
INTRODUCTION
Lung cancer is a highly incident disease, commonly associated to tobacco exposure, and currently represents the most frequent cause of cancer mortality. More than 80% of the cases present non-small cell lung cancer (NSCLC) histology, including adenocarcinoma and squamous cell carcinoma [1, 2]. Many molecular analyses have shown the complexity driving the carcinogenesis of the clinically relevant lung cancer phenotypes [3]. This includes DNA methylation patterns that undergo complex changes in cancer [4]. This process is important for gene expression and genome stability, and its disruption is thought to play a key role in the initiation and progression of lung cancer [5].
According to classical Mendelian laws, the majority of genes in a human cell are inherited in two functionally equivalent parental copies. However, there is a small subset of genes with one turned-off copy in a parent-of-origin-dependent manner[6]. This phenomenon, known as genomic imprinting, is an epigenetic process that involves monoallelic expression. Some genes are imprinted across the complete genome sequence, but the vast majority of imprinted genes are clustered [7, 8]. Genomic imprinting occurs in many diseases, such as neurological disorders, cancer and many others [9–11]. Loss of imprinting is commonly observed in human tumors [12], suggesting the presence of the imbalanced expression of imprinted tumor-suppressor genes or oncogenes. The IGF2 and P73 genes have been pointed out as examples of imprinted oncogene and tumor suppressor gene, respectively, in several tumors [11, 13–20].
The DLK1-DIO3 imprinted cluster is located on chromosome 14q32.2. This region includes protein-coding genes (DLK1, RTL1 and DIO3), long non-coding RNAs (MEG3, MEG8, MEG9 and LINC00524), two large clusters of miRNAs, two families of small nucleolar RNAs (SNORD113 and SNORD114) and several pseudogenes [21]. Aberrations involving some components of the DLK1-DIO3 cluster have been linked to pathological processes [22]. Increased DLK1 levels may become oncogenic in NSCLC by upregulating cell cycle machinery, which has been proposed to be highly associated with tumor invasion [23]. Similarly, overexpression of the RTL1 gene promotes hepatocarcinogenesis [24]. miRNAs located in the DLK1-DIO3 cluster have been shown to be involved in the development of different tumors such as lymphomas, glioblastomas, gastric tumors, and many others [22]. At the same time, it has been demonstrated that SNORD113 expression is linked to decreased survival in patients with hepatocellular carcinoma [25]. Concerning lung cancer, Valdmanis et al. recently described that the expression of a cluster of ~53 microRNAs and mRNAs at the DLK1-DIO3 locus on mouse chromosome 12qF1 was markedly increased in tumors compared to non-tumoral tissue, and they have proposed a role for the cluster in the origin of cancer cells [26]. Altogether, the aforementioned data suggest aberrant epigenetic regulation in genomic imprinting may promote imbalanced growth, thus leading to human tumorigenesis. To elucidate the role of the DLK1-DIO3 cluster in the tumorigenesis of non-small cell lung cancer, we have analyzed the methylation pattern in lung tumors as compared to non-tumoral lung tissue.
RESULTS
DNA methylation pattern of the DLK1-DIO3 cluster in lung cancer
To evaluate the potential role of the DLK1-DIO3 cluster in lung cancer, we analyzed the methylation status of the cluster in human lung tissue. The methylation profile of DLK1-DIO3 was evaluated in human tumor samples and compared to non-tumoral tissue using the Illumina Infinium Human Methylation 450 BeadChip. The methylation levels in lung cancer versus non-tumoral tissue are represented in Figure 1A and Supplementary Table S1. Patients with lung cancer showed three deregulated protein-coding genes: one was hypermethylated (DIO3) and two were hypomethylated (DLK1 and RTL1). Statistically significant differences (adjusted p-value < 0.05) were also detected in two different families of SNORDs (SNORD113 and SNORD114), with the exception of SNORD114-30 that showed a similar but non-significant pattern of DNA hypomethylation (p=0.07), (Table 1). Similarly, two miRNA clusters and four lncRNAs (MEG3, MEG8, MEG9 and LINC00524) were found to be significantly less methylated in tumors as compared to non-tumoral tissue (Figure 1B and Table 1). To validate these findings we used methylation data from an independent cohort obtained from TCGA database, and a different method of analysis. Consistently significant differences in methylation were observed again for the DLK1-DIO3 cluster (Figure 2). All genes included in the cluster show a hypomethylated pattern with the exception of DIO3 that was hypermethylated in tumor tissue. In order to investigate methylation-dependent RNA expression changes, some components of the DLK1-DIO3 cluster, such as DIO3, SNORD113-5, SNORD113-7, SNORD114-9, and miR-889, were randomly selected to analyze their expression levels in matched normal-tumor samples. The results confirmed that the methylation levels were inversely associated with the expression levels, i.e., hypomethylation of SNORD113-5, SNORD113-7, SNORD114-9, and miR-889 correlated with high gene expression in tumor samples relative to non-tumoral tissue. In the case of DIO3, we found lower levels of mRNA expression in lung tumor tissue (Figure 3).
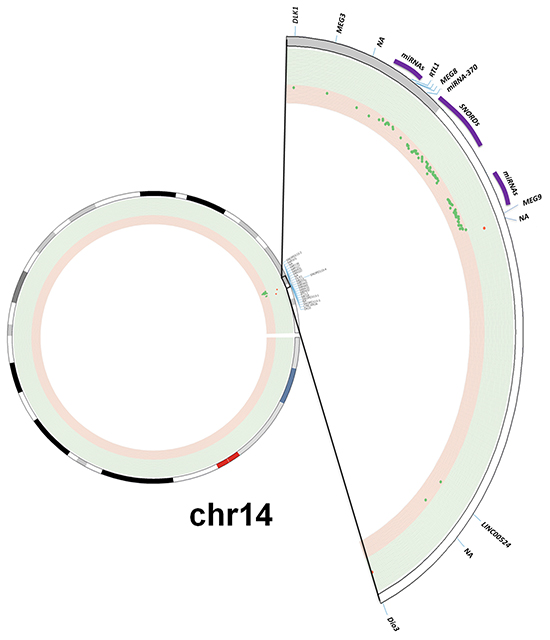
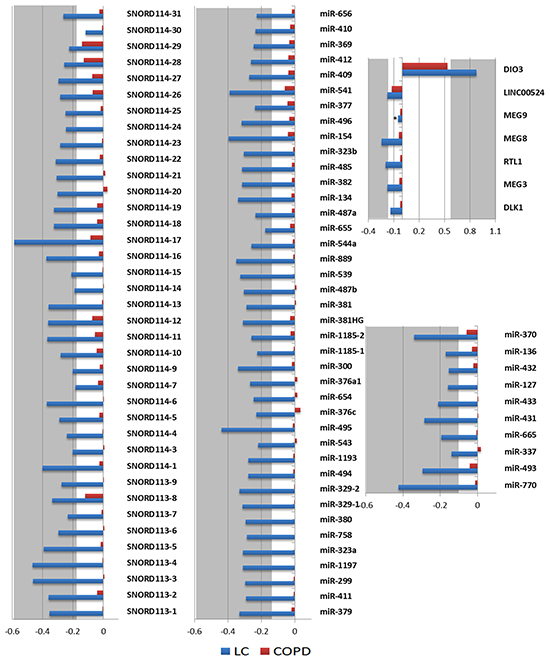
Figure 1: Methylation profile of the DLK1-DIO3 cluster in lung cancer. A. The DLK1-DIO3 cluster is located on chromosome 14 (14q32). Using a Circos plot, we plotted the methylation levels on chromosome 14. From inside to outside: methylation levels, ideogram and gene labels. Hypermethylation (red dots and green background) and hypomethylation events (green dots and red background) in patients with lung cancer versus non-tumoral samples. B. Observed methylation changes (log2 ratio). Relative levels of methylation in patients with lung cancer relative to the control group are represented in blue bars, whereas methylation levels of COPD patients with respect to the control group are represented by red bars. A grey background or asterisks represent statistically significant differences (adjusted p-value < 0.05) of methylation levels with respect to the control group. NA: Not available.
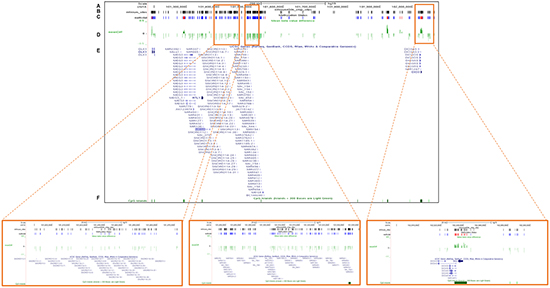
Figure 2: Validation of the DNA methylation status of the DLK1-DIO3 cluster using TCGA data. The plot shows the methylation analysis of the DLK1-DIO3 cluster in lung cancer versus non-tumoral tissue (100 vs. 32 samples) of current and former smokers visualized by UCSC browser. Zoomed-in views of the SNORD114-9, miR-889 and DIO3 regions are highlighted in orange squares. A. Chromosome 14-scale. B. CpG sites included in the Illumina Infinium Human Methylation 450 BeadChip. C. Differential DNA methylation in lung cancer versus non-tumoral tissue of current and former smokers. Bars represent hypermethylated (red) and hypomethylated (blue) probes in lung cancer tissue with respect to normal tissue. D. ß-value differences between lung cancer and non-tumoral tissue. E. Gene names of the members of the DLK1-DIO3 cluster, and F. the position of the CpG islands present in the DLK1-DIO3 cluster (green).
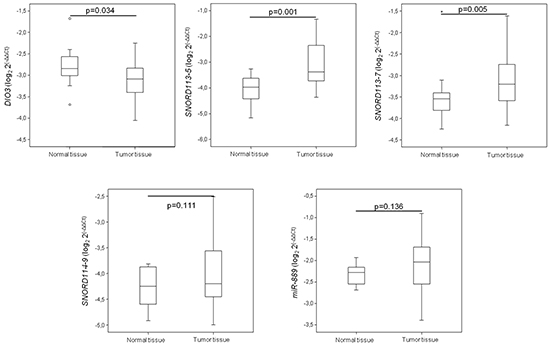
Figure 3: mRNA expression levels of some DLK1-DIO3 cluster components. Expression of some DLK1-DIO3 cluster components, which was validated by real-time PCR. mRNA expression levels were determined in tumor samples and paired normal lung tissue from lung cancer patients. Data derived from RT-qPCR are presented as the log2 2-ΔΔCt values.
Table 1: Statistical differences between miRNAs and SNORDs in human tumor samples compared to non-tumoral tissue
Genes |
Adjusted p-value |
|
|---|---|---|
miRNAs |
miR-770 |
2.51232E-14 |
miR-493 |
5.81898E-11 |
|
miR-337 |
7.41923E-06 |
|
miR-665 |
1.16877E-08 |
|
miR-431 |
7.32285E-13 |
|
miR-433 |
2.10219E-11 |
|
miR-127 |
1.32018E-08 |
|
miR-432 |
2.10017E-08 |
|
miR-136 |
6.70757E-09 |
|
miR-370 |
6.16842E-11 |
|
miR-379 |
2.90118E-17 |
|
miR-411 |
3.10121E-17 |
|
miR-299 |
7.69421E-17 |
|
miR-1197 |
1.37059E-15 |
|
miR-323A |
1.73804E-15 |
|
miR-758 |
1.45179E-14 |
|
miR-380 |
1.13522E-14 |
|
miR-329-1 |
6.75807E-14 |
|
miR-329-2 |
1.64162E-13 |
|
miR-494 |
3.65725E-12 |
|
miR-1193 |
3.65725E-12 |
|
miR-543 |
9.62007E-09 |
|
miR-495 |
6.14013E-16 |
|
miR-376C |
2.39664E-11 |
|
miR-654 |
3.35623E-12 |
|
miR-376A1 |
8.48877E-13 |
|
miR-300 |
1.65112E-14 |
|
miR-1185-1 |
1.86087E-11 |
|
miR-1185-2 |
6.87418E-14 |
|
miR-381HG |
1.17148E-14 |
|
miR-381 |
7.43382E-11 |
|
miR-487B |
3.24504E-12 |
|
miR-539 |
4.29749E-14 |
|
miR-889 |
2.92232E-16 |
|
miR-544A |
1.15212E-12 |
|
miR-655 |
1.48893E-07 |
|
miR-487A |
8.934E-13 |
|
miR-382 |
1.90249E-14 |
|
miR-485 |
1.90249E-14 |
|
miR-323B |
1.05772E-13 |
|
miR-154 |
3.35275E-17 |
|
miR-496 |
2.18282E-14 |
|
miR-377 |
1.07483E-09 |
|
miR-541 |
3.62941E-15 |
|
miR-409 |
1.59813E-13 |
|
miR-412 |
4.93041E-13 |
|
miR-369 |
2.73461E-12 |
|
miR-410 |
9.88918E-12 |
|
miR-656 |
5.71628E-12 |
|
SNORDs |
SNORD113-1 |
2.85848E-13 |
SNORD113-2 |
1.06116E-08 |
|
SNORD113-3 |
2.81624E-08 |
|
SNORD113-4 |
8.3473E-11 |
|
SNORD113-5 |
1.37034E-10 |
|
SNORD113-6 |
5.34803E-09 |
|
SNORD113-7 |
0.003019259 |
|
SNORD113-8 |
1.63653E-07 |
|
SNORD113-9 |
1.80412E-09 |
|
SNORD114-1 |
5.89536E-14 |
|
SNORD114-3 |
0.001522221 |
|
SNORD114-4 |
7.88509E-06 |
|
SNORD114-5 |
1.21425E-08 |
|
SNORD114-6 |
4.64279E-06 |
|
SNORD114-7 |
9.64877E-06 |
|
SNORD114-9 |
0.000312165 |
|
SNORD114-10 |
6.11146E-06 |
|
SNORD114-11 |
4.40691E-09 |
|
SNORD114-12 |
1.6391E-09 |
|
SNORD114-13 |
3.02053E-09 |
|
SNORD114-14 |
1.02738E-07 |
|
SNORD114-15 |
1.36835E-07 |
|
SNORD114-16 |
1.36239E-10 |
|
SNORD114-17 |
8.38123E-14 |
|
SNORD114-18 |
6.61886E-14 |
|
SNORD114-19 |
6.61886E-14 |
|
SNORD114-20 |
0.009700679 |
|
SNORD114-21 |
1.49467E-07 |
|
SNORD114-22 |
1.06972E-08 |
|
SNORD114-23 |
1.45598E-08 |
|
SNORD114-24 |
8.99314E-10 |
|
SNORD114-25 |
4.74915E-08 |
|
SNORD114-26 |
2.01146E-06 |
|
SNORD114-27 |
6.35641E-09 |
|
SNORD114-28 |
2.50354E-07 |
|
SNORD114-29 |
9.6575E-05 |
|
SNORD114-30 |
0.071177541 |
|
SNORD114-31 |
9.91748E-07 |
|
SNORD114-31 |
9.91748E-07 |
|
SNORD114-31 |
9.91748E-07 |
DLK1-DIO3 methylation profile by smoking characteristics
We evaluated variations of the DLK1-DIO3 methylation profile of lung tissues, tumoral and non-tumoral, according to the prior smoking exposure of patients. We found no differences on cluster DLK1-DIO3 methylation between non-tumoral and tumoral lung tissue among never smokers (Figure 4A). The methylation status of DIO3, miR-889 and SNORD114-9 was further validated by 454 bisulfite sequencing using pooled non-tumor and tumor samples from non-smokers (Figure 4B). In current and former smokers with lung cancer, the DLK1-DIO3 cluster showed higher methylation in non-tumoral tissue as compared to tumor. In the latter group of patients, statistically significant differences in methylation were identified in all components of the DLK1-DIO3 cluster (adjusted p-value <0.001) (Figure 4C).
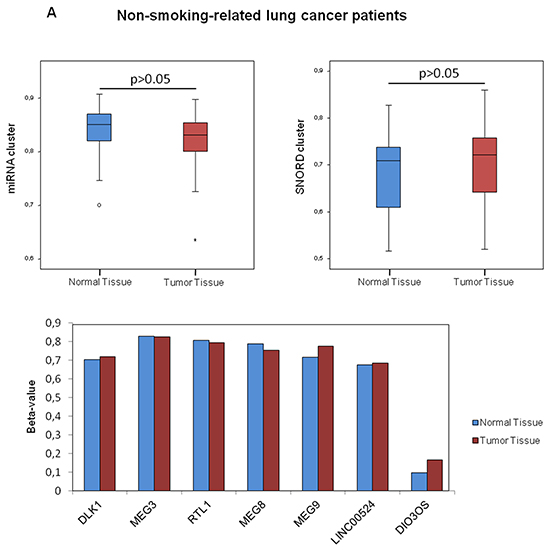
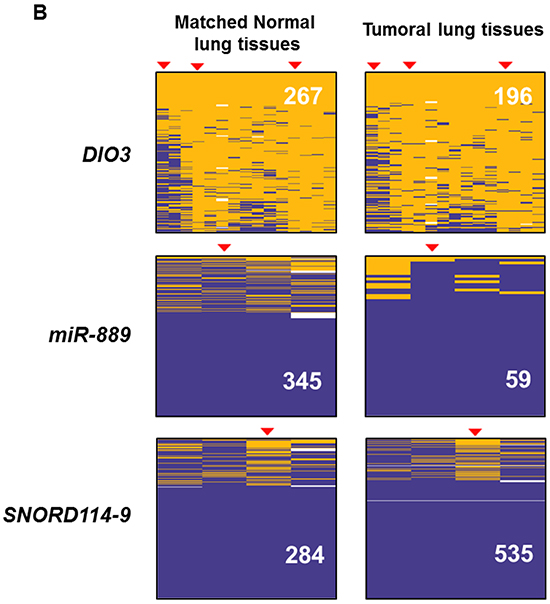
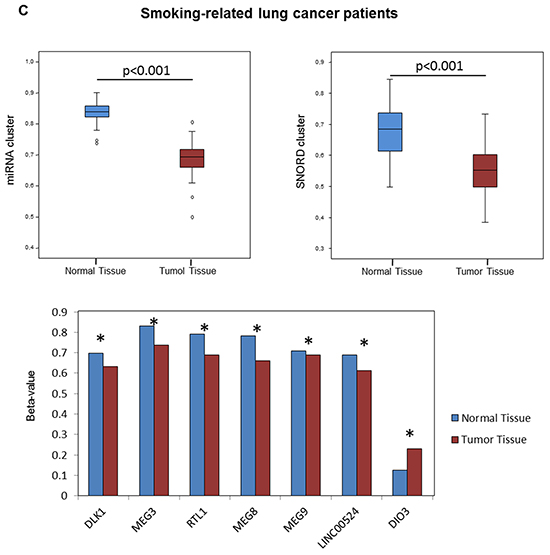
Figure 4: Association between smoking behavior and methylation profile of the DLK1-DIO3 cluster. A. DLK1-DIO3 cluster methylation level differences between non-tumoral and tumoral tissue in non-smoking-related lung cancer patients. B. Validation of the DNA methylation status of DIO3, miR-889 and SNORD114-9 by 454 bisulfite sequencing using pools of 10 non-tumoral and tumoral tissue samples in non-smoking-related lung cancer patients. Each column shows the methylation status of an individual CpG and the rows (and also the white numbers) indicate the amount of reads. The red arrowheads highlight the position of the CpG represented on the Illumina Infinium Human Methylation 450 BeadChip. Methylated CpGs are displayed in purple, unmethylated CpGs in orange and gaps in white. C. DLK1-DIO3 cluster methylation level differences between normal tissue and tumoral tissue in smoking-related lung cancer patients, including current and former smokers. *: adjusted p-value<0.001. β-value: sum of methylated and unmethylated probe intensities.
We then next analyzed whether the DNA methylation pattern in non-tumoral lung parenchyma of current and former smokers with COPD differed from that of current and former smokers without COPD and found no statistical differences (Figure 1B). Of note, an increase in the DNA promoter methylation of DIO3 was noticeable in COPD lung tissue although not statistically significant (log2 ratio COPD+/COPD-LC- was 0.535 and the adjusted p-value was 0.840) (Supplementary Table S1). In addition, we have found that the DLK1-DIO3 methylation profile of non-malignant tissue from lung cancer patients and control subjects was similar (data not shown).
DLK1-DIO3 methylation profile by histological subtypes
Due to its specific global hypomethylation in lung cancer, we have evaluated the methylation of the DLK1-DIO3 cluster in the two main histological subtypes of NSCLC, squamous cell carcinoma (SCC) and adenocarcinoma, as compared to non-tumoral tissue. The DLK1-DIO3 imprinted cluster hypomethylation was consistently evident in both histological subtypes with respect to non-tumoral tissue (Supplementary Table S1). We found that SCC samples showed significantly higher hypomethylation levels in the SNORD cluster than in lung adenocarcinoma (Figure 5A). As with SNORD114-30 for general lung cancer, we did not find any statistically significant differences for lung adenocarcinoma or SCC (adjusted p-value=0.494 and 0.095, respectively). SNORD113-7 and SNORD114-3 were only statistically significant in the SCC group (log2 ratio SCC+/ non-tumoral tissue = -0.309; adjusted p-value= 0.025 and log2 ratio SCC+/ non-tumoral tissue= -0.234; adjusted p-value = 0.031, respectively) (Supplementary Table S1). MicroRNA clusters were also notably more hypomethylated in SCC (Figure 5B). RTL1, MEG8 and LINC00524 were hypomethylated in both histological subgroups. Moreover, DLK1 and MEG9 were significantly hypomethylated in patients with SCC (p<0.001 and p=0.01, respectively), while in lung adenocarcinoma, DLK1 was very similar to the control group (p=0.127). DIO3 was significantly hypermethylated in patients with lung adenocarcinoma (p<0.001) (Figure 5C).
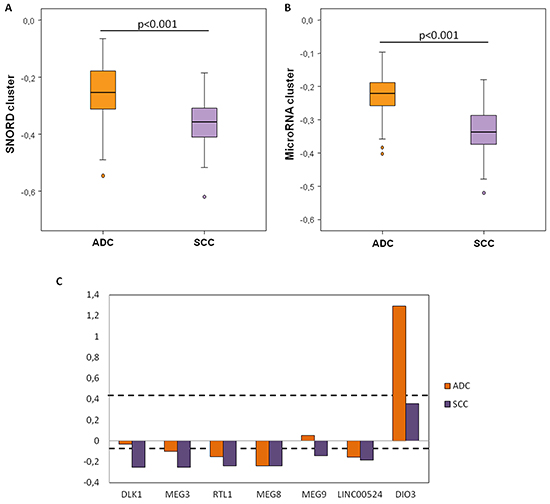
Figure 5: Methylation levels of the DLK1-DIO3 cluster components according to the histological subtype of lung cancer. A. Relative SNORD methylation levels in patients with squamous cell carcinoma (SCC) relative to the control group is represented in purple bars, whereas methylation levels in patients with adenocarcinoma (ADC) with respect to the control group are represented by orange bars. B. Relative microRNA methylation levels in patients with SCC (purple) and ADC (orange) relative to the control group. C. Relative methylation levels of DIO3, LINC00524, MEG9, MEG8, RTL1, MEG3 and DLK1 in patients with SCC (purple) and ADC (orange) with respect to the control group. Black broken lines represent statistically significant differences (adjusted p-value < 0.05) of methylation levels with respect to the control group.
Transcriptional mapping of the DLK1-DIO3 cluster
To analyze genomic features associated with different mechanisms of the transcriptional regulation of the DLK1-DIO3 cluster, we used the Wash U Epigenome Browser to display the epigenomic mapping of the DLK1-DIO3 cluster (14q32) to the human genome reference (hg19; chr14: 101.140.120–102.044.779 genomic coordinates) (Figure 6A). We observed the distribution of several transcriptional mechanisms (direct regulators and structural determinants) along the DLK1-DIO3 cluster (Figure 6B). Multiple enhancers are distributed in the length of the cluster (yellow arrows), with the exception of the region located between 101380-10530Kb on chromosome 14. The SNORD113 and SNORD114 families and a miRNA cluster are located in this region without enhancers. In this same region, we found few effects of polycomb protein-mediated epigenetic regulation (grey arrows). In addition, we identified a heterochromatin rich domain situated approximately 101277Kb distal to the 5’ region (purple arrows). Therefore, following with MEG9 lncRNA (101536Kb), we found a highly condensed region before it reached the 5′ region of the DIO3 gene. Interestingly, the 5’ flanking region of the MEG3 lncRNA (~101292 kb) showed activity in transcription initiation, where there is an active transcriptional start site (TSS) marked by a red arrow. No other region of the DLK1-DIO3 cluster showed active TSS. From the active TSS, we found an approximately 240Kb a domain with strong transcription (green arrows) (Figure 6B). However, we identified several CpG islands located near genes throughout this region, including lncRNAs, miRNAs, SNORDs and pseudogenes (crimson lines) (Figure 6C). However, the CpG sites were distributed along the DLK1-DIO3 cluster (white lines) (Figure 6D). The GC-content was reduced by 30% from MEG3 to MEG9 (Figure 6E).
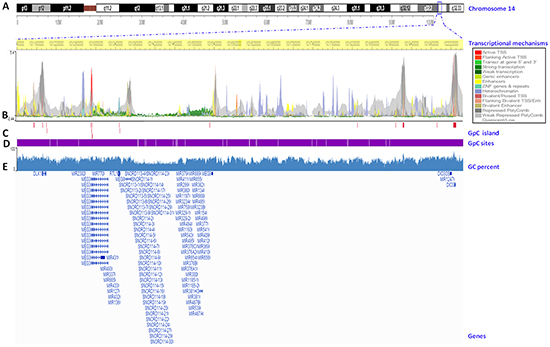
Figure 6: Transcriptional mapping of the DLK1-DIO3 cluster to the human reference genome hg19. A. Chromosome 14 ideogram. The DLK1-DIO3 cluster is located on 14q32. The exact position of the cluster in the region is marked with a blue square. B. Chromosome position. Base pairs of the DLK1-DIO3 cluster on chromosome 14 (highlighted in yellow). The transcriptional mechanisms underlying the expression of the DLK1-DIO3 cluster, such as the transcription start sites (TSS), enhancer regions (Enh), zinc finger (ZNF), packed form of DNA, and polycomb group proteins. C. CpG islands in the DLK1-DIO3 cluster (crimson lines). D. The genomic distribution of CpG sites in the DLK1-DIO3 cluster (white lines). E. GC percent in the DLK1-DIO3 cluster (blue). Finally, the sequence references of members of the DLK1-DIO3 cluster are represented.
Experimentally validated target interactions of the DLK1-DIO3 miRNA cluster
To study the functional relevance of the DLK1-DIO3 miRNA cluster in the regulation of validated mRNAs as a potential tumor suppressor, we used the miRWalk algorithm [27]. A total of 538 genes were found to be validated targets of the miRNAs included in this cluster. These targets were located on all chromosomes except the Y chromosome (Figure 7). Several miRNAs were complementary with more than one validated gene, and some targets were regulated by more than one miRNA. We classified validated targets by gene ontology (GO) molecular function and biological processes using the PANTHER program. The main GO molecular functions were binding (GO:0005488) (38.9%), catalytic activity (GO:0003824) (27.4%), nucleic acid binding transcription factor activity (GO:0001071) (13.5%), and receptor activity (GO:0004872) (10.4%). The primary binding types were protein binding (GO:0005515) and nucleic acid binding (GO:0003676). In the case of catalytic activity, we primarily found activities for hydrolase (GO:0016787), transferase (GO:0016740) and enzyme regulators (GO:0007154). Validated targets displayed six main biological processes: metabolic processes (GO:0008152) (21.2%), such as cell communication (GO:0030234) and cell cycle (GO:0007049); cellular process (GO:0009987) (20.2%), including system development (GO:0048731), anatomical structure and morphogenesis (GO:0009653) and death (GO:0016265); developmental processes (GO:0032502) (12.4%); response to stimulus (GO:0050896) (5.6%); and apoptotic processes (GO:0006915) (5.3%) (Supplementary Figure S1). Finally, we found that 26 out of 538 validated genes (AXIN2, BRCA1, CCNB1, CCND2, CDKN1A, CDKN2A, CSNK2A1, FOXO1, GRB2, HDAC6, NFKB1, NTRK3, PPP2R4, PPP2R1B, PPP2R2A, PTEN, RARA, RASA1, RB1, SOCS3, TGFB1, TGFB2, TGFBR2, TNF, TP53, and TP73) were targets of miRNAs included in the DLK1-DIO3 cluster, which have been proposed to have a functional role as tumor suppressors (Supplementary Table S2).
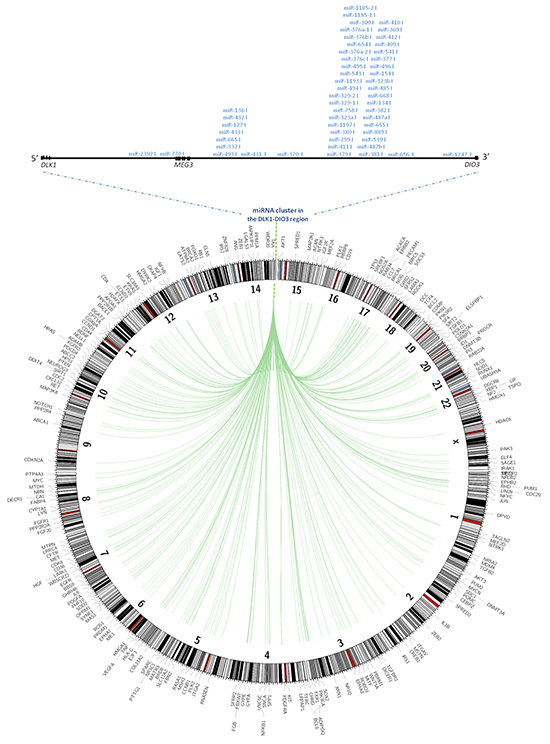
Figure 7: Validated targets of miRNAs contained in the DLK1-DIO3 cluster. Plot showing with position of validated miRNA targets in the whole human genome. The validated targets are connected to the miRNAs within the DLK1-DIO3 region on chromosome 14 by green lines.
DISCUSSION
In the present study we firstly identified characteristic methylation pattern of the DLK1-DIO3 cluster in human lung cancer, as compared, to lung parenchyma. Specifically, we identified that all components of the afore-mentioned cluster are hypomethylated, with the exception of DIO3 that is hypermethylated. Importantly this specific methylation signature of the imprinted cluster is restricted to smoking induced lung cancer since it is absent in lung tumors from never smokers. In addition, this locus was more aberrantly regulated in SCC, typically exposed to heavier smoking history, than in lung adenocarcinoma. In a murine lung carcinoma model with mutated KRAS, it has been observed that the DLK1-DIO3 miRNAs cluster on chromosome on 12qF1 was substantially overexpressed, suggesting a role as an oncogenic driver of lung cancer in these circumstances [26]. Those findings are consistent with our results, particularly considering KRAS is the most frequently mutated proto-oncogenes among smokers with lung carcinoma [28, 29].
Imprinted genes are particularly more susceptible to epigenetic regulatory changes due to having one specifically silenced allele [6]. Therefore, monoallelic changes in methylation status may have deleterious consequences. The DLK1-DIO3 cluster includes protein-coding genes, lncRNAs, miRNAs, SNORDs and pseudogenes [21]. DLK1, RTL1 and DIO3 are expressed only from the paternal allele. However, large and small noncoding RNA genes and pseudogenes come from the maternal allele [30]. Our data showed that the paternally derived DLK1 and RTL1 genes are hypomethylated and that the DIO3 gene is hypermethylated. DLK1 has been reported aberrantly expressed in various types of tumors, such as hepatocellular carcinoma, glioma, renal cell carcinoma and lung cancer [31-34]. In addition, Li et al. recently identified in an in vitro lung model that overexpression of DLK1 enhanced extracellular matrix invasion by Notch signaling in a dependent and independent manner [23]. In the case of the RTL1 gene, it has been proposed to act as an oncogene in hepatocarcinoma. In vivo RTL1 overexpression in the liver contributed to tumor growth [24]. Accordingly, the hypomethylation of these genes can play an important role in the pathogenesis of lung cancer. The hypermethylation of DIO3 has been reported in hematologic neoplasms [35]. We also identified higher levels of global DNA methylation in lung cancer versus normal lung tissue. Moreover, the MEG3 lncRNA has been described as a tumor suppressor; MEG3 levels were significantly downregulated in patients with NSCLC [36]. However, our study showed that the methylation level was significantly higher in human lung cancer relative to non-tumoral tissue. These differences could be due to the differences between the subjects recruited for each study. Lu et al. reported lower MEG3 expression in patients with an advanced pathological stage [36], while in our study, almost 80% of patients were at clinical stage I or II. Otherwise, the loss of expression of this tumor suppressor could also be due to micro-deletion at the MEG3-DMR locus or deletion of a transcription factor-binding site on the MEG3 promoter [37, 38]. In the case of MEG8 and MEG9 lncRNAs, their functions in cancer remain unknown.
In our study, we observed an important pattern of DNA hypomethylation in two SNORD families and two miRNA clusters in patients with lung cancer. In addition, we visualized less CpG and strong transcription in this region. MicroRNAs are post-transcriptional modulators of many biological processes. The dysregulation of their physiological roles may contribute to the initiation and progression of cancer [39]. Along with miRNAs, other types of small regulatory non-protein-coding RNAs were detected, such as small nucleolar RNAs (snoRNAs). It has been reported that alterations in snoRNA expression levels may lead to various diseases, including cancer [40, 41]. Valleron et al. found that several variants of SNORDs of the DLK1-DIO3 cluster were overexpressed in patients with acute promyelocytic leukemia (APL) [42]. In addition, it has been shown that in vitro SNORD114-1 overexpression promoted cell growth by cell cycle modulation [42]. The same results have been obtained in a large cohort of of acute promyelocytic leukemia (APL) patients, suggesting that increased expression of SNORD114-3 is a good biomarker of APL [43]. In our study, we identified that the increased expression of SNORD114-3 was only statistically significant in the SCC group although a tendency was observed in the lung adenocarcinoma group. The DLK1-DIO3 cluster contains also 53 miRNAs. The deregulation of some of them has been associated with enhanced tumorigenicity [22]. MicroRNA-154, miR-379 and miR-409 were highly expressed in metastatic prostate cancer cells [44]. MicroRNA-127 was overexpressed in the serum of patients with esophageal squamous cell carcinoma [45]. Haller et al. revealed the deregulation of miRNA-329, miR-370, miR-376c and miR409 expression based on the location of the tumor in gastrointestinal stromal tumors (GISTs); higher expression occurred in intestinal compared to gastric tumors [46]. Indeed, it has even been proposed that miR-376c can promote cell proliferation and invasion [47, 48]. Recently, miRNAs of the DLK1-DIO3 cluster have been identified as being upregulated in a mouse model of lung adenocarcinoma [26]. The results obtained in a mouse model are in line with those obtained from the patient cohort of our study. We found a pattern of DNA hypomethylation in the DLK1-DIO3 microRNA cluster in current and former smoker patients with lung cancer. This profile was representative of both SCC and lung adenocarcinoma histological subtypes. On the other hand, it has been found that hepatocellular carcinoma patients with overexpression of the DLK1-DIO3 miRNA cluster had significantly poorer overall survival [49]. In addition, 3 miRNAs of the DLK1-DIO3 cluster have been associated with poor survival in patients with surgically resected lung adenocarcinoma [50]. These data warrant a validation of the DLK1-DIO3 cluster methylation as a prognostic tool in a prospective cohort study.
We identified experimentally validated targets and predicted their molecular function. We found that they may be involved in catalytic activity and the binding of nucleic acids to transcription factors. The biological process categories were mostly metabolic, cellular, developmental, response to stimulus and apoptotic processes. In addition, we identified 26 validated targets, including AXIN2, BRCA1, CCNB1, CCND2, CDKN1A, CDKN2A, CSNK2A1, FOXO1, GRB2, HDAC6, NFKB1, NTRK3, PPP2R4, PPP2R1B, PPP2R2A, PTEN, RARA, RASA1, RB1, SOCS3, TGFB1, TGFB2, TGFBR2, TNF, TP53, and TP73, which can act as tumor suppressors. Thus, the hypomethylation of the DLK1-DIO3 miRNA cluster may lead to its activation, promoting the downregulation of these relevant tumor suppressors in lung cancer.
In conclusion, we found the DLK1-DIO3 cluster to be hypomethylated in current and former smoker patients with NSCLC. This pattern suggests that aberrant expression may contribute to tumorigenesis in the lung. Moreover, given that some components of the DLK1-DIO3 cluster may be responsible of the regulation of tumor suppressors in lung cancer, they could have great potential as novel biomarkers and therapeutic targets. The present analysis suggests that future studies on this topic are warranted.
MATHERIALS AND METHODS
Patients and clinical specimens
The present study was performed in 70 subjects fromVirgen del Rocio University Hospital (Seville). The samples were divided into two cohorts. A first group of samples, from 47 patients who had undergone surgical resection for clinical early stage NSCLC. During the surgical procedure, the tumor and matched non-tumoral tissue samples were collected from all patients and then immediately snap-frozen to −80°C until further use. The clinical features of patients with NSCLC are summarized in Supplementary Table S3. A second group from 23 subjects was used as control cohort. The control cohort without lung cancer was constituted of COPD patients and non-COPD subjects. A description of this cohort can be found on Supplementary Table S4. A written consent form was obtained from all participants. The study protocol and the use of human samples were approved by the Ethical Committee of the Virgen del Rocio University Hospital.
DNA samples
Genomic DNA was extracted from tumor and matched non-tumoral tissue samples by the QIAamp DNA mini kit (QIAGEN, Valencia, CA, USA). DNA was quantified using the QuantiFluor dsDNA system (Promega, Madison, WI, USA) according to the manufacturers’ instructions.
Illumina 450 K methylation
The Illumina Infinium Human Methylation 450 BeadChip (Illumina Inc., San Diego, CA) was used to interrogate 485,000 methylation sites across the genome per sample at single-nucleotide resolution. It covers 96% of the CpG islands, with additional coverage in island shores and the flanking regions. We treated 500 ng of DNA with sodium bisulfate using the EZ DNA Methylation™ Kit and cleaned the DNA with the ZR-96 DNA Clean-up Kit™ (EZ DNA, Zymo Research, Irvine, CA) before standard Illumina amplification, hybridization, and imaging steps. The resulting intensity files were analyzed with Illumina’s GenomeStudio, which generated β-scores (i.e., the proportion of total signal from the methylation-specific probe or color channel).
Methylome data processing
Methylome data were processed using the RnBeads R package [51]. After a quality check, the probe median intensity was normalized with the SWAN method [52] and converted to beta values. The probes were tested for differential methylation with the limma method, a linear model followed by empirical Bayes methods for the comparisons of interest [53]. The Benjamini–Hochberg method was used to adjust the p-values and ensure that the false discovery rate (FDR) was lower than 0.05. The CpG status (hypo- versus hyper-methylated) and CpG chromosomal location were realized using the Circos data visualization software [54]. DNA methylation data were visualized by the Wash U Epigenome Browser [55]. Validated mRNA targets of miRNAs by previous research located in the DLK1-DIO3 cluster were identified with the miRWalk algorithm [27]. The molecular function and proposed biological process of experimentally verified mRNAs were determined using the PANTHER program [56].
Validation of DNA methylation status
Smoking-related samples from lung cancer patients in TCGA database (https://tcga-data.nci.nih.gov) were selected to interrogate the DLK1-DIO3 cluster methylation status. Their characteristics are summarized in Table S5. Differences on the DLK1-DIO3 methylation profile between tumor and normal tissues from current and former smoker lung cancer patients were identified with a method previously described [57]. Non-smoking habit of lung cancer patients in TCGA database were often incomplete. For this reason, 454 bisulfite sequencing (Roche, Mannheim, Germany) was used to validate the DNA methylation status of DLK1, miR-889 and SNORD114-9 [58]. Primer sequences are listed in Supplementary table S6 and the genomic features of regions selected for 454 bisulfite sequencing are represented in Supplementary Figure S2.
Expression levels of some DLK1-DIO3 cluster components
Expression of some randomly selected DLK1-DIO3 cluster components (DIO3, SNORD113-5, SNORD113-7, SNORD114-9, and miR-889) was analyzed by qPCR. For genes, 100 ng/µL of total RNA was converted into cDNA by the reverse transcriptase reaction which was performed by sequential incubation at 25°C for 10 min, 37°C for 2 h and 85°C for 5 min. For the miR-889 expression levels, 2 ng/µL of total RNA was converted into cDNA by the reverse transcriptase reaction, which was performed by sequential incubation at 16°C for 30 min, 42°C for 30 min and 85°C for 5 min. The PCR reaction mixture was initially incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 seconds and 60°C for 60 seconds. The expression of miR-889 was normalized to the expression of RNU48 and B2M for the normalization of different genes. Gene expression data of tumor versus normal tissue were compared by a t-test. The results were processed and analyzed as previously described [59, 60].
ACKNOWLEDGMENTS
We thank R. Melendez for their continuous technical support. The authors thank the donors and the HUVR-IBiS Biobank (Andalusian Public Health System Biobank and ISCIII-Red de Biobancos PT13/0010/0056) for the human specimens used in this study.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
SMP is funded by Instituto de Salud Carlos III (PI17/00033) and co-funded by European Union (ERDF/ESF, “Investing in your future”), Consejería de Salud y Bienestar Social (PI2009-0224 and PI-0046-2012), and Fundación Mutua Madrileña (2014). LPA is funded by Instituto de Salud Carlos III (PI1102688, 1401964, R12/0036/0028 and CB16/12/00442). AC lab was supported by grants to from the Spanish Ministry of Economy and Competitivity, Plan Nacional de I+D+I 2008-2011, Plan Estatal de I+D+I 2013-2016, ISCIII (Fis: PI12/00137, PI15/00045, RTICC: RD12/0036/0028, CB16/12/00275) co-funded by FEDER from Regional Development European Funds (European Union), Consejeria de Ciencia e Innovacion (CTS-6844 and CTS-1848) and Consejeria de Salud of the Junta de Andalucia (PI-0135-2010 and PI-0306-2012). CC and EJL are supported by grants from PN I+D+I 2008-2011, Spain, Instituto de Salud Carlos III, (PI12/02838), Subdirección General de Redes y Centros de Investigación Cooperativa, Red Temática de Investigación Cooperativa en Cáncer (RD12/0036/0025). This work has been also possible thanks to the Plan Estatal de I+D+i 2013-2016, Grant PIE13/0004 co-funded by the ISCIII and FEDER funds. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: a cancer journal for clinicians. 2015; 65:5-29.
2. Travis WD, Travis LB, Devesa SS. Lung cancer. Cancer. 1995; 75:191-202.
3. Perez-Soler R. Individualized therapy in non-small-cell lung cancer: future versus current clinical practice. Oncogene. 2009; 28:S38-45.
4. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010; 31:27-36.
5. Mehta A, Dobersch S, Romero-Olmedo AJ, Barreto G. Epigenetics in lung cancer diagnosis and therapy. Cancer Metastasis Rev. 2015.
6. Bartolomei MS. Genomic imprinting: employing and avoiding epigenetic processes. Genes Dev. 2009; 23:2124-2133.
7. Brannan CI, Bartolomei MS. Mechanisms of genomic imprinting. Curr Opin Genet Dev. 1999; 9:164-170.
8. Court F, Tayama C, Romanelli V, Martin-Trujillo A, Iglesias-Platas I, Okamura K, Sugahara N, Simon C, Moore H, Harness JV, Keirstead H, Sanchez-Mut JV, Kaneki E, et al. Genome-wide parent-of-origin DNA methylation analysis reveals the intricacies of human imprinting and suggests a germline methylation-independent mechanism of establishment. Genome Res. 2014; 24:554-569.
9. Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005; 6:597-610.
10. Horsthemke B. In brief: genomic imprinting and imprinting diseases. J Pathol. 2014; 232:485-487.
11. Jelinic P, Shaw P. Loss of imprinting and cancer. J Pathol. 2007; 211:261-268.
12. Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006; 7:21-33.
13. Zhang M, Wu CH, Zhu XL, Wang YJ. Loss of imprinting of insulin-like growth factor 2 is associated with increased risk of primary lung cancer in the central China region. Asian Pac J Cancer Prev. 2014; 15:7799-7803.
14. Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, Ferrara P, McKeon F, Caput D. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997; 90:809-819.
15. Mai M, Yokomizo A, Qian C, Yang P, Tindall DJ, Smith DI, Liu W. Activation of p73 silent allele in lung cancer. Cancer Res. 1998; 58:2347-2349.
16. Mai M, Qian C, Yokomizo A, Tindall DJ, Bostwick D, Polychronakos C, Smith DI, Liu W. Loss of imprinting and allele switching of p73 in renal cell carcinoma. Oncogene. 1998; 17:1739-1741.
17. Kang MJ, Park BJ, Byun DS, Park JI, Kim HJ, Park JH, Chi SG. Loss of imprinting and elevated expression of wild-type p73 in human gastric adenocarcinoma. Clin Cancer Res. 2000; 6:1767-1771.
18. Martinez-Delgado B, Melendez B, Cuadros M, Jose Garcia M, Nomdedeu J, Rivas C, Fernandez-Piqueras J, Benitez J. Frequent inactivation of the p73 gene by abnormal methylation or LOH in non-Hodgkin’s lymphomas. Int J Cancer. 2002; 102:15-19.
19. Cai YC, Yang GY, Nie Y, Wang LD, Zhao X, Song YL, Seril DN, Liao J, Xing EP, Yang CS. Molecular alterations of p73 in human esophageal squamous cell carcinomas: loss of heterozygosity occurs frequently; loss of imprinting and elevation of p73 expression may be related to defective p53. Carcinogenesis. 2000; 21:683-689.
20. Cui H. Loss of imprinting of IGF2 as an epigenetic marker for the risk of human cancer. Dis Markers. 2007; 23:105-112.
21. Irving MD, Buiting K, Kanber D, Donaghue C, Schulz R, Offiah A, Mohammed SN, Oakey RJ. Segmental paternal uniparental disomy (patUPD) of 14q32 with abnormal methylation elicits the characteristic features of complete patUPD14. Am J Med Genet A. 2010; 152A:1942-1950.
22. Benetatos L, Hatzimichael E, Londin E, Vartholomatos G, Loher P, Rigoutsos I, Briasoulis E. The microRNAs within the DLK1-DIO3 genomic region: involvement in disease pathogenesis. Cell Mol Life Sci. 2013; 70:795-814.
23. Li L, Tan J, Zhang Y, Han N, Di X, Xiao T, Cheng S, Gao Y, Liu Y. DLK1 promotes lung cancer cell invasion through upregulation of MMP9 expression depending on Notch signaling. PLoS One. 2014; 9:e91509.
24. Riordan JD, Keng VW, Tschida BR, Scheetz TE, Bell JB, Podetz-Pedersen KM, Moser CD, Copeland NG, Jenkins NA, Roberts LR, Largaespada DA, Dupuy AJ. Identification of rtl1, a retrotransposon-derived imprinted gene, as a novel driver of hepatocarcinogenesis. PLoS Genet. 2013; 9:e1003441.
25. Xu G, Yang F, Ding CL, Zhao LJ, Ren H, Zhao P, Wang W, Qi ZT. Small nucleolar RNA 113-1 suppresses tumorigenesis in hepatocellular carcinoma. Mol Cancer. 2014; 13:216.
26. Valdmanis PN, Roy-Chaudhuri B, Kim HK, Sayles LC, Zheng Y, Chuang CH, Caswell DR, Chu K, Zhang Y, Winslow MM, Sweet-Cordero EA, Kay MA. Upregulation of the microRNA cluster at the Dlk1-Dio3 locus in lung adenocarcinoma. Oncogene. 2015; 34:94-103.
27. Dweep H, Sticht C, Pandey P, Gretz N. miRWalk--database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J Biomed Inform. 2011; 44:839-847.
28. Wistuba II, Mao L, Gazdar AF. Smoking molecular damage in bronchial epithelium. Oncogene. 2002; 21:7298-7306.
29. Riely GJ, Marks J, Pao W. KRAS mutations in non-small cell lung cancer. Proc Am Thorac Soc. 2009; 6:201-205.
30. da Rocha ST, Edwards CA, Ito M, Ogata T, Ferguson-Smith AC. Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet. 2008; 24:306-316.
31. Yin D, Xie D, Sakajiri S, Miller CW, Zhu H, Popoviciu ML, Said JW, Black KL, Koeffler HP. DLK1: increased expression in gliomas and associated with oncogenic activities. Oncogene. 2006; 25:1852-1861.
32. Liu Y, Tan J, Li L, Li S, Zou S, Zhang Y, Zhang X, Ling B, Han N, Guo S, Gao Y. [Study on the molecular mechanisms of dlk1 stimulated lung cancer cell proliferation]. Zhongguo Fei Ai Za Zhi. 2010; 13:923-927.
33. Huang J, Zhang X, Zhang M, Zhu JD, Zhang YL, Lin Y, Wang KS, Qi XF, Zhang Q, Liu GZ, Yu J, Cui Y, Yang PY, et al. Up-regulation of DLK1 as an imprinted gene could contribute to human hepatocellular carcinoma. Carcinogenesis. 2007; 28:1094-1103.
34. Kawakami T, Chano T, Minami K, Okabe H, Okada Y, Okamoto K. Imprinted DLK1 is a putative tumor suppressor gene and inactivated by epimutation at the region upstream of GTL2 in human renal cell carcinoma. Hum Mol Genet. 2006; 15:821-830.
35. Martin-Subero JI, Ammerpohl O, Bibikova M, Wickham-Garcia E, Agirre X, Alvarez S, Bruggemann M, Bug S, Calasanz MJ, Deckert M, Dreyling M, Du MQ, Durig J, et al. A comprehensive microarray-based DNA methylation study of 367 hematological neoplasms. PLoS One. 2009; 4:e6986.
36. Lu KH, Li W, Liu XH, Sun M, Zhang ML, Wu WQ, Xie WP, Hou YY. Long non-coding RNA MEG3 inhibits NSCLC cells proliferation and induces apoptosis by affecting p53 expression. BMC Cancer. 2013; 13:461.
37. Zhao J, Zhang X, Zhou Y, Ansell PJ, Klibanski A. Cyclic AMP stimulates MEG3 gene expression in cells through a cAMP-response element (CRE) in the MEG3 proximal promoter region. Int J Biochem Cell Biol. 2006; 38:1808-1820.
38. Kagami M, O’Sullivan MJ, Green AJ, Watabe Y, Arisaka O, Masawa N, Matsuoka K, Fukami M, Matsubara K, Kato F, Ferguson-Smith AC, Ogata T. The IG-DMR and the MEG3-DMR at human chromosome 14q32.2: hierarchical interaction and distinct functional properties as imprinting control centers. PLoS Genet. 2010; 6:e1000992.
39. Garzon R, Calin GA, Croce CM. MicroRNAs in Cancer. Annu Rev Med. 2009; 60:167-179.
40. Thorenoor N, Slaby O. Small nucleolar RNAs functioning and potential roles in cancer. Tumour Biol. 2015; 36:41-53.
41. Bratkovic T, Rogelj B. The many faces of small nucleolar RNAs. Biochim Biophys Acta. 2014; 1839:438-443.
42. Valleron W, Laprevotte E, Gautier EF, Quelen C, Demur C, Delabesse E, Agirre X, Prosper F, Kiss T, Brousset P. Specific small nucleolar RNA expression profiles in acute leukemia. Leukemia. 2012; 26:2052-2060.
43. Liuksiala T, Teittinen KJ, Granberg K, Heinaniemi M, Annala M, Maki M, Nykter M, Lohi O. Overexpression of SNORD114-3 marks acute promyelocytic leukemia. Leukemia. 2014; 28:233-236.
44. Gururajan M, Josson S, Chu GC, Lu CL, Lu YT, Haga CL, Zhau HE, Liu C, Lichterman J, Duan P, Posadas EM, Chung LW. miR-154* and miR-379 in the DLK1-DIO3 microRNA mega-cluster regulate epithelial to mesenchymal transition and bone metastasis of prostate cancer. Clin Cancer Res. 2014; 20:6559-6569.
45. Zhang C, Wang C, Chen X, Yang C, Li K, Wang J, Dai J, Hu Z, Zhou X, Chen L, Zhang Y, Li Y, Qiu H, et al. Expression profile of microRNAs in serum: a fingerprint for esophageal squamous cell carcinoma. Clin Chem. 2010; 56:1871-1879.
46. Haller F, von Heydebreck A, Zhang JD, Gunawan B, Langer C, Ramadori G, Wiemann S, Sahin O. Localization- and mutation-dependent microRNA (miRNA) expression signatures in gastrointestinal stromal tumours (GISTs), with a cluster of co-expressed miRNAs located at 14q32.31. J Pathol. 2010; 220:71-86.
47. Fu G, Ye G, Nadeem L, Ji L, Manchanda T, Wang Y, Zhao Y, Qiao J, Wang YL, Lye S, Yang BB, Peng C. MicroRNA-376c impairs transforming growth factor-beta and nodal signaling to promote trophoblast cell proliferation and invasion. Hypertension. 2013; 61:864-872.
48. Ye G, Fu G, Cui S, Zhao S, Bernaudo S, Bai Y, Ding Y, Zhang Y, Yang BB, Peng C. MicroRNA 376c enhances ovarian cancer cell survival by targeting activin receptor-like kinase 7: implications for chemoresistance. J Cell Sci. 2011; 124:359-368.
49. Luk JM, Burchard J, Zhang C, Liu AM, Wong KF, Shek FH, Lee NP, Fan ST, Poon RT, Ivanovska I, Philippar U, Cleary MA, Buser CA, et al. DLK1-DIO3 genomic imprinted microRNA cluster at 14q32.2 defines a stemlike subtype of hepatocellular carcinoma associated with poor survival. The Journal of biological chemistry. 2011; 286:30706-30713.
50. Nadal E, Zhong J, Lin J, Reddy RM, Ramnath N, Orringer MB, Chang AC, Beer DG, Chen G. A MicroRNA cluster at 14q32 drives aggressive lung adenocarcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014; 20:3107-3117.
51. Assenov Y, Muller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nature methods. 2014; 11:1138-1140.
52. Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012; 13:R44.
53. Ritchie ME, Diyagama D, Neilson J, van Laar R, Dobrovic A, Holloway A, Smyth GK. Empirical array quality weights in the analysis of microarray data. BMC Bioinformatics. 2006; 7:261.
54. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639-1645.
55. Zhou X, Li D, Lowdon RF, Costello JF, Wang T. methylC Track: visual integration of single-base resolution DNA methylation data on the WashU EpiGenome Browser. Bioinformatics. 2014; 30:2206-2207.
56. Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013; 41:D377-386.
57. Geyh S, Rodriguez-Paredes M, Jager P, Khandanpour C, Cadeddu RP, Gutekunst J, Wilk CM, Fenk R, Zilkens C, Hermsen D, Germing U, Kobbe G, Lyko F, et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia. 2016; 30:683-691.
58. Gronniger E, Weber B, Heil O, Peters N, Stab F, Wenck H, Korn B, Winnefeld M, Lyko F. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS genetics. 2010; 6:e1000971.
59. Molina-Pinelo S, Gutierrez G, Pastor MD, Hergueta M, Moreno-Bueno G, Garcia-Carbonero R, Nogal A, Suarez R, Salinas A, Pozo-Rodriguez F, Lopez-Rios F, Agullo-Ortuno MT, Ferrer I, et al. MicroRNA-dependent regulation of transcription in non-small cell lung cancer. PloS one. 2014; 9:e90524.
60. Molina-Pinelo S, Pastor MD, Suarez R, Romero-Romero B, Gonzalez De la Pena M, Salinas A, Garcia-Carbonero R, De Miguel MJ, Rodriguez-Panadero F, Carnero A, Paz-Ares L. MicroRNA clusters: dysregulation in lung adenocarcinoma and COPD. Eur Respir J. 2014; 43:1740-1749.