
INTRODUCTION
Oleandrin is a monomeric compound extracted from leaves and seeds of Nerium oleander, with a molecular weight of 576.73 and molecular formula of C32H48O9. As a kind of cardiac glycosides, it was well known for its effect in treating congestive heart failure [1]. Not only that, in the past decades, more and more studies have revealed that oleandrin may be a potential anti-tumor agent, as oleandrin can effectively inhibit proliferation of various cancer cells and induce apoptosis [2-5]. Besides that, oleandrin can also enhance the efficiency of radiotherapy [6]. Interestingly, the anti-tumor role of oleandrin seemed to be selective, as oleandrin can kill certain human tumor cells but not murine tumor cells [7, 8]. In a phase I study, oleandrin was used to remedy patients with refractory solid tumors, where oleandrin was found to be well tolerated and only few adverse events were reported [9]. Up to now, various studies on many possible pathways have been made to elucidate the anti-cancer role of oleandrin. Some argues that oleandrin’s ability to inhibit cancer cells proliferation were because of the decrease in levels of Na, K-ATPase [10]. Mitochondrial injury caused by the generation of reactive oxygen species (ROS) was also taken into account [11]. Others suggest that activation of caspase-3 by oleandrin may be a cause of tumor cells apoptosis [6]. It had been reported that cancer cells were arrested in G2/M cell cycle by oleandrin [12], suggesting activation of DNA damage checkpoint. However, detailed mechanisms of the anti-tumor role of oleandrin are still not fully understood.
As we know, Genomic instability is one of the main features of cancer cells, it can be the combined effect of DNA damage and tumour-specific DNA repair defects [13], and plays important roles during tumorigenesis. At the present, many chemotherapy agents were designed to target DNA damage repair to induce cancer cell apoptosis. Here, we investigated roles of oleandrin in induction of cancer cell apoptosis as well as its impact on DNA damage response.
RESULTS
Oleandrin Induces cell death in multiple cancer cell lines
To better understand how oleandrin induces cancer cell apoptosis, A549 cells were treated with oleandrin, followed by detection of apoptosis by flow cytometry (FCM). Increased concentrations of oleandrin (0.01ug/ml, 0.02ug/ml, 0.04ug/ml) were incubated with A549 cells for 24 hours, where as low as 0.02 ug/ml was found to be sufficient in apoptosis induction (Figure 1A, 1B). Compared with the control group, apoptosis of A549 cells with treatment of oleandrin (0.02ug/ml, 0.04ug/ml) groups showed a statistically significant increase. Furthermore, apoptosis was induced by oleandrin (0.02ug/ml) in a time-dependent manner (Figure 1C, 1D). Similar experiments were performed in additional two cell lines, including HBE (a human bronchial epithelial cell line) and H1299 (a human non-small cell lung carcinoma cell line). Interestingly, the two cancer cell lines were more sensitive to oleandrin treatment, while HBE cells showed little toxicity (Figure 2A-2C).
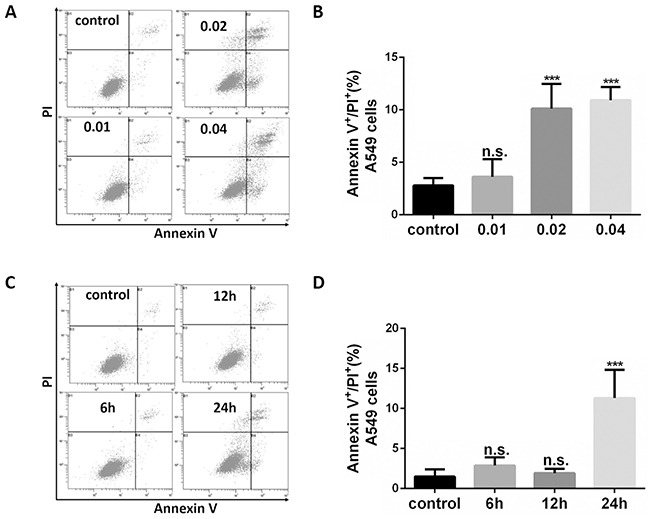
Figure 1: Oleandrin induced apoptosis in A549 cells. A. Representative FACS profiles of A549 cells treated with different concentrations of oleandrin for 24 hours. B. Quantification of Annexin V positive and PI positive A549 cells following treatment of oleandrin at indicated concentrations were shown. C. Representative FACS profiles of A549 cells treated with oleandrin (0.02 ug/ml) for different hours (6h, 12h, 24h). D. Quantification of Annexin V positive and PI positive A549 cells following treatment of oleandrin for indicated hours were shown. All data represent mean ± SEM (n=3); n.s. not significant, ***P<0.001, compared with the controls.
In addition, we also detected the toxicity of oleandrin in A549 cells and ATDC5 cells(a normal cartilage cell line) with the method of CCK-8 assay, found that the cell viability of A549 cells were largely limited, while it did no harmful to ATDC5 cells (Figure 2D, 2E).
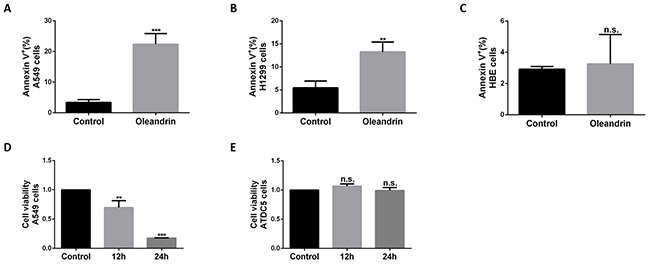
Figure 2: Oleandrin effectively induced cell death in A549 and H1299 cells, but not in HBE and ATDC5 cells. A. Quantification of cell death in A549 cells treated with oleandrin (0.02ug/ml) for 24 hours, detected by FACS. B. Quantification of cell death in H1299 cells treated with oleandrin (0.02ug/ml) for 24 hours, detected by FACS. C. Quantification of cell death in HBE cells treated with oleandrin (0.02ug/ml) for 24 hours, detected by FACS. D, E. Quantification of cell viability in A549 cells and ATDC5 cells treated with oleandrin (0.02ug/ml) for 24 hours, detected by CCK-8 assay. All data represent mean±SEM(n=3); n.s. not siginificant; **P<0.01; ***P<0.001, compared with controls.
Oleandrin induces DNA damage response in lung cancer cells
RPA is a ubiquitous eukaryotic single-stranded DNA (ssDNA) binding protein that serves to protect every generated ssDNA from degradation [14]. With the method of immunofluorescence, we measured localization of RPA in A549 and H1299 cells following treatment with oleandrin (0.02 ug/ml). It was obvious that oleandrin could significantly increase levels of RPA foci formation at 12 hours (Figure 3A-3D), at which point no significant cell death was yet detected (Figure 1).
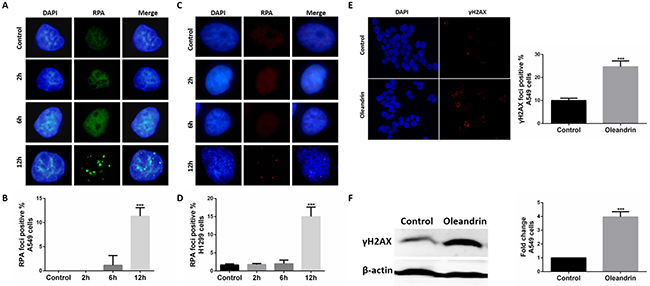
Figure 3: Oleandrin induced RPA and gH2AX foci in lung cancer cells. A. Immunofluorescence staining of RPA in A549 cells treated with oleandrin (0.02ug/ml) for 2hours, 6hours, 12hours. B. Quantification of (A). C. Immunofluorescence staining of RPA in H1299 cells treated with oleandrin (0.02ug/ml) for 2hours, 6hours, 12hours. D. Quantification of (C). E. Immunofluorescence staining of γH2AX in A549 cells treated with oleandrin (0.02ug/ml) for 24hours. F. A549 cells treated with oleandrin (0.02ug/ml) for 24hours, γH2AX was analyzed by western blot. All data represent mean±SEM (n=3). ***P<0.001, compared with controls.
To investigate whether oleandrin could induce DNA damage in cancer cells, we measured localization of γH2AX, a marker for DNA double-strand breaks (DSBs), in A549 cells following treatment with oleandrin (0.02 ug/ml) for 24 hours. It was obvious that oleandrin could significantly induce γH2AX foci formation (Figure 3E). To confirm this, we also detected the expression of γH2AX with the method of western blot, and the results (Figure 3F) were consistent with those in immunofluorescence.
Oleandrin down-regulates expression of HR protein RAD51
There are two major DSB repair pathways in higher eukaryotes, HR and non-homologous end joining (NHEJ). And, HR plays an important role in DSB repair. To ensure whether the DNA breaks-induced by oleandrin were repaired by HR pathway. We measured the expression of RAD51 by western blot. In fact, the expression level of RAD51 reduced significantly in A549 and H1299 cells following treatment with oleandrin (Figure 4A-4B). XRCC1-mediated single-strand breaks repair(SSBR) plays a compensatory role to RAD51-mediated HR pathway. We therefore measured levels of XRCC1 to test whether the DSBs induced by oleandrin were repaired through SSBR pathway. Not surprisingly, cells treated with oleandrin showed increased expression levels of XRCC1 protein (Figure 4C). As HR is usually cell cycle dependent. To rule out the possibility of cell cycle arrest-induced down-regulation of RAD51, we measured cell cycle profile of oleandrin-treated cells, and did not detect any significant alteration of G2/M population (Figure 4D).
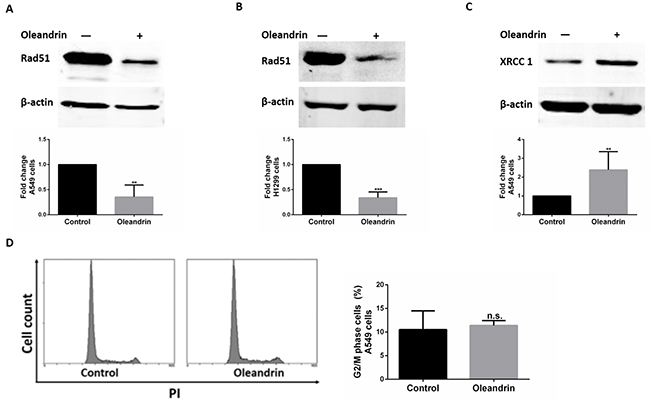
Figure 4: Expression of DNA damage repair proteins in cancer cell lines following treatment with oleandrin. A. Western blot analysis of RAD51 and Actin in A549 cells. Quantification of RAD51 expression level was performed by densitometric analysis. B. Western blot analysis of RAD51 and Actin in H1299 cells. Quantification of RAD51 expression level was performed by densitometric analysis. C. Western blot analysis of XRCC1 in A549 cells. Quantification of XRCC1 expression level was performed by densitometric analysis. D. Cell cycle of A549 cells treated with oleandrin (0.02ug/ml) for 24 hours. Percentage of cells stalled in G2/M phase were quantified. All data represent mean±SEM (n=3). n.s. not significant; **P<0.01; ***P<0.001, compared with controls.
XRCC1 is required for cell survival against oleandrin-induced DNA damage
We have now shown that Rad51 was significantly decreased, while XRCC1 expression level was elevated by oleandrin. In order to explore potential roles of XRCC1 in repair against oleandrin-induced DNA damage, we knocked down XRCC1 by siRNA strategy with satisfactory efficiency (Figure 5A). Interestingly, depletion of XRCC1 was found to significantly sensitize cells to oleandrin-induced cell death (Figure 5B-5E), suggesting that XRCC1 may play an essential role in promoting cell survival following oleandrin treatment.
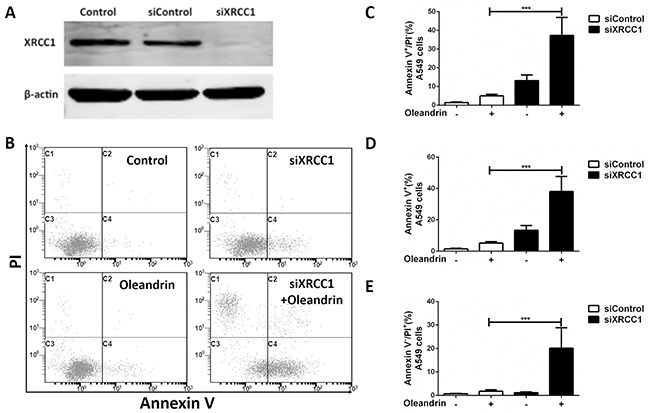
Figure 5: Loss of XRCC1 sensitized A549 cells to oleandrin-induced cell death. A. Western blot analysis of XRCC1 and Actin in A549 cells following indicated siRNA transfection. B. A549 cells were incubated with oleandrin (0.02ug/ml) for 12 hours, 24 hours after the transfection of XRCC1 siRNA. Cell death was detected with FACS analysis. C, D, E. Quantification of (B) for indicated values. All data represent mean±SEM (n=3). ***P<0.001.
DISCUSSION
Oleandrin is a lipid-soluble cardiac glycoside, with a molecular weight of 576.73 and molecular formula of C32H48O9, the main cardiac glycoside in Nerium oleander L. (Apocynaceae). Consistent with previous report [12], oleandrin was able to induce cancer cell death. We found that oleandrin at a low concentration of 0.02ug/ml could significantly induce death of A549 following a 24h treatment, indicating that oleandrin may be a potential effective anti-tumor agent. Interestingly, the anti-tumor role of oleandrin seemed to have some selectivity, as it was more harmful to lung cancer cells than normal bronchial epithelial cells. And in ATDC5 cells, it also presented little toxicity by our results. As we know, it had not been reported at one time. We thought the possible reason to explain the different roles of oleandrin between cancer cells and normal cells may be DNA damage. DNA damage is one reason of tumorigenesis, and most of tumors have genetic DNA defects, while normal cells usually does not have these problems. In fact, many strategies have been made to treat tumor target DNA damage, take Olaparib(a DNA damage repair inhibitor) for example, it was approved by FDA for its benefit in BRCA deficiency cancers [15], Olaparib was particular effective to HR pathway deficiency tumors. Our results also presented that oleandrin could reduce RAD51 expression and may inhibit HR pathway, this may increase the sensitivity of cancer cells to oleandrin. However, we can’t deny that oleandrin is a kind of poison agent, and the selectivity of its toxicity was bound. And the detailed mechanisms of this were still unknown and further study need to be done to elucidate it.
In our study, RPA foci was significantly increased by oleandrin. As a major single-stranded DNA binding protein, RPA plays crucial roles in almost all aspects of eukaryotic DNA metabolism include DNA replication, DNA repair, recombination, cell cycle and DNA damage checkpoints [16]. It is abundant in eukaryotic cells, and can bind to single-stranded DNA with sub-nanomolar affinity [17]. So, any single-stranded region formed in DNA will be immediately bound by RPA. The essential role of RPA is to preserve genome stability [18]. It contributed to the early stages of DNA damage signaling cascade and DNA repair [16, 19]. So the increased level of RPA foci indicated increased levels of single-stranded DNA region in genomic DNA. If these single-stranded DNA were not repaired in time, they are likely to be transferred into DNA double-strand break, and the expression of γH2AX may be detected. According to our results, oleandrin induce the increased foci of RPA at an early time of 12 hours, and at this time little toxicity was presented until 24 hours. All this may reflect the potential role of RPA in the death of tumor cells, and increased risk of genetic instability induced by oleandrin.
γH2AX is a sensitive marker of DSBs [20, 21]. DSBs are considered to be one of the most severe forms of DNA damage to a cell, as DSBs can lead to genomic instability and cell apoptosis if not repaired in proper way [22]. When a DSB happened, the histone 2A (H2A)X was rapidly phosphorylated at ser139 by PI3K- like kinase, including ATM (ataxia telangiectasia mutated)/ATR (ataxia telangiectasia and Rad3-related) and DNA-dependent protein kinase [23, 24]. And the phosphorylated variant H2AX is referred as γH2AX. It is also an important player in preserving genome integrity, it attaches to chromatin near DSBs and induces the repair of damaged DNA [25]. Then, it recruits DNA damage repair proteins to the sites of DSBs and initiates the DNA damage signal transduction, and determines whether the damaged DNA will be repaired. When the DNA damage was repaired, γH2AX would be dephosphorylated to H2AX, and enter normal cell cycle [26, 27]. Usually, the presence of γH2AX indicating that some of the DNA damage remains unrepaired. In our study, a lot more γH2AX foci were seen in the oleandrin-induced A549 cells than that in the control group, and western blot assay also confirmed this. Taken together, we think oleandrin induces DNA damage in cancer cells. As mentioned above, high level of DNA damage induced by oleandrin may not all be repaired in a proper way, and may contribute to the death of cancer cells.
HR and NHEJ are two major DNA breaks repair pathways in higher eukaryotes. And HR is the most conservative pathway to repair DSBs and usually happens during S and G2 phases of cell cycle, it requires highly homology for the repair of DSBs [28, 29]. While NHEJ, using limited or no homology for end joining, has a minimal role to repair DSBs. Compared to HR pathway, NHEJ is a “quick and dirty” process, but less accurate [30-32]. However, it provides a compensatory way to DSBs repair when the HR pathway was blocked. NHEJ pathway is considered to be a possible way that enables cells to maximize their chances of survival [32]. In this study, to elucidate the mechanism of DNA repair in oleandrin induced anti-tumor effect, we measured expression of DNA damage repair proteins RAD51 and XRCC1 in oleandrin-treated cancer cells.
If the DNA breaks-induced by oleandrin were mainly repaired by HR pathway, an increased level of RAD51 expression might be observed. However, the expression level of RAD51 in oleandrin-treated cancer cells showed a significant reduction. RAD51 is a key protein in HR and plays an important role in cellular proliferation by repairing DNA damage. It had been reported that the inhibitor of RAD51 weakens HR pathway and sensitizes cancer cells to anti-tumor therapy [33]. To rule out the possibility of cell cycle arrest-induced down-regulation of RAD51, we measured cell cycle profile of oleandrin-treated cells, and did not detect any significant alteration of G2/M population. Taken together, the decreased level of RAD51 induced by oleandrin may contribute to the inhibition of HR pathway, and lead to the death of cancer cells.
As we know, XRCC1 plays an important role in single strand break repair, and backup nonhomologous end-joining pathway. XRCC1 mediated SSBR also plays a compensatory role to RAD51-mediated HR pathway. We therefore measured the level of XRCC1 to test whether the DSBs induced by oleandrin were repaired through SSBR pathway. As expected, A549 cells treated with oleandrin showed an increased level of XRCC1 expression. XRCC1 is a critical factor in SSBR, it functions as a molecular scaffold protein and involved in SSBR by interacting with DNA glycosylases, apurinic/apyrimidinic endonulcease (APE1), PARP-1, polynulceotide kinase (PNK), and ligase III [34, 35]. In our study, the increased level of XRCC1 expression indicated the activation of XRCC1 induced DNA repair pathway such as SSBR, and this may be due to the inhibition of HR pathway, and to activate downstream repair machinery in response to oleandrin-induced DNA strand breaks. We also did a further study that A549 cells were more sensitive to oleandrin when XRCC1 was knocked down, suggesting that XRCC1 may berequired for cell survival against oleandrin-induced DNA damage.
In addition, as presented in Fiugre 5E, there is a small part of cancer cells were stained with PI. Annexin V represent the status of early apoptosis; while PI would emerge in late apoptosis or necrosis cells. So we think the death of cancer cells induced by oleandrin mainly through apoptosis, but apoptosis was not the only, necrosis may also existed.
CONCLUSIONS
In conclusion, oleandrin could effectively induce the death of cancer cells at a very low concentration; the anti-tumor role of oleandrin may be related with DNA damage repair; and oleandrin may be a novel HR inhibitor by suppressing the expression of Rad51.
MATERIALS AND METHODS
Cell culture
The lung cancer cell lines A549 and H1299 were grown in DMEM/ high glucose medium (HyClone) with 10% Fetal Bovine Serum at 37 °C under an atmosphere containing 5% CO2. The normal epithelial cell line HBE was grown in RPMI Medium 1640 basic (1x) (Gibco by life technologies) with 10% Fetal Bovine Serum at 37 °C under an atmosphere containing 5% CO2.
Annexin V/PI stain, CCK-8 assay and RNA interference
Oleandrin (purchased from Shanghai Yaji Biological Technology Co., Ltd) was dissolved in DMSO at an original concentration of 5ug/ul, and then diluted into proper concentration in demand. A549, H1299, HBE cells were plated in triplicate into six-well plates at proper concentration overnight before the addition of indicated treatment, then oleandrin was added. Annexin V/PI apoptosis kit (purchased from Multi Sciences Biotech Co., Ltd.) was used to detect the apoptosis rate.
CCK-8 assay kit was purchased from Bomai Meditech. The test was carried on following the manufacturer’s protocol.
The XRCC1 siRNA was purchased from the GenePharma (siXRCC1: 5’-CUGGUCACCUCAUCUU UCATT-3’; siControl: 5’-UUCUCCGAACGUGUCA CGUTT-3’), transfection reagent (RNAiMax) was purchased from Invitrogen. The siRNA transfection was carried on following the manufacturer’s protocol. A549 cells were transfected for 24hours, and then treated with oleandrin for 12 hours. FCM was used to detect the apoptosis rate.
Immunofluorescence
Following indicated treatment, cells were fixed and stained. The primary antibody used was mouse monoclonal antibody against RPA (Abcam2175) and γH2AX (Abcam81299). The secondary antibodies used were Alexa Fluor 488-conjugated goat anti-mouse IgG (Molecular Probes) and Alexa Fluor 555-conjugated goat anti-mouse IgG (Molecular Probes). DNA was counterstained with DAPI. More than 300 cells were counted from random fields for each condition, and cells with more than 3 nuclear foci were calculated as positive staining.
Cell cycle
A549 cells were planted in six-well plates, treated with oleandrin 24 hours, and cell cycle staining kit (purchased from MultiSciences) was used to detect the cell cycle following the manufacturer’s protocol.
Western blotting
Cells were lysed in RIPA buffer in the presence of 1 × protease inhibitor cocktail (Sigma). An aliquot of 50 µg total protein was run on an SDS-PAGE gel and transferred to PVDF(polyvinylidene fluoride) membrane (Millipore). This membrane was immunoblotted with mouse monoclonal antibody against XRCC1 (Abcam 1838), rabbit polyclonal antibodies against RAD51 (Santa Cruz 8349) in 5% Milk overnight. Immunoreactive proteins were visualized following the manufacturer’s instructions. Densitometric analysis was performed using ImageJ 1.46 software to normalize the expression of target protein with the corresponding loading control.
Statistical analysis
Data were presented in the form of means ± SEM, analysed with GraphPad Prism 6.0 (GraphPad software, San Diego, California, USA). Differences between groups were analysed using the One-way ANOVA (and nonparametric) (n.s. not significant; * p < 0.05; **p < 0.01; ***p < 0.001).
ACKNOWLEDGMENTS
This work was supported by the National 1000 Talents Program for Young Scholars, the National Natural Science Foundation of China (81422031), Zhejiang University K.P.Chao’s High Technology Development Foundation, and the Zhejiang Provincial Natural Science Foundation of China (LR14H160001). We are grateful to undergraduate students Lingjian Zhang, Lisha Ye, Xiao Chen, Xuezhen Lu, Xiaoxiao Chen, Kai Xv, Dongcai Jin, and Xicheng Zhang for their technical assistance.
REFERENCES
1. Kjeldsen K, Norgaard A and Gheorghiade M. Myocardial Na,K-ATPase: the molecular basis for the hemodynamic effect of digoxin therapy in congestive heart failure. Cardiovascular research. 2002; 55:710-713.
2. Lin Y, Ho DH and Newman RA. Human tumor cell sensitivity to oleandrin is dependent on relative expression of Na+, K+-ATPase subunitst. Journal of experimental therapeutics & oncology. 2009; 8:271-286.
3. Turan N, Akgun-Dar K, Kuruca SE, Kilicaslan-Ayna T, Seyhan VG, Atasever B, Mericli F and Carin M. Cytotoxic effects of leaf, stem and root extracts of Nerium oleander on leukemia cell lines and role of the p-glycoprotein in this effect. J Exp Ther Oncol. 2006; 6:31-38.
4. Manna SK, Sah NK, Newman RA, Cisneros A and Aggarwal BB. Oleandrin suppresses activation of nuclear transcription factor-kappaB, activator protein-1, and c-Jun NH2-terminal kinase. Cancer research. 2000; 60:3838-3847.
5. McConkey DJ, Lin Y, Nutt LK, Ozel HZ and Newman RA. Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer research. 2000; 60:3807-3812.
6. Nasu S, Milas L, Kawabe S, Raju U and Newman RA. Enhancement of radiotherapy by oleandrin is a caspase-3 dependent process. Cancer letters. 2002; 185:145-151.
7. Pathak S, Multani AS, Narayan S, Kumar V and Newman RA. Anvirzel, an extract of Nerium oleander, induces cell death in human but not murine cancer cells. Anti-cancer drugs. 2000; 11:455-463.
8. Raghavendra PB, Sreenivasan Y and Manna SK. Oleandrin induces apoptosis in human, but not in murine cells: dephosphorylation of Akt, expression of FasL, and alteration of membrane fluidity. Molecular immunology. 2007; 44:2292-2302.
9. Mekhail T, Kaur H, Ganapathi R, Budd GT, Elson P and Bukowski RM. Phase 1 trial of Anvirzel™ in patients with refractory solid tumors. Investigational new drugs. 2006; 24:423-427.
10. Yang P, Menter DG, Cartwright C, Chan D, Dixon S, Suraokar M, Mendoza G, Llansa N and Newman RA. Oleandrin-mediated inhibition of human tumor cell proliferation: Importance of Na, K-ATPase α subunits as drug targets. Molecular cancer therapeutics. 2009; 8:2319-2328.
11. Newman RA, Yang P, Hittelman WN, Lu T, Ho DH, Ni D, Chan D, Vijjeswarapu M, Cartwright C and Dixon S. Oleandrin-mediated oxidative stress in human melanoma cells. J Exp Ther Oncol. 2006; 5:167-181.
12. Newman RA, Kondo Y, Yokoyama T, Dixon S, Cartwright C, Chan D, Johansen M and Yang P. Autophagic cell death of human pancreatic tumor cells mediated by oleandrin, a lipid-soluble cardiac glycoside. Integrative cancer therapies. 2007; 6:354-364.
13. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
14. Chen J, Le S, Basu A, Chazin WJ and Yan J. Mechanochemical regulations of RPA’s binding to ssDNA. Sci Rep. 2015; 5:9296.
15. Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, Chiu HJ, Ricks TK, Palmby T, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clinical cancer research. 2015; 21:4257-4261.
16. Zou Y, Liu Y, Wu X and Shell SM. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. Journal of cellular physiology. 2006; 208:267-273.
17. Kumaran S, Kozlov AG and Lohman TM. Saccharomyces cerevisiae replication protein A binds to single-stranded DNA in multiple salt-dependent modes. Biochemistry. 2006; 45:11958-11973.
18. Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J and Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013; 155:1088-1103.
19. Zou L, Liu D and Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proceedings of the National Academy of Sciences of the United States of America. 2003; 100:13827-13832.
20. Rothkamm K and Löbrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proceedings of the National Academy of Sciences. 2003; 100:5057-5062.
21. Mah LJ, El-Osta A and Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010; 24:679-686.
22. Rich T, Allen RL and Wyllie AH. Defying death after DNA damage. Nature. 2000; 407:777-783.
23. Fernandez-Capetillo O, Lee A, Nussenzweig M and Nussenzweig A. H2AX: the histone guardian of the genome. DNA repair. 2004; 3:959-967.
24. Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Löbrich M and Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer research. 2004; 64:2390-2396.
25. van Attikum H and Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends in cell biology. 2009; 19:207-217.
26. Gavrilov B, Vezhenkova I, Firsanov D, Solovjeva L, Svetlova M, Mikhailov V and Tomilin N. Slow elimination of phosphorylated histone gamma-H2AX from DNA of terminally differentiated mouse heart cells in situ. Biochemical and biophysical research communications. 2006; 347:1048-1052.
27. Benson LJ, Gu Y, Yakovleva T, Tong K, Barrows C, Strack CL, Cook RG, Mizzen CA and Annunziato AT. Modifications of H3 and H4 during chromatin replication, nucleosome assembly, and histone exchange. The Journal of biological chemistry. 2006; 281:9287-9296.
28. Rothkamm K, Krüger I, Thompson LH and Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Molecular and cellular biology. 2003; 23:5706-5715.
29. Pâques F and Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiology and molecular biology reviews. 1999; 63:349-404.
30. Wang C and Lees-Miller SP. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. International Journal of Radiation Oncology Biology Physics. 2013; 86:440-449.
31. Hall EJ and Giaccia AJ. (2006). Radiobiology for the Radiologist: (Lippincott Williams & Wilkins).
32. Wouters BG and Begg AC. Irradiation-induced damage and the DNA damage response. Basic Clinical Radiobiology (MC Joiner and A van der Kogel, Eds). 2009:11-26.
33. Alagpulinsa DA, Ayyadevara S and Shmookler Reis RJ. A Small-Molecule Inhibitor of RAD51 Reduces Homologous Recombination and Sensitizes Multiple Myeloma Cells to Doxorubicin. Frontiers in oncology. 2014; 4:289.
34. Ladiges W. Mouse models of XRCC1 DNA repair polymorphisms and cancer. Oncogene. 2006; 25:1612-1619.
35. Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA and Wilson SH. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell research. 2008; 18:48-63.