, Haug J, Wellik L, Fonseca R, Greipp PR and Rajkumar SV. Expression of VEGF and its receptors by myeloma cells. Leukemia. 2003; 17:2025-2031.
29. Manning G, Whyte DB, Martinez R, Hunter T and Sudarsanam S. The protein kinase complement of the human genome. Science (New York, NY). 2002; 298:1912-1934.
30. Fleuren ED, Zhang L, Wu J and Daly RJ. The kinome ‘at large’ in cancer. Nature reviews Cancer. 2016; 16:83-98.
31. Rask-Andersen M, Zhang J, Fabbro D and Schioth HB. Advances in kinase targeting: current clinical use and clinical trials. Trends in pharmacological sciences. 2014; 35:604-620.
32. Sprynski AC, Hose D, Caillot L, Reme T, Shaughnessy JD, Jr., Barlogie B, Seckinger A, Moreaux J, Hundemer M, Jourdan M, Meissner T, Jauch A, Mahtouk K, et al. The role of IGF-1 as a major growth factor for myeloma cell lines and the prognostic relevance of the expression of its receptor. Blood. 2009; 113:4614-4626.
33. Kuhn DJ, Berkova Z, Jones RJ, Woessner R, Bjorklund CC, Ma W, Davis RE, Lin P, Wang H, Madden TL, Wei C, Baladandayuthapani V, Wang M, et al. Targeting the insulin-like growth factor-1 receptor to overcome bortezomib resistance in preclinical models of multiple myeloma. Blood. 2012; 120:3260-3270.
34. Ullrich A, Gray A, Tam AW, Yang-Feng T, Tsubokawa M, Collins C, Henzel W, Le Bon T, Kathuria S, Chen E and et al. Insulin-like growth factor I receptor primary structure: comparison with insulin receptor suggests structural determinants that define functional specificity. The EMBO journal. 1986; 5:2503-2512.
35. Clemmons DR. Involvement of insulin-like growth factor-I in the control of glucose homeostasis. Current opinion in pharmacology. 2006; 6:620-625.
36. Yin D, Sleight B, Alvey C, Hansson AG and Bello A. Pharmacokinetics and pharmacodynamics of figitumumab, a monoclonal antibody targeting the insulin-like growth factor 1 receptor, in healthy participants. Journal of clinical pharmacology. 2013; 53:21-28.
37. Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, Atkins C, Dumble M, Yang J, Anderson K, Kruger RG, Gontarek RR, Maksimchuk KR, et al. GSK1838705A inhibits the insulin-like growth factor-1 receptor and anaplastic lymphoma kinase and shows antitumor activity in experimental models of human cancers. Molecular cancer therapeutics. 2009; 8:2811-2820.
38. Manara MC, Landuzzi L, Nanni P, Nicoletti G, Zambelli D, Lollini PL, Nanni C, Hofmann F, Garcia-Echeverria C, Picci P and Scotlandi K. Preclinical in vivo study of new insulin-like growth factor-I receptor—specific inhibitor in Ewing’s sarcoma. Clinical cancer research. 2007; 13:1322-1330.
39. Dispenzieri A, Gertz MA, Lacy MQ, Geyer SM, Greipp PR, Rajkumar SV, Kimlinger T, Lust JA, Fonseca R, Allred J and Witzig TE. A phase II trial of imatinib in patients with refractory/relapsed myeloma. Leukemia & lymphoma. 2006; 47:39-42.
40. Derksen PW, de Gorter DJ, Meijer HP, Bende RJ, van Dijk M, Lokhorst HM, Bloem AC, Spaargaren M and Pals ST. The hepatocyte growth factor/Met pathway controls proliferation and apoptosis in multiple myeloma. Leukemia. 2003; 17:764-774.
41. Mahtouk K, Tjin EP, Spaargaren M and Pals ST. The HGF/MET pathway as target for the treatment of multiple myeloma and B-cell lymphomas. Biochimica et biophysica acta. 2010; 1806:208-219.
42. Zaman S, Shentu S, Yang J, He J, Orlowski RZ, Stellrecht CM and Gandhi V. Targeting the pro-survival protein MET with tivantinib (ARQ 197) inhibits growth of multiple myeloma cells. Neoplasia (New York, NY). 2015; 17:289-300.
43. Phillip CJ, Zaman S, Shentu S, Balakrishnan K, Zhang J, Baladandayuthapani V, Taverna P, Redkar S, Wang M, Stellrecht CM and Gandhi V. Targeting MET kinase with the small-molecule inhibitor amuvatinib induces cytotoxicity in primary myeloma cells and cell lines. Journal of hematology & oncology. 2013; 6:92.
44. Ferrucci A, Moschetta M, Frassanito MA, Berardi S, Catacchio I, Ria R, Racanelli V, Caivano A, Solimando AG, Vergara D, Maffia M, Latorre D, Rizzello A, et al. A HGF/cMET autocrine loop is operative in multiple myeloma bone marrow endothelial cells and may represent a novel therapeutic target. Clinical cancer research. 2014; 20:5796-5807.
45. Katoh M and Nakagama H. FGF receptors: cancer biology and therapeutics. Medicinal research reviews. 2014; 34:280-300.
46. Chell V, Balmanno K, Little AS, Wilson M, Andrews S, Blockley L, Hampson M, Gavine PR and Cook SJ. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene. 2013; 32:3059-3070.
47. Smadja NV, Bastard C, Brigaudeau C, Leroux D and Fruchart C. Hypodiploidy is a major prognostic factor in multiple myeloma. Blood. 2001; 98:2229-2238.
48. Keats JJ, Reiman T, Maxwell CA, Taylor BJ, Larratt LM, Mant MJ, Belch AR and Pilarski LM. In multiple myeloma, t(4;14)(p16;q32) is an adverse prognostic factor irrespective of FGFR3 expression. Blood. 2003; 101:1520-1529.
49. Chesi M and Bergsagel PL. Advances in the pathogenesis and diagnosis of multiple myeloma. International journal of laboratory hematology. 2015; 37 Suppl 1:108-114.
50. Grand EK, Chase AJ, Heath C, Rahemtulla A and Cross NC. Targeting FGFR3 in multiple myeloma: inhibition of t(4;14)-positive cells by SU5402 and PD173074. Leukemia. 2004; 18:962-966.
51. Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F and Dammacco F. Bone marrow angiogenesis and progression in multiple myeloma. British journal of haematology. 1994; 87:503-508.
52. Brito AB, Lourenco GJ, Oliveira GB, De Souza CA, Vassallo J and Lima CS. Associations of VEGF and VEGFR2 polymorphisms with increased risk and aggressiveness of multiple myeloma. Annals of hematology. 2014; 93:1363-1369.
53. Weis SM and Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nature medicine. 2011; 17:1359-1370.
54. Medinger M and Passweg J. Role of tumour angiogenesis in haematological malignancies. Swiss medical weekly. 2014; 144:w14050.
55. Giuliani N, Storti P, Bolzoni M, Palma BD and Bonomini S. Angiogenesis and multiple myeloma. Cancer microenvironment. 2011; 4:325-337.
56. Klein B, Zhang XG, Lu ZY and Bataille R. Interleukin-6 in human multiple myeloma. Blood. 1995; 85:863-872.
57. Ferrajoli A, Faderl S, Ravandi F and Estrov Z. The JAK-STAT pathway: a therapeutic target in hematological malignancies. Current cancer drug targets. 2006; 6:671-679.
58. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS and Jove R. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999; 10:105-115.
59. Quintanilla-Martinez L, Kremer M, Specht K, Calzada-Wack J, Nathrath M, Schaich R, Hofler H and Fend F. Analysis of signal transducer and activator of transcription 3 (Stat 3) pathway in multiple myeloma: Stat 3 activation and cyclin D1 dysregulation are mutually exclusive events. The American journal of pathology. 2003; 162:1449-1461.
60. de Weers M, Mensink RG, Kraakman ME, Schuurman RK and Hendriks RW. Mutation analysis of the Bruton’s tyrosine kinase gene in X-linked agammaglobulinemia: identification of a mutation which affects the same codon as is altered in immunodeficient xid mice. Human molecular genetics. 1994; 3:161-166.
61. Liu Y, Dong Y, Jiang QL, Zhang B and Hu AM. Bruton’s tyrosine kinase: potential target in human multiple myeloma. Leukemia & lymphoma. 2014; 55:177-181.
62. Eda H, Santo L, Cirstea DD, Yee AJ, Scullen TA, Nemani N, Mishima Y, Waterman PR, Arastu-Kapur S, Evans E, Singh J, Kirk CJ, Westlin WF, et al. A novel Bruton’s tyrosine kinase inhibitor CC-292 in combination with the proteasome inhibitor carfilzomib impacts the bone microenvironment in a multiple myeloma model with resultant antimyeloma activity. Leukemia. 2014; 28:1892-1901.
63. Sulzmaier FJ, Jean C and Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nature reviews Cancer. 2014; 14:598-610.
64. Wang SY, Hao HL, Deng K, Li Y, Cheng ZY, Lv C, Liu ZM, Yang J and Pan L. Expression levels of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and focal adhesion kinase in patients with multiple myeloma and their relationship to clinical stage and extramedullary infiltration. Leukemia & lymphoma. 2012; 53:1162-1168.
65. Ko BS, Chang TC and Liou JY. Focal adhesion kinase as a therapeutic target of bortezomib. Anti-cancer agents in medicinal chemistry. 2010; 10:747-752.
66. Zhang J, Ai L, Lv T, Jiang X and Liu F. Asiatic acid, a triterpene, inhibits cell proliferation through regulating the expression of focal adhesion kinase in multiple myeloma cells. Oncology letters. 2013; 6:1762-1766.
67. Zhang Y, Moschetta M, Huynh D, Tai YT, Zhang Y, Zhang W, Mishima Y, Ring JE, Tam WF, Xu Q, Maiso P, Reagan M, Sahin I, et al. Pyk2 promotes tumor progression in multiple myeloma. Blood. 2014; 124:2675-2686.
68. Meads MB, Fang B, Mathews L, Gemmer J, Nong L, Rosado-Lopez I, Nguyen T, Ring JE, Matsui W, MacLeod AR, Pachter JA, Hazlehurst LA, Koomen JM, et al. Targeting PYK2 mediates microenvironment-specific cell death in multiple myeloma. Oncogene. 2015; doi:10.1038/onc.2015.334:1-12.
69. Coluccia AM, Cirulli T, Neri P, Mangieri D, Colanardi MC, Gnoni A, Di Renzo N, Dammacco F, Tassone P, Ribatti D, Gambacorti-Passerini C and Vacca A. Validation of PDGFRbeta and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood. 2008; 112:1346-1356.
70. de la Puente P, Azab F, Muz B, Luderer M, Arbiser J and Azab AK. Tris DBA palladium overcomes hypoxia mediated drug resistance in multiple myeloma. Leukemia & lymphoma. 2015:1-22.
71. Wildes TM, Procknow E, Gao F, Dipersio JF and Vij R. Dasatinib in relapsed or plateau-phase multiple myeloma. Leukemia & lymphoma. 2009; 50:137-140.
72. Jungkunz-Stier I, Zekl M, Stuhmer T, Einsele H and Seggewiss-Bernhardt R. Modulation of natural killer cell effector functions through lenalidomide/dasatinib and their combined effects against multiple myeloma cells. Leukemia & lymphoma. 2014; 55:168-176.
73. Mocsai A, Ruland J and Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nature reviews Immunology. 2010; 10:387-402.
74. Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, Stilgenbauer S, Jumaa H, Veelken H and Zirlik K. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood. 2010; 115:4497-4506.
75. Koerber RM, Held SA, Heine A, Kotthoff P, Daecke SN, Bringmann A and Brossart P. Analysis of the anti-proliferative and the pro-apoptotic efficacy of Syk inhibition in multiple myeloma. Experimental hematology & oncology. 2015; 4:21.
76. Kubiczkova L, Sedlarikova L, Hajek R and Sevcikova S. TGF-beta - an excellent servant but a bad master. Journal of translational medicine. 2012; 10:183.
77. Takeuchi K, Abe M, Hiasa M, Oda A, Amou H, Kido S, Harada T, Tanaka O, Miki H, Nakamura S, Nakano A, Kagawa K, Yata K, et al. Tgf-Beta inhibition restores terminal osteoblast differentiation to suppress myeloma growth. PloS one. 2010; 5:e9870.
78. Hayashi T, Hideshima T, Nguyen AN, Munoz O, Podar K, Hamasaki M, Ishitsuka K, Yasui H, Richardson P, Chakravarty S, Murphy A, Chauhan D, Higgins LS, et al. Transforming growth factor beta receptor I kinase inhibitor down-regulates cytokine secretion and multiple myeloma cell growth in the bone marrow microenvironment. Clinical cancer research. 2004; 10:7540-7546.
79. Wu P and Hu YZ. PI3K/Akt/mTOR pathway inhibitors in cancer: a perspective on clinical progress. Current medicinal chemistry. 2010; 17:4326-4341.
80. Rosean TR, Tompkins VS, Tricot G, Holman CJ, Olivier AK, Zhan F and Janz S. Preclinical validation of interleukin 6 as a therapeutic target in multiple myeloma. Immunologic research. 2014; 59:188-202.
81. Lai K, Killingsworth MC and Lee CS. Gene of the month: PIK3CA. Journal of clinical pathology. 2015; 68:253-257.
82. Kok K, Geering B and Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends in biochemical sciences. 2009; 34:115-127.
83. Ikeda H, Hideshima T, Fulciniti M, Perrone G, Miura N, Yasui H, Okawa Y, Kiziltepe T, Santo L, Vallet S, Cristea D, Calabrese E, Gorgun G, et al. PI3K/p110{delta} is a novel therapeutic target in multiple myeloma. Blood. 2010; 116:1460-1468.
84. Wong KK, Engelman JA and Cantley LC. Targeting the PI3K signaling pathway in cancer. Current opinion in genetics & development. 2010; 20:87-90.
85. Ismail SI, Mahmoud IS, Msallam MM and Sughayer MA. Hotspot mutations of PIK3CA and AKT1 genes are absent in multiple myeloma. Leukemia research. 2010; 34:824-826.
86. Thorpe LM, Yuzugullu H and Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nature reviews Cancer. 2015; 15:7-24.
87. Pandey R and Kapur R. Targeting phosphatidylinositol-3-kinase pathway for the treatment of Philadelphia-negative myeloproliferative neoplasms. Molecular cancer. 2015; 14:118.
88. Zhu J, Hou T and Mao X. Discovery of selective phosphatidylinositol 3-kinase inhibitors to treat hematological malignancies. Drug discovery today. 2015; 20:988-994.
89. Tang J, Zhu J, Yu Y, Zhang Z, Chen G, Zhou X, Qiao C, Hou T and Mao X. A virtual screen identified C96 as a novel inhibitor of phosphatidylinositol 3-kinase that displays potent preclinical activity against multiple myeloma in vitro and in vivo. Oncotarget. 2014; 5:3836-3848. doi:10.18632/oncotarget.1657.
90. Zhu J, Wang M, Yu Y, Qi H, Han K, Tang J, Zhang Z, Zeng Y, Cao B, Qiao C, Zhang H, Hou T and Mao X. A novel PI3K inhibitor PIK-C98 displays potent preclinical activity against multiple myeloma. Oncotarget. 2015; 6:185-195. doi:10.18632/oncotarget.2688.
91. Munugalavadla V, Mariathasan S, Slaga D, Du C, Berry L, Del Rosario G, Yan Y, Boe M, Sun L, Friedman LS, Chesi M, Leif Bergsagel P and Ebens A. The PI3K inhibitor GDC-0941 combines with existing clinical regimens for superior activity in multiple myeloma. Oncogene. 2014; 33:316-325.
92. Glauer J, Pletz N, Schon M, Schneider P, Liu N, Ziegelbauer K, Emmert S, Wulf GG and Schon MP. A novel selective small-molecule PI3K inhibitor is effective against human multiple myeloma in vitro and in vivo. Blood cancer journal. 2013; 3:e141.
93. Qian C, Lai CJ, Bao R, Wang DG, Wang J, Xu GX, Atoyan R, Qu H, Yin L, Samson M, Zifcak B, Ma AW, DellaRocca S, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clinical cancer research. 2012; 18:4104-4113.
94. Younes A, Berdeja JG, Patel MR, Flinn I, Gerecitano JF, Neelapu SS, Kelly KR, Copeland AR, Akins A, Clancy MS, Gong L, Wang J, Ma A, et al. Safety, tolerability, and preliminary activity of CUDC-907, a first-in-class, oral, dual inhibitor of HDAC and PI3K, in patients with relapsed or refractory lymphoma or multiple myeloma: an open-label, dose-escalation, phase 1 trial. The lancet oncology. 2016; 17:622-631.
95. Gan ZY, Fitter S, Vandyke K, To LB, Zannettino AC and Martin SK. The effect of the dual PI3K and mTOR inhibitor BEZ235 on tumour growth and osteolytic bone disease in multiple myeloma. European journal of haematology. 2014; 94:343-354.
96. Baumann P, Schneider L, Mandl-Weber S, Oduncu F and Schmidmaier R. Simultaneous targeting of PI3K and mTOR with NVP-BGT226 is highly effective in multiple myeloma. Anti-cancer drugs. 2012; 23:131-138.
97. Glassford J, Kassen D, Quinn J, Stengel C, Kallinikou K, Khwaja A and Yong KL. Inhibition of cell cycle progression by dual phosphatidylinositol-3-kinase and mTOR blockade in cyclin D2 positive multiple myeloma bearing IgH translocations. Blood cancer journal. 2012; 2:e50.
98. Pearce LR, Komander D and Alessi DR. The nuts and bolts of AGC protein kinases. Nature reviews Molecular cell biology. 2010; 11:9-22.
99. Song G, Ouyang G and Bao S. The activation of Akt/PKB signaling pathway and cell survival. Journal of cellular and molecular medicine. 2005; 9:59-71.
100. Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP and Brandhuber BJ. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PloS one. 2010; 5:e12913.
101. Keane NA, Glavey SV, Krawczyk J and O’Dwyer M. AKT as a therapeutic target in multiple myeloma. Expert opinion on therapeutic targets. 2014:1-19.
102. Lin K, Lin J, Wu WI, Ballard J, Lee BB, Gloor SL, Vigers GP, Morales TH, Friedman LS, Skelton N and Brandhuber BJ. An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Science signaling. 2012; 5:ra37.
103. Vadlakonda L, Dash A, Pasupuleti M, Anil Kumar K and Reddanna P. The Paradox of Akt-mTOR Interactions. Frontiers in oncology. 2013; 3:165.
104. Voorhees P, Spencer A, Sutherland H, O’Dwyer M, Huang S, Stewart K, Chari A, Rosenzwieg M, Nooka A, Rosenbaum C, Hofmeister C, Smith D, Antal J, et al. (2013). Novel AKT inhibitor afuresertib in combination with bortezomib and dexamethasone demonstrates favorable safety profile and significant clinical activity in patients with relapsed/refractory multiple myeloma. Abstract 283, Proceedings of the 55th Annual Meeting of the American Society of Hematology (New Orleans, LA: ASH).
105. Tolcher AW, Patnaik A, Papadopoulos KP, Rasco DW, Becerra CR, Allred AJ, Orford K, Aktan G, Ferron-Brady G, Ibrahim N, Gauvin J, Motwani M and Cornfeld M. Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer chemotherapy and pharmacology. 2015; 75:183-189.
106. Calleja V, Laguerre M, Parker PJ and Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS biology. 2009; 7:e17.
107. Ramakrishnan V, Kimlinger T, Haug J, Painuly U, Wellik L, Halling T, Rajkumar SV and Kumar S. Anti-myeloma activity of Akt inhibition is linked to the activation status of PI3K/Akt and MEK/ERK pathway. PloS one. 2012; 7:e50005.
108. Balasis ME, Forinash KD, Chen YA, Fulp WJ, Coppola D, Hamilton AD, Cheng JQ and Sebti SM. Combination of farnesyltransferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clinical cancer research. 2011; 17:2852-2862.
109. Berndt N, Yang H, Trinczek B, Betzi S, Zhang Z, Wu B, Lawrence NJ, Pellecchia M, Schonbrunn E, Cheng JQ and Sebti SM. The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell death and differentiation. 2010; 17:1795-1804.
110. Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA and Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Molecular cancer therapeutics. 2003; 2:1093-1103.
111. Chiarini F, Del Sole M, Mongiorgi S, Gaboardi GC, Cappellini A, Mantovani I, Follo MY, McCubrey JA and Martelli AM. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia. 2008; 22:1106-1116.
112. Fensterle J, Aicher B, Seipelt I, Teifel M and Engel J. Current view on the mechanism of action of perifosine in cancer. Anti-cancer agents in medicinal chemistry. 2014; 14:629-635.
113. Jakubowiak AJ, Richardson PG, Zimmerman T, Alsina M, Kaufman JL, Kandarpa M, Kraftson S, Ross CW, Harvey C, Hideshima T, Sportelli P, Poradosu E, Gardner L, et al. Perifosine plus lenalidomide and dexamethasone in relapsed and relapsed/refractory multiple myeloma: a Phase I Multiple Myeloma Research Consortium study. British journal of haematology. 2012; 158:472-480.
114. Richardson PG, Eng C, Kolesar J, Hideshima T and Anderson KC. Perifosine, an oral, anti-cancer agent and inhibitor of the Akt pathway: mechanistic actions, pharmacodynamics, pharmacokinetics, and clinical activity. Expert opinion on drug metabolism & toxicology. 2012; 8:623-633.
115. Raimondi C and Falasca M. Targeting PDK1 in cancer. Current medicinal chemistry. 2011; 18:2763-2769.
116. Chinen Y, Kuroda J, Shimura Y, Nagoshi H, Kiyota M, Yamamoto-Sugitani M, Mizutani S, Sakamoto N, Ri M, Kawata E, Kobayashi T, Matsumoto Y, Horiike S, et al. Phosphoinositide protein kinase PDPK1 is a crucial cell signaling mediator in multiple myeloma. Cancer research. 2014; 74:7418-7429.
117. Schonthal AH, Chen TC, Hofman FM, Louie SG and Petasis NA. Celecoxib analogs that lack COX-2 inhibitory function: preclinical development of novel anticancer drugs. Expert opinion on investigational drugs. 2008; 17:197-208.
118. Zhang S, Suvannasankha A, Crean CD, White VL, Johnson A, Chen CS and Farag SS. OSU-03012, a novel celecoxib derivative, is cytotoxic to myeloma cells and acts through multiple mechanisms. Clinical cancer research. 2007; 13:4750-4758.
119. Guertin DA and Sabatini DM. Defining the role of mTOR in cancer. Cancer cell. 2007; 12:9-22.
120. Frost P, Moatamed F, Hoang B, Shi Y, Gera J, Yan H, Frost P, Gibbons J and Lichtenstein A. In vivo antitumor effects of the mTOR inhibitor CCI-779 against human multiple myeloma cells in a xenograft model. Blood. 2004; 104:4181-4187.
121. Farag SS, Zhang S, Jansak BS, Wang X, Kraut E, Chan K, Dancey JE and Grever MR. Phase II trial of temsirolimus in patients with relapsed or refractory multiple myeloma. Leukemia research. 2009; 33:1475-1480.
122. Ghobrial IM, Weller E, Vij R, Munshi NC, Banwait R, Bagshaw M, Schlossman R, Leduc R, Chuma S, Kunsman J, Laubach J, Jakubowiak AJ, Maiso P, et al. Weekly bortezomib in combination with temsirolimus in relapsed or relapsed and refractory multiple myeloma: a multicentre, phase 1/2, open-label, dose-escalation study. The lancet oncology. 2011; 12:263-272.
123. Yee AJ, Hari P, Marcheselli R, Mahindra AK, Cirstea DD, Scullen TA, Burke JN, Rodig SJ, Hideshima T, Laubach JP, Ghobrial IM, Schlossman RL, Munshi NC, et al. Outcomes in patients with relapsed or refractory multiple myeloma in a phase I study of everolimus in combination with lenalidomide. British journal of haematology. 2014; 166:401-409.
124. Li J, Zhu J, Cao B and Mao X. The mTOR signaling pathway is an emerging therapeutic target in multiple myeloma. Current pharmaceutical design. 2014; 20:125-135.
125. Hoang B, Frost P, Shi Y, Belanger E, Benavides A, Pezeshkpour G, Cappia S, Guglielmelli T, Gera J and Lichtenstein A. Targeting TORC2 in multiple myeloma with a new mTOR kinase inhibitor. Blood. 2010; 116:4560-4568.
126. Maiso P, Liu Y, Morgan B, Azab AK, Ren P, Martin MB, Zhang Y, Liu Y, Sacco A, Ngo H, Azab F, Quang P, Rodig SJ, et al. Defining the role of TORC1/2 in multiple myeloma. Blood. 2011; 118:6860-6870.
127. Cirstea D, Santo L, Hideshima T, Eda H, Mishima Y, Nemani N, Mahindra A, Yee A, Gorgun G, Hu Y, Ohguchi H, Suzuki R, Cottini F, et al. Delineating the mTOR kinase pathway using a dual TORC1/2 inhibitor, AZD8055, in multiple myeloma. Molecular cancer therapeutics. 2014; 13:2489-2500.
128. Swords R, Kelly K, Carew J, Nawrocki S, Mahalingam D, Sarantopoulos J, Bearss D and Giles F. The Pim kinases: new targets for drug development. Current drug targets. 2011; 12:2059-2066.
129. Bullock AN, Debreczeni JE, Fedorov OY, Nelson A, Marsden BD and Knapp S. Structural basis of inhibitor specificity of the human protooncogene proviral insertion site in moloney murine leukemia virus (PIM-1) kinase. Journal of medicinal chemistry. 2005; 48:7604-7614.
130. Lu J, Zavorotinskaya T, Dai Y, Niu XH, Castillo J, Sim J, Yu J, Wang Y, Langowski JL, Holash J, Shannon K and Garcia PD. Pim2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood. 2013; 122:1610-1620.
131. Malumbres M. Physiological relevance of cell cycle kinases. Physiological reviews. 2011; 91:973-1007.
132. Kollareddy M, Dzubak P, Zheleva D and Hajduch M. Aurora kinases: structure, functions and their association with cancer. Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, Czechoslovakia. 2008; 152:27-33.
133. Farag SS. The potential role of Aurora kinase inhibitors in haematological malignancies. British journal of haematology. 2011; 155:561-579.
134. Malumbres M and Perez de Castro I. Aurora kinase A inhibitors: promising agents in antitumoral therapy. Expert opinion on therapeutic targets. 2014; 18:1377-1393.
135. Carmena M, Wheelock M, Funabiki H and Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nature reviews Molecular cell biology. 2012; 13:789-803.
136. Perrera C, Colombo R, Valsasina B, Carpinelli P, Troiani S, Modugno M, Gianellini L, Cappella P, Isacchi A, Moll J and Rusconi L. Identification of Myb-binding protein 1A (MYBBP1A) as a novel substrate for aurora B kinase. The Journal of biological chemistry. 2010; 285:11775-11785.
137. Choudary I, Barr PM and Friedberg J. Recent advances in the development of Aurora kinases inhibitors in hematological malignancies. Therapeutic advances in hematology. 2015; 6:282-294.
138. Malumbres M and Barbacid M. Mammalian cyclin-dependent kinases. Trends in biochemical sciences. 2005; 30:630-641.
139. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M, et al. The molecular classification of multiple myeloma. Blood. 2006; 108:2020-2028.
140. De Azevedo WF, Leclerc S, Meijer L, Havlicek L, Strnad M and Kim SH. Inhibition of cyclin-dependent kinases by purine analogues: crystal structure of human cdk2 complexed with roscovitine. European journal of biochemistry / FEBS. 1997; 243:518-526.
141. de Azevedo WF, Jr., Canduri F and da Silveira NJ. Structural basis for inhibition of cyclin-dependent kinase 9 by flavopiridol. Biochemical and biophysical research communications. 2002; 293:566-571.
142. Dhariwala FA and Rajadhyaksha MS. An unusual member of the Cdk family: Cdk5. Cellular and molecular neurobiology. 2008; 28:351-369.
143. Arif A. Extraneuronal activities and regulatory mechanisms of the atypical cyclin-dependent kinase Cdk5. Biochemical pharmacology. 2012; 84:985-993.
144. Levacque Z, Rosales JL and Lee KY. Level of cdk5 expression predicts the survival of relapsed multiple myeloma patients. Cell cycle (Georgetown, Tex). 2012; 11:4093-4095.
145. Zhu YX, Tiedemann R, Shi CX, Yin H, Schmidt JE, Bruins LA, Keats JJ, Braggio E, Sereduk C, Mousses S and Stewart AK. RNAi screen of the druggable genome identifies modulators of proteasome inhibitor sensitivity in myeloma including CDK5. Blood. 2011; 117:3847-3857.
146. Raje N, Kumar S, Hideshima T, Roccaro A, Ishitsuka K, Yasui H, Shiraishi N, Chauhan D, Munshi NC, Green SR and Anderson KC. Seliciclib (CYC202 or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates activity via down-regulation of Mcl-1 in multiple myeloma. Blood. 2005; 106:1042-1047.
147. Alagpulinsa DA, Ayyadevara S, Yaccoby S and Shmookler Reis RJ. A cyclin-dependent kinase inhibitor, dinaciclib, impairs homologous recombination and sensitizes multiple myeloma cells to PARP inhibition. Molecular cancer therapeutics. 2016; 15:241-250.
148. Wesierska-Gadek J, Maurer M, Zulehner N and Komina O. Whether to target single or multiple CDKs for therapy? That is the question. Journal of cellular physiology. 2011; 226:341-349.
149. Swords R, Mahalingam D, O’Dwyer M, Santocanale C, Kelly K, Carew J and Giles F. Cdc7 kinase - a new target for drug development. European journal of cancer. 2010; 46:33-40.
150. Montagnoli A, Moll J and Colotta F. Targeting cell division cycle 7 kinase: a new approach for cancer therapy. Clinical cancer research. 2010; 16:4503-4508.
151. Bonte D, Lindvall C, Liu H, Dykema K, Furge K and Weinreich M. Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia (New York, NY). 2008; 10:920-931.
152. Natoni A, Coyne MR, Jacobsen A, Rainey MD, O’Brien G, Healy S, Montagnoli A, Moll J, O’Dwyer M and Santocanale C. Characterization of a dual cdc7/cdk9 inhibitor in multiple myeloma cellular models. Cancers. 2013; 5:901-918.
153. Menichincheri M, Bargiotti A, Berthelsen J, Bertrand JA, Bossi R, Ciavolella A, Cirla A, Cristiani C, Croci V, D’Alessio R, Fasolini M, Fiorentini F, Forte B, et al. First Cdc7 kinase inhibitors: pyrrolopyridinones as potent and orally active antitumor agents. 2. Lead discovery. Journal of medicinal chemistry. 2009; 52:293-307.
154. Craig SN, Wyatt MD and McInnes C. Current assessment of polo-like kinases as anti-tumor drug targets. Expert opinion on drug discovery. 2014; 9:773-789.
155. Eckerdt F, Yuan J and Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005; 24:267-276.
156. Lee KS, Burke TR, Jr., Park JE, Bang JK and Lee E. Recent advances and new strategies in targeting plk1 for anticancer therapy. Trends in pharmacological sciences. 2015; 36:858-877.
157. Evans RP, Dueck G, Sidhu R, Ghosh S, Toman I, Loree J, Bahlis N, Klimowicz AC, Fung J, Jung M, Lai R, Pilarski LM, Belch AR, et al. Expression, adverse prognostic significance and therapeutic small molecule inhibition of Polo-like kinase 1 in multiple myeloma. Leukemia research. 2011; 35:1637-1643.
158. Stewart HJ, Kishikova L, Powell FL, Wheatley SP and Chevassut TJ. The polo-like kinase inhibitor BI 2536 exhibits potent activity against malignant plasma cells and represents a novel therapy in multiple myeloma. Experimental hematology. 2011; 39:330-338.
159. Zhang G, Zhang Z and Liu Z. Scytonemin inhibits cell proliferation and arrests cell cycle through downregulating Plk1 activity in multiple myeloma cells. Tumour biology. 2013; 34:2241-2247.
160. Finlay MR and Griffin RJ. Modulation of DNA repair by pharmacological inhibitors of the PIKK protein kinase family. Bioorganic & medicinal chemistry letters. 2012; 22:5352-5359.
161. Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, Proszek PZ, Johnson DC, Kaiser MF, Melchor L, Aronson LI, Scales M, Pawlyn C, et al. Mutational spectrum, copy number changes, and outcome: results of a sequencing study of patients with newly diagnosed myeloma. Journal of clinical oncology. 2015; 33:3911-3920.
162. Landau HJ, McNeely SC, Nair JS, Comenzo RL, Asai T, Friedman H, Jhanwar SC, Nimer SD and Schwartz GK. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Molecular cancer therapeutics. 2012; 11:1781-1788.
163. Cremona CA and Behrens A. ATM signalling and cancer. Oncogene. 2014; 33:3351-3360.
164. Weber AM and Ryan AJ. ATM and ATR as therapeutic targets in cancer. Pharmacology & therapeutics. 2015; 149:124-138.
165. Demel HR, Feuerecker B, Piontek G, Seidl C, Blechert B, Pickhard A and Essler M. Effects of topoisomerase inhibitors that induce DNA damage response on glucose metabolism and PI3K/Akt/mTOR signaling in multiple myeloma cells. American journal of cancer research. 2015; 5:1649-1664.
166. Newton AC. Protein kinase C: poised to signal. American journal of physiology Endocrinology and metabolism. 2010; 298:E395-402.
167. Podar K, Raab MS, Chauhan D and Anderson KC. The therapeutic role of targeting protein kinase C in solid and hematologic malignancies. Expert opinion on investigational drugs. 2007; 16:1693-1707.
168. Neri A, Marmiroli S, Tassone P, Lombardi L, Nobili L, Verdelli D, Civallero M, Cosenza M, Bertacchini J, Federico M, De Pol A, Deliliers GL and Sacchi S. The oral protein-kinase C beta inhibitor enzastaurin (LY317615) suppresses signalling through the AKT pathway, inhibits proliferation and induces apoptosis in multiple myeloma cell lines. Leukemia & lymphoma. 2008; 49:1374-1383.
169. Jourdan E, Leblond V, Maisonneuve H, Benhadji KA, Hossain AM, Nguyen TS, Wooldridge JE and Moreau P. A multicenter phase II study of single-agent enzastaurin in previously treated multiple myeloma. Leukemia & lymphoma. 2013; 55:2013-2017.
170. Sharkey J, Khong T and Spencer A. PKC412 demonstrates JNK-dependent activity against human multiple myeloma cells. Blood. 2007; 109:1712-1719.
171. Cooper BW, Kindwall-Keller TL, Craig MD, Creger RJ, Hamadani M, Tse WW and Lazarus HM. A phase I study of midostaurin and azacitidine in relapsed and elderly AML patients. Clinical lymphoma, myeloma & leukemia. 2015; 15:428-432.e422.
172. Ni H, Ergin M, Tibudan SS, Denning MF, Izban KF and Alkan S. Protein kinase C-delta is commonly expressed in multiple myeloma cells and its downregulation by rottlerin causes apoptosis. British journal of haematology. 2003; 121:849-856.
173. Zadra G, Batista JL and Loda M. Dissecting the dual role of AMPK in cancer: from experimental to human studies. Molecular cancer research. 2015; 13:1059-1072.
174. Baumann P, Mandl-Weber S, Emmerich B, Straka C and Schmidmaier R. Activation of adenosine monophosphate activated protein kinase inhibits growth of multiple myeloma cells. Experimental cell research. 2007; 313:3592-3603.
175. Baumann P, Mandl-Weber S, Emmerich B, Straka C and Schmidmaier R. Inhibition of adenosine monophosphate-activated protein kinase induces apoptosis in multiple myeloma cells. Anti-cancer drugs. 2007; 18:405-410.
176. Castillo JJ, Mull N, Reagan JL, Nemr S and Mitri J. Increased incidence of non-Hodgkin lymphoma, leukemia, and myeloma in patients with diabetes mellitus type 2: a meta-analysis of observational studies. Blood. 2012; 119:4845-4850.
177. Wu W, Merriman K, Nabaah A, Seval N, Seval D, Lin H, Wang M, Qazilbash MH, Baladandayuthapani V, Berry D, Orlowski RZ, Lee MH and Yeung SC. The association of diabetes and anti-diabetic medications with clinical outcomes in multiple myeloma. British journal of cancer. 2014; 111:628-636.
178. Zi FM, He JS, Li Y, Wu C, Yang L, Yang Y, Wang LJ, He DH, Zhao Y, Wu WJ, Zheng GF, Han XY, Huang H, et al. Metformin displays anti-myeloma activity and synergistic effect with dexamethasone in in vitro and in vivo xenograft models. Cancer letters. 2015; 356:443-453.
179. Platanias LC. Map kinase signaling pathways and hematologic malignancies. Blood. 2003; 101:4667-4679.
180. Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, Sougnez C, Knoechel B, Gould J, Saksena G, Cibulskis K, McKenna A, Chapman MA, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer cell. 2014; 25:91-101.
181. Andrulis M, Lehners N, Capper D, Penzel R, Heining C, Huellein J, Zenz T, von Deimling A, Schirmacher P, Ho AD, Goldschmidt H, Neben K and Raab MS. Targeting the BRAF V600E mutation in multiple myeloma. Cancer discovery. 2013; 3:862-869.
182. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, Anderson KC, Ardlie KG, Auclair D, et al. Initial genome sequencing and analysis of multiple myeloma. Nature. 2011; 471:467-472.
183. Sharman JP, Chmielecki J, Morosini D, Palmer GA, Ross JS, Stephens PJ, Stafl J, Miller VA and Ali SM. Vemurafenib response in 2 patients with posttransplant refractory BRAF V600E-mutated multiple myeloma. Clinical lymphoma, myeloma & leukemia. 2014; 14:e161-163.
184. David JP, Mehic D, Bakiri L, Schilling AF, Mandic V, Priemel M, Idarraga MH, Reschke MO, Hoffmann O, Amling M and Wagner EF. Essential role of RSK2 in c-Fos-dependent osteosarcoma development. The Journal of clinical investigation. 2005; 115:664-672.
185. Kang S, Elf S, Lythgoe K, Hitosugi T, Taunton J, Zhou W, Xiong L, Wang D, Muller S, Fan S, Sun SY, Marcus AI, Gu TL, et al. p90 ribosomal S6 kinase 2 promotes invasion and metastasis of human head and neck squamous cell carcinoma cells. The Journal of clinical investigation. 2010; 120:1165-1177.
186. Kang S, Elf S, Dong S, Hitosugi T, Lythgoe K, Guo A, Ruan H, Lonial S, Khoury HJ, Williams IR, Lee BH, Roesel JL, Karsenty G, et al. Fibroblast growth factor receptor 3 associates with and tyrosine phosphorylates p90 RSK2, leading to RSK2 activation that mediates hematopoietic transformation. Molecular and cellular biology. 2009; 29:2105-2117.
187. Kang S, Dong S, Gu TL, Guo A, Cohen MS, Lonial S, Khoury HJ, Fabbro D, Gilliland DG, Bergsagel PL, Taunton J, Polakiewicz RD and Chen J. FGFR3 activates RSK2 to mediate hematopoietic transformation through tyrosine phosphorylation of RSK2 and activation of the MEK/ERK pathway. Cancer cell. 2007; 12:201-214.
188. Cuadrado A and Nebreda AR. Mechanisms and functions of p38 MAPK signalling. The Biochemical journal. 2010; 429:403-417.
189. He J, Liu Z, Zheng Y, Qian J, Li H, Lu Y, Xu J, Hong B, Zhang M, Lin P, Cai Z, Orlowski RZ, Kwak LW, et al. p38 MAPK in myeloma cells regulates osteoclast and osteoblast activity and induces bone destruction. Cancer research. 2012; 72:6393-6402.
190. Liu H, He J and Yang J. Tumor cell p38 MAPK: A trigger of cancer bone osteolysis. Cancer cell & microenvironment. 2015; 2:e464.
191. Ishitsuka K, Hideshima T, Neri P, Vallet S, Shiraishi N, Okawa Y, Shen Z, Raje N, Kiziltepe T, Ocio EM, Chauhan D, Tassone P, Munshi N, et al. p38 mitogen-activated protein kinase inhibitor LY2228820 enhances bortezomib-induced cytotoxicity and inhibits osteoclastogenesis in multiple myeloma; therapeutic implications. British journal of haematology. 2008; 141:598-606.
192. Navas TA, Nguyen AN, Hideshima T, Reddy M, Ma JY, Haghnazari E, Henson M, Stebbins EG, Kerr I, O’Young G, Kapoun AM, Chakravarty S, Mavunkel B, et al. Inhibition of p38alpha MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating Hsp27, Bcl-X(L), Mcl-1 and p53 levels in vitro and inhibits tumor growth in vivo. Leukemia. 2006; 20:1017-1027.
193. Fuchs O. Transcription factor NF-kappaB inhibitors as single therapeutic agents or in combination with classical chemotherapeutic agents for the treatment of hematologic malignancies. Current molecular pharmacology. 2010; 3:98-122.
194. Ni H, Ergin M, Huang Q, Qin JZ, Amin HM, Martinez RL, Saeed S, Barton K and Alkan S. Analysis of expression of nuclear factor kappa B (NF-kappa B) in multiple myeloma: downregulation of NF-kappa B induces apoptosis. British journal of haematology. 2001; 115:279-286.
195. Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harbor perspectives in biology. 2010; 2:a000109.
196. Gardam S and Beyaert R. The kinase NIK as a therapeutic target in multiple myeloma. Expert opinion on therapeutic targets. 2011; 15:207-218.
197. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, Montalto G, D’Assoro AB, Libra M, Nicoletti F, Maestro R, Basecke J, Rakus D, et al. GSK-3 as potential target for therapeutic intervention in cancer. Oncotarget. 2014; 5:2881-2911. doi:10.18632/oncotarget.2037.
198. Piazza F, Manni S and Semenzato G. Novel players in multiple myeloma pathogenesis: role of protein kinases CK2 and GSK3. Leukemia research. 2013; 37:221-227.
199. Piazza F, Manni S, Tubi LQ, Montini B, Pavan L, Colpo A, Gnoato M, Cabrelle A, Adami F, Zambello R, Trentin L, Gurrieri C and Semenzato G. Glycogen synthase kinase-3 regulates multiple myeloma cell growth and bortezomib-induced cell death. BMC cancer. 2010; 10:526.
200. Gunn WG, Krause U, Lee N and Gregory CA. Pharmaceutical inhibition of glycogen synthetase kinase-3beta reduces multiple myeloma-induced bone disease in a novel murine plasmacytoma xenograft model. Blood. 2011; 117:1641-1651.
201. Salvi M, Sarno S, Cesaro L, Nakamura H and Pinna LA. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochimica et biophysica acta. 2009; 1793:847-859.
202. Chon HJ, Bae KJ, Lee Y and Kim J. The casein kinase 2 inhibitor, CX-4945, as an anti-cancer drug in treatment of human hematological malignancies. Frontiers in pharmacology. 2015; 6:70.
203. Sarno S, Papinutto E, Franchin C, Bain J, Elliott M, Meggio F, Kazimierczuk Z, Orzeszko A, Zanotti G, Battistutta R and Pinna LA. ATP site-directed inhibitors of protein kinase CK2: an update. Current topics in medicinal chemistry. 2011; 11:1340-1351.
204. Zhao M, Ma J, Zhu HY, Zhang XH, Du ZY, Xu YJ and Yu XD. Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Molecular cancer. 2011; 10:104.
205. McDonald PC, Fielding AB and Dedhar S. Integrin-linked kinase—essential roles in physiology and cancer biology. Journal of cell science. 2008; 121:3121-3132.
206. Wang X, Zhang Z and Yao C. Targeting integrin-linked kinase increases apoptosis and decreases invasion of myeloma cell lines and inhibits IL-6 and VEGF secretion from BMSCs. Medical oncology (Northwood, London, England). 2011; 28:1596-1600.
207. Venkata JK, An N, Stuart R, Costa LJ, Cai H, Coker W, Song JH, Gibbs K, Matson T, Garrett-Mayer E, Wan Z, Ogretmen B, Smith C, et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood. 2014; 124:1915-1925.
208. Cuvillier O, Ader I, Bouquerel P, Brizuela L, Malavaud B, Mazerolles C and Rischmann P. Activation of sphingosine kinase-1 in cancer: implications for therapeutic targeting. Current molecular pharmacology. 2010; 3:53-65.
209. Newton J, Lima S, Maceyka M and Spiegel S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Experimental cell research. 2015; 333:195-200.
210. Heffernan-Stroud LA and Obeid LM. Sphingosine kinase 1 in cancer. Advances in cancer research. 2013; 117:201-235.
211. Alshaker H, Sauer L, Monteil D, Ottaviani S, Srivats S, Bohler T and Pchejetski D. Therapeutic potential of targeting SK1 in human cancers. Advances in cancer research. 2013; 117:143-200.
212. Tsukamoto S, Huang Y, Kumazoe M, Lesnick C, Yamada S, Ueda N, Suzuki T, Yamashita S, Kim YH, Fujimura Y, Miura D, Kay NE, Shanafelt TD, et al. Sphingosine kinase-1 protects multiple myeloma from apoptosis driven by cancer-specific inhibition of RTKs. Molecular cancer therapeutics. 2015; 14:2303-2312.
213. Neubauer HA and Pitson SM. Roles, regulation and inhibitors of sphingosine kinase 2. The FEBS journal. 2013; 280:5317-5336.
214. Liao A, Hu R, Zhao Q, Li J, Li Y, Yao K, Zhang R, Wang H, Yang W and Liu Z. Autophagy induced by FTY720 promotes apoptosis in U266 cells. European journal of pharmaceutical sciences. 2012; 45:600-605.
215. Bernales S, Papa FR and Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006; 22:487-508.
216. Ali MM, Bagratuni T, Davenport EL, Nowak PR, Silva-Santisteban MC, Hardcastle A, McAndrews C, Rowlands MG, Morgan GJ, Aherne W, Collins I, Davies FE and Pearl LH. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. The EMBO journal. 2011; 30:894-905.
217. Newbatt Y, Hardcastle A, McAndrew PC, Strover JA, Mirza A, Morgan GJ, Burke R, Davies FE, Collins I and van Montfort RL. Identification of autophosphorylation inhibitors of the inositol-requiring enzyme 1 alpha (IRE1alpha) by high-throughput screening using a DELFIA assay. Journal of biomolecular screening. 2013; 18:298-308.
218. Axten JM, Romeril SP, Shu A, Ralph J, Medina JR, Feng Y, Li WH, Grant SW, Heerding DA, Minthorn E, Mencken T, Gaul N, Goetz A, et al. Discovery of GSK2656157: an optimized PERK inhibitor selected for preclinical development. ACS medicinal chemistry letters. 2013; 4:964-968.
219. Bagratuni T, Mavrianoun N, Kastritis E, Liakos C, Erpos E and Dimopoulos MA. Characterization of a PERK kinase Inhibitor with anti-myeloma activity. Proceedings of the 57th Annual Meeting of the American Society of Hematology (Orlando, FL: ASH). 2015.
220. Rask-Andersen M, Masuram S and Schioth HB. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annual review of pharmacology and toxicology. 2014; 54:9-26.
221. Gentile M, Recchia AG, Mazzone C, Lucia E, Vigna E and Morabito F. Perspectives in the treatment of multiple myeloma. Expert opinion on biological therapy. 2013; 13 Suppl 1:S1-22.
222. Catley L, Tai YT, Chauhan D and Anderson KC. Perspectives for combination therapy to overcome drug-resistant multiple myeloma. Drug resistance updates. 2005; 8:205-218.
223. Niesvizky R, Badros AZ, Costa LJ, Ely SA, Singhal SB, Stadtmauer EA, Haideri NA, Yacoub A, Hess G, Lentzsch S, Spicka I, Chanan-Khan AA, Raab MS, et al. Phase 1/2 study of CDK4/6 inhibitor palbociclib (PD-0332991) with bortezomib and dexamethasone in relapsed/refractory multiple myeloma. Leukemia & lymphoma. 2015:1-21.
224. Richardson P, Nagler A, Ben-Yehuda D, Badros A, Hari P, Hajek R, Spicka I, Kaya H, Le Blanc R, Yoon S, Kim K, Martinez-Lopez J and Mittelman M. (2013). Randomized placebo-controlled phase III study of perifosine combined with bortezomib and dexamethasone in relapsed, refractory multiple myeloma patients previously treated with bortezomib. Abstract 3189, Proceedings of the 55th annual meeting of the American Society of Hematology (New Orleans, LA: ASH).
225. Cohen P and Alessi DR. Kinase drug discovery—what’s next in the field? ACS chemical biology. 2013; 8:96-104.
226. Bubici C and Papa S. JNK signalling in cancer: in need of new, smarter therapeutic targets. British journal of pharmacology. 2014; 171:24-37.
227. Krejci P, Murakami S, Prochazkova J, Trantirek L, Chlebova K, Ouyang Z, Aklian A, Smutny J, Bryja V, Kozubik A and Wilcox WR. NF449 is a novel inhibitor of fibroblast growth factor receptor 3 (FGFR3) signaling active in chondrocytes and multiple myeloma cells. The Journal of biological chemistry. 2010; 285:20644-20653.
228. Keane NA, Glavey SV, Krawczyk J and O’Dwyer M. AKT as a therapeutic target in multiple myeloma. Expert opinion on therapeutic targets. 2014; 18:897-915.
229. Duong-Ly KC and Peterson JR. The human kinome and kinase inhibition. Current protocols in pharmacology; SJ Enna (editor-in-chief). 2013; Chapter 2:Unit 2.9.
230. Garuti L, Roberti M and Bottegoni G. Non-ATP competitive protein kinase inhibitors. Current medicinal chemistry. 2010; 17:2804-2821.
231. Sabbatini P, Rowand JL, Groy A, Korenchuk S, Liu Q, Atkins C, Dumble M, Yang J, Anderson K, Wilson BJ, Emmitte KA, Rabindran SK and Kumar R. Antitumor activity of GSK1904529A, a small-molecule inhibitor of the insulin-like growth factor-I receptor tyrosine kinase. Clinical cancer research. 2009; 15:3058-3067.
232. Liang SB, Yang XZ, Trieu Y, Li Z, Zive J, Leung-Hagesteijn C, Wei E, Zozulya S, Coss CC, Dalton JT, Fantus IG and Trudel S. Molecular target characterization and antimyeloma activity of the novel, insulin-like growth factor 1 receptor inhibitor, GTx-134. Clinical cancer research. 2011; 17:4693-4704.
233. Mulvihill MJ, Cooke A, Rosenfeld-Franklin M, Buck E, Foreman K, Landfair D, O’Connor M, Pirritt C, Sun Y, Yao Y, Arnold LD, Gibson NW and Ji QS. Discovery of OSI-906: a selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future medicinal chemistry. 2009; 1:1153-1171.
234. Meyer AN, McAndrew CW and Donoghue DJ. Nordihydroguaiaretic acid inhibits an activated fibroblast growth factor receptor 3 mutant and blocks downstream signaling in multiple myeloma cells. Cancer research. 2008; 68:7362-7370.
235. Hewish M, Chau I and Cunningham D. Insulin-like growth factor 1 receptor targeted therapeutics: novel compounds and novel treatment strategies for cancer medicine. Recent patents on anti-cancer drug discovery. 2009; 4:54-72.
236. Mitsiades CS, Hayden PJ, Anderson KC and Richardson PG. From the bench to the bedside: emerging new treatments in multiple myeloma. Best practice & research Clinical haematology. 2007; 20:797-816.
237. Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Akiyama M, Hideshima T, Chauhan D, Joseph M, Libermann TA, Garcia-Echeverria C, Pearson MA, Hofmann F, et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer cell. 2004; 5:221-230.
238. Girnita A, Girnita L, del Prete F, Bartolazzi A, Larsson O and Axelson M. Cyclolignans as inhibitors of the insulin-like growth factor-1 receptor and malignant cell growth. Cancer research. 2004; 64:236-242.
239. Heinrich MC, Griffith DJ, Druker BJ, Wait CL, Ott KA and Zigler AJ. Inhibition of c-kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood. 2000; 96:925-932.
240. Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J and Lydon NB. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nature medicine. 1996; 2:561-566.
241. Dubreuil P, Letard S, Ciufolini M, Gros L, Humbert M, Casteran N, Borge L, Hajem B, Lermet A, Sippl W, Voisset E, Arock M, Auclair C, et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PloS one. 2009; 4:e7258.
242. You WK, Sennino B, Williamson CW, Falcon B, Hashizume H, Yao LC, Aftab DT and McDonald DM. VEGF and c-Met blockade amplify angiogenesis inhibition in pancreatic islet cancer. Cancer research. 2011; 71:4758-4768.
243. Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, Orf J, You A, Laird AD, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Molecular cancer therapeutics. 2011; 10:2298-2308.
244. Wang X, Le P, Liang C, Chan J, Kiewlich D, Miller T, Harris D, Sun L, Rice A, Vasile S, Blake RA, Howlett AR, Patel N, et al. Potent and selective inhibitors of the Met [hepatocyte growth factor/scatter factor (HGF/SF) receptor] tyrosine kinase block HGF/SF-induced tumor cell growth and invasion. Molecular cancer therapeutics. 2003; 2:1085-1092.
245. Munshi N, Jeay S, Li Y, Chen CR, France DS, Ashwell MA, Hill J, Moussa MM, Leggett DS and Li CJ. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Molecular cancer therapeutics. 2010; 9:1544-1553.
246. Trudel S, Li ZH, Wei E, Wiesmann M, Chang H, Chen C, Reece D, Heise C and Stewart AK. CHIR-258, a novel, multitargeted tyrosine kinase inhibitor for the potential treatment of t(4;14) multiple myeloma. Blood. 2005; 105:2941-2948.
247. Scheid C, Reece D, Beksac M, Spencer A, Callander N, Sonneveld P, Kalimi G, Cai C, Shi M, Scott JW and Stewart AK. Phase 2 study of dovitinib in patients with relapsed or refractory multiple myeloma with or without t(4;14) translocation. European journal of haematology. 2015; 95:316-324.
248. Zhao G, Li WY, Chen D, Henry JR, Li HY, Chen Z, Zia-Ebrahimi M, Bloem L, Zhai Y, Huss K, Peng SB and McCann DJ. A novel, selective inhibitor of fibroblast growth factor receptors that shows a potent broad spectrum of antitumor activity in several tumor xenograft models. Molecular cancer therapeutics. 2011; 10:2200-2210.
249. de Brito LR, Batey MA, Zhao Y, Squires MS, Maitland H, Leung HY, Hall AG, Jackson G, Newell DR and Irving JA. Comparative pre-clinical evaluation of receptor tyrosine kinase inhibitors for the treatment of multiple myeloma. Leukemia research. 2011; 35:1233-1240.
250. Mohammadi M, Froum S, Hamby JM, Schroeder MC, Panek RL, Lu GH, Eliseenkova AV, Green D, Schlessinger J and Hubbard SR. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. The EMBO journal. 1998; 17:5896-5904.
251. Vatsveen TK, Brenne AT, Dai HY, Waage A, Sundan A and Borset M. FGFR3 is expressed and is important for survival in INA-6, a human myeloma cell line without a t(4;14). European journal of haematology. 2009; 83:471-476.
252. Sun L, Tran N, Liang C, Tang F, Rice A, Schreck R, Waltz K, Shawver LK, McMahon G and Tang C. Design, synthesis, and evaluations of substituted 3-[(3- or 4-carboxyethylpyrrol-2-yl)methylidenyl]indolin-2-ones as inhibitors of VEGF, FGF, and PDGF receptor tyrosine kinases. Journal of medicinal chemistry. 1999; 42:5120-5130.
253. Ochiiwa H, Fujita H, Itoh K, Sootome H, Hashimoto A, Fujioka Y, Nakatsuru Y, Oda N, Yonekura K, Hirai H and Utsugi T. TAS-120, a highly potent and selective irreversible FGFR inhibitor, is effective in tumors harboring various FGFR gene abnormalities. Molecular cancer therapeutics. 2014; 12:A270-A270.
254. Podar K, Catley LP, Tai YT, Shringarpure R, Carvalho P, Hayashi T, Burger R, Schlossman RL, Richardson PG, Pandite LN, Kumar R, Hideshima T, Chauhan D, et al. GW654652, the pan-inhibitor of VEGF receptors, blocks the growth and migration of multiple myeloma cells in the bone marrow microenvironment. Blood. 2004; 103:3474-3479.
255. Huh JI, Calvo A, Stafford J, Cheung M, Kumar R, Philp D, Kleinman HK and Green JE. Inhibition of VEGF receptors significantly impairs mammary cancer growth in C3(1)/Tag transgenic mice through antiangiogenic and non-antiangiogenic mechanisms. Oncogene. 2005; 24:790-800.
256. Kropff M, Kienast J, Bisping G, Berdel WE, Gaschler-Markefski B, Stopfer P, Stefanic M and Munzert G. An open-label dose-escalation study of BIBF 1120 in patients with relapsed or refractory multiple myeloma. Anticancer research. 2009; 29:4233-4238.
257. Hilberg F, Roth GJ, Krssak M, Kautschitsch S, Sommergruber W, Tontsch-Grunt U, Garin-Chesa P, Bader G, Zoephel A, Quant J, Heckel A and Rettig WJ. BIBF 1120: triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer research. 2008; 68:4774-4782.
258. Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, Davis-Ward RG, Epperly AH, Hinkle KW, Hunter RN, 3rd, Johnson JH, Knick VB, Laudeman CP, Luttrell DK, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. Journal of medicinal chemistry. 2008; 51:4632-4640.
259. Podar K, Tonon G, Sattler M, Tai YT, Legouill S, Yasui H, Ishitsuka K, Kumar S, Kumar R, Pandite LN, Hideshima T, Chauhan D and Anderson KC. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:19478-19483.
260. Fong TA, Shawver LK, Sun L, Tang C, App H, Powell TJ, Kim YH, Schreck R, Wang X, Risau W, Ullrich A, Hirth KP and McMahon G. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer research. 1999; 59:99-106.
261. Srkalovic G, Hussein MA, Hoering A, Zonder JA, Popplewell LL, Trivedi H, Mazzoni S, Sexton R, Orlowski RZ and Barlogie B. A phase II trial of BAY 43-9006 (sorafenib) (NSC-724772) in patients with relapsing and resistant multiple myeloma: SWOG S0434. Cancer medicine. 2014; 3:1275-1283.
262. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer research. 2004; 64:7099-7109.
263. Sun L, Liang C, Shirazian S, Zhou Y, Miller T, Cui J, Fukuda JY, Chu JY, Nematalla A, Wang X, Chen H, Sistla A, Luu TC, et al. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. Journal of medicinal chemistry. 2003; 46:1116-1119.
264. Ikezoe T, Nishioka C, Tasaka T, Yang Y, Komatsu N, Togitani K, Koeffler HP and Taguchi H. The antitumor effects of sunitinib (formerly SU11248) against a variety of human hematologic malignancies: enhancement of growth inhibition via inhibition of mammalian target of rapamycin signaling. Molecular cancer therapeutics. 2006; 5:2522-2530.
265. Sanda T, Kuwano T, Nakao S, Iida S, Ishida T, Komatsu H, Shudo K, Kuwano M, Ono M and Ueda R. Antimyeloma effects of a novel synthetic retinoid Am80 (Tamibarotene) through inhibition of angiogenesis. Leukemia. 2005; 19:901-909.
266. Kovacs MJ, Reece DE, Marcellus D, Meyer RM, Mathews S, Dong RP and Eisenhauer E. A phase II study of ZD6474 (Zactima), a selective inhibitor of VEGFR and EGFR tyrosine kinase in patients with relapsed multiple myeloma—NCIC CTG IND.145. Investigational new drugs. 2006; 24:529-535.
267. Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Chester R, Jackson JA, Boffey SJ, Valentine PJ, Curwen JO, Musgrove HL, Graham GA, Hughes GD, Thomas AP, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer research. 2002; 62:4645-4655.
268. Wood JM, Bold G, Buchdunger E, Cozens R, Ferrari S, Frei J, Hofmann F, Mestan J, Mett H, O’Reilly T, Persohn E, Rosel J, Schnell C, et al. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer research. 2000; 60:2178-2189.
269. Neri P, Tassone P, Shammas M, Yasui H, Schipani E, Batchu RB, Blotta S, Prabhala R, Catley L, Hamasaki M, Hideshima T, Chauhan D, Jacob GS, et al. Biological pathways and in vivo antitumor activity induced by atiprimod in myeloma. Leukemia. 2007; 21:2519-2526.
270. Hedvat M, Huszar D, Herrmann A, Gozgit JM, Schroeder A, Sheehy A, Buettner R, Proia D, Kowolik CM, Xin H, Armstrong B, Bebernitz G, Weng S, et al. The JAK2 inhibitor AZD1480 potently blocks Stat3 signaling and oncogenesis in solid tumors. Cancer cell. 2009; 16:487-497.
271. Scuto A, Krejci P, Popplewell L, Wu J, Wang Y, Kujawski M, Kowolik C, Xin H, Chen L, Wang Y, Kretzner L, Yu H, Wilcox WR, et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia. 2011; 25:538-550.
272. Liu Y, Chen XQ, Liang HX, Zhang FX, Zhang B, Jin J, Chen YL, Cheng YX and Zhou GB. Small compound 6-O-angeloylplenolin induces mitotic arrest and exhibits therapeutic potentials in multiple myeloma. PloS one. 2011; 6:e21930.
273. Park S, Lee HJ, Jeong SJ, Song HS, Kim M, Lee HJ, Lee EO, Kim DH, Ahn KS and Kim SH. Inhibition of JAK1/STAT3 signaling mediates compound K-induced apoptosis in human multiple myeloma U266 cells. Food and chemical toxicology. 2011; 49:1367-1372.
274. Rhee YH, Jeong SJ, Lee HJ, Lee HJ, Koh W, Jung JH, Kim SH and Sung-Hoon K. Inhibition of STAT3 signaling and induction of SHP1 mediate antiangiogenic and antitumor activities of ergosterol peroxide in U266 multiple myeloma cells. BMC cancer. 2012; 12:28.
275. Lee JH, Kim C, Kim SH, Sethi G and Ahn KS. Farnesol inhibits tumor growth and enhances the anticancer effects of bortezomib in multiple myeloma xenograft mouse model through the modulation of STAT3 signaling pathway. Cancer letters. 2015; 360:280-293.
276. Zhu S, Wang Z, Li Z, Peng H, Luo Y, Deng M, Li R, Dai C, Xu Y, Liu S and Zhang G. Icaritin suppresses multiple myeloma, by inhibiting IL-6/JAK2/STAT3. Oncotarget. 2015; 6:10460-10472. doi:10.18632/oncotarget.3399.
277. Burger R, Le Gouill S, Tai YT, Shringarpure R, Tassone P, Neri P, Podar K, Catley L, Hideshima T, Chauhan D, Caulder E, Neilan CL, Vaddi K, et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Molecular cancer therapeutics. 2009; 8:26-35.
278. Koppikar P, Abdel-Wahab O, Hedvat C, Marubayashi S, Patel J, Goel A, Kucine N, Gardner JR, Combs AP, Vaddi K, Haley PJ, Burn TC, Rupar M, et al. Efficacy of the JAK2 inhibitor INCB16562 in a murine model of MPLW515L-induced thrombocytosis and myelofibrosis. Blood. 2010; 115:2919-2927.
279. Li J, Favata M, Kelley JA, Caulder E, Thomas B, Wen X, Sparks RB, Arvanitis A, Rogers JD, Combs AP, Vaddi K, Solomon KA, Scherle PA, et al. INCB16562, a JAK1/2 selective inhibitor, is efficacious against multiple myeloma cells and reverses the protective effects of cytokine and stromal cell support. Neoplasia (New York, NY). 2010; 12:28-38.
280. Pardanani A, Lasho T, Smith G, Burns CJ, Fantino E and Tefferi A. CYT387, a selective JAK1/JAK2 inhibitor: in vitro assessment of kinase selectivity and preclinical studies using cell lines and primary cells from polycythemia vera patients. Leukemia. 2009; 23:1441-1445.
281. Monaghan KA, Khong T, Burns CJ and Spencer A. The novel JAK inhibitor CYT387 suppresses multiple signalling pathways, prevents proliferation and induces apoptosis in phenotypically diverse myeloma cells. Leukemia. 2011; 25:1891-1899.
282. Alas S and Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin’s lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clinical cancer research. 2003; 9:316-326.
283. Schmeel FC, Schmeel LC, Kim Y and Schmidt-Wolf IG. Piceatannol exhibits selective toxicity to multiple myeloma cells and influences the Wnt/ beta-catenin pathway. Hematological oncology. 2014; 32:197-204.
284. Schmeel LC, Schmeel FC, Kim Y, Endo T, Lu D and Schmidt-Wolf IG. Targeting the Wnt/beta-catenin pathway in multiple myeloma. Anticancer research. 2013; 33:4719-4726.
285. Lucet IS, Fantino E, Styles M, Bamert R, Patel O, Broughton SE, Walter M, Burns CJ, Treutlein H, Wilks AF and Rossjohn J. The structural basis of Janus kinase 2 inhibition by a potent and specific pan-Janus kinase inhibitor. Blood. 2006; 107:176-183.
286. Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J, Bornmann W and Bromberg J. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer research. 2006; 66:9714-9721.
287. Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010; 115:3109-3117.
288. Yang Y, Groshong JS, Matta H, Gopalakrishnan R, Yi H and Chaudhary PM. Constitutive NF-kappaB activation confers interleukin 6 (IL6) independence and resistance to dexamethasone and Janus kinase inhibitor INCB018424 in murine plasmacytoma cells. The Journal of biological chemistry. 2011; 286:27988-27997.
289. Zhang Z, Mao H, Du X, Zhu J, Xu Y, Wang S, Xu X, Ji P, Yu Y, Cao B, Han K, Hou T, Xu Z, et al. A novel small molecule agent displays potent anti-myeloma activity by inhibiting the JAK2-STAT3 signaling pathway. Oncotarget. 2016; 7:9296-9308. doi:10.18632/oncotarget.6974.
290. Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, Finke C, Mak CC, Mesa R, Zhu H, Soll R, Gilliland DG and Tefferi A. TG101209, a small molecule JAK2-selective kinase inhibitor potently inhibits myeloproliferative disorder-associated JAK2V617F and MPLW515L/K mutations. Leukemia. 2007; 21:1658-1668.
291. Ramakrishnan V, Kimlinger T, Haug J, Timm M, Wellik L, Halling T, Pardanani A, Tefferi A, Rajkumar SV and Kumar S. TG101209, a novel JAK2 inhibitor, has significant in vitro activity in multiple myeloma and displays preferential cytotoxicity for CD45+ myeloma cells. American journal of hematology. 2010; 85:675-686.
292. Sagawa M, Tabayashi T, Kimura Y, Tomikawa T, Nemoto T, Watanabe R, Tokuhira M, Ri M, Hashimoto Y, Iida S and Kizaki M. TM-233, a novel analog of ACA, induces cell death in myeloma cells by inhibiting both JAK/STAT and proteasome activities. Cancer science. 2015; 106:438-446.
293. Meydan N, Grunberger T, Dadi H, Shahar M, Arpaia E, Lapidot Z, Leeder JS, Freedman M, Cohen A, Gazit A, Levitzki A and Roifman CM. Inhibition of acute lymphoblastic leukaemia by a Jak-2 inhibitor. Nature. 1996; 379:645-648.
294. Luo C and Laaja P. Inhibitors of JAKs/STATs and the kinases: a possible new cluster of drugs. Drug discovery today. 2004; 9:268-275.
295. Kim HJ, Kim SM, Park KR, Jang HJ, Na YS, Ahn KS, Kim SH and Ahn KS. Decursin chemosensitizes human multiple myeloma cells through inhibition of STAT3 signaling pathway. Cancer letters. 2011; 301:29-37.
296. Jang J, Jeong SJ, Kwon HY, Jung JH, Sohn EJ, Lee HJ, Kim JH, Kim SH, Kim JH and Kim SH. Decursin and doxorubicin are in synergy for the induction of apoptosis via STAT3 and/or mTOR pathways in human multiple myeloma cells. Evidence-based complementary and alternative medicine. 2013; 2013:506324.
297. Nam D, Song J, Kim SM, Chiang SY, Kim JS, Chung WS, Jang HJ, Jung SH, Na YS, Kim SH, Shim BS and Ahn KS. 8-Hydrocalamenene, derived from Reynoutria elliptica, suppresses constitutive STAT3 activation, inhibiting proliferation and enhancing chemosensitization of human multiple myeloma cells. Journal of medicinal food. 2014; 17:365-373.
298. Sandur SK, Pandey MK, Sung B and Aggarwal BB. 5-hydroxy-2-methyl-1,4-naphthoquinone, a vitamin K3 analogue, suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase, SHP-1: potential role in chemosensitization. Molecular cancer research. 2010; 8:107-118.
299. Wu J, Zhang M and Liu D. Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. Journal of hematology & oncology. 2016; 9:21.
300. Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT, Witowski SR, Lounsbury H, Chaturvedi P, Mazdiyasni H, Zhu Z, Nacht M, Freed MI, et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. The Journal of pharmacology and experimental therapeutics. 2013; 346:219-228.
301. Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, Li S, Pan Z, Thamm DH, Miller RA and Buggy JJ. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:13075-13080.
302. Tanjoni I, Walsh C, Uryu S, Tomar A, Nam JO, Mielgo A, Lim ST, Liang C, Koenig M, Sun C, Patel N, Kwok C, McMahon G, et al. PND-1186 FAK inhibitor selectively promotes tumor cell apoptosis in three-dimensional environments. Cancer biology & therapy. 2010; 9:764-777.
303. Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, Coleman K, Yao L, Martinez-Alsina L, et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer research. 2008; 68:1935-1944.
304. Yamamoto N, Takeshita K, Shichijo M, Kokubo T, Sato M, Nakashima K, Ishimori M, Nagai H, Li YF, Yura T and Bacon KB. The orally available spleen tyrosine kinase inhibitor 2-[7-(3,4-dimethoxyphenyl)-imidazo[1,2-c]pyrimidin-5-ylamino]nicotinamide dihydrochloride (BAY 61-3606) blocks antigen-induced airway inflammation in rodents. The Journal of pharmacology and experimental therapeutics. 2003; 306:1174-1181.
305. Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, Qu K, Herlaar E, Lau A, Young C, Wong BR, Lovell S, Sun T, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. The Journal of pharmacology and experimental therapeutics. 2006; 319:998-1008.
306. Azab F, Vali S, Abraham J, Potter N, Muz B, de la Puente P, Fiala M, Paasch J, Sultana Z, Tyagi A, Abbasi T, Vij R and Azab AK. PI3KCA plays a major role in multiple myeloma and its inhibition with BYL719 decreases proliferation, synergizes with other therapies and overcomes stroma-induced resistance. British journal of haematology. 2014; 165:89-101.
307. Juric D, Rodon J, Gonzalez-Angulo A, Burris H, Bendell J, Berlin J, Middleton M, Bootle D, Boehm M, Schmitt A, Rouyrre N, Quadt C and Baselga J. (2012). BYL719, a next generation PI3K alpha specific inhibitor: Preliminary safety, PK, and efficacy results from the first- in-human study. Abstract CT-01 AACR 103rd annual meeting (Chicago, IL).
308. Han K, Xu X, Chen G, Zeng Y, Zhu J, Du X, Zhang Z, Cao B, Liu Z and Mao X. Identification of a promising PI3K inhibitor for the treatment of multiple myeloma through the structural optimization. Journal of hematology & oncology. 2014; 7:9.
309. Markman B, Tabernero J, Krop I, Shapiro GI, Siu L, Chen LC, Mita M, Melendez Cuero M, Stutvoet S, Birle D, Anak O, Hackl W and Baselga J. Phase I safety, pharmacokinetic, and pharmacodynamic study of the oral phosphatidylinositol-3-kinase and mTOR inhibitor BGT226 in patients with advanced solid tumors. Annals of oncology. 2012; 23:2399-2408.
310. Zheng Y, Yang J, Qian J, Zhang L, Lu Y, Li H, Lin H, Lan Y, Liu Z, He J, Hong S, Thomas S, Shah J, et al. Novel phosphatidylinositol 3-kinase inhibitor NVP-BKM120 induces apoptosis in myeloma cells and shows synergistic anti-myeloma activity with dexamethasone. Journal of molecular medicine (Berlin, Germany). 2012; 90:695-706.
311. Zhu J, Wang M, Cao B, Hou T and Mao X. Targeting the phosphatidylinositol 3-kinase/AKT pathway for the treatment of multiple myeloma. Current medicinal chemistry. 2014; 21:3173-3187.
312. Martin SK, Gan ZY, Fitter S, To LB and Zannettino AC. The effect of the PI3K inhibitor BKM120 on tumour growth and osteolytic bone disease in multiple myeloma. Leukemia research. 2015; 39:380-387.
313. Sahin I, Azab F, Mishima Y, Moschetta M, Tsang B, Glavey SV, Manier S, Zhang Y, Sacco A, Roccaro AM, Azab AK and Ghobrial IM. Targeting survival and cell trafficking in multiple myeloma and Waldenstrom macroglobulinemia using pan-class I PI3K inhibitor, buparlisib. American journal of hematology. 2014; 89:1030-1036.
314. Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, Frazier K, Ng S, Smith A, et al. Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS medicinal chemistry letters. 2011; 2:774-779.
315. Liu N, Rowley BR, Bull CO, Schneider C, Haegebarth A, Schatz CA, Fracasso PR, Wilkie DP, Hentemann M, Wilhelm SM, Scott WJ, Mumberg D and Ziegelbauer K. BAY 80-6946 is a highly selective intravenous PI3K inhibitor with potent p110alpha and p110delta activities in tumor cell lines and xenograft models. Molecular cancer therapeutics. 2013; 12:2319-2330.
316. Wu CP, Hsieh YJ, Hsiao SH, Su CY, Li YQ, Huang YH, Huang CW, Hsieh CH, Yu JS and Wu YS. Human ATP-Binding Cassette transporter ABCG2 confers resistance to CUDC-907, a dual inhibitor of histone deacetylase and phosphatidylinositol 3-kinase. Molecular pharmaceutics. 2016; 13:784-794.
317. McMillin DW, Ooi M, Delmore J, Negri J, Hayden P, Mitsiades N, Jakubikova J, Maira SM, Garcia-Echeverria C, Schlossman R, Munshi NC, Richardson PG, Anderson KC, et al. Antimyeloma activity of the orally bioavailable dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235. Cancer research. 2009; 69:5835-5842.
318. Baumann P, Mandl-Weber S, Oduncu F and Schmidmaier R. The novel orally bioavailable inhibitor of phosphoinositol-3-kinase and mammalian target of rapamycin, NVP-BEZ235, inhibits growth and proliferation in multiple myeloma. Experimental cell research. 2009; 315:485-497.
319. Maira SM, Stauffer F, Brueggen J, Furet P, Schnell C, Fritsch C, Brachmann S, Chene P, De Pover A, Schoemaker K, Fabbro D, Gabriel D, Simonen M, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Molecular cancer therapeutics. 2008; 7:1851-1863.
320. Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. 2011; 117:591-594.
321. Tong Y, Zhu W, Huang X, You L, Han X, Yang C and Qian W. PI3K inhibitor LY294002 inhibits activation of the Akt/mTOR pathway induced by an oncolytic adenovirus expressing TRAIL and sensitizes multiple myeloma cells to the oncolytic virus. Oncology reports. 2014; 31:1581-1588.
322. Asano J, Nakano A, Oda A, Amou H, Hiasa M, Takeuchi K, Miki H, Nakamura S, Harada T, Fujii S, Kagawa K, Endo I, Yata K, et al. The serine/threonine kinase Pim-2 is a novel anti-apoptotic mediator in myeloma cells. Leukemia. 2011; 25:1182-1188.
323. Chaussade C, Rewcastle GW, Kendall JD, Denny WA, Cho K, Gronning LM, Chong ML, Anagnostou SH, Jackson SP, Daniele N and Shepherd PR. Evidence for functional redundancy of class IA PI3K isoforms in insulin signalling. The Biochemical journal. 2007; 404:449-458.
324. Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, Hayes A, Hancox TC, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. Journal of medicinal chemistry. 2008; 51:5522-5532.
325. Harvey RD and Lonial S. PI3 kinase/AKT pathway as a therapeutic target in multiple myeloma. Future oncology (London, England). 2007; 3:639-647.
326. De P, Dey N, Terakedis B, Bergsagel PL, Li ZH, Mahadevan D, Garlich JR, Trudel S, Makale MT and Durden DL. An integrin-targeted, pan-isoform, phosphoinositide-3 kinase inhibitor, SF1126, has activity against multiple myeloma in vivo. Cancer chemotherapy and pharmacology. 2013; 71:867-881.
327. Mahadevan D, Chiorean EG, Harris WB, Von Hoff DD, Stejskal-Barnett A, Qi W, Anthony SP, Younger AE, Rensvold DM, Cordova F, Shelton CF, Becker MD, Garlich JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. European journal of cancer. 2012; 48:3319-3327.
328. Mao X, Cao B, Wood TE, Hurren R, Tong J, Wang X, Wang W, Li J, Jin Y, Sun W, Spagnuolo PA, MacLean N, Moran MF, et al. A small-molecule inhibitor of D-cyclin transactivation displays preclinical efficacy in myeloma and leukemia via phosphoinositide 3-kinase pathway. Blood. 2011; 117:1986-1997.
329. Dumble M, Crouthamel MC, Zhang SY, Schaber M, Levy D, Robell K, Liu Q, Figueroa DJ, Minthorn EA, Seefeld MA, Rouse MB, Rabindran SK, Heerding DA, et al. Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with the MEK inhibitor. PloS one. 2014; 9:e100880.
330. Spencer A, Yoon SS, Harrison SJ, Morris SR, Smith DA, Brigandi RA, Gauvin J, Kumar R, Opalinska JB and Chen C. The novel AKT inhibitor afuresertib shows favorable safety, pharmacokinetics, and clinical activity in multiple myeloma. Blood. 2014; 124:2190-2195.
331. Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS and Kotani H. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Molecular cancer therapeutics. 2010; 9:1956-1967.
332. Gursel DB, Connell-Albert YS, Tuskan RG, Anastassiadis T, Walrath JC, Hawes JJ, Amlin-Van Schaick JC and Reilly KM. Control of proliferation in astrocytoma cells by the receptor tyrosine kinase/PI3K/AKT signaling axis and the use of PI-103 and TCN as potential anti-astrocytoma therapies. Neuro-oncology. 2011; 13:610-621.
333. Bai LY, Weng JR, Tsai CH, Sargeant A, Lin CW and Chiu CF. OSU-03012 sensitizes TIB-196 myeloma cells to imatinib mesylate via AMP-activated protein kinase and STAT3 pathways. Leukemia research. 2010; 34:816-820.
334. Zhu J, Huang JW, Tseng PH, Yang YT, Fowble J, Shiau CW, Shaw YJ, Kulp SK and Chen CS. From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors. Cancer research. 2004; 64:4309-4318.
335. Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer research. 2010; 70:288-298.
336. Bendell JC, Kelley RK, Shih KC, Grabowsky JA, Bergsland E, Jones S, Martin T, Infante JR, Mischel PS, Matsutani T, Xu S, Wong L, Liu Y, et al. A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer. 2015; 121:3481-3490.
337. Sedrani R, Cottens S, Kallen J and Schuler W. Chemical modification of rapamycin: the discovery of SDZ RAD. Transplantation proceedings. 1998; 30:2192-2194.
338. Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM and Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nature chemical biology. 2008; 4:691-699.
339. Han K, Xu X, Xu Z, Chen G, Zeng Y, Zhang Z, Cao B, Kong Y, Tang X and Mao X. SC06, a novel small molecule compound, displays preclinical activity against multiple myeloma by disrupting the mTOR signaling pathway. Scientific reports. 2015; 5:12809.
340. Shor B, Zhang WG, Toral-Barza L, Lucas J, Abraham RT, Gibbons JJ and Yu K. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer research. 2008; 68:2934-2943.
341. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS and Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009; 137:873-886.
342. Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM and Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. The Journal of biological chemistry. 2009; 284:8023-8032.
343. Stengel C, Cheung CW, Quinn J, Yong K and Khwaja A. Optimal induction of myeloma cell death requires dual blockade of phosphoinositide 3-kinase and mTOR signalling and is determined by translocation subtype. Leukemia. 2012; 26:1761-1770.
344. Yu K, Toral-Barza L, Shi C, Zhang WG, Lucas J, Shor B, Kim J, Verheijen J, Curran K, Malwitz DJ, Cole DC, Ellingboe J, Ayral-Kaloustian S, et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer research. 2009; 69:6232-6240.
345. Johrer K, Obkircher M, Neureiter D, Parteli J, Zelle-Rieser C, Maizner E, Kern J, Hermann M, Hamacher F, Merkel O, Wacht N, Zidorn C, Scheideler M, et al. Antimyeloma activity of the sesquiterpene lactone cnicin: impact on Pim-2 kinase as a novel therapeutic target. Journal of molecular medicine (Berlin, Germany). 2012; 90:681-693.
346. Garcia PD, Langowski JL, Wang Y, Chen M, Castillo J, Fanton C, Ison M, Zavorotinskaya T, Dai Y, Lu J, Niu XH, Basham S, Chan J, et al. Pan-PIM kinase inhibition provides a novel therapy for treating hematologic cancers. Clinical cancer research. 2014; 20:1834-1845.
347. Keane NA, Reidy M, Natoni A, Raab MS and O’Dwyer M. Targeting the Pim kinases in multiple myeloma. Blood cancer journal. 2015; 5:e325.
348. Tian Z, Zhao JJ, Tai YT, Amin SB, Hu Y, Berger AJ, Richardson P, Chauhan D and Anderson KC. Investigational agent MLN9708/2238 targets tumor-suppressor miR33b in MM cells. Blood. 2012; 120:3958-3967.
349. Chen LS, Redkar S, Taverna P, Cortes JE and Gandhi V. Mechanisms of cytotoxicity to Pim kinase inhibitor, SGI-1776, in acute myeloid leukemia. Blood. 2011; 118:693-702.
350. Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, Wysong DR, Janowick DA, Hyer ML, et al. Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clinical cancer research. 2011; 17:7614-7624.
351. Kelly KR, Shea TC, Goy A, Berdeja JG, Reeder CB, McDonagh KT, Zhou X, Danaee H, Liu H, Ecsedy JA, Niu H, Benaim E and Padmanabhan Iyer S. Phase I study of MLN8237-investigational Aurora A kinase inhibitor-in relapsed/refractory multiple myeloma, Non-Hodgkin lymphoma and chronic lymphocytic leukemia. Investigational new drugs. 2014; 32:489-499.
352. Gorgun G, Calabrese E, Hideshima T, Ecsedy J, Perrone G, Mani M, Ikeda H, Bianchi G, Hu Y, Cirstea D, Santo L, Tai YT, Nahar S, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010; 115:5202-5213.
353. Rosenthal A, Kumar S, Hofmeister C, Laubach J, Vij R, Dueck A, Gano K and Stewart AK. A Phase Ib study of the combination of the Aurora Kinase inhibitor alisertib (MLN8237) and bortezomib in relapsed multiple myeloma. British journal of haematology. 2015:[Epub ahead of print; available on March 25, <2017].
354. Santo L, Hideshima T, Cirstea D, Bandi M, Nelson EA, Gorgun G, Rodig S, Vallet S, Pozzi S, Patel K, Unitt C, Squires M, Hu Y, et al. Antimyeloma activity of a multitargeted kinase inhibitor, AT9283, via potent Aurora kinase and STAT3 inhibition either alone or in combination with lenalidomide. Clinical cancer research. 2011; 17:3259-3271.
355. Howard S, Berdini V, Boulstridge JA, Carr MG, Cross DM, Curry J, Devine LA, Early TR, Fazal L, Gill AL, Heathcote M, Maman S, Matthews JE, et al. Fragment-based discovery of the pyrazol-4-yl urea (AT9283), a multitargeted kinase inhibitor with potent aurora kinase activity. Journal of medicinal chemistry. 2009; 52:379-388.
356. Evans RP, Naber C, Steffler T, Checkland T, Maxwell CA, Keats JJ, Belch AR, Pilarski LM, Lai R and Reiman T. The selective Aurora B kinase inhibitor AZD1152 is a potential new treatment for multiple myeloma. British journal of haematology. 2008; 140:295-302.
357. Wilkinson RW, Odedra R, Heaton SP, Wedge SR, Keen NJ, Crafter C, Foster JR, Brady MC, Bigley A, Brown E, Byth KF, Barrass NC, Mundt KE, et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clinical cancer research. 2007; 13:3682-3688.
358. Carpinelli P, Ceruti R, Giorgini ML, Cappella P, Gianellini L, Croci V, Degrassi A, Texido G, Rocchetti M, Vianello P, Rusconi L, Storici P, Zugnoni P, et al. PHA-739358, a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer. Molecular cancer therapeutics. 2007; 6:3158-3168.
359. Meulenbeld HJ, Mathijssen RH, Verweij J, de Wit R and de Jonge MJ. Danusertib, an aurora kinase inhibitor. Expert opinion on investigational drugs. 2012; 21:383-393.
360. Zhang S and Farag SS. From cell biology to therapy: ENMD-2076 in the treatment of multiple myeloma. Expert opinion on investigational drugs. 2011; 20:1015-1028.
361. How J and Yee K. ENMD-2076 for hematological malignancies. Expert opinion on investigational drugs. 2012; 21:717-732.
362. Fletcher GC, Brokx RD, Denny TA, Hembrough TA, Plum SM, Fogler WE, Sidor CF and Bray MR. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Molecular cancer therapeutics. 2011; 10:126-137.
363. Wang X, Sinn AL, Pollok K, Sandusky G, Zhang S, Chen L, Liang J, Crean CD, Suvannasankha A, Abonour R, Sidor C, Bray MR and Farag SS. Preclinical activity of a novel multiple tyrosine kinase and aurora kinase inhibitor, ENMD-2076, against multiple myeloma. British journal of haematology. 2010; 150:313-325.
364. Hose D, Reme T, Meissner T, Moreaux J, Seckinger A, Lewis J, Benes V, Benner A, Hundemer M, Hielscher T, Shaughnessy JD, Jr., Barlogie B, Neben K, et al. Inhibition of aurora kinases for tailored risk-adapted treatment of multiple myeloma. Blood. 2009; 113:4331-4340.
365. Cheetham GM, Charlton PA, Golec JM and Pollard JR. Structural basis for potent inhibition of the Aurora kinases and a T315I multi-drug resistant mutant form of Abl kinase by VX-680. Cancer letters. 2007; 251:323-329.
366. Hofmeister CC, Poi M, Bowers MA, Zhao W, Phelps MA, Benson DM, Kraut EH, Farag S, Efebera YA, Sexton J, Lin TS, Grever M and Byrd JC. A phase I trial of flavopiridol in relapsed multiple myeloma. Cancer chemotherapy and pharmacology. 2014; 73:249-257.
367. Senderowicz AM. The cell cycle as a target for cancer therapy: basic and clinical findings with the small molecule inhibitors flavopiridol and UCN-01. The oncologist. 2002; 7 Suppl 3:12-19.
368. Squires MS, Feltell RE, Wallis NG, Lewis EJ, Smith DM, Cross DM, Lyons JF and Thompson NT. Biological characterization of AT7519, a small-molecule inhibitor of cyclin-dependent kinases, in human tumor cell lines. Molecular cancer therapeutics. 2009; 8:324-332.
369. Santo L, Vallet S, Hideshima T, Cirstea D, Ikeda H, Pozzi S, Patel K, Okawa Y, Gorgun G, Perrone G, Calabrese E, Yule M, Squires M, et al. AT7519, A novel small molecule multi-cyclin-dependent kinase inhibitor, induces apoptosis in multiple myeloma via GSK-3beta activation and RNA polymerase II inhibition. Oncogene. 2010; 29:2325-2336.
370. Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, Seghezzi W, Paruch K, Dwyer MP, Doll R, Nomeir A, Windsor W, Fischmann T, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Molecular cancer therapeutics. 2010; 9:2344-2353.
371. Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, Fruth B, Roy V, Erlichman C and Stewart AK. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood. 2015; 125:443-448.
372. McMillin DW, Delmore J, Negri J, Buon L, Jacobs HM, Laubach J, Jakubikova J, Ooi M, Hayden P, Schlossman R, Munshi NC, Lengauer C, Richardson PG, et al. Molecular and cellular effects of multi-targeted cyclin-dependent kinase inhibition in myeloma: biological and clinical implications. British journal of haematology. 2011; 152:420-432.
373. Manohar SM, Rathos MJ, Sonawane V, Rao SV and Joshi KS. Cyclin-dependent kinase inhibitor, P276-00 induces apoptosis in multiple myeloma cells by inhibition of Cdk9-T1 and RNA polymerase II-dependent transcription. Leukemia research. 2011; 35:821-830.
374. Raje N, Hideshima T, Mukherjee S, Raab M, Vallet S, Chhetri S, Cirstea D, Pozzi S, Mitsiades C, Rooney M, Kiziltepe T, Podar K, Okawa Y, et al. Preclinical activity of P276-00, a novel small-molecule cyclin-dependent kinase inhibitor in the therapy of multiple myeloma. Leukemia. 2009; 23:961-970.
375. Altenburg JD and Farag SS. The potential role of PD0332991 (Palbociclib) in the treatment of multiple myeloma. Expert opinion on investigational drugs. 2015; 24:261-271.
376. Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, Albassam M, Zheng X, Leopold WR, Pryer NK and Toogood PL. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Molecular cancer therapeutics. 2004; 3:1427-1438.
377. Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C, Patton V, Alzani R, Ciavolella A, et al. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nature chemical biology. 2008; 4:357-365.
378. Cirstea D, Hideshima T, Santo L, Eda H, Mishima Y, Nemani N, Hu Y, Mimura N, Cottini F, Gorgun G, Ohguchi H, Suzuki R, Loferer H, et al. Small-molecule multi-targeted kinase inhibitor RGB-286638 triggers P53-dependent and -independent anti-multiple myeloma activity through inhibition of transcriptional CDKs. Leukemia. 2013; 27:2366-2375.
379. Josefsberg Ben-Yehoshua L, Beider K, Shimoni A, Ostrovsky O, Samookh M, Peled A and Nagler A. Characterization of cyclin E expression in multiple myeloma and its functional role in seliciclib-induced apoptotic cell death. PloS one. 2012; 7:e33856.
380. Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG and Moulinoux JP. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. European journal of biochemistry / FEBS. 1997; 243:527-536.
381. Tong WG, Chen R, Plunkett W, Siegel D, Sinha R, Harvey RD, Badros AZ, Popplewell L, Coutre S, Fox JA, Mahadocon K, Chen T, Kegley P, et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. Journal of clinical oncology. 2010; 28:3015-3022.
382. Conroy A, Stockett DE, Walker D, Arkin MR, Hoch U, Fox JA and Hawtin RE. SNS-032 is a potent and selective CDK 2, 7 and 9 inhibitor that drives target modulation in patient samples. Cancer chemotherapy and pharmacology. 2009; 64:723-732.
383. Alvarez-Fernandez S, Ortiz-Ruiz MJ, Parrott T, Zaknoen S, Ocio EM, San Miguel J, Burrows FJ, Esparis-Ogando A and Pandiella A. Potent antimyeloma activity of a novel ERK5/CDK inhibitor. Clinical cancer research. 2013; 19:2677-2687.
384. Aleem E and Arceci RJ. Targeting cell cycle regulators in hematologic malignancies. Frontiers in cell and developmental biology. 2015; 3:16.
385. Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Current biology. 2007; 17:316-322.
386. Stevenson CS, Capper EA, Roshak AK, Marquez B, Eichman C, Jackson JR, Mattern M, Gerwick WH, Jacobs RS and Marshall LA. The identification and characterization of the marine natural product scytonemin as a novel antiproliferative pharmacophore. The Journal of pharmacology and experimental therapeutics. 2002; 303:858-866.
387. Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ and Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer research. 2004; 64:9152-9159.
388. Zabludoff SD, Deng C, Grondine MR, Sheehy AM, Ashwell S, Caleb BL, Green S, Haye HR, Horn CL, Janetka JW, Liu D, Mouchet E, Ready S, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Molecular cancer therapeutics. 2008; 7:2955-2966.
389. Yang C, Betti C, Singh S, Toor A and Vaughan A. Impaired NHEJ function in multiple myeloma. Mutation research. 2009; 660:66-73.
390. Veuger SJ, Curtin NJ, Richardson CJ, Smith GC and Durkacz BW. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer research. 2003; 63:6008-6015.
391. Sharman JP, Chmielecki J, Morosini D, Palmer GA, Ross JS, Stephens PJ, Stafl J, Miller VA and Ali SM. Vemurafenib response in 2 patients with posttransplant refractory BRAF V600E-mutated multiple myeloma. Clinical lymphoma, myeloma & leukemia. 2014; 14:e161-163.
392. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010; 467:596-599.
393. Sebolt-Leopold JS, Dudley DT, Herrera R, Van Becelaere K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR and Saltiel AR. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nature medicine. 1999; 5:810-816.
394. Lunghi P, Giuliani N, Mazzera L, Lombardi G, Ricca M, Corradi A, Cantoni AM, Salvatore L, Riccioni R, Costanzo A, Testa U, Levrero M, Rizzoli V, et al. Targeting MEK/MAPK signal transduction module potentiates ATO-induced apoptosis in multiple myeloma cells through multiple signaling pathways. Blood. 2008; 112:2450-2462.
395. Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR, Przybranowski SA, Sebolt-Leopold J, Van Becelaere K, et al. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorganic & medicinal chemistry letters. 2008; 18:6501-6504.
396. Kim K, Kong SY, Fulciniti M, Li X, Song W, Nahar S, Burger P, Rumizen MJ, Podar K, Chauhan D, Hideshima T, Munshi NC, Richardson P, et al. Blockade of the MEK/ERK signalling cascade by AS703026, a novel selective MEK1/2 inhibitor, induces pleiotropic anti-myeloma activity in vitro and in vivo. British journal of haematology. 2010; 149:537-549.
397. Pei XY, Dai Y, Youssefian LE, Chen S, Bodie WW, Takabatake Y, Felthousen J, Almenara JA, Kramer LB, Dent P and Grant S. Cytokinetically quiescent (G0/G1) human multiple myeloma cells are susceptible to simultaneous inhibition of Chk1 and MEK1/2. Blood. 2011; 118:5189-5200.
398. Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, Marlow A, Hurley B, Lyssikatos J, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clinical cancer research. 2007; 13:1576-1583.
399. Yamaguchi T, Kakefuda R, Tajima N, Sowa Y and Sakai T. Antitumor activities of JTP-74057 (GSK1120212), a novel MEK1/2 inhibitor, on colorectal cancer cell lines in vitro and in vivo. International journal of oncology. 2011; 39:23-31.
400. Liu S, Ma Z, Cai H, Li Q, Rong W and Kawano M. Inhibitory effect of baicalein on IL-6-mediated signaling cascades in human myeloma cells. European journal of haematology. 2010; 84:137-144.
401. Shimura Y, Kuroda J, Ri M, Nagoshi H, Yamamoto-Sugitani M, Kobayashi T, Kiyota M, Nakayama R, Mizutani S, Chinen Y, Sakamoto N, Matsumoto Y, Horiike S, et al. RSK2(Ser227) at N-terminal kinase domain is a potential therapeutic target for multiple myeloma. Molecular cancer therapeutics. 2012; 11:2600-2609.
402. Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P and Alessi DR. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. The Biochemical journal. 2007; 401:29-38.
403. Miller RM, Paavilainen VO, Krishnan S, Serafimova IM and Taunton J. Electrophilic fragment-based design of reversible covalent kinase inhibitors. Journal of the American Chemical Society. 2013; 135:5298-5301.
404. Zhu YX, Yin H, Bruins LA, Shi CX, Jedlowski P, Aziz M, Sereduk C, Kortuem KM, Schmidt JE, Champion M, Braggio E and Keith Stewart A. RNA interference screening identifies lenalidomide sensitizers in multiple myeloma, including RSK2. Blood. 2015; 125:483-491.
405. Maloney DJ and Hecht SM. Synthesis of a potent and selective inhibitor of p90 Rsk. Organic letters. 2005; 7:1097-1099.
406. Doudican NA, Mazumder A, Kapoor S, Sultana Z, Kumar A, Talawdekar A, Basu K, Agrawal A, Aggarwal A, Shetty K, Singh NK, Kumar C, Tyagi A, et al. Predictive simulation approach for designing cancer therapeutic regimens with novel biological mechanisms. Journal of Cancer. 2014; 5:406-416.
407. Bennett BL, Sasaki DT, Murray BW, O’Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM and Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:13681-13686.
408. Mader M, de Dios A, Shih C, Bonjouklian R, Li T, White W, Lopez de Uralde B, Sanchez-Martinez C, del Prado M, Jaramillo C, de Diego E, Martin Cabrejas LM, Dominguez C, et al. Imidazolyl benzimidazoles and imidazo[4,5-b]pyridines as potent p38alpha MAP kinase inhibitors with excellent in vivo antiinflammatory properties. Bioorganic & medicinal chemistry letters. 2008; 18:179-183.
409. Gaul L, Mandl-Weber S, Baumann P, Emmerich B and Schmidmaier R. Bendamustine induces G2 cell cycle arrest and apoptosis in myeloma cells: the role of ATM-Chk2-Cdc25A and ATM-p53-p21-pathways. Journal of cancer research and clinical oncology. 2008; 134:245-253.
410. Davies SP, Reddy H, Caivano M and Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. The Biochemical journal. 2000; 351:95-105.
411. Wen J, Cheng HY, Feng Y, Rice L, Liu S, Mo A, Huang J, Zu Y, Ballon DJ and Chang CC. P38 MAPK inhibition enhancing ATO-induced cytotoxicity against multiple myeloma cells. British journal of haematology. 2008; 140:169-180.
412. Lali FV, Hunt AE, Turner SJ and Foxwell BM. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. The Journal of biological chemistry. 2000; 275:7395-7402.
413. Medicherla S, Protter AA, Ma JY, Mangadu R, Almirez R, Koppelman B, Kerr I, Navas TA, Movius F, Reddy M, Liu YW, Luedtke G, Perumattam J, et al. Preventive and therapeutic potential of p38 alpha-selective mitogen-activated protein kinase inhibitor in nonobese diabetic mice with type 1 diabetes. The Journal of pharmacology and experimental therapeutics. 2006; 318:99-107.
414. Medicherla S, Reddy M, Ying J, Navas TA, Li L, Nguyen AN, Kerr I, Hanjarappa N, Protter AA and Higgins LS. p38alpha-selective MAP kinase inhibitor reduces tumor growth in mouse xenograft models of multiple myeloma. Anticancer research. 2008; 28:3827-3833.
415. Murali Dhar TG, Wrobleski ST, Lin S, Furch JA, Nirschl DS, Fan Y, Todderud G, Pitt S, Doweyko AM, Sack JS, Mathur A, McKinnon M, Barrish JC, et al. Synthesis and SAR of p38alpha MAP kinase inhibitors based on heterobicyclic scaffolds. Bioorganic & medicinal chemistry letters. 2007; 17:5019-5024.
416. Hideshima T, Akiyama M, Hayashi T, Richardson P, Schlossman R, Chauhan D and Anderson KC. Targeting p38 MAPK inhibits multiple myeloma cell growth in the bone marrow milieu. Blood. 2003; 101:703-705.
417. Brown KK, Heitmeyer SA, Hookfin EB, Hsieh L, Buchalova M, Taiwo YO and Janusz MJ. P38 MAP kinase inhibitors as potential therapeutics for the treatment of joint degeneration and pain associated with osteoarthritis. Journal of inflammation (London, England). 2008; 5:22.
418. Tagoug I, Sauty De Chalon A and Dumontet C. Inhibition of IGF-1 signalling enhances the apoptotic effect of AS602868, an IKK2 inhibitor, in multiple myeloma cell lines. PloS one. 2011; 6:e22641.
419. Frelin C, Imbert V, Griessinger E, Loubat A, Dreano M and Peyron JF. AS602868, a pharmacological inhibitor of IKK2, reveals the apoptotic potential of TNF-alpha in Jurkat leukemic cells. Oncogene. 2003; 22:8187-8194.
420. Rauert-Wunderlich H, Siegmund D, Maier E, Giner T, Bargou RC, Wajant H and Stuhmer T. The IKK inhibitor Bay 11-7082 induces cell death independent from inhibition of activation of NFkappaB transcription factors. PloS one. 2013; 8:e59292.
421. Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T and Gerritsen ME. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. The Journal of biological chemistry. 1997; 272:21096-21103.
422. Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y and Zusi FC. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. The Journal of biological chemistry. 2003; 278:1450-1456.
423. Chen G, Han K, Xu X, Du X, Zhang Z, Tang J, Shi M, Wang M, Li J, Cao B and Mao X. An anti-leishmanial thiadiazine agent induces multiple myeloma cell apoptosis by suppressing the nuclear factor kappaB signalling pathway. British journal of cancer. 2014; 110:63-70.
424. Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, Dave S, Hurt EM, Tan B, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer cell. 2007; 12:115-130.
425. Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J and Anderson KC. NF-kappa B as a therapeutic target in multiple myeloma. The Journal of biological chemistry. 2002; 277:16639-16647.
426. Demchenko YN, Brents LA, Li Z, Bergsagel LP, McGee LR and Kuehl MW. Novel inhibitors are cytotoxic for myeloma cells with NFkB inducing kinase-dependent activation of NFkB. Oncotarget. 2014; 5:4554-4566. doi:10.18632/oncotarget.2128.