
INTRODUCTION
Congenital Vertebral Malformations(CVM) represent a group of serious birth defects, which may present as congenital scoliosis, kyphosis, Klippel Feil syndrome(OMIM 118100) [1] (short neck and low posterior hairline in association with cervical vertebral fusion) and may occur in conjunction with other birth defects or as part of an underlying genetic syndrome. The prevalence of CVM is approximately 0.5-1.0 per 1000 persons [2]. Some patients’ lack of overt deformity may lead to a delayed or missed diagnosis, thus the actual proportion of CVM may be greater [3]. Other syndromes associated with CVM include: Alagille syndrome (OMIM 118450: intrahepatic cholestatis, pulmonic stenosis, embryotoxon and butterfly vertebral anomaly in association with distinct facial features) [4], spondylocostal dysostosis (OMIM 122600 : contiguous vertebral malformations throughout the spine with a characteristic “pebble beach sign” in conjunction with rib malformations)(SCD) [5, 6], VACTERL syndrome [7] (OMIM 314390: V = vertebral malformations; A = anal abnormalities, C = cardiac malformations; TE = tracheoesophageal fistula and L = Limb malformations), Goldenhar syndrome(OMIM 164210 ) [8, 9] and spondylothoracic dystrophy (STD-vertebral malformations with ribs emanating from a common point of origin) [10, 11] and others reviewed in.
CVMs occur as a result in altered development of the paraxial mesoderm, somites or axial skeleton [12]. In vertebrate embryogenesis, the paraxial mesoderm lies adjacent to the neural tube and develops from the anterior portion of the embryo into somites during a specific process which is called somitogenesis. This process is mainly regulated by WNT, FGF and Notch signaling pathways. Each somite is later subdivided into the ventromedial sclerotome (which the vertebral body is derived from) and the dorsolateral dermomyotome (from which body skeletal muscles and dorsal dermis arise). Any defect of paraxial mesoderm or somite formation may contribute to CVM [13, 14]. CVM represent a complex condition with multiple causes which remain to be elucidated.
THE ESSENTIAL ROLE OF TBX6 IN SOMITOGENESIS
TBX6 gene is known as T-box 6, a member of the T-box family, and encodes a transcription factor which plays an important role in the regulation of development process [15]. TBX6 has been localized to 16p11.2, with a 6,095bp in size, and contains 8 exons. Tbx6, the TBX6 homologous gene, is located in the chromosome 7 in mouse genome [15]. In the zebrafish genome, tbx24 gene in chromosome 12 is suggested to be the homologous gene with mice Tbx6 and human TBX6 [16].
Tbx6 gene codes for a 1.9-kb transcript which could be detected in the embryonic part of the egg cylinder at 7.0 days post coitus (dpc) [17]. The Tbx6 transcripts are in a stripe which is equivalent to the early primitive streak. At 7.5 dpc, the expression of Tbx6 was detected in the primitive streak, extending caudally along the allantois from the node to the base as well as in the paraxial mesoderm surrounding the streak. At 8.5 dpc, the expression of Tbx6 transcripts was identified in the presomitic mesoderm (PSM) of the tail portion (unsegmented), surrounding the caudal end of the neural plate. The expression of Tbx6 in tailbud was positive until 12.5 dpc and become negative after 13.5 dpc. Thus, the expression of Tbx6 in the primitive streak, tailbud and PSM has an important influence on the development process. Additionally, Tbx6 mutation affects the differentiation of paraxial mesoderm [18]. Tbx6 gene was mutated through homologous recombination in an embryonic stem, which deleted the initiating methionine of Tbx6 and established the mutant allele Tbx6tm1Pa. The Tbx6/ Tbx6tm1Pa were viable, and the offspring were detected without obvious abnormalities as well as homozygotes. The histological segmentation abnormalities were detected firstly at embryonic day of development (E)8.5. At E9.5 and E10.5, an enlarged tail bud was observed that was full of large numbers of undifferentiated mesenchymal cells. In the expanded tail bud and abnormal somites, three mesoderm markers, Delta-like gene 1 (Dll1), paraxis and Mox-1 remained negative. These observations supported the important role Tbx6 plays during somitogenesis.
Sex determining region Y-box 2 (Sox2) regulated by Tbx6 is essential for the specification of paraxial mesoderm from the axial stem cells [19]. The enhancer N1 of Sox2 is activated in the caudal lateral epiblast (CLE), to maintain the cells in the superficial layer of the neural plate sustaining the activation of N1 and Sox2 expression. The cells which developed into mesoderm activate Tbx6 to turn off enhancer N1 before the migration into the paraxial mesoderm. In contrast to this, Tbx6 mutants display persistence of enhancer N1 activity in the paraxial mesoderm, eliciting ectopic Sox2 activation which results in transformation of the paraxial mesoderm into neural tube. Introduction of the N1-specific deletion mutation into Tbx6 mutants prevented the development of neural tube formation due to the prevention of ectopic Sox2 activation in the mesodermal compartment, indicating that Tbx6 regulated Sox2 through enhancer N1. Additionally, Tbx6-dependent repression of Wnt3a was also involved in this process. After the paraxial mesoderm-specific misexpression of a Sox2 transgene, ectopic development of neural tubes was formed in the wild type embryos. This is in consistent with the findings that Wnt signaling pathway regulated the differentiation of paraxial mesoderm through the axial stem cells [20].
MECHANISM OF THE SOMITOGENESIS AND THE CLOCK-WAVEFRONT MODEL
Embryonic development of somite is regulated by a variety of factors. The most widely accepted model of somitogenesis is the clock-wavefront model [21, 22]. Cooke et al [23] proposed the clock and the wavefront model in 1976 for the first time. Pourquie et al [24] further verified the existence of segmentation clock and identified Notch signaling pathway which plays an important role in the formation of somites. Expression of cyclic genes is periodic to form one wave of expression passes through the PSM during the formation of one somite [25-28]. Notch signaling is essential for the separation of intermingled PSM cells belonging to adjacent segments and for intrasomitic anterior-posterior patterning [29-31]. In mice and zebrafish, mutations of Notch pathway genes may lead to the loss of periodic expression patterns. Some mutations in Notch pathway components will lead to defects in somitogenesis, the expression of cyclic genes [27, 28, 32], as well as syndromes contain CVM such as spondylocostal dysostosis in humans [33-35]. Disruption of WNT signaling also affects somitogenesis, whereas cyclic Axin2 expression is maintained when Notch signaling is impaired [36]. Fgf8 is proposed to encode wavefront activity because experimental manipulation of Fgf8 levels caused corresponding shifts in the position of the determination front in cultured chick and zebrafish embryos [37, 38]. FGF4 and FGF8 comprise the wavefront activity in the process of somitogenesis [39].
In the clock-wavefront model, the PSM progressively segmented into repetitive somites, which was driven by the action of Notch signal pathways. Segmentation clock and the wavefront are consisted of a series of related genes and signaling pathway products. The segmentation clock receives oscillatory expression of the segmentation HAIY1 gene and a series of oscillators, with several genes expressed periodically representing the “clock” portion of model. The wavefront portion of the model corresponds to mRNA regulated FGF and WNT gradient. In the paraxial mesoderm, the formation of segmentation clock depends on both the periodic expression of related genes, and genes having clock topologies acting though negative feedback loop mechanisms [14]. For instance, in the Wnt pathway, Wnt3a stimulates the expression of downstream β-cat, which actives the expression of the downstream the scaffolding protein (AXIN2) and the soluble inhibitor Dickkopf-related protein 1(DKK1). Both the AXIN2 and DKK1 proteins are negative-feedback inhibitors of β-cat, forming a cyclical change [40, 41]. Similarly, FGF4 and FGF8 genes in the FGF pathway stimulate the expression of phosphorylated ERK (pERK) as well. This initiated the expression of downstream genes that encoded dual specificity protein phosphatase 4 (DUSP4), dual specificity protein phosphatase 6 (DUSP6) and Sprouty 2 (SPRY2), which are negative-feedback inhibitors of the FGF pathway [42]. In the Notch pathway, activation of the Notch ligand Delta-like gene 1 (DLL1) results in expression of the downstream transcriptional effector Notch intracellular domain (NICD), thereby activing the expression of Notch targets lunatic fringe (LFNG) and Notch-regulated ankyrin repeat protein (NRARP), which are negative-feedback inhibitors to NICD [43, 44]. In addition, pERK and NICD have been also reported in the stimulation of HES7 gene which is a negative-feedback inhibitor of pERK and NICD as well [45, 46].
When the paraxial mesoderm cells receive a clock signal, MESP2 is activated by NICD (Notch signalling) and TBX6. This action can be repressed by pERK. MESP2 is initially expressed in a restricted area (one segment length) and subsequently with Ripply1/2 expresses in the posterior half region, thus defining future segment boundaries [47]. The downstream target gene-RIPPLY2 is activated, which is thought to be a negative-feedback inhibitor of MESP2 and TBX6. The process contributes to the definition of the anterior boundary of the newly-formed segment [48]. Inactivation of MESP1 and MESP2 results in failure of paraxial mesoderm formation, indicating their importance in somitogenesis. The main components are summarized as follows (Figure 1) [14, 41, 49-53].
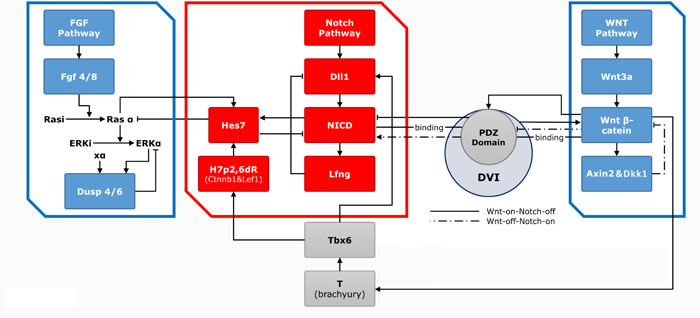
Figure 1: The three main pathways with related genes and effectors in the somitogenesis. The clock (red frame)-wavefront (blue frame) model and components are shown. The clock is mainly consisted of Notch pathway and series of genes and effectors. Dll1 and its downstream NICD and Lfng expressed periodically through a feed-back loop to form the “clock” model. FGF and WNT signal gradients constitute the wavefront. In the Wnt pathway, Wnt3a induces the expression of downstream β-cat and the Axin2. The AXIN2 and DKK1 proteins are both negative-feedback inhibitors of β-cat, thereby forming a cyclical change. Wnt3a induces the expression of Tbx6, which further activates the expression of Dll1 and connects with NICD through Hes7, thus establishing communication rapport of Notch and WNT pathway. In the FGF pathway, Fgf4 and Ff8 genes induce the expression of pERK, which further prompts the expression of downstream Dusp4 and Dusp6. The FGF signal oscillation is also based on a feed-back mechanism.
THE IMPORTANCE OF TBX6 AND ITS RELATED GENES IN THE CLOCK-WAVEFRONT MODEL
It has been reported that the interaction between TBX6 and the clock-wavefront model-related genes or TBX6 itself will result in abnormal formation of somites, contributing to CVM [14, 41, 54].
In the first stage of PSM formation, the Wnt3a, FGF receptor 1(Fgfr1) and the T-box transcription factor brachyury(T) play key role during the epithelial-mesenchymal transition. Any mutants lacking of these factors would lead to an early arrest of trunk elongation at approximately the 8- to 12-somite stage [55-58]. Later, the mesogenin 1(Msgn1) plays a key role in the process of PSM maturation. As is mentioned in early papers, T is a target gene of WNT signaling [59, 60], which is essential for the early stage of mesoderm formation and migration [61], and acts its upstream gene Tbx6 [62]. The Msgn1 is a direct target of the synergism of WNT signaling and Tbx6 [63]. The Msgn1 expression is detected only in a subset of mesodermal cells with the levels of Wnt3a messenger RNA are reduced in the tailbud of such embryos at E9.5-E11.5 [63, 64]. Using the Tbx6-knockout allele described previously [18], the expression of Msgn1 is detected strongly downregulated in Tbx6-null mutant embryos, which indicts that Tbx6 acts upstream of Msgn1 [63]. Msgn1 is described previously that it is essential for the specification and maturation of the paraxial mesoderm downstream of Wnt3a [65, 66]. The Msgn1−/− mutant was found to contribute to a kinked neural tube, lack of paraxial mesoderm and an expanded tail region. The Msgn1−/− and Tbx6−/− double mutants also lead to a severely kinked neural tube, loss of posterior somites, severely aberrant anterior somites, loss of paraxial mesoderm and an expanded tail region [67]. These findings suggest that Tbx6 is in a close association with Msgn1 during the somitogenesis, and any mutant of the two factors will contribute to severe CVM.
Wnt3a connects its downstream AXIN2 and DVI protein that plays a key role in vertebral symmetry [52]. The interaction between WNT3a and TBX6 may affect the development of somites during embryogenesis. It has been reported that both Wnt3a and Tbx6 mutants could lead to the formation of ectopic neural tubes concomitant with the absence of paraxial mesoderm or posterior somites [17, 68]. The Wnt3a, Msgn1 and Tbx6 mutant mice possessed a complete absence of paraxial mesoderm posterior to 6/7 somite, and abnormal morphogenesis of the 1-6/7 somites [67]. At the headfold stage, Tbx6 expressed predominantly the wings of nascent mesoderm, and in a small portion of epiblast cells at the site of cell ingression where it overlapped with Wnt3a, indicating the close interaction during the somitogenesis. Of the three mutants, the Tbx6−/− mutant was found to contribute to the most severe somite segmentation defect, with an aberrant anterior somites, an absence of posterior somites, and a slightly kinked neural tube as well as an expanded tail region. The Wnt3a−/− mutant showed the deficiency of posterior somites and a reduced tail region. The Wnt3a−/− and Tbx6−/− double mutants also lead to the most severe phenotypes (somites segmentation defects). This includes severely kinked neural tube, no notochord, a reduced tail region as well as lacking most anterior somites, which were small and lacked the stereotypical organization.
Dll1 is synergistically regulated by WNT signaling and Tbx6, and is essential for the activation of Notch gene homologue 1(Notch1) in the PSM [62, 69]. Dll1 is a downstream target gene of Tbx6 in the paraxial mesoderm [51]. Rib-vertebrae is a hypomorphic allele of the Tbx6 gene [70], and Dll1 expression is described previously reduced significantly in the PSM of rib-vertebrae mutant embryos [71]. Hofmann et al [72] identified that Dll1 expression in Tbx6-null mutant embryos was severely down-regulated on E8, and Dll1 transcripts were not detected on E8.5. Dll1 and Dll3, tow Notch ligands, are known to be involved in R-C(rostral and caudal) somite patterning [73, 74]. White et al [75] found that although Dll1 expression is not detected in the tail mesoderm as normally of the Tbx6-null embryos, Dll1 expression is restore in the Tbx6tm1Pa/Tbx6tm1Pa Tg46/+( a permanent transgenic line used for rescue) embryos. These findings suggest the close relationship between Tbx6 and Dll1 during somotogenesis, any mutant in the factors may lead to severe CVM.
HES7 is a key gene in somitogenesis because of its connection with all the three major pathways [76]. By gradually creating smaller promoter reporters, the X-gal staining of the PSM was lost, confirming that 400 bp Hes7 promoter region plays a key role in Hes7 promoter activity [50]. This 400 bp region is strongly conserved in humans and comprises binding sites for remarkable transcription factors such as Lef1 (Wnt pathway) [77], Ets (FGF pathway) [78] and Tbx6 [18]. Cooperation between Tbx6 and the Wnt pathway was hypothesized to upregulate Hes7, while Tbx6 alone did not. The findings suggest a closely connection between Tbx6 and Hes7 in the somitogenesis, and contribute to the occurrence of severe CVM.
In the somitogenesis, somite segmentation is a key step to form the normal spine. TBX6 and MESP2 (activated by TBX6) and its downstream RIPPLY2 are one of the pivotal factors in somite segmentation [14]. Takahashi et al [79] compared Ripply1 null mutant, Ripply2 null mutant and Ripply1/2 double-null mutant mice with wild-type mice, to observe the region and quantity of Tbx6, Mesp2 and RipplyY1/2 expression during somite segmentation. In wild-type mice, Mesp2 firstly expresses in a restricted area (a one-somite-length fashion), later with the Ripply1/2 expression in the posterior half region. Under the interaction among the three factors, Mesp2 expression is dependent on the Tbx6 transcription factor and Notch signaling [80]. Mesp2 leads to ubiquitin-dependent degradation of Tbx6 proteins [81], and along with Ripply1/2 contribute to degradation of Tbx6 and Mesp2. Thus, Mesp2 expresses in the anterior top of the Tbx6 expression domain and form a next segmental border through the degradation of Tbx6. The Tbx6 and Mesp2 domain was expanded in Ripply1 or Ripply2 null mutant mice, while the posterior half region suffered segmentation defects. The segmentation defects become more severe in Ripply1/2 double-null mutant. This process is summarized in Figure 2 [79, 81, 82].
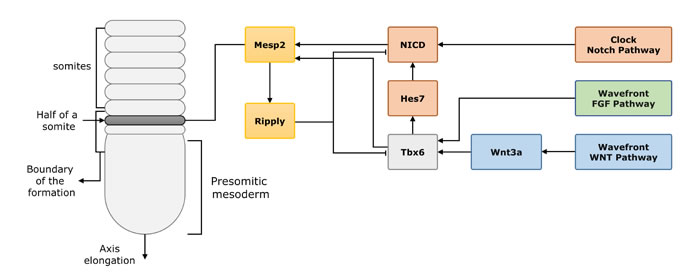
Figure 2: The process of vertebrate segmentation and the interaction of the related genes. In the process of vertebrate segmentation, Tbx6, Mesp2 and downstream Ripply1/2 are important genes involved in somite segmentation. Tbx6 stimulated by the Wnt and FGF pathway signaling, later induces the expression of NICD (also induced by Notch signal) and Mesp2. Mesp2 initially expressed in a restricted area (one segment length), later with the Ripply1/2, (a feed-back inhibitor to Tbx6 and Mesp2) expression in the posterior half region, which defines the future segment boundaries.
CLINICAL REPORTS OF TBX6 MUTATIONS CONTRIBUTING TO CVM
Several studies have reported that mutations in TBX6, such as single-nucleotide variation (SNV), may contribute to CVM (Table 1).
Ghebranious et al [83] hypothesized that mutations in T and/or TBX6 gene may lead to the occurrence of CVM. They reported fifty CVM persons, and sequenced the complete T and TBX6 coding regions, splice junctions, and 500bp of the promoter region. Three unrelated patients harbored a same c.1013C > T substitution, resulting in a predicted Ala338Val missense alteration in exon 8 of the T gene while no variation was identified inTBX6 sequence. Duncan B. Sparrow et al [84] used whole-exome sequencing to investigate a three-generation Macedonian family who is consisted of three SCD-phenotyped individuals. In the five members of the family, three of whom had clinical features and radiographic evidence of SCD, and two were clinically unaffected. The three affected individuals were confirmed to be heterozygous for the TBX6 mutation, although unaffected individuals were homozygous wild-type indicating altered penetrance for the TBX6 mutation. It is believed that the sequence variant disrupts the natural stop codon (c.1311A.T) and results in the addition of 81 nonsense amino acids to the C-terminus (p.*437Cext*81).
It has been reported single-nucleotide polymorphisms (SNPs) of TBX6 were associated with CVM. Qi et al [87] genotyped two known SNPs in TBX6 among 254 Chinese Han subjects(comprising of 127 congenital scoliosis patients and 127 controls). For the single SNP analysis, rs2289292 (SNP1, exon 8) and rs3809624 (SNP2, 5’ untranslated region) are significantly different between cases and controls (P = 0.017 and 0.033 respectively). The haplotypic analysis showed a significant association between SNP1/SNP2 and CS cases (P = 0.017), with the G-A haplotype more frequently observed in controls (odds ratio, 0.71; 95% confidence interval, 0.51-0.99).
Recently, there has been a focus on determining the relationship between copy number variations (CNVs) and CVM. Several articles reported CNVs in 16p11.2 region which harbors the TBX6 gene may contribute to CVM. Shimojima et al [85] reported a 3-year-old boy with developmental delay, inguinal hernia, hemivertebrae of T10, T12, and L3, a missing right twelfth rib and hypoplasia of the left twelfth rib. The patient had a 593-kb interstitial deletion of 16p11.2 and the mother had the same deletion from the results of array comparative genome hybridization (aCGH). Al-Kateb et al [86] analyzed radiologic data obtained from 10 patients with 16p11.2 CNV (nine with deletions and one with duplication), and found that all the patients are affected with scoliosis, eight of them having congenital scoliosis (present at birth, a combination of scoliosis and vertebral deformities such as incomplete formation of vertebrae, failure of separation of vertebrae and a mixed one) and the remaining 2 having idiopathic scoliosis(cause unknown, sub-classified as infantile, juvenile, adolescent, or adult, according to when onset occurred, patients only appear as scoliosis). They additionally reviewed 5 patients reported previously with 16p11.2 rearrangement and similar skeletal abnormalities, concluding that two of them were affected with congenital scoliosis while the others had idiopathic scoliosis. Although there have been many reports on the association of 16p11.2 CNV with CVM, the exact mechanism was still unclear. Recently, Wu et al [88] elaborated the TBX6 null variants and a TBX6 common hypomorphic allele may contribute to CS with a compound inheritance model. They firstly enrolled 161 Han Chinese persons with sporadic congenital scoliosis, and 166 Han Chinese as controls. The CNV analysis identified 17 heterozygous TBX6 null mutations in the 161 persons. This included 12 instances of a 16p11.2 deletion affecting TBX6 and single-nucleotide variants (1 nonsense and 4 frame-shift mutations), and no such mutation was identified in the control group. Then they took analysis of 2 pedigrees, of which several unaffected family members showed a 16p11.2 deletion, and hypothesized that a heterozygous TBX6 null mutation is insufficient to cause congenital scoliosis. They subsequently identified a common TBX6 haplotype as the second risk allele in all 17 carriers of TBX6 null mutations. The following replication studies in both congenital scoliosis cohorts and 16p11.2 CNV cohorts confirmed these findings.
Table 1: Clinical reports of mutants of TBX6 or T gene contribute to CVM.
Authors |
Year |
Mutation types |
Variants |
Number of patients with mutations/total Number of patients |
Vertebral Malformations |
Other clinical manifestations |
|
Ghebranious et al.[83] |
2008 |
missense |
T gene: c.1013C>T p.Ala338Val |
3/50 |
Hemivertebrae, Butterfly vertebrae, Vertebrae fusion, C4 hypoplasia, Absent of S3-S5 |
Aortic stenosis, Bicuspid valve, Abnormalities of 1st and 2nd rib, Multiple left-sided rib fusions, Adducent left thumb, Conus terminates at L1 |
|
Duncan B. Sparrow et al.[84] |
2013 |
stoploss |
TBX6 gene: c.1311A>T p.*437Cysext |
3/5 |
Scoliosis, Hemivertebrae, Fused vertebral blocks vertebral blocks |
Short stature |
|
Shimojima et al.[85] |
2008 |
CNV |
a 593-kb interstitial deletion of 16p11.2 |
2/3 |
Hemivertebrae of T10, T12, and L3* |
A missing right twelfth rib, hypoplasia of the left twelfth rib |
|
Al-Kateb et al.[86] |
2014 |
CNV |
deletion and duplication of 16p11.2 region |
15 |
Congenital scoliosis, Idiopathic Scoliosis |
Autism,spectrum, disorders,seizures, Behavioral abnormalities,developmental delay |
|
Qi Fei et al.[87] |
2010 |
SNP |
rs2289292 and rs3809624 |
127 |
Congenital scoliosis |
Deformities in syndrome associated with CVM |
|
Wu et al.[88] |
2015 |
CNV |
deletion of 16p11.2 region + T-C-A# |
17/237 |
Butterfly, Hemivertebrae, thoracic wedge vertebra |
Rib abnormalities(missing ribs and bifurcation of ribs) |
|
frameshift |
TBX6: c.1169_1170insC + T-C-A |
1/237 |
Left L2 hemivertebra, T8 butterfly vertebra |
None |
|||
frameshift |
TBX6: c.1250_1251insT + T-C-A |
1/237 |
Left lumbar hemivertebra, atrial septal defect |
Missing bilateral 12th ribs |
|||
frameshift |
TBX6: c.266_267insC + T-C-A |
1/237 |
Left hemivertebra between T12 and L1 |
Missing right 12h rib |
|||
frameshift |
TBX6: c.704_705insG + T-C-A |
1/237 |
Left T12 hemivertebra |
Missing right 12th rib |
|||
frameshift |
TBX6: c.1179_1180delAG + T-C-A |
1/237 |
Right T12 hemivertebra |
Missing left 12th rib |
|||
nonsense |
TBX6: c.844C>T (p.R282X) + T-C-A |
1/237 |
T1 and L3 butterfly vertebrae, Right T4 hemivertebra, Left T7 hemivertebra |
None |
|||
CNV: copy number variation; SNP: Single nucleotide polymorphism
#T-C-A (rs2289292, rs3809624 and rs3809627) is risk haplotype in other allele of TBX6
*The proband and his mother share the same deletion, while his mother is a normal phenotype. The “Vertebral Malformations” and “Other clinical manifestations” refer to the proband’s deformities.
PROSPECT
CVM is a polygenic disease. In the somite development process, three major signaling pathways (Notch, FGF and Wnt pathway) make up the “segmentation clock - wavefront” model and many related genes are associated with TBX6. Therefore, mutations of TBX6 and its interrelated genes may lead to the defects in somitogenesis resulting in CVM.
To date, the TBX6 compound inheritance model shed new lights on the maximum ratio of CVM. The TBX6 related mechanisms need further elucidation to provide more etiology explanation of CVM and potential targets for CVM prevention and intervention clinically.
Although clock-wavefront is the most popular model in studying somitogenesis, there are still other models. Further studies needed to be carried out to check whether TBX6 still plays a key role during somitogenesis, and how it works to cause CVM. Besides, it is confirmed that signaling network underlying the clock is markedly vary among species [89]. Researches for the mechanism of TBX6 lead to CVM are based on animal models at present. However, some animal models do not contain an exact TBX6 homologous gene, such as zebra fish, which has tbx6, tbx6l and tbx16 as the TBX6 homologous genes [90]. More researches needed to be carried out to verify whether results from these animal models are the same in human.
Currently, the most accepted methods to identify genetic etiology of CVM are based on DNA analysis of SNP arrays or the next generation sequencing technology. However, there is no evidence whether DNA extracted from peripheral blood cells could restore the scene of vertebral development during somitogenesis. To achieve this, a much deeper research analyzing the deformed and normal tissues could be applied.
Actually, CVM is a complex disease which means several genes may involve in the pathogenesis. Indeed TBX6 plays an important role during the somitogenesis, we cannot ignore roles of other genes and environment. For the further studies, we are also looking forward to see more findings revealed the roles of other genes and environment factors in the contribution to CVM.
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interests.
GRANT SUPPORT
This work was supported by National Natural Science Foundation of China (81501852, 81472046, 81271942, 81130034, 81472045), the Distinguished Young Scholars of Peking Union Medical College Hospital (JQ201506), Beijing nova program of science and technology (2016), and The Central Level Public Interest Program for Scientific Research Institute (No.13, 2015).
References
1. Tawk RG, Ondra SL, Jorge AM, Morrison T and Ganju A. Hypersegmentation, Klippel-Feil syndrome, and hemivertebra in a scoliotic patient. J Am Coll Surg. 2002; 195(4):570-571.
2. Giampietro PF, Dunwoodie SL, Kusumi K, Pourquie O, Tassy O, Offiah AC, Cornier AS, Alman BA, Blank RD, Raggio CL, Glurich I and Turnpenny PD. Progress in the understanding of the genetic etiology of vertebral segmentation disorders in humans. Ann N Y Acad Sci. 2009; 1151:38-67.
3. Brand MC. Examination of the newborn with congenital scoliosis: focus on the physical. Adv Neonatal Care. 2008; 8(5):265-273, 274-275.
4. Dayem-Quere M, Giuliano F, Wagner-Mahler K, Massol C, Crouzet-Ozenda L, Lambert JC and Karmous-Benailly H. Delineation of a region responsible for panhypopituitarism in 20p11.2. Am J Med Genet A. 2013; 161A(7):1547-1554.
5. Bulman MP, Kusumi K, Frayling TM, McKeown C, Garrett C, Lander ES, Krumlauf R, Hattersley AT, Ellard S and Turnpenny PD. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet. 2000; 24(4):438-441.
6. Gucev ZS, Tasic V, Pop-Jordanova N, Sparrow DB, Dunwoodie SL, Ellard S, Young E and Turnpenny PD. Autosomal dominant spondylocostal dysostosis in three generations of a Macedonian family: Negative mutation analysis of DLL3, MESP2, HES7, and LFNG. Am J Med Genet A. 2010; 152A(6):1378-1382.
7. Avcu S, Akgun C, Temel H, Arslan S, Akbayram S and Unal O. Report of a girl with vacterl syndrome and right pulmonary agenesis. Genet Couns. 2009; 20(4):379-383.
8. Al KA, Ben CF, Ganger R, Klaushofer K and Grill F. Distinctive spine abnormalities in patients with Goldenhar syndrome: tomographic assessment. Eur Spine J. 2015; 24(3):594-599.
9. Callier P, Faivre L, Thauvin-Robinet C, Marle N, Mosca AL, D’Athis P, Guy J, Masurel-Paulet A, Joly L, Guiraud S, Teyssier JR, Huet F and Mugneret F. Array-CGH in a series of 30 patients with mental retardation, dysmorphic features, and congenital malformations detected an interstitial 1p22.2-p31.1 deletion in a patient with features overlapping the Goldenhar syndrome. Am J Med Genet A. 2008; 146A(16):2109-2115.
10. Cornier AS, Staehling-Hampton K, Delventhal KM, Saga Y, Caubet J, Sasaki N, Ellard S, Young E, Ramirez N, Carlo SE, Torres J, Emans JB, Turnpenny PD and Pourquié O. Mutations in the MESP2 Gene Cause Spondylothoracic Dysostosis/Jarcho-Levin Syndrome. The American Journal of Human Genetics. 2008; 82(6):1334-1341.
11. Giampietro PF. Genetic aspects of congenital and idiopathic scoliosis. Scientifica (Cairo). 2012; 2012:152365.
12. Giampietro PF, Raggio CL, Blank RD, McCarty C, Broeckel U and Pickart MA. Clinical, Genetic and Environmental Factors Associated with Congenital Vertebral Malformations. Molecular Syndromology. 2012.
13. McMaster MJ. Congenital scoliosis caused by a unilateral failure of vertebral segmentation with contralateral hemivertebrae. Spine (Phila Pa 1976). 1998; 23(9):998-1005.
14. Hubaud A and Pourquie O. Signalling dynamics in vertebrate segmentation. Nat Rev Mol Cell Biol. 2014; 15(11):709-721.
15. Papapetrou C, Putt W, Fox M and Edwards YH. The human TBX6 gene: cloning and assignment to chromosome 16p11.2. Genomics. 1999; 55(2):238-241.
16. Ahn D, You KH and Kim CH. Evolution of the tbx6/16 subfamily genes in vertebrates: insights from zebrafish. Mol Biol Evol. 2012; 29(12):3959-3983.
17. Chapman DL, Agulnik I, Hancock S, Silver LM and Papaioannou VE. Tbx6, a mouse T-Box gene implicated in paraxial mesoderm formation at gastrulation. Dev Biol. 1996; 180(2):534-542.
18. Chapman DL and Papaioannou VE. Three neural tubes in mouse embryos with mutations in the T-box gene Tbx6. Nature. 1998; 391(6668):695-697.
19. Takemoto T, Uchikawa M, Yoshida M, Bell DM, Lovell-Badge R, Papaioannou VE and Kondoh H. Tbx6-dependent Sox2 regulation determines neural or mesodermal fate in axial stem cells. Nature. 2011; 470(7334):394-398.
20. Martin BL and Kimelman D. Canonical Wnt signaling dynamically controls multiple stem cev Cell. 2012; 22(1):223-232.
21. Baker RE, Schnell S and Maini PK. A mathematical investigation of a Clock and Wavefront model for somitogenesis. J Math Biol. 2006; 52(4):458-482.
22. Baker RE, Schnell S and Maini PK. A clock and wavefront mechanism for somite formation. Dev Biol. 2006; 293(1):116-126.
23. Cooke J and Zeeman EC. A clock and wavefront model for control of the number of repeated structures during animal morphogenesis. J Theor Biol. 1976; 58(2):455-476.
24. Pourquie O. The vertebrate segmentation clock. J ANAT. 2001; 199(Pt 1-2):169-175.
25. Palmeirim I, Henrique D, Ish-Horowicz D and Pourquie O. Avian hairy gene expression identifies a molecular clock linked to vertebrate segmentation and somitogenesis. Cell. 1997; 91(5):639-648.
26. Forsberg H, Crozet F and Brown NA. Waves of mouse Lunatic fringe expression, in four-hour cycles at two-hour intervals, precede somite boundary formation. Curr Biol. 1998; 8(18):1027-1030.
27. Jiang YJ, Aerne BL, Smithers L, Haddon C, Ish-Horowicz D and Lewis J. Notch signalling and the synchronization of the somite segmentation clock. Nature. 2000; 408(6811):475-479.
28. Jouve C, Palmeirim I, Henrique D, Beckers J, Gossler A, Ish-Horowicz D and Pourquie O. Notch signalling is required for cyclic expression of the hairy-like gene HES1 in the presomitic mesoderm. Development. 2000; 127(7):1421-1429.
29. Conlon RA, Reaume AG and Rossant J. Notch1 is required for the coordinate segmentation of somites. Development. 1995; 121(5):1533-1545.
30. Hrabe DAM, McIntyre JN and Gossler A. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature. 1997; 386(6626):717-721.
31. Sato Y, Yasuda K and Takahashi Y. Morphological boundary forms by a novel inductive event mediated by Lunatic fringe and Notch during somitic segmentation. Development. 2002; 129(15):3633-3644.
32. Dunwoodie SL, Clements M, Sparrow DB, Sa X, Conlon RA and Beddington RS. Axial skeletal defects caused by mutation in the spondylocostal dysplasia/pudgy gene Dll3 are associated with disruption of the segmentation clock within the presomitic mesoderm. Development. 2002; 129(7):1795-1806.
33. Whittock NV, Sparrow DB, Wouters MA, Sillence D, Ellard S, Dunwoodie SL and Turnpenny PD. Mutated MESP2 causes spondylocostal dysostosis in humans. AM J Hum Genet. 2004; 74(6):1249-1254.
34. Sparrow DB, Chapman G, Wouters MA, Whittock NV, Ellard S, Fatkin D, Turnpenny PD, Kusumi K, Sillence D and Dunwoodie SL. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. J Hum Genet. 2006; 78(1):28-37.
35. Bulman MP, Kusumi K, Frayling TM, McKeown C, Garrett C, Lander ES, Krumlauf R, Hattersley AT, Ellard S and Turnpenny PD. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat Genet. 2000; 24(4):438-441.
36. Aulehla A and Herrmann BG. Segmentation in vertebrates: clock and gradient finally joined. Genes Dev. 2004; 18(17):2060-2067.
37. Sawada A, Shinya M, Jiang YJ, Kawakami A, Kuroiwa A and Takeda H. Fgf/MAPK signalling is a crucial positional cue in somite boundary formation. Development. 2001; 128(23):4873-4880.
38. Dubrulle J, McGrew MJ and Pourquie O. FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell. 2001; 106(2):219-232.
39. Naiche LA, Holder N and Lewandoski M. FGF4 and FGF8 comprise the wavefront activity that controls somitogenesis. Proceedings of the National Academy of Sciences. 2011; 108(10):4018-4023.
40. Dequeant ML, Glynn E, Gaudenz K, Wahl M, Chen J, Mushegian A and Pourquie O. A complex oscillating network of signaling genes underlies the mouse segmentation clock. Seience. 2006; 314(5805):1595-1598.
41. Aulehla A, Wehrle C, Brand-Saberi B, Kemler R, Gossler A, Kanzler B and Herrmann BG. Wnt3a plays a major role in the segmentation clock controlling somitogenesis. DEV Cell. 2003; 4(3):395-406.
42. Niwa H. How is pluripotency determined and maintained? Development. 2007; 134(4):635-646.
43. Huppert SS, Ilagan MXG, De Strooper B and Kopan R. Analysis of Notch Function in Presomitic Mesoderm Suggests a γ-Secretase-Independent Role for Presenilins in Somite Differentiation. Dev Cell. 2005; 8(5):677-688.
44. Morimoto M, Takahashi Y, Endo M and Saga Y. The Mesp2 transcription factor establishes segmental borders by suppressing Notch activity. Nature. 2005; 435(7040):354-359.
45. Bessho Y. Periodic repression by the bHLH factor Hes7 is an essential mechanism for the somite segmentation clock. Gene Dev. 2003; 17(12):1451-1456.
46. Giudicelli F, Ozbudak EM, Wright GJ and Lewis J. Setting the tempo in development: an investigation of the zebrafish somite clock mechanism. Plos Biol. 2007; 5(6):e150.
47. Takahashi Y, Koizumi K, Takagi A, Kitajima S, Inoue T, Koseki H and Saga Y. Mesp2 initiates somite segmentation through the Notch signalling pathway. Nat Genet. 2000; 25(4):390-396.
48. Zhao W, Ajima R, Ninomiya Y and Saga Y. Segmental border is defined by Ripply2-mediated Tbx6 repression independent of Mesp2. Dev Biol. 2015; 400(1):105-117.
49. Goldbeter A and Pourquie O. Modeling the segmentation clock as a network of coupled oscillations in the Notch, Wnt and FGF signaling pathways. J Theor Biol. 2008; 252(3):574-585.
50. Gonzalez A, Manosalva I, Liu T and Kageyama R. Control of Hes7 expression by Tbx6, the Wnt pathway and the chemical Gsk3 inhibitor LiCl in the mouse segmentation clock. Plos One 2013; 8(1):e53323.
51. White PH and Chapman DL. Dll1 is a downstream target of Tbx6 in the paraxial mesoderm. Genesis 2005; 42(3):193-202.
52. Nakaya MA, Biris K, Tsukiyama T, Jaime S, Rawls JA and Yamaguchi TP. Wnt3a links left-right determination with segmentation and anteroposterior axis elongation. Development. 2005; 132(24):5425-5436.
53. Dunty WJ, Biris KK, Chalamalasetty RB, Taketo MM, Lewandoski M and Yamaguchi TP. Wnt3a/beta-catenin signaling controls posterior body development by coordinating mesoderm formation and segmentation. Development. 2008; 135(1):85-94.
54. Hirata H, Bessho Y, Kokubu H, Masamizu Y, Yamada S, Lewis J and Kageyama R. Instability of Hes7 protein is crucial for the somite segmentation clock. Nat Genet. 2004; 36(7):750-754.
55. Ciruna BG, Schwartz L, Harpal K, Yamaguchi TP and Rossant J. Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: a role for FGFR1 in morphogenetic movement through the primitive streak. Development. 1997; 124(14):2829-2841.
56. Kispert A, Koschorz B and Herrmann BG. The T protein encoded by Brachyury is a tissue-specific transcription factor. Embo J. 1995; 14(19):4763-4772.
57. Wilkinson DG, Bhatt S and Herrmann BG. Expression pattern of the mouse T gene and its role in mesoderm formation. Nature. 1990; 343(6259):657-659.
58. Yoshikawa Y, Fujimori T, McMahon AP and Takada S. Evidence that absence of Wnt-3a signaling promotes neuralization instead of paraxial mesoderm development in the mouse. DevBiol. 1997; 183(2):234-242.
59. Arnold SJ, Stappert J, Bauer A, Kispert A, Herrmann BG and Kemler R. Brachyury is a target gene of the Wnt/beta-catenin signaling pathway. Mech Dev. 2000; 91(1-2):249-258.
60. Yamaguchi TP, Takada S, Yoshikawa Y, Wu N and McMahon AP. T (Brachyury) is a direct target of Wnt3a during paraxial mesoderm specification. Genes Dev. 1999; 13(24):3185-3190.
61. Wilson V and Beddington R. Expression of T protein in the primitive streak is necessary and sufficient for posterior mesoderm movement and somite differentiation. Dev Biol. 1997; 192(1):45-58.
62. Hofmann M, Schuster-Gossler K, Watabe-Rudolph M, Aulehla A, Herrmann BG and Gossler A. WNT signaling, in synergy with T/TBX6, controls Notch signaling by regulating Dll1 expression in the presomitic mesoderm of mouse embryos. Genes Dev. 2004; 18(22):2712-2717.
63. Wittler L, Shin EH, Grote P, Kispert A, Beckers A, Gossler A, Werber M and Herrmann BG. Expression of Msgn1 in the presomitic mesoderm is controlled by synergism of WNT signalling and Tbx6. Embo Rep. 2007; 8(8):784-789.
64. Greco TL, Takada S, Newhouse MM, McMahon JA, McMahon AP and Camper SA. Analysis of the vestigial tail mutation demonstrates that Wnt-3a gene dosage regulates mouse axial development. Genes Dev. 1996; 10(3):313-324.
65. Yoon JK, Moon RT and Wold B. The bHLH class protein pMesogenin1 can specify paraxial mesoderm phenotypes. Dev Biol. 2000; 222(2):376-391.
66. Aulehla A, Wehrle C, Brand-Saberi B, Kemler R, Gossler A, Kanzler B and Herrmann BG. Wnt3a plays a major role in the segmentation clock controlling somitogenesis. DEV Cell. 2003; 4(3):395-406.
67. Nowotschin S, Ferrer-Vaquer A, Concepcion D, Papaioannou VE and Hadjantonakis A. Interaction of Wnt3a, Msgn1 and Tbx6 in neural versus paraxial mesoderm lineage commitment and paraxial mesoderm differentiation in the mouse embryo. Dev Biol. 2012; 367(1):1-14.
68. Yoshikawa Y, Fujimori T, McMahon AP and Takada S. Evidence that absence of Wnt-3a signaling promotes neuralization instead of paraxial mesoderm development in the mouse. Dev BiolL. 1997; 183(2):234-242.
69. Galceran J, Sustmann C, Hsu SC, Folberth S and Grosschedl R. LEF1-mediated regulation of Delta-like1 links Wnt and Notch signaling in somitogenesis. Genes Dev. 2004; 18(22):2718-2723.
70. Watabe-Rudolph M, Schlautmann N, Papaioannou VE and Gossler A. The mouse rib-vertebrae mutation is a hypomorphic Tbx6 allele. Mech Dev. 2002; 119(2):251-256.
71. Beckers J, Schlautmann N and Gossler A. The mouse rib-vertebrae mutation disrupts anterior-posterior somite patterning and genetically interacts with a Delta1 null allele. Mech Dev. 2000; 95(1-2):35-46.
72. Hofmann M, Schuster-Gossler K, Watabe-Rudolph M, Aulehla A, Herrmann BG and Gossler A. WNT signaling, in synergy with T/TBX6, controls Notch signaling by regulating Dll1 expression in the presomitic mesoderm of mouse embryos. Genes Dev. 2004; 18(22):2712-2717.
73. Barrantes IB, Elia AJ, Wunsch K, Hrabe DAM, Mak TW, Rossant J, Conlon RA, Gossler A and de la Pompa JL. Interaction between Notch signalling and Lunatic fringe during somite boundary formation in the mouse. CurrBIOL. 1999; 9(9):470-480.
74. Dunwoodie SL, Henrique D, Harrison SM and Beddington RS. Mouse Dll3: a novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development. 1997; 124(16):3065-3076.
75. White PH. Defective somite patterning in mouse embryos with reduced levels of Tbx6. Development. 2003; 130(8):1681-1690.
76. Pourquie O. Vertebrate segmentation: from cyclic gene networks to scoliosis. Cell. 2011; 145(5):650-663.
77. Galceran J, Hsu SC and Grosschedl R. Rescue of a Wnt mutation by an activated form of LEF-1: regulation of maintenance but not initiation of Brachyury expression. Proc Natl Acad Sci U S A. 2001; 98(15):8668-8673.
78. Znosko WA, Yu S, Thomas K, Molina GA, Li C, Tsang W, Dawid IB, Moon AM and Tsang M. Overlapping functions of Pea3 ETS transcription factors in FGF signaling during zebrafish development. DEV BIOL. 2010; 342(1):11-25.
79. Takahashi J, Ohbayashi A, Oginuma M, Saito D, Mochizuki A, Saga Y and Takada S. Analysis of Ripply1/2-deficient mouse embryos reveals a mechanism underlying the rostro-caudal patterning within a somite. Dev Biol. 2010; 342(2):134-145.
80. Yasuhiko Y, Haraguchi S, Kitajima S, Takahashi Y, Kanno J and Saga Y. Tbx6-mediated Notch signaling controls somite-specific Mesp2 expression. Proc Natl Acad Sci U S A. 2006; 103(10):3651-3656.
81. Oginuma M, Niwa Y, Chapman DL and Saga Y. Mesp2 and Tbx6 cooperatively create periodic patterns coupled with the clock machinery during mouse somitogenesis. Development. 2008; 135(15):2555-2562.
82. Zhao W, Ajima R, Ninomiya Y and Saga Y. Segmental border is defined by Ripply2-mediated Tbx6 repression independent of Mesp2. Dev Biol. 2015; 400(1):105-117.
83. Ghebranious N, Blank RD, Raggio CL, Staubli J, McPherson E, Ivacic L, Rasmussen K, Jacobsen FS, Faciszewski T, Burmester JK, Pauli RM, Boachie-Adjei O, Glurich I and Giampietro PF. A missense T (Brachyury) mutation contributes to vertebral malformations. J Bone Miner Res. 2008; 23(10):1576-1583.
84. Sparrow DB, McInerney-Leo A, Gucev ZS, Gardiner B, Marshall M, Leo PJ, Chapman DL, Tasic V, Shishko A, Brown MA, Duncan EL and Dunwoodie SL. Autosomal dominant spondylocostal dysostosis is caused by mutation in TBX6. Hum Mol Genet. 2013; 22(8):1625-1631.
85. Shimojima K, Inoue T, Fujii Y, Ohno K and Yamamoto T. A familial 593-kb microdeletion of 16p11.2 associated with mental retardation and hemivertebrae. Eur J Med Genet. 2009; 52(6):433-435.
86. Al-Kateb H, Khanna G, Filges I, Hauser N, Grange DK, Shen J, Smyser CD, Kulkarni S and Shinawi M. Scoliosis and vertebral anomalies: additional abnormal phenotypes associated with chromosome 16p11.2 rearrangement. AM J Med Genet. 2014; 164A(5):1118-1126.
87. Fei Q, Wu Z, Wang H, Zhou X, Wang N, Ding Y, Wang Y and Qiu G. The association analysis of TBX6 polymorphism with susceptibility to congenital scoliosis in a Chinese Han population. Spine (Phila Pa 1976). 2010; 35(9):983-988.
88. Wu N, Ming X, Xiao J, Wu Z, Chen X, Shinawi M, Shen Y, Yu G, Liu J, Xie H, Gucev ZS, Liu S, Yang N, Al-Kateb H, Chen J and Zhang J, et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N Engl J Med. 2015; 372(4):341-350.
89. Krol AJ, Roellig D, Dequeant ML, Tassy O, Glynn E, Hattem G, Mushegian A, Oates AC and Pourquie O. Evolutionary plasticity of segmentation clock networks. Development. 2011; 138(13):2783-2792.
90. Ruvinsky I, Silver LM and Ho RK. Characterization of the zebrafish tbx16 gene and evolution of the vertebrate T-box family. Dev Genes Evol. 1998; 208(2):94-99.